

Abstract
In a population-based genome-wide association (GWA) study of n-back working memory task-related brain activation, we extracted the average percent BOLD signal change (2-back minus 0-back) from 46 regions-of-interest (ROIs) in functional MRI scans from 863 healthy twins and siblings. ROIs were obtained by creating spheres around group random effects analysis local maxima, and by thresholding a voxel-based heritability map of working memory brain activation at 50%. Quality control for test-retest reliability and heritability of ROI measures yielded 20 reliable (r>0.7) and heritable (h2>20%) ROIs. For GWA analysis, the cohort was divided into a discovery (n=679) and replication (n=97) sample. No variants survived the stringent multiple-testing-corrected genome-wide significance threshold (p<4.5×10−9), or were replicated (p<0.0016), but several genes were identified that are worthy of further investigation. A search of 529,379 genomic markers resulted in discovery of 31 independent single nucleotide polymorphisms (SNPs) associated with BOLD signal change at a discovery level of p<1×10−5. Two SNPs (rs7917410 and rs7672408) were associated at a significance level of p<1×10−7. Only one, most strongly affecting BOLD signal change in the left supramarginal gyrus (R2=5.5%), had multiple SNPs associated at p<1×10−5 in linkage disequilibrium with it, all located in and around the BANK1 gene. BANK1 encodes a B-cell-specific scaffold protein and has been shown to negatively regulate CD40-mediated AKT activation. AKT is part of the dopamine-signaling pathway, suggesting a mechanism for the involvement of BANK1 in the BOLD response to working memory. Variants identified here may be relevant to (the susceptibility to) common disorders affecting brain function.
Keywords: Genome-wide association study, n-back, working memory, functional MRI, BOLD signal, region-of-interest
1 Introduction
Working memory task-related brain activation is altered, showing mostly increased activation, in several neuropsychiatric disorders, such as schizophrenia (Bor et al., 2011; Callicott et al., 2003b), bipolar disorder (Drapier et al., 2008), and major depressive disorder (Matsuo et al., 2007), as well as the healthy, at-genetic-risk, siblings of patients for some of these disorders (e.g., Callicott et al., 2003a; Drapier et al., 2008; Winterer et al., 2003). These disorders are highly heritable, but their onset and trajectory are thought to be influenced by a large number of genetic polymorphisms, each with a small effect, as well as environmental factors. Because abnormal working memory (WM) brain activation is implicated in brain disorders, factors that influence the blood oxygen level–dependent (BOLD) response in human populations are of great interest. In our prior voxel-wise analyses of heritability, up to 65% (averaging ∼33%) of the variation in WM task-related cerebral activation (Blokland et al., 2011) and up to 75% (averaging ∼36%) of the variance in WM task-related cerebellar activation (Blokland et al., 2014) was attributed to genetic factors. While these studies showed that human brain function is under substantial genetic control, specific genetic variants influencing individual differences are largely unknown. Genes that contribute to brain function are important to identify, as several known examples confer protection or risk for brain disorders. Carriers of the ‘disrupted in schizophrenia 1’ (DISC1) risk haplotype, for example, have a fivefold increased risk for schizophrenia (Zhang et al., 2006).
Candidate gene studies of WM brain function have provided mechanistic support for the implication of certain genetic variants associated with neuropsychiatric disorders. For example, healthy individuals carrying the ZNF804A schizophrenia and bipolar disorder risk allele (O'Donovan et al., 2008), showed changes in functional connectivity of the right dorsolateral prefrontal cortex (DLPFC) during WM that resembled those observed in schizophrenia (Esslinger et al., 2009). Their findings were partly replicated in an independent sample (Paulus et al., 2011). During rest and during an emotional task, a pattern of reduced inter-hemispheric prefrontal connectivity with increasing number of risk alleles similar to that during WM has been demonstrated, suggesting a state-independent influence of the gene variant on inter-hemispheric processing (Esslinger et al., 2011). Other studies have found genetic associations between the schizophrenia-risk gene neuroregulin1 (NRG1) (Li et al., 2006; Stefansson et al., 2003; Stefansson et al., 2002; Zhao et al., 2004) and WM brain activation in healthy individuals (Krug et al., 2008). While there were no effects of genetic status on behavioral task performance, the number of NRG1 risk alleles had a linear effect on hyperactivation of the superior frontal gyrus. Nicodemus et al. (2010b) showed similar inefficient processing in carriers of risk-associated genotypes, but in the DLPFC instead. Other candidate genes—mainly risk genes for schizophrenia and bipolar disorder—that have been associated with WM brain activation, include genes related to dopaminergic function, such as COMT (Bertolino et al., 2006a; Bertolino et al., 2006b; Egan et al., 2001; Pomarol-Clotet et al., 2010), the dopamine transporter gene (DAT1) (Bertolino et al., 2006a; Stollstorff et al., 2010; Tan et al., 2007), dopamine receptor genes DRD1, DRD2 and DRD4 (Bertolino et al., 2009; Bertolino et al., 2010; Herrmann et al., 2007; Tura et al., 2008); genes related to serotonergic function and glutamatergic action, such as MAOA, DAOA and GRM3 (Cerasa et al., 2008; Nixon et al., 2011; Tan et al., 2007); in addition to several genes involved in various other neuronal functions, such as CACNA1C (Bigos et al., 2010; Paulus et al., 2014), CYP2D6 (Stingl et al., 2012), AKT1 (Nicodemus et al., 2010b; Tan et al., 2008), and BDNF (Cerasa et al., 2010).
These candidate gene studies have been somewhat helpful in improving our understanding of the neurobiology underlying neuropsychiatric disorders, but the genes they studied only explain a small proportion (4-10% variance explained; (Bertolino et al., 2009; Egan et al., 2001; Munafò et al., 2008)) of the heritability of brain activation (up to 65%, averaging 33%; Blokland et al., 2011) and/or of the disorders themselves. Furthermore, these studies were generally limited by small sample sizes, and the findings would be more credible if verified in larger samples. Genome-wide association (GWA) studies using quantitative traits relevant to brain function or disorders have the potential to improve our understanding of the etiology of these processes even further by identifying genes whose relationship with the phenotype has not previously been hypothesized. Using GWA analysis of WM performance, a genetic polymorphism within SCN1A (encoding a subunit of the type I voltage-gated sodium channel) was replicated in three independent populations (n=1699) (Papassotiropoulos et al., 2011). In a subsequent candidate gene fMRI study, SCN1A allele-dependent activation differences during an n-back WM task were detected (Papassotiropoulos et al., 2011). However, very few GWA studies have been carried out that use brain activation as a quantitative phenotype. To the best of our knowledge only two GWA studies have investigated brain activation in response to a WM task. Potkin et al. (2009b) studied activation (mean BOLD signal in the DLPFC) during the Sternberg Item Recognition Paradigm in n=64 schizophrenia patients and n=74 matched controls, and identified 6 genes or chromosomal regions involved in neurodevelopment and response to stress (ROBO1-ROBO2, TNIK, CTXN3-SLC12A2 POU3F2, TRAF, and GPC1) with single nucleotide polymorphisms (SNPs) significant at p<10−6 for the interaction between BOLD response and schizophrenia diagnosis. Potkin et al. (2009a) extended this study to n=82 schizophrenia patients and n=91 controls and identified 2 different genes worthy of further study, RSRC1 and ARHGAP18. However, these 2 studies were carried out in a patient-control sample. It would be of great interest to identify gene variants associated with brain function in healthy individuals as this may improve our understanding of normal brain function.
Here, we used an unbiased genome-wide search to identify common genetic variants associated with variations in the fMRI BOLD response to an n-back WM task in a healthy young adult twin-sibling cohort, the Queensland Twin Imaging Study (n=679). We incorporated prior knowledge from a voxel-wise study about the total genetic influence on the BOLD response to WM (Blokland et al., 2011), to reduce the number of phenotypes being tested to a manageable number, while maximizing the quality of the functional quantitative trait. A few studies have attempted to carry out voxel-wise GWA analyses on imaging phenotypes (e.g. Hibar et al., 2011; Stein et al., 2010), but with the enormous number of statistical tests performed in voxel-wise GWA analyses (≤ 200,000 voxels × ≤ 1,000,000 SNPs), and the stringent multiple testing corrections needed to account for this, it is almost impossible to find significant results. Additionally, in a previous ROI-based study on the heritability of WM brain activation (Blokland et al., 2008), we discovered that using mean BOLD signal across anatomically defined ROIs might obscure the genetic variance, as it is possible that not the entire anatomical region is reliably activated and heritable. Here we carried out several strict quality control steps on the functional phenotype before proceeding to GWA analyses. We enforced a genome-wide statistical threshold, and used two independent samples, a large discovery sample of young adult twins and siblings (n=679) and a smaller replication sample (n=97), to verify any associations and help diagnose false-positive findings.
2 Materials and Methods
2.1 Participants
This study uses data from participants in the Queensland Twin Imaging Study (QTIMS). Most of these twins and siblings, between 16 and 30 years of age, had previously participated in the Brisbane Adolescent Twin study (Wright and Martin, 2004), so measures of cognitive functioning, birth information, and parental socio-economic status (SES) were available for most participants, in addition to the imaging data (details described elsewhere; Blokland et al., 2011). Of n=2,645 individuals that were initially approached for the study by letter, 520 declined participation (19.7%), 677 were unable to participate (e.g. moved out of state) or could not be contacted by phone for further screening (25.6%). Prior to inclusion in QTIMS, twins were assessed for handedness using the Annett Handedness Questionnaire (6 questions) (Annett, 1970; Wright and Martin, 2004), and screened (by self-report) for their suitability for imaging. Of n=1,420 individuals that had been screened for inclusion in the imaging study at the time this paper was written, 2.2% were excluded because they were left-handed, and a further 22.5% were excluded because they had a history of significant medical, psychiatric or neurological conditions, including head injuries, MRI contra-indicators, a current or past diagnosis of substance abuse, or current use of medication that could affect cognition.
A sample of n=1,070 twins and singleton siblings met the inclusion criteria and n=1,060 (99%) completed the study. Subsequently, 43 individuals were excluded from analysis due to head motion or abnormal findings on their structural scans, 70 individuals were excluded due to head motion on their functional scans, and an additional 84 individuals were excluded due to insufficient task performance (<40% accuracy on 0-back condition; <30% accuracy on 2-back condition). After excluding these individuals, n=863 twins and siblings remained for analysis. As data acquisition was ongoing when we published our heritability studies, a subset of the current sample was included in our previous voxel-wise studies on the heritability of WM brain activation in the cerebrum (285 of 863, 33.0%) and the cerebellum (353 of 863, 40.9%) (Blokland et al., 2014; Blokland et al., 2011).
There were no significant differences in sex distribution (χ2 = 0.02, df = 1, p = 0.89), ancestry distribution (χ2 = 0.0006, df = 1, p = 0.98) or SES (t = 0.65, df = 4.03, p = 0.55) between individuals who met inclusion criteria who completed the imaging study and those who did not, nor were there differences in ancestry distribution between individuals who met inclusion criteria and individuals who did not (χ2 = 1.57, df = 1, p = 0.21), but there was an increased proportion of females among individuals who met inclusion criteria (62%) compared to excluded individuals (47%) (χ2 = 20.66, df = 1, p = 5.48×10−6) and slightly higher SES in the included group (t = -2.44, df = 259.8, p = 0.01).
All twins (n=760; 110 MZ pairs and 138 DZ pairs), aged 22±3 years (mean±s.d.; range 16–30 years), were included in our phenotypic and heritability analyses. Due to the low number of complete twin pairs with a singleton sibling, siblings were not included in the heritability analyses. Zygosity was determined by genotyping of 8–10 independent highly polymorphic DNA markers (PIC > 0.7) with a 99.99% probability of correct zygosity assignment (Wright and Martin, 2004) and where available (n=776 of 863, 89.9%) confirmed by genome-wide SNP genotyping (see paragraph on Genotyping).
A date cut-off was set for inclusion in the GWAS discovery and replication samples. The GWAS discovery sample (sample 1) consisted of n=679 participants. The GWAS replication sample (sample 2) consisted of n=97 participants. Table 1 provides a detailed sample composition. Independent samples t-tests compared demographic sample means.
Table 1.
Sample compositions for the variance components, reliability, and genome-wide association (GWA) discovery and replication analyses.
| Variance components | Test-retest reliability | GWA discovery | GWA replication | |
|---|---|---|---|---|
| Monozygotic pairs | 110 | 10 | 81 | 7 |
| Monozygotic pairs plus one singleton sibling | 0 | 0 | 6 | 0 |
| Dizygotic pairs | 138 | 12 | 93 | 20 |
| Dizygotic (trizygotic) triplets | 0 | 0 | 1 | 0 |
| Dizygotic pairs plus one singleton sibling | 0 | 0 | 7 | 0 |
| Dizygotic pairs plus two singleton siblings | 0 | 0 | 1 | 0 |
| Unpaired twins | 264 | 10 | 187 | 28 |
| Single non-twin siblings | 0 | 4 | 37 | 3 |
| Two non-twin siblings | 0 | 0 | 29 | 6 |
| Three non-twin siblings | 0 | 0 | 1 | 0 |
|
| ||||
| Total n | 760 | 58 | 679 | 97 |
| (males/females) | (287/473) | (26/32) | (247/432) | (40/57) |
Human Research Ethics Committees of the Queensland Institute of Medical Research, University of Queensland, and Uniting Health Care approved the study. Written informed consent was obtained from each participant, and each participant received a $100 gift voucher in appreciation of their time.
2.2 Experimental procedure
Imaging was conducted on a 4 Tesla Bruker Medspec whole body scanner (Bruker, Germany) in Brisbane, Australia. Functional images were acquired using a T2*-weighted gradient echo planar imaging (EPI) sequence, sensitive to BOLD contrast (interleaved; repetition time, TR = 2100 ms; echo time, TE = 30 ms; flip angle = 90°; field of view, FOV = 230×230 mm), and using a radio-frequency receive-transmit transverse electromagnetic head coil (Vaughan, 1999). Geometric distortions in the EPI images caused by magnetic field inhomogeneities at high-field were corrected using a point-spread mapping approach (Zeng and Constable, 2002). Over a continuous imaging run, we acquired 127 axial brain volumes, one volume every 2.1 s, with 36 coronal slices of 3 mm thickness (64×64 matrix; voxel size 3.6×3.6×3.0 mm), and with a 0.6 mm slice gap. In addition to the functional scans, 3D T1-weighted images were acquired (MPRAGE; TR = 1500 ms; TE = 3.35 ms; TI = 700 ms; pulse angle = 8°; coronal orientation; FOV = 230 mm; 256×256×256 matrix; slice thickness = 0.9 mm).
During functional imaging the participants performed the 0-back and 2-back versions of a block design spatial, numerical n-back working memory task (Callicott et al., 2003a; 1998). In this task, a number (1–4, randomized) was presented in a fixed position in one of four white circles, positioned at the corners of a diamond-shaped square. Participants pressed one of the four buttons on a response box in the same configuration as the stimuli to match the target stimulus. For n = 0 (i.e., 0-back), a simple button press in response to the number displayed was required. For n = 2 (i.e., 2-back), participants pressed the button corresponding to the number presented two trials before the current one. Participants were scanned through 16 alternating blocks of the 0-back and 2-back conditions, for a total experimental length of 4 min and 16 s. A detailed explanation of the task paradigm can be found elsewhere (Blokland et al., 2008; 2011). Task performance was measured as the percentage of correct responses (accuracy) and average response time (RT; across correct trials) for each of the task conditions separately.
2.3 Image pre-processing
Images were processed and analyzed using Statistical Parametric Mapping software (SPM8, Wellcome Trust Centre for Neuroimaging, London, UK) implemented in MATLAB (The MathWorks Inc.). The first five EPI volumes were discarded to ensure that steady state tissue magnetization was reached. Time-series volumes were realigned and unwarped using a robust rigid-body transformation procedure (Freire et al., 2002). A mean image generated during realignment was then co-registered with the participant's 3D T1 image. The T1-weighted image was subsequently segmented using the “New Segment” procedure in SPM8. The “DARTEL” toolbox (Ashburner, 2007) was then employed to create a custom group template from the grey and white matter images and individual flow fields that were used to normalize the realigned fMRI volumes to the Montreal Neurological Institute (MNI) atlas T1 template. The resulting images were then resampled to 3×3×3 mm voxels and smoothed with an 8×8×8-mm full width half maximum (FWHM) isotropic Gaussian kernel. Global signal effects were estimated and removed using a voxel-level general linear model (Macey et al., 2004). High pass (cut-off: 128 s) and low pass (AR1 model) filtering were applied to discard signals of no interest.
2.4 Image analysis
Image analysis was conducted in two stages: First, block design fixed effects models were fitted at the single-subject level. Separate regressors were constructed for the 0- and 2-back conditions comprising a boxcar reference waveform convolved with a canonical hemodynamic response function. Second, the resulting single-subject 2-back > 0-back t-contrast images were entered into a second-level group random effects model (one-sample t-test), irrespective of zygosity (p < 0.05, FWE-corrected, extent threshold 25 voxels).
A region of interest (ROI) analysis was performed using two independent methods of defining ROIs. Firstly, sphere ROIs (diameter 9 mm) were created around the local maxima from the group random effects analysis of individual 2>0 back contrast images, including individuals with >50% performance accuracy on 0-back and 2-back (sample 1). Secondly, ROIs were created from the voxel-by-voxel heritability map from our prior study on heritability of working memory brain activation (Blokland et al., 2011), selecting clusters of ≥ 3 voxels with >50% heritability (i.e. >50% of the total phenotypic variance is explained by additive genetic factors, h2). We then employed the method used by Matthews et al. (2007) to extract, for each participant and each task condition, the average percent BOLD signal across all voxels in each of the ROIs using the MarsBaR Toolbox for SPM (Brett et al., 2002). The average BOLD signal change percentages in the ROIs (2-back minus 0-back) were used as phenotypes for further analysis.
2.5 Reproducibility
The number of ROIs was reduced based on their test-retest reliability according to intra-class correlations (ICC) (Shrout and Fleiss, 1979). For further analysis we retained only those ROIs which had good test-retest reliability (ICC>0.70) in a subsample of 58 individuals (from sample 1) rescanned approximately 3 months after their initial MRI scan (range 35-291 days; mean±SD = 113±55 days). The test-retest sample consisted of 10 MZ pairs, 12 DZ pairs, 10 unpaired twins, and 4 singleton siblings – 32 females and 26 males (See Table 1).
2.6 Genetic modeling
The number of ROIs was reduced further based on a (re-) estimation of their heritability. Heritability of the functional ROI measures was determined through structural equation modeling with Mx (Neale et al., 2002). Genetic twin modeling was used to determine the relative influences of genetic and environmental factors on the functional ROI measures, making use of the differences in genetic similarity between MZ (who share all their genes) and DZ (who share, on average, 50% of their genes) twins. Depending on the pattern of intra-pair twin correlations, it is customary to fit a model that includes additive genetic (A), common or shared environmental (C), and unique or unshared environmental (E) factors (ACE model, if the DZ twin correlation is more than half the MZ twin correlation), or a model that includes additive genetic (A), dominance genetic (D), and unique or unshared environmental (E) factors (ADE model, if the DZ twin correlation is less than half the MZ twin correlation) (Falconer and Mackay, 1996). Model fit is determined by the consecutive dropping of parameters, to see if this results in a significant (p < 0.05) reduction in model fit, as indexed by the chi-squared (χ2) fit statistic. The full ACE model was compared with an AE model, a CE model, and an E model; the full ADE model was compared with an AE and E model, to arrive at the most parsimonious model.
2.7 Genotyping
DNA was obtained from blood samples (as part of a different project), in accordance with standard protocols. SNP genotyping for all DZ twin pairs, a co-twin of each MZ pair, and all singleton siblings of twins was performed using the Illumina Human610-Quad BeadChip (San Diego, CA, USA) by deCODE Genetics. Non-genotyped MZ twins with a genotyped co-twin were assigned their co-twin's genotype. Standard Quality Control (QC) criteria were applied to these genotype data (Medland et al., 2009). Families were excluded from analysis if they were ancestry outliers (non-European ancestry) or had an abnormally high Mendelian error rate. SNPs were excluded from analyses if they had: (1) call rate per SNP <90%; (2) Minor Allele Frequency (MAF) <1%; and (3) significant violation of Hardy-Weinberg equilibrium test of p<10−6. After quality control, 529,379 SNPs remained.
2.8 Genome-wide association analyses
Genome-wide association to find common variants that contribute to the heritability of WM brain activation was conducted using the family based association test (Chen and Abecasis, 2007) in MERLIN (Abecasis et al., 2002), which takes family relationships into account. The best guess genotype at each SNP was tested for association with each of the ROIs that met all the QC criteria. The additive genetic effect for each SNP was computed by modeling the genotypic mean of the heterozygote (Aa) as the average of the two homozygotes (AA, aa). Sex, age, 3 principal components for ancestry to correct for population stratification (McEvoy et al., 2009), and n-back performance accuracy (0-back and 2-back) were included as covariates.
The generally accepted genome-wide significance level for the association between SNP and phenotype at α = 0.05 is p<7.2×10–8, correcting for the total number of independent SNP tests (Dudbridge and Gusnanto, 2008). Given that we conducted 20 distinct genome-wide scans for SNP associations, we corrected the generic genome-wide significance level accordingly by estimating the effective number of independent variables through matrix spectrum decomposition of the phenotypic correlation matrix (Nyholt, 2004). The effective number of independent variables was estimated at 16 (Li and Ji, 2005), resulting in a very stringent corrected genome-wide significance level of 4.5×10−9 (7.2×10−8 divided by 16). However, because we applied a two-stage process to identify interesting SNPs to carry forward to a second stage in which they can be replicated, we used a less stringent search criterion of p<1×10−5 to select SNPs that were associated in the larger discovery sample, in keeping with Welcome Trust Case Control Consortium recommendations (Burton et al., 2007). Association analysis was then repeated in the replication sample for the most strongly associated SNPs identified in the discovery sample (p<1×10−5). We set a corrected p-value threshold of p<0.0016 (0.05 divided by 31 independent SNPs) for significant replication of a SNP association.
The genetic annotation was performed with WGA Viewer software: Package of Post Association Genomic Annotation, Version 1.25N (Duke University, 2008).
2.9 Brain expression analysis
Brain expression data from the Stanford Brain RNA-Seq database ((Zhang et al., 2014); http://web.stanford.edu/group/barres_lab/brain_rnaseq.html), the Allen Brain Atlas ((Sunkin et al., 2013); http://human.brain-map.org), the Genotype-Tissue Expression project ((GTEx Consortium, 2013, 2015); http://www.gtexportal.org), and the Human Brain Transcriptome project ((Kang et al., 2011; Pletikos et al., 2014); http://hbatlas.org/) were evaluated to validate the association findings and aid in the interpretation of the results. The expression levels from the Allen Brain Atlas were averaged across the 6 brain tissue samples and up to 6 probes per gene.
3 Results
3.1 Demographics and task performance
Sample 1 and sample 2 differed significantly on mean age (sample 1: 22.6±3.2 vs. sample 2: 19.3±0.9; d = 1.40, p < 0.001), mean gestational age (sample 1: 37.7±3.0 weeks vs. sample 2: 36.9±3.0 weeks; d = 0.27, p < 0.05), and mean reaction time on 0-back (sample 1: 448.0±61.2 ms vs. sample 2: 424.7±55.1 ms; d = 0.40, p < 0.001) and 2-back (sample 1: 248.9±114.0 vs. sample 2: 221.3±118.3; d = 0.24, p < 0.05). The samples did not differ on birth weight (2557.7±541.3 grams), parental socio-economic status (54.1±24.7), FIQ (115.2±12.1), task performance accuracy on 0-back (86.8±11.2) and 2-back (71.2±18.9), or gender distribution (sample 1: 36.4% males; sample 2: 41.2% males). Task performance was consistent with our prior reports on the n-back task (Blokland et al., 2014; Blokland et al., 2011).
3.2 Regions of interest
The group random effects analysis identified 30 cerebral local maxima, consistent with our previous reports (Blokland et al., 2008; Blokland et al., 2011). Height and cluster-thresholding of the voxel-wise heritability map at 50% and ≥3 voxels yielded 16 heritability clusters. A few ROIs partially overlapped. Means and ranges for spherical and cluster ROIs are shown in Table 2. The means showed that there were considerable variations in activation intensity between the different ROIs. On average BOLD signal was higher in the sphere ROIs than in the cluster ROIs. Samples 1 and 2 had significant mean differences for BOLD signal in 15 of the 46 ROIs. Generally, means were higher for sample 1, suggesting that the slightly older sample had less efficient processing requiring greater activation. This is in line with our prior observation that there are significant age effects on working memory brain activation, even within this narrow age range (Blokland et al., 2011), and was addressed by including age as a covariate in the genetic analyses.
Table 2.
Descriptive statistics for average BOLD percent signal change (2-back minus 0-back) in spherical and cluster regions-of-interest (ROIs).
| Area | MNI Coordinates | Size ROI | Discovery (Sample 1) mean (±s.d.) | Replication (Sample 2) mean (±s.d.) | Total range (n=863) | h2 (95% CI) | ICC (95% CI) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| x | y | z | k voxels | mm3 | |||||||
| Sphere ROIs | L thalamus | −15 | −21 | 18 | 14 | 384 | .03 (±.06)*** | .01 (±.05) | −.32; .36 | 0 | .29 (.04;.51)* |
| L SFG | −15 | 9 | 63 | 14 | 384 | .05 (±.05) | .05 (±.06) | −.21; .22 | 21 (5;36) | .69 (.53; .81)*** | |
| L hippocampus | −24 | −36 | 9 | 14 | 384 | .04 (±.05)* | .03 (±.04) | −.27; .22 | 24 (8;39) | .20 −(.06;.43) | |
| L MFG | −27 | 6 | 54 | 14 | 384 | .10 (±.06) | .09 (±.08) | −.19; .37 | 29 (13;44) | .69 (.53;.81)*** | |
| L IPL | −33 | −45 | 39 | 14 | 384 | .11 (±.06) | .10 (±.08) | −.21; .38 | 48 (34;59) | .75 (.61;.84)*** | |
| L insula | −33 | 18 | 3 | 14 | 384 | .09 (±.06) | .09 (±.08) | −.20; .41 | 41 (26;54) | .56 (.35;.71)*** | |
| L insula | −33 | 24 | 0 | 14 | 384 | .10 (±.06) | .10 (±.09) | −.18; .40 | 0 | .67 (.50;.79)*** | |
| L IPL | −39 | −42 | 39 | 14 | 384 | .11 (±.06) | .10 (±.08) | −.10; .35 | 47 (32;59) | .82 (.71;.89)*** | |
| L insula | −39 | 15 | −3 | 14 | 384 | .09 (±.06)* | .07 (±.09) | −.27; .46 | 28 (14;42) | .49 (.27;.67)*** | |
| L PCG | −42 | 3 | 33 | 14 | 384 | .10 (±.07) | .10 (±.08) | −.13; .56 | 0 | .82 (.71;.89)*** | |
| L IFG (PTr) | −45 | 27 | 27 | 14 | 384 | .11 (±.09) | .09 (±.09) | −.18; .55 | 42 (27;54) | .75 (.61;.84)*** | |
| L IFG (POp; BA 44) | −48 | 6 | 18 | 14 | 384 | .07 (±.06) | .07 (±.07) | −.15; .29 | 41 (27;53) | .80 (.68;.87)*** | |
| L MTG | −51 | −48 | 12 | 14 | 384 | .05 (±.05) | .03 (±.08) | −.27; .25 | 0 | .77 (.63;.85)*** | |
| L IFG (POp) | −51 | 9 | 9 | 14 | 384 | .05 (±.05) | .04 (±.07) | −.32; .29 | 27 (10;43) | .78 (.66;.86)*** | |
| R thalamus | 12 | −6 | 9 | 14 | 384 | .03 (±.05) | .03 (±.04) | −.30; .21 | 0 | .57 (.37;.72)*** | |
| R SMA (BA 6) | 15 | 9 | 63 | 14 | 384 | .08 (±.05)** | .06 (±.07) | −.29; .32 | 0 | .73 (.59;.83)*** | |
| R caudate nucleus | 18 | 6 | 15 | 14 | 384 | .05 (±.04)* | .04 (±.05) | −.14; .23 | 0 | .44 (.21;.63)*** | |
| R IPL | 27 | −54 | 42 | 14 | 384 | .06 (±.05)** | .04 (±.07) | −.34; .32 | 0 | .61 (.42;.75)*** | |
| R IPL | 27 | −57 | 36 | 14 | 384 | .06 (±.05) | .04 (±.10) | −.35; .72 | 0 | .71 (.55;.81)*** | |
| R MFGs | 27 | 9 | 51 | 14 | 384 | .13 (±.06)* | .11 (±.08) | −.15; .35 | 38 (23;50) | .86 (.78;.92)*** | |
| R insula | 36 | 21 | 3 | 14 | 384 | .12 (±.07) | .11 (±.08) | −.12; .41 | 41 (25;54) | .49 (.27;.67)*** | |
| R SPL | 39 | −39 | 39 | 14 | 384 | .13 (±.07) | .11 (±.08) | −.11; .41 | 0 | .78 (.65;.86)*** | |
| L SMG | 3 | 24 | 42 | 14 | 384 | .16 (±.08)* | .13 (±.12) | −.28; .49 | 0 | .62 (.43;.75)*** | |
| R MFG | 42 | 39 | 24 | 14 | 384 | .14 (±.09)*** | .09 (±.13) | −.92; .48 | 38 (23;50) | .76 (.63;.85)*** | |
| R MTG | 45 | −54 | 9 | 14 | 384 | .04 (±.05) | .03 (±.04) | −.09; .22 | 0 | .72 (.57;.83)*** | |
| R IFG (POp) | 45 | 12 | 27 | 14 | 384 | .13 (±.08)*** | .09 (±.09) | −.10; .45 | 0 | .78 (.66;.87)*** | |
| R insula | 45 | 15 | −6 | 14 | 384 | .09 (±.07)*** | .06 (±.10) | −.61; .34 | 0 | .41 (.18;.61)** | |
| R IPL | 48 | −42 | 18 | 14 | 384 | .06 (±.06) | .06 (±.06) | −.10; .34 | 39 (25;52) | .75 (.60;.84)*** | |
| R IFG (POp; BA 44) | 51 | 12 | 15 | 14 | 384 | .11 (±.07) | .10 (±.08) | −.12; .44 | 30 (15;44) | .73 (.59;.83)*** | |
| R PCUN | 9 | −45 | 48 | 14 | 384 | .07 (±.06)*** | .03 (±.09) | −.34; .30 | 0 | .74 (.59;.84)*** | |
| Cluster ROIs | L SOG | −22 | −67 | 36 | 17 | 459 | .05 (±.04) | .05 (±.07) | −.18; .49 | 38 (23;51) | .75 (.61;.84)*** |
| L SPL | −25 | −52 | 47 | 5 | 135 | .06 (±.06) | .06 (±.08) | −.20; .38 | 35 (18;50) | .69 (.52;.80)*** | |
| L PCUN | −2 | −64 | 46 | 42 | 1134 | .07 (±.10) | .08 (±.11) | −.24; .42 | 54 (40;65) | .81 (.69;.88)*** | |
| L AG | −33 | −57 | 34 | 9 | 243 | .04 (±.05) | .04 (±.07) | −.27; .26 | 0 | .78 (.65;.86)*** | |
| L PCG | −40 | −2 | 52 | 4 | 107 | .06 (±.07) | .06 (±.08) | −.27; .40 | 29 (13;43) | .85 (.75;.91)*** | |
| L IFG (PTr) | −43 | 27 | 26 | 17 | 459 | .09 (±.08) | .08 (±.08) | −.15; .39 | 44 (30;57) | .74 (.59;.84)*** | |
| L PCUN | −4 | −46 | 54 | 8 | 216 | .05 (±.06)* | .03 (±.07) | −.25; .28 | 38 (23;51) | .64 (.47;.77)*** | |
| L SMA | 0 | 23 | 51 | 3 | 81 | .12 (±.10) | .12 (±.11) | −.14; .55 | 40 (22;55) | .76 (.62;.85)*** | |
| L SMG | 0 | 36 | 34 | 20 | 540 | .07 (±.09)*** | .04 (±.10) | −.51; .33 | 53 (42;63) | .73 (.59;.83)*** | |
| R PCUN | 15 | −51 | 58 | 8 | 216 | .06 (±.06) | .04 (±.18) | −1.35; .55 | 39 (25;51) | .79 (.67;.87)*** | |
| R PCUN | 19 | −70 | 46 | 7 | 189 | .05 (±.06) | .05 (±.13) | −.52; .68 | 51 (37;63) | .73 (.58;.83)*** | |
| R SFG | 22 | 23 | 50 | 5 | 135 | .08 (±.07) | .07 (±.07) | −.14; .31 | 36 (21;50) | .73 (.59;.83)*** | |
| R IPL(BA 2) | 32 | −43 | 54 | 14 | 378 | .04 (±.06) | .04 (±.09) | −.19; .51 | 29 (13;43) | .59 (.40;.74)*** | |
| R IPL / MOG | 42 | −70 | 30 | 8 | 216 | .05 (±.07) | .04 (±.09) | −.35; .36 | 37 (22;50) | .75 (.62;.85)*** | |
| R IFG (PTr; BA 45) | 48 | 23 | 22 | 8 | 216 | .10 (±.10)*** | .05 (±.09) | −.21; .59 | 42 (26;55) | .72 (.57;.83)*** | |
| R IFG (POp; BA 44) | 60 | 12 | 15 | 3 | 81 | .05 (±.07) | .06 (±.09) | −.16; .49 | 29 (14;43) | .52 (.31;.69)*** | |
Mean differences between discovery and replication samples (samples 1 and 2) were evaluated using independent samples t-tests. P-values:
p < 0.05;
p < 0.01;
p < 0.001.
Heritability (h2) estimates are the estimates under the best fitting model (see Supplementary Table 1 for full model fitting results). Intra-class correlations (ICC) are for the 3-month test-retest reliability (95% confidence interval).
Abbreviations: AG, angular gyrus; BA, Brodmann area; IFG, inferior frontal gyrus; IPL, inferior parietal lobule; L, left; MFG, middle frontal gyrus; MNI, Montréal Neurological Institute; MOG, middle occipital gyrus; MTG, middle temporal gyrus; PCG, precentral gyrus; PCUN, precuneus; POp, pars opercularis; PTr, pars triangularis; R, right; s.d., standard deviation; SFG, superior frontal gyrus; SMA, supplementary motor area; SMG, superior medial gyrus; SOG, superior occipital gyrus; SPL, superior parietal lobule.
3.3 Test-retest reliability, twin correlations and heritability estimates
Test-retest intra-class correlations for the subsample (n = 58), and twin correlations corrected for sex, age, and task performance accuracy, with confidence intervals, are shown in Supplementary Table 1. Intra-class correlations are also shown in Table 2. The majority of DZ correlations were low (range −0.08–0.33, averaging 0.14) and about half the MZ correlations (range 0.09–0.55, averaging 0.32), indicating few common environmental (C) or genetic dominance (D) influences. As can be seen in Supplementary Table 1, the majority of C-influences were non-significant. A few ROIs showed indications of D-influences, however, none of the D-influences were significant in ADE modeling (Supplementary Table 2). Parameter estimates for the best fitting model are also shown in Table 2. The genetic modeling results confirm the usefulness of the cluster ROIs obtained from voxel-wise heritability analyses, as overall, these ROIs have higher heritability and reliability than the sphere local maxima ROIs. Consistent with the fact that reliability places an upper limit on heritability, there is high agreement between heritability estimates and test-retest reliability. The poorest agreement was for 2 sphere ROIs in the insula, that had relatively high heritability at 41%, but test-retest ICCs <= 0.56. This may be due to the location, which is more prone to physiological noise (Di et al., 2013). Quality control identified a total of 20 ROIs with high test-retest reliability (>0.70), significant MZ correlations, and significant heritability (>20%). Test-retest reliability for these ROIs ranged between 0.72 and 0.86, averaging 0.77. Heritability for these ROIs ranged between 27% and 54%, averaging 41%. These 9 sphere ROIs and 11 cluster ROIs (highlighted in bold font) were included in subsequent GWA analyses. The location of these regions is shown in Figure 1.
Figure 1.
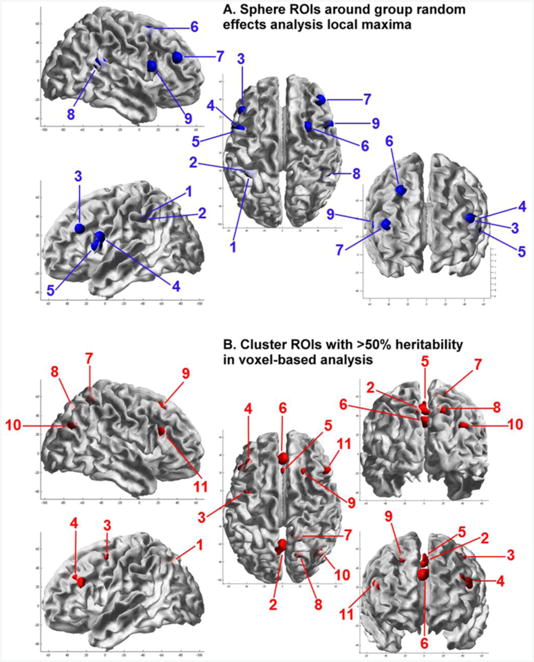
Localisation of (A) spherical regions of interest around group random effects analysis local maxima, and (B) cluster regions of interest with >50% heritability in voxel-based analysis. ROIs are displayed on the pial surface of the MNI template using the SRender Toolbox for SPM8, authored by John Ashburner.
(A) Sphere ROIs: 1 = L IPL [−33; −45; 39]; 2 = L IPL [−39; −42; 39]; 3 = L IFG (PTr) [−45; 27; 27]; 4 = L IFG (POp; BA 44) [−48; 6; 18]; 5 = L IFG (POp) [−51; 9; 9]; 6 = R MFG [27; 9; 51]; 7 = R MFG [42; 39; 24]; 8 = R IPL [48; −42; 18]; 9 = R IFG (POp; BA 44) [51; 12; 15].
(B) Cluster ROIs: 1 = L SOG [−22; −67; 36]; 2 = L PCUN [−2; −64; 46]; 3 = L PCG [−40; −2; 52]; 4 = L IFG (PTr) [−43; 27; 26]; 5 = L SMA [0; 23; 51]; 6 = L SMG [0; 36; 34]; 7 = R PCUN [15; −51; 58]; 8 = R PCUN [19; −70; 46]; 9 = R SFG [22; 23; 50]; 10 = R IPL / MOG [42; −70; 30]; 11 = R IFG (PTr; BA 45) [48; 23; 22].
ROI abbreviations: AG, angular gyrus; BA, Brodmann area; IFG, inferior frontal gyrus; IPL, inferior parietal lobule; L, left; MFG, middle frontal gyrus; MNI, Montréal Neurological Institute; MOG, middle occipital gyrus; MTG, middle temporal gyrus; PCG, precentral gyrus; PCUN, precuneus; POp, pars opercularis; PTr, pars triangularis; R, right; SFG, superior frontal gyrus; SMA, supplementary motor area; SMG, superior medial gyrus; SOG, superior occipital gyrus; SPL, superior parietal lobule.
3.4 Phenotypic correlations
Pearson phenotypic correlations (2-tailed) between the ROIs included in the GWA analyses are shown in Supplementary Table 3, ranging between 0 and 0.76, respectively. Correlations were highest between areas that were anatomically close. A few ROIs partially overlapped. Phenotypic correlations were higher between sphere ROIs (group activation) than between cluster ROIs (heritability). Phenotypic correlations were of similar magnitude in the discovery and replication samples, so correlations shown here combine both samples. Through matrix spectrum decomposition of the phenotypic correlation matrix (Li and Ji, 2005; Nyholt, 2004), the effective number of independent ROI phenotypes was estimated at 16.
3.5 Genome-wide association analyses
Here, 529,379 SNPs were tested for association with 9 spherical ROIs and 11 cluster ROIs, correcting for sex, age, 3 ancestry principal components, and task performance accuracy (0-back and 2-back). The SNP with the lowest p-value (8.1×10−8) for a spherical ROI, in the left inferior frontal gyrus (pars opercularis, Brodmann area 44) [−48; 6; 18], was on chromosome 10 (rs7917410), while the SNP with the lowest p-value (9.9×10−8) for a cluster ROI, in the left supramarginal gyrus [0; 36; 34], was on Chromosome 4 (rs7672408) (Table 3). The SNPs accounted for 5.4% and 5.5% of the trait variance, respectively. Figure 2a, showing the results of these two association analyses in Manhattan plots, indicates that there were no genome-wide significant association signals (p<4.5×10−9). The association between the observed versus the expected p-value of the autosomal association (under the null-hypothesis of no association) for the two variables is shown in the Quantile-Quantile (QQ) plots (Figure 2b). The QQ and Manhattan plots for the spherical ROI in the left inferior frontal gyrus (pars opercularis, Brodmann area 44) [−48; 6; 18] suggest that this may be a false positive finding, whereas the QQ and Manhattan plots for the cluster ROI in the left supramarginal gyrus [0; 36; 34] suggest it may be a true association. This association peak was located in and around the BANK1 gene. A detailed view of the locus showing suggestive association (p=9.9×10−8) with average BOLD percent signal change in the left supramarginal gyrus [0; 36; 34] is shown in Figure 3. Neither of the top two SNPs associated with BOLD signal was significantly associated with 0-back or 2-back accuracy or reaction time (p>0.05) and no SNPs had p-values <1×10−5, so none were carried forward to replication. The top SNP associations for task performance (p<1×10−4) are shown in Supplementary Table 4.
Table 3.
Genetic markers showing strongest association with regional average BOLD response to an n-back working memory task, in spherical ROIs based on group activation local maxima and cluster ROIs with >50% heritability in voxel-based analysis (independent markers within top SNPs with p<1×10−5).
| SNP | Chr | BP | p | h2 | β | SE | SNPs in LD | A1 | MAF | ROI(s) | ROI type | Gene | SNP type | REPL p | REPL h2 | REPL β | REPL SE |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs7917410 | 10 | 4322824 | 8.2 × 10−8 | 5.38 | 0.023 | 0.001 | C | 0.26 | L IFG (POp; BA 44) [-48; 6; 18] | sphere | intergenic | 0.699 | 0.23 | 0.005 | 0.013 | ||
| 1.0 × 10−4 | 2.85 | 0.012 | 0.001 | L SOG [-22; -67; 36] | cluster | 0.149 | 3.30 | -0.017 | |||||||||
| 1.4 × 10−3 | 1.95 | 0.014 | 0.001 | L IPL [-33; -45; 39] | sphere | 0.886 | 0.03 | -0.002 | |||||||||
| 3.3 × 10−3 | 1.60 | 0.010 | 0.001 | L IFG (POp) [-51; 9; 9] | sphere | 0.597 | 0.43 | 0.007 | |||||||||
| 7.1 × 10−3 | 1.35 | 0.013 | 0.001 | L PCG [-40; -2; 52] | cluster | 0.582 | 0.48 | 0.007 | |||||||||
| 7.5 × 10−3 | 1.34 | 0.012 | 0.001 | L IPL [-39; -42; 39] | sphere | 0.461 | 0.86 | 0.011 | |||||||||
|
| |||||||||||||||||
| rs7672408 | 4 | 103220464 | 9.9 × 10−8 | 5.49 | -0.029 | 0.001 | 4 | C | 0.39 | L SMG [0; 36; 34] | cluster | BANK1 | intergenic | 0.338 | 1.53 | -0.014 | 0.015 |
| 1.3 × 10−3 | 1.94 | -0.012 | 0.001 | L IFG (POp) [-51; 9; 9] | sphere | 0.661 | 0.33 | -0.005 | |||||||||
| 1.5 × 10−3 | 1.91 | -0.019 | 0.010 | L SMA [0; 23; 51] | cluster | 0.762 | 0.16 | -0.006 | |||||||||
|
| |||||||||||||||||
| rs2118263 | 6 | 1197894 | 2.2 × 10−7 | 4.63 | -0.028 | 0.001 | A | 0.39 | L IFG (PTr) [-45; 27; 27] | sphere | intergenic | 0.075 | 5.83 | -0.033 | 0.018 | ||
| 9.8 × 10−6 | 3.37 | -0.021 | 0.001 | L IFG (PTr) [-43; 27; 26] | cluster | 0.031 | 8.59 | -0.036 | |||||||||
|
| |||||||||||||||||
| rs7148741 | 14 | 20962005 | 2.4 × 10−7 | 4.39 | -0.033 | 0.010 | G | 0.23 | L SMA [0; 23; 51] | cluster | CHD8 | intronic | 0.192 | 2.55 | 0.029 | 0.022 | |
| 1.1 × 10−3 | 1.80 | -0.019 | 0.010 | L SMG [0; 36; 34] | cluster | ||||||||||||
| 7.3 × 10−3 | 1.18 | -0.011 | 0.001 | L IFG (POp; BA 44) [-48; 6; 18] | sphere | BCL2L14 | |||||||||||
|
| |||||||||||||||||
| rs2160523 | 12 | 12134077 | 2.5 × 10−7 | 4.92 | -0.030 | 0.010 | T | 0.49 | R IFG (PTr; BA 45) [48; 23; 22] | cluster | intronic | 0.424 | 1.16 | -0.012 | 0.015 | ||
| 6.5 × 10−3 | 1.38 | -0.014 | 0.001 | R MFG [42; 39; 24] | sphere | KIAA1609 | |||||||||||
|
| |||||||||||||||||
| rs11641157 | 16 | 83114818 | 2.8 × 10−7 | 4.54 | 0.019 | 0.001 | 1 | G | 0.48 | L IPL [-39; -42; 39] | intergenic | 0.408 | 1.70 | -0.014 | 0.017 | ||
| 6.2 × 10−5 | 2.80 | 0.015 | 0.001 | L IPL [-33; -45; 39] | sphere | ||||||||||||
| 9.9 × 10−3 | 1.11 | 0.01 | 0.001 | R IFG (PTr; BA 45) [48; 23; 22] | cluster | ||||||||||||
|
| |||||||||||||||||
| rs874941 | 15 | 48499359 | 2.9 × 10−7 | 4.81 | -0.034 | 0.010 | C | 0.31 | L PCUN [-2; -64; 46] | cluster | USP8 | intergenic | 0.446 | 1.07 | -0.015 | 0.019 | |
|
| |||||||||||||||||
| rs1954482 | 14 | 61588636 | 4.6 × 10−7 | 4.55 | 0.020 | 0.001 | A | 0.32 | L IPL [-39; -42; 39] | sphere | SYT16 | intronic | 0.478 | 0.79 | -0.010 | 0.014 | |
| 6.7 × 10−5 | 2.88 | 0.016 | 0.001 | L IPL [-33; -45; 39] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs7242427 | 18 | 20649845 | 8.0 × 10−7 | 4.62 | -0.034 | 0.010 | G | 0.25 | R IFG (PTr; BA 45) [48; 23; 22] | cluster | intergenic | 0.524 | 0.95 | -0.012 | 0.019 | ||
|
| |||||||||||||||||
| rs10497853 | 2 | 201183402 | 9.1 × 10−7 | 4.11 | -0.036 | 0.010 | 1 | G | 0.07 | R SFG [22; 23; 50] | cluster | AOX1 | intronic | 0.947 | 0.01 | 0.002 | 0.024 |
|
| |||||||||||||||||
| rs2061443 | 4 | 120134118 | 1.2 × 10−6 | 4.70 | -0.017 | 0.001 | C | 0.35 | R PCUN [15; -51; 58]: | cluster | SYNPO2 | intronic | 0.425 | 1.53 | -0.029 | 0.037 | |
|
| |||||||||||||||||
| rs6689092 | 1 | 35074213 | 2.0 × 10−6 | 4.43 | 0.022 | 0.001 | 1 | T | 0.45 | L IFG (PTr) [-43; 27; 26] | cluster | C1orf212 | intergenic | 0.669 | 0.27 | -0.006 | 0.015 |
| 6.0 × 10−6 | 4.02 | 0.025 | 0.010 | L IFG (PTr) [-45; 27; 27] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs2607120 | 15 | 49783055 | 2.1 × 10−6 | 4.18 | -0.018 | 0.001 | T | 0.19 | L IFG (POp) [-51; 9; 9] | sphere | SCG3 | intronic | 0.445 | 1.25 | -0.013 | 0.017 | |
| 5.5 × 10−3 | 1.45 | -0.013 | 0.001 | L IFG (POp; BA 44) [-48; 6; 18] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs3760790 | 19 | 53538288 | 2.2 × 10−6 | 4.09 | -0.024 | 0.001 | A | 0.16 | R MFG [27; 9; 51] | sphere | TMEM143 | intronic | 0.618 | 0.30 | 0.007 | 0.014 | |
|
| |||||||||||||||||
| rs6897982 | 5 | 159899502 | 2.3 × 10−6 | 3.97 | -0.037 | 0.010 | G | 0.12 | R MFG [42; 39; 24] | sphere | ATP10B | intergenic | 0.696 | 0.37 | -0.013 | 0.033 | |
| 8.0 × 10−3 | 1.26 | -0.015 | 0.010 | R MFG [27; 9; 51] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs2830255 | 21 | 26787849 | 2.4 × 10−6 | 4.25 | -0.027 | 0.010 | A | 0.13 | L IPL [-33; -45; 39] | sphere | CYYR1 | intronic | 0.627 | 0.79 | 0.013 | 0.026 | |
| 1.7 × 10−4 | 2.65 | -0.021 | 0.010 | L IPL [-39; -42; 39] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs3213191 | 15 | 89041048 | 2.6 × 10−6 | 4.14 | 0.019 | 0.001 | 1 | C | 0.30 | L IPL [-33; -45; 39] | sphere | BLM | intergenic | 0.939 | 0.01 | 0.001 | 0.014 |
| 1.7 × 10−3 | 1.82 | 0.013 | 0.001 | L IPL [-39; -42; 39] | sphere | ||||||||||||
| 5.5 × 10−3 | 1.43 | 0.008 | 0.001 | L SOG [-22; -67; 36] | cluster | ||||||||||||
|
| |||||||||||||||||
| rs1559804 | 18 | 70268238 | 2.7 × 10−6 | 4.17 | 0.018 | 0.001 | T | 0.39 | R PCUN [19; -70; 46] | cluster | FAM69C | intronic | 0.352 | 1.56 | -0.022 | 0.024 | |
| 3.2 × 10−3 | 1.65 | 0.008 | 0.001 | L SOG [-22; -67; 36] | cluster | ||||||||||||
|
| |||||||||||||||||
| rs7231579 | 18 | 71730019 | 2.8 × 10−6 | 4.16 | -0.048 | 0.010 | 2 | A | 0.07 | R MFG [42; 39; 24] | sphere | intergenic | 0.258 | 3.81 | -0.052 | 0.046 | |
| 4.1 × 10−3 | 1.60 | -0.022 | 0.010 | L IPL [-33; -45; 39] | sphere | ||||||||||||
| 7.1 × 10−3 | 1.41 | -0.028 | 0.010 | L SMG [0; 36; 34] | cluster | ||||||||||||
| 7.9 × 10−3 | 1.34 | -0.020 | 0.010 | R MFG [27; 9; 51] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs12667261 | 7 | 25598718 | 2.8 × 10−6 | 4.46 | -0.026 | 0.010 | A | 0.15 | R MFG [27; 9; 51] | sphere | intergenic | 0.105 | 6.73 | -0.034 | 0.021 | ||
| 6.0 × 10−3 | 1.52 | -0.021 | 0.010 | R MFG [42; 39; 24] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs10207939 | 2 | 61106919 | 2.9 × 10−6 | 3.91 | -0.022 | 0.001 | G | 0.22 | R IPL / MOG [42; -70; 30] | cluster | PEX13 | intronic | 0.711 | 0.22 | -0.007 | 0.018 | |
|
| |||||||||||||||||
| rs4661327 | 1 | 15421260 | 3.0 × 10−6 | 3.72 | -0.026 | 0.010 | A | 0.12 | L IPL [-39; -42; 39] | sphere | TMEM51 | intergenic | 0.177 | 2.17 | -0.024 | 0.018 | |
| 4.6 × 10−4 | 2.10 | -0.019 | 0.010 | L IPL [-33; -45; 39] | sphere | ||||||||||||
| 5.7 × 10−3 | 1.30 | -0.010 | 0.001 | L SOG [-22; -67; 36] | cluster | ||||||||||||
| 7.8 × 10−3 | 1.22 | -0.024 | 0.010 | L PCUN [-2; -64; 46] | cluster | ||||||||||||
| 9.7 × 10−3 | 1.14 | -0.014 | 0.001 | R PCUN [19; -70; 46] | cluster | ||||||||||||
|
| |||||||||||||||||
| rs1298730 | 14 | 68869767 | 3.0 × 10−6 | 4.20 | -0.029 | 0.010 | T | 0.47 | L PCUN [-2; -64; 46] | cluster | GALNTL1 | intronic | 0.895 | 0.03 | -0.002 | 0.017 | |
|
| |||||||||||||||||
| rs1354537 | 7 | 42655789 | 3.3 × 10−6 | 3.96 | -0.026 | 0.010 | G | 0.13 | R MFG [27; 9; 51] | sphere | intergenic | 0.684 | 0.24 | 0.007 | 0.017 | ||
| 4.4 × 10−4 | 2.25 | -0.020 | 0.010 | R SFG [22; 23; 50] | cluster | ||||||||||||
| 1.0 × 10−2 | 1.21 | -0.021 | 0.010 | L IFG (PTr) [-45; 27; 27] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs1547258 | 9 | 6513056 | 3.3 × 10−6 | 3.80 | -0.013 | 0.001 | T | 0.29 | L SOG [-22; -67; 36] | cluster | GLDC | intergenic | 0.634 | 0.63 | -0.007 | 0.015 | |
| 2.6 × 10−3 | 1.57 | -0.009 | 0.001 | L IFG (POp) [-51; 9; 9] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs1504121 | 18 | 24369641 | 3.4 × 10−6 | 3.89 | -0.025 | 0.001 | 1 | C | 0.46 | L IFG (PTr) [-45; 27; 27] | sphere | intergenic | 0.478 | 0.94 | 0.013 | 0.018 | |
| 2.1 × 10−5 | 3.27 | -0.019 | 0.001 | L IFG (PTr) [-43; 27; 26] | cluster | ||||||||||||
|
| |||||||||||||||||
| rs4668521 | 2 | 7161780 | 3.5 × 10−6 | 3.53 | -0.019 | 0.001 | 2 | C | 0.32 | R IPL / MOG [42; -70; 30] | cluster | RNF144A | intergenic | 0.757 | 0.20 | -0.006 | 0.018 |
| 6.1 × 10−3 | 1.25 | -0.010 | 0.001 | R PCUN [19; -70; 46] | cluster | ||||||||||||
|
| |||||||||||||||||
| rs7542551 | 1 | 175946654 | 3.6 × 10−6 | 3.79 | 0.025 | 0.001 | A | 0.34 | L SMG [0; 36; 34] | cluster | intergenic | 0.518 | 0.88 | 0.011 | 0.017 | ||
| 1.5 × 10−3 | 1.74 | 0.013 | 0.001 | R SFG [22; 23; 50] | cluster | ||||||||||||
| 4.3 × 10−3 | 1.38 | 0.011 | 0.001 | R IPL / MOG [42; -70; 30] | cluster | ||||||||||||
|
| |||||||||||||||||
| rs10946888 | 6 | 27059657 | 3.6 × 10−6 | 3.77 | 0.018 | 0.001 | T | 0.42 | R SFG [22; 23; 50] | cluster | NCRNA00240 | non-coding | 0.194 | 4.08 | -0.017 | 0.013 | |
|
| |||||||||||||||||
| rs2432560 | 16 | 57091210 | 3.8 × 10−6 | 4.06 | 0.018 | 0.001 | G | 0.38 | L IPL [-33; -45; 39] | sphere | NDRG4 | intronic | 0.096 | 6.31 | -0.025 | 0.015 | |
| 2.2 × 10−5 | 3.39 | 0.017 | 0.001 | L IPL [-39; -42; 39] | sphere | ||||||||||||
|
| |||||||||||||||||
| rs1478272 | 8 | 4455581 | 3.8 × 10−6 | 4.03 | 0.043 | 0.010 | G | 0.12 | L PCUN [-2; -64; 46] | cluster | CSMD1 | intronic | 0.398 | 0.73 | 0.017 | 0.020 | |
Genetic markers are ranked by p-value. Independent markers were those more than 500 kb apart and in LD of r2 < 0.70. In groups of non-independent markers, the most significant SNP is shown, and SNPs in LD shows the number of correlated SNPs that are in the top SNPs with p<1×10−5.
SNP = single nucleotide polymorphism; Chr = chromosome; BP = base pair positions obtained from the HapMapI + II (b36r22) CEU legend files; β = effect size, indicating the mean increase in average percent BOLD signal (2-back minus 0-back) per added reference allele controlling for sex, age, principal components for ancestry, and n-back task performance accuracy; SE = standard error; LD = linkage disequilibrium; A1 = minor allele or reference allele; MAF = Minor Allele Frequency; ROI(s) = all region-of-interest phenotypes where this SNP was in the top 50; Gene = name of gene if the SNP is located in a known gene or within 50 kb distance from a gene, obtained from WGA Viewer using release 57, cells are empty if no gene is within ±50 kb; REPL = replication sample.
ROI Abbreviations: AG, angular gyrus; BA, Brodmann area; IFG, inferior frontal gyrus; IPL, inferior parietal lobule; L, left; MFG, middle frontal gyrus; MNI, Montréal Neurological Institute; MOG, middle occipital gyrus; MTG, middle temporal gyrus; PCG, precentral gyrus; PCUN, precuneus; POp, pars opercularis; PTr, pars triangularis; R, right; ROI, region of interest; SFG, superior frontal gyrus; SMA, supplementary motor area; SMG, superior medial gyrus; SOG, superior occipital gyrus; SPL, superior parietal lobule.
Figure 2.
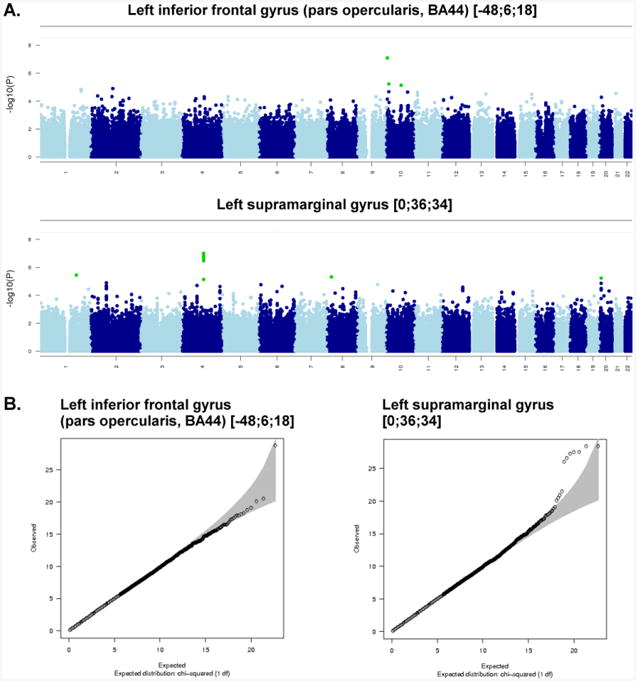
Manhattan plots (A) and Quantile-Quantile (Q-Q) plots (B) for the two region of interest BOLD measures with the strongest SNP associations: Left Inferior Frontal Gyrus (Pars Opercularis; BA 44) [−48; 6; 18], and Left Supramarginal Gyrus [0; 36; 34].
(A) Each point in the Manhattan plot is a SNP laid out across the human chromosomes from left to right, and the heights correspond to the strength of the association to working memory task-related cortical activation. The horizontal line on the Manhattan plots shows the genome-wide significance level, which has been corrected from the generic GWAS significance level (p<7.2×10−8) by estimating the number of independent variables in the data (Li and Ji, 2005; Nyholt, 2004). We estimated that the required significance level to account for multiple testing was p<1.9×10−9. (B) The Q-Q plot is used to assess the number and magnitude of observed associations compared with the expectations under no association. The nature of deviations from the identity line provide clues whether the observed associations are true associations or may be due to for example population stratification or cryptic relatedness.
Figure 3.
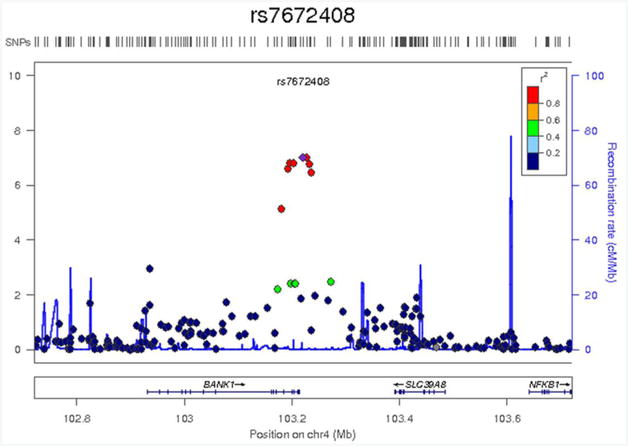
Detailed view of the locus showing suggestive association (p = 9.9×10−8) with average BOLD percent signal change in the left supramarginal gyrus [0; 36; 34]. Genetic markers are represented as circles. Markers are placed at their position on chromosome 4 (x-axis) and graphed based on the −log10(p-values) of their association to the phenotype (y-axis). The level of linkage disequilibrium to the most associated SNP (rs7672408) is represented in colour using the CEU panel from HapMap Phase II. The location of genes is shown below the plots. Images were created using LocusZoom (http://csg.sph.umich.edu/locuszoom/).
Table 3 shows that the top SNPs are approximately evenly distributed amongst sphere (15) and cluster ROIs (16). The ROIs which show up most often in the top independent SNPs, i.e., the ROIs with the most p-values <1×10−5 are the left inferior parietal lobule [−39; −42; 39], the left precuneus [−2; −64; 46], the right middle frontal gyrus [27; 9; 51], and the left inferior parietal lobule [−33; −45; 39], with 3 SNPs each.
The last 4 columns of Table 3 show the replication results for the most strongly associated SNPs in the discovery sample. No SNPs reached significance in the replication sample at the corrected p<0.0016 level, or at the nominal significance level of p<0.05. The SNP with the lowest replication p-value with consistent direction of effect was rs2118263 (p=0.075).
3.6 Brain expression analysis
Supplementary Table 5 summarizes the brain expression data for the genes listed in Table 3 derived from four databases. Supplementary Table 6 shows the full expression data derived from the Allen Brain Atlas. Supplementary Figure 1 shows the life expression course in 6 brain tissue types derived from the Human Brain Transcriptome Project for the genes with p<1×10−6. Brain expression for the BANK1 gene, one of the top two association findings, is highest in the cerebellar cortex, as indicated by the Allen Brain Atlas, GTEx, and HBT (Supplementary Table 5), and slightly below the average brain expression in the supramarginal gyrus where the strongest association is found (Allen Brain Atlas; Supplementary Table 6). The Stanford Brain RNA-Seq database shows expression among seven brain cell types is highest in microglia (Supplementary Table 5).
4 Discussion
Here we report on a GWA study of WM brain activation that was carried out in a population-based family cohort, consisting of MZ and DZ twins, and their singleton siblings (n=679). We made use of prior knowledge from a voxel-based twin study on the heritability of WM brain activation about which areas of the cerebral cortex are most strongly activated and which areas are most highly heritable to define our functional phenotype. We then searched genome-wide for genetic variants influencing the normal brain response to WM. We tested >500,000 SNPs for association with each of the 20 BOLD phenotypes that met all quality control criteria and identified several genetic variants of interest for BOLD response to WM, though none reached the genome-wide significance level.
Strict quality control criteria of test-retest reliability (r>0.7) and significant heritability reduced the number of phenotypes to test for genetic association from 46 to 20. Heritability of average BOLD percent signal change in the 20 ROIs included in the GWA analyses ranged between 27% and 54%, averaging 41%. The heritability estimates for BOLD response agree with our prior voxel-based fMRI twin study, in which heritability across significant voxels averaged ∼33% (Blokland et al., 2011). Areas with low heritability generally had low variances, low twin correlations, and low test-retest reliability. As expected, since they were defined based on voxel-wise heritability estimates from a subsample, overall, average BOLD response in the less strongly activated cluster ROIs was more heritable than in the highly activated sphere local maxima ROIs. However, BOLD response in several sphere local maxima ROIs was strongly heritable as well.
The most strongly associated SNP (p=8.2×10−8) was located in an intergenic region on chromosome 10 (rs7917410), between the KLF6 gene (distance>500kb), and non-coding RNA LOC100216001 (distance>350kb). Genetic association was found for a left inferior frontal gyrus sphere ROI [–48; 6; 18], with the SNP accounting for 5.4% of the trait variance. However, no other SNPs in this genomic region were associated with the phenotype, the association did not replicate, and the Manhattan and QQ plots suggest this may be a false positive finding.
The gene with the most SNPs (five) with p<1×10−6 is BANK1, on Chromosome 4. The top SNP (rs7672408; p=9.9×10−8) accounted for 5.5% of the trait variance in the left supramarginal gyrus [0; 36; 34]. BANK1, encoding a B-cell-specific scaffold protein, has been primarily associated with autoimmune diseases (Fan et al., 2011; Guo et al., 2009; Orozco et al., 2009; Rueda et al., 2010), but more recently the BANK1 locus was shown to be moderately (p<5×10−5) associated with schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). BANK1 has been shown to negatively regulate CD40-mediated AKT1 activation (Aiba et al., 2006). The interaction of the CD40 receptor and its ligand is necessary for amyloid-beta-induced microglial activation, and thus is thought to be an early event in Alzheimer disease pathogenesis (Laporte et al., 2008). AKT1, also known as Protein Kinase B, plays a key role in multiple cellular processes such as glucose metabolism, apoptosis, cell proliferation, transcription and cell migration. The AKT1/GSK-3 signaling pathway has been associated with several human diseases, including schizophrenia. Studies in preclinical models have demonstrated that impaired AKT1 signaling affects neuronal connectivity and neuromodulation and have identified AKT1 as a key signaling intermediary downstream of dopamine receptor 2 (DRD2), the best-established target of antipsychotic drugs (Arguello and Gogos, 2008; Beaulieu et al., 2005). This AKT1/GSK-3 pathway influences the expression of dopamine-associated psychomotor behaviors that, in transgenic models, have been predictably modulated by dopaminergic agonists and antagonists (Alimohamad et al., 2005). AKT1-knockout mice showed evidence of poorer WM performance under dopaminergic agonist challenge (Emamian et al., 2004) as well as concurrent changes in prefrontal pyramidal dendritic ultrastructure, possibly mediated by downstream alterations in the expression of genes controlling neuronal development in prefrontal cortex (Lai et al., 2006). This represents a means by which D2 receptor signaling and associated cognitive and neuropsychiatric effects could be mediated (Beaulieu et al., 2005; Beaulieu et al., 2004; Lai et al., 2006). AKT1 has also recently been found to indirectly have an impact on dopamine signaling by regulating the trafficking of presynaptic dopamine transporters, which remove dopamine from extracortical synapses (Wei et al., 2007). Moreover, in prior candidate gene imaging studies, AKT1 has been shown to be associated with WM brain activation, regional cortical grey matter density, and verbal memory (Nicodemus et al., 2010b; Pietilainen et al., 2009; Tan et al., 2012; Tan et al., 2008). Together, these studies explain a possible mechanism by which BANK1 has an effect on BOLD response to WM.
On the other hand, brain expression data for BANK1 revealed that the gene is most highly expressed in the cerebellar cortex, and to a lesser degree in the cerebral cortex and subcortical structures. Analysis of expression in specific brain cell types indicated that BANK1 is mostly expressed in microglia. Microglia act as the first and main form of active immune defense in the central nervous system, but also have been shown to play a role in synaptic pruning (Schafer and Stevens, 2015). Recently, Zhan et al. (2014) showed in Cx3cr1 knockout mice, who exhibit a transient reduction of microglia during the early postnatal period and consequent deficit in synaptic pruning, that deficient synaptic pruning is associated with weak synaptic transmission, and decreased functional brain connectivity. This suggests that BANK1 might have an indirect effect on brain activation through its effect on neuron-microglia signaling. The direct role of microglia in the BOLD response is not well known; although there is support for the role of other glial cells in neurovascular coupling (Iadecola and Nedergaard, 2007; Metea and Newman, 2006).
As for the brain location of the association, the supramarginal gyrus activation is thought to be related to visuospatial strategy used to maintain information in working memory (Salmon et al., 1996), but the fact that the association is strongest for this region is likely because the BOLD signal increase in this ROI was most heritable/reliable. The observation that multiple ROIs are moderately associated with the same SNP, not just for this SNP, but also for many others, suggests there are genes that are more generalized in their function. These may be genes not specific to working memory function, but for instance genes that are important to the BOLD response or brain development in general. Although the results in the discovery sample are promising, the BANK1 association also did not replicate in the smaller sample.
Interestingly, the SNP association that came closest to replication (p<0.1) is for an intergenic SNP (rs2118263) close to the FOXQ1 gene (distance=59.78kb). FOXQ1, forkhead box Q1, encodes a Winged Helix/Forkhead Transcription Factor. Recently, FOXQ1 was identified in a schizophrenia and bipolar disorder case-control GWA study, where it showed an epistatic interaction with SUMO1P1 for psychomotor speed on the Grooved Pegboard test (LeBlanc et al., 2012).
Although these replications did not reach significance here, likely due to the limited statistical power in the relatively small replication sample, the results are strong enough to warrant further investigation in larger samples with different demographic characteristics and MRI acquisition parameters. Several studies advocate examining multiple cohorts where the spectrum of observable variation is larger than that in the general population, particularly in the discovery phase. However, the fact that we found such low p-values with the present sample size underlines the power of the family study, which reduces the possible impact of population stratification. Also, here we analyzed a population-based sample that consisted solely of healthy individuals, whereas other GWA imaging studies have most often used patient-control samples. The advantage of this is that it provides information on the genetics of healthy brain function, which may provide new clues for investigations of brain disorders.
Age-specific gene effects could also explain a lack of replication. Even though the discovery and replication samples originated from the same cohort, the Queensland Twin Imaging Study, they differed significantly on mean age, with the replication sample being younger on average, and this age difference was apparent in their average percentages BOLD signal change in the ROIs. In our previous study (Blokland et al., 2011) we showed that there are significant effects of age on the brain activation phenotype and therefore included age as a covariate in the genetic association analyses, but not in the group random effects analysis. This may have affected the number of significant activation clusters and therefore the number of ROIs. However, it is possible that there are age-dependent gene effects on WM brain activation, especially in this cohort aged 16-30 years as many of the brain regions involved in WM have been shown to continue developing into adulthood (Fuster, 2002; Giedd et al., 1999). Certain genes can impact BOLD response more strongly during a different phase of life (see e.g. Nichols et al., 2012). Not regressing out age effects before the genetic analyses will allow us to investigate possible gene-by-age interaction effects in future analyses. Age differences could also explain differences between our results and those of other studies. All individuals in the QTIMS cohort are between 16 and 30 years of age, whereas most other association studies had wider age ranges, and/or only included adults.
Remarkably, here we found none of the genes that Potkin and colleagues reported in their GWA analysis on the mean percent BOLD signal change in the DLPFC (Potkin et al., 2009a; Potkin et al., 2009b). This could be due to their use of a patient-control sample, or other demographic sample characteristics, including age, and their selection of functional quantitative phenotype. However, we also did not detect association with any of the candidate genes that have been associated with WM brain activation previously, such as NRG1, DAT1, dopamine receptor genes, COMT, BDNF, and AKT1, suggesting some of these earlier findings may have been false positives (Bertolino et al., 2006a; Bertolino et al., 2006b; Bertolino et al., 2009; Bertolino et al., 2010; Egan et al., 2001; Herrmann et al., 2007; Krug et al., 2008; Li et al., 2006; Nicodemus et al., 2010b; Pomarol-Clotet et al., 2010; Stefansson et al., 2003; Stefansson et al., 2002; Stollstorff et al., 2010; Tan et al., 2007; Tan et al., 2008; Tura et al., 2008; Zhao et al., 2004).
Finally, while the SNPs with the lowest p-values explained a considerable proportion of the trait variance (∼4–6%) in both the discovery and replication samples, much of the genetic variance in WM brain activation is still unaccounted for. This suggests that many genes of small effect are influencing WM brain activation. Missing heritability might be attributed to low power, rare variants, un-genotyped variants, epistatic interactions or epigenetic contributions to heritability (Manolio et al., 2009). It is likely that much of the genetic variance is due to epistatic interactions, as several epistatic interactions have been reported with regards to some of the aforementioned candidate genes and their influence on WM brain activation (Nicodemus et al., 2010a; Nicodemus et al., 2010b; Nixon et al., 2011; Tan et al., 2007). Also, it is important to mention that this GWA analysis looked at additive genetic variance; i.e., it estimated whether having more (zero, one, or two) risk alleles has a linear effect on the phenotype. In our genetic modeling, we found indication that a considerable part of the genetic variance in BOLD response to a WM task might be due to genetic dominance. This is in line with our prior findings in our voxel-based analyses (Blokland et al., 2011), although we did not have the statistical power to disentangle the additive and dominance genetic variance. As we applied an additive genetic model in our GWA, it may not have captured all the genetic processes underlying WM brain activation. This will be an interesting venture for the future.
In summary, in one of the largest functional imaging genetics studies ever performed (n=863), we identified specific genetic variations associated with BOLD response to a WM task. To the best of our knowledge this is the first GWA scan to map the genetic loci that affect normal variation in WM task-related brain activation. Though not genome-wide significant, our results highlight a region of the genome that may provide a stronger understanding of BOLD signal neurobiology, human brain function and susceptibility to the development of common psychiatric and neurological disorders affecting WM brain activation. Although replication will be needed, the present results are encouraging. The fact that we found such low p-values and large effect sizes in a sample that is much smaller in size than those used in some current GWA studies strongly suggests that ROI-based measures of BOLD response to WM are powerful, genetically informative tools with which to search the genome and may be used successfully to find genetic variants in multi-site genetic meta-analyses. Future meta-analyses in even larger samples may be sufficiently powered to relate genetic differences in brain function to observable differences in cognition or risk for the disorders in which WM is implicated.
Supplementary Material
Highlights.
We used voxel-level BOLD heritability to define region-of-interest phenotypes
We performed genome-wide association on 20 n-back functional MRI BOLD phenotypes
A BANK1 SNP explained 5.5% of variance in supramarginal gyrus n-back BOLD signal
31 independent SNPs were associated with BOLD signal change at p<1×10−5
Acknowledgments
This study was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (RO1 HD050735), USA, and National Health and Medical Research Council (NHMRC; Project Grants 496682 and 628952), Australia. The collection of IQ data and zygosity typing was supported by the Australian Research Council (ARC; A7960034, A79906588, A79801419, DP0212016). G.A.M.B. was supported by an ANZ Trustees PhD Scholarship in Medical Research, Queensland, Australia (CT-10803, CT-12643). G.I.Z. was supported by an ARC Future Fellowship (FT0991634). The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, The National Institutes of Health, NHMRC, or ARC. We are very grateful to the twins for their generosity of time and willingness to participate in our studies. We thank research nurses Marlene Grace and Ann Eldridge for twin recruitment, research assistants Lachlan Strike, Kori Johnson, Aaron Quiggle, and Natalie Garden and radiographers Matthew Meredith, Peter Hobden, Kate Borg, Aiman Al Najjar, and Anita Burns for data acquisition, and David Butler and Daniel Park for IT support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Aiba Y, Yamazaki T, Okada T, Gotoh K, Sanjo H, Ogata M, Kurosaki T. BANK negatively regulates Akt activation and subsequent B cell responses. Immunity. 2006;24:259–268. doi: 10.1016/j.immuni.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Alimohamad H, Rajakumar N, Seah YH, Rushlow W. Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol Psychiatry. 2005;57:533–542. doi: 10.1016/j.biopsych.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Annett M. A classification of hand preference by association analysis. Br J Psychol. 1970;61:303–321. doi: 10.1111/j.2044-8295.1970.tb01248.x. [DOI] [PubMed] [Google Scholar]
- Arguello PA, Gogos JA. A signaling pathway AKTing up in schizophrenia. J Clin Invest. 2008;118:2018–2021. doi: 10.1172/JCI35931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino A, Blasi G, Latorre V, Rubino V, Rampino A, Sinibaldi L, Caforio G, Petruzzella V, Pizzuti A, Scarabino T, Nardini M, Weinberger DR, Dallapiccola B. Additive effects of genetic variation in dopamine regulating genes on working memory cortical activity in human brain. J Neurosci. 2006a;26:3918–3922. doi: 10.1523/JNEUROSCI.4975-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino A, Caforio G, Petruzzella V, Latorre V, Rubino V, Dimalta S, Torraco A, Blasi G, Quartesan R, Mattay VS, Callicott JH, Weinberger DR, Scarabino T. Prefrontal dysfunction in schizophrenia controlling for COMT Val158Met genotype and working memory performance. Psychiatry Res. 2006b;147:221–226. doi: 10.1016/j.pscychresns.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Fazio L, Di Giorgio A, Blasi G, Romano R, Taurisano P, Caforio G, Sinibaldi L, Ursini G, Popolizio T, Tirotta E, Papp A, Dallapiccola B, Borrelli E, Sadee W. Genetically determined interaction between the dopamine transporter and the D2 receptor on prefronto-striatal activity and volume in humans. J Neurosci. 2009;29:1224–1234. doi: 10.1523/JNEUROSCI.4858-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino A, Taurisano P, Pisciotta NM, Blasi G, Fazio L, Romano R, Gelao B, Lo Bianco L, Lozupone M, Di Giorgio A, Caforio G, Sambataro F, Niccoli-Asabella A, Papp A, Ursini G, Sinibaldi L, Popolizio T, Sadee W, Rubini G. Genetically determined measures of striatal D2 signaling predict prefrontal activity during working memory performance. PLoS ONE. 2010;5:e9348. doi: 10.1371/journal.pone.0009348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigos KL, Mattay VS, Callicott JH, Straub RE, Vakkalanka R, Kolachana B, Hyde TM, Lipska BK, Kleinman JE, Weinberger DR. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch Gen Psychiatry. 2010;67:939–945. doi: 10.1001/archgenpsychiatry.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokland GAM, McMahon KL, Hoffman J, Zhu G, Meredith M, Martin NG, Thompson PM, de Zubicaray GI, Wright MJ. Quantifying the heritability of task-related brain activation and performance during the N-back working memory task: A twin fMRI study. Biol Psychol. 2008;79:70–79. doi: 10.1016/j.biopsycho.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokland GAM, McMahon KL, Thompson PM, Hickie IB, Martin NG, de Zubicaray GI, Wright MJ. Genetic effects on the cerebellar role in working memory: same brain, different genes? Neuroimage. 2014;86:392–403. doi: 10.1016/j.neuroimage.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokland GAM, McMahon KL, Thompson PM, Martin NG, de Zubicaray GI, Wright MJ. Heritability of working memory brain activation. J Neurosci. 2011;31:10882–10890. doi: 10.1523/JNEUROSCI.5334-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bor J, Brunelin J, Sappey-Marinier D, Ibarrola D, d'Amato T, Suaud-Chagny MF, Saoud M. Thalamus abnormalities during working memory in schizophrenia. An fMRI study. Schizophr Res. 2011;125:49–53. doi: 10.1016/j.schres.2010.10.018. [DOI] [PubMed] [Google Scholar]
- Brett M, Anton JL, Valabregue R, Poline JB. Region of interest analysis using an SPM toolbox, 8th International Conference on Functional Mapping of the Human Brain, Sendai, Japan. NeuroImage. 2002;16 abstract 497. [Google Scholar]
- Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, Todd JA, Donnelly P, Barrett JC, Davison D, Easton D, Evans D, Leung HT, Marchini JL, Morris AP, Spencer CCA, Tobin MD, Attwood AP, Boorman JP, Cant B, Everson U, Hussey JM, Jolley JD, Knight AS, Koch K, Meech E, Nutland S, Prowse CV, Stevens HE, Taylor NC, Walters GR, Walker NM, Watkins NA, Winzer T, Jones RW, McArdle WL, Ring SM, Strachan DP, Pembrey M, Breen G, St Clair D, Caesar S, Gordon-Smith K, Jones L, Fraser C, Green EK, Grozeva D, Hamshere ML, Holmans PA, Jones IR, Kirov G, Moskvina V, Nikolov I, O'Donovan MC, Owen MJ, Collier DA, Elkin A, Farmer A, Williamson R, McGuffin P, Young AH, Ferrier IN, Ball SG, Balmforth AJ, Barrett JH, Bishop DT, Iles MM, Maqbool A, Yuldasheva N, Hall AS, Braund PS, Dixon RJ, Mangino M, Stevens S, Thompson JR, Bredin F, Tremelling M, Parkes M, Drummond H, Lees CW, Nimmo ER, Satsangi J, Fisher SA, Forbes A, Lewis CM, Onnie CM, Prescott NJ, Sanderson J, Mathew CG, Barbour J, Mohiuddin MK, Todhunter CE, Mansfield JC, Ahmad T, Cummings FR, Jewell DP, Webster J, Brown MJ, Lathrop GM, Connell J, Dominiczak A, Marcano CAB, Burke B, Dobson R, Gungadoo J, Lee KL, Munroe PB, Newhouse SJ, Onipinla A, Wallace C, Xue MZ, Caulfield M, Farrall M, Barton A, Bruce IN, Donovan H, Eyre S, Gilbert PD, Hider SL, Hinks AM, John SL, Potter C, Silman AJ, Symmons DPM, Thomson W, Worthington J, Dunger DB, Widmer B, Frayling TM, Freathy RM, Lango H, Perry JRB, Shields BM, Weedon MN, Hattersley AT, Elliott KS, Groves CJ, Lindgren CM, Rayner NW, Timpson NJ, Zeggini E, Newport M, Sirugo G, Lyons E, Vannberg F, Brown MA, Franklyn JA, Heward JM, Simmonds MJ, Hill AVS, Bradbury LA, Farrar C, Pointon JJ, Wordsmith P, Gough SCL, Seal S, Stratton MR, Rahman N, Ban M, Goris A, Sawcer SJ, Compston A, Conway D, Jallow M, Bumpstead SJ, Chaney A, Downes K, Ghori MJR, Gwilliam R, Inouye M, Keniry A, King E, McGinnis R, Potter S, Ravindrarajah R, Whittaker P, Withers D, Pereira-Gale J, Hallgrimsdottir IB, Howie BN, Su Z, Teo YY, Vukcevic D, Bentley D, Wellcome Trust Case Control Consortium. Biol RAG, Genom S Syndicate; Breast Canc Susceptib C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callicott JH, Egan MF, Mattay VS, Bertolino A, Bone AD, Verchinksi B, Weinberger DR. Abnormal fMRI response of the dorsolateral prefrontal cortex in cognitively intact siblings of patients with schizophrenia. Am J Psychiatry. 2003a;160:709–719. doi: 10.1176/appi.ajp.160.4.709. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR. Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry. 2003b;160:2209–2215. doi: 10.1176/appi.ajp.160.12.2209. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Ramsey NF, Tallent K, Bertolino A, Knable MB, Coppola R, Goldberg T, van Gelderen P, Mattay VS, Frank JA, Moonen CT, Weinberger DR. Functional magnetic resonance imaging brain mapping in psychiatry: methodological issues illustrated in a study of working memory in schizophrenia. Neuropsychopharmacology. 1998;18:186–196. doi: 10.1016/S0893-133X(97)00096-1. [DOI] [PubMed] [Google Scholar]
- Cerasa A, Gioia MC, Fera F, Passamonti L, Liguori M, Lanza P, Muglia M, Magariello A, Quattrone A. Ventro-lateral prefrontal activity during working memory is modulated by MAO A genetic variation. Brain Res. 2008;1201:114–121. doi: 10.1016/j.brainres.2008.01.048. [DOI] [PubMed] [Google Scholar]
- Cerasa A, Tongiorgi E, Fera F, Gioia MC, Valentino P, Liguori M, Manna I, Zito G, Passamonti L, Nistico R, Quattrone A. The effects of BDNF Val66Met polymorphism on brain function in controls and patients with multiple sclerosis: An imaging genetic study. Behav Brain Res. 2010;207:377–386. doi: 10.1016/j.bbr.2009.10.022. [DOI] [PubMed] [Google Scholar]
- Chen WM, Abecasis GR. Family-based association tests for genomewide association scans. Am J Hum Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di X, Kannurpatti SS, Rypma B, Biswal BB. Calibrating BOLD fMRI activations with neurovascular and anatomical constraints. Cereb Cortex. 2013;23:255–263. doi: 10.1093/cercor/bhs001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapier D, Surguladze S, Marshall N, Schulze K, Fern A, Hall MH, Walshe M, Murray RM, McDonald C. Genetic liability for bipolar disorder is characterized by excess frontal activation in response to a working memory task. Biol Psychiatry. 2008;64:513–520. doi: 10.1016/j.biopsych.2008.04.038. [DOI] [PubMed] [Google Scholar]
- Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32:227–234. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- Esslinger C, Kirsch P, Haddad L, Mier D, Sauer C, Erk S, Schnell K, Arnold C, Witt SH, Rietschel M, Cichon S, Walter H, Meyer-Lindenberg A. Cognitive state and connectivity effects of the genome-wide significant psychosis variant in ZNF804A. Neuroimage. 2011;54:2514–2523. doi: 10.1016/j.neuroimage.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Esslinger C, Walter H, Kirsch P, Erk S, Schnell K, Arnold C, Haddad L, Mier D, Opitz von Boberfeld C, Raab K, Witt SH, Rietschel M, Cichon S, Meyer-Lindenberg A. Neural mechanisms of a genome-wide supported psychosis variant. Science. 2009;324:605. doi: 10.1126/science.1167768. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4th. Longmans Green; Harlow, Essex, UK: 1996. [Google Scholar]
- Fan Y, Tao JH, Zhang LP, Li LH, Ye DQ. The association between BANK1 and TNFAIP3 gene polymorphisms and systemic lupus erythematosus: A meta-analysis. Int J Immunogenet. 2011;38:151–159. doi: 10.1111/j.1744-313X.2010.00990.x. [DOI] [PubMed] [Google Scholar]
- Freire L, Roche A, Mangin JF. What is the best similarity measure for motion correction in fMRI time series? IEEE Trans Med Imaging. 2002;21:470–484. doi: 10.1109/TMI.2002.1009383. [DOI] [PubMed] [Google Scholar]
- Fuster JM. Frontal lobe and cognitive development. Journal of neurocytology. 2002;31:373–385. doi: 10.1023/a:1024190429920. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Blumenthal J, Jeffries NO, Castellanos FX, Liu H, Zijdenbos A, Paus T, Evans AC, Rapoport JL. Brain development during childhood and adolescence: a longitudinal MRI study. Nat Neurosci. 1999;2:861–863. doi: 10.1038/13158. [DOI] [PubMed] [Google Scholar]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Deshmukh H, Lu R, Vidal GS, Kelly JA, Kaufman KM, Dominguez N, Klein W, Kim-Howard X, Bruner GR, Scofield RH, Moser KL, Gaffney PM, Dozmorov IM, Gilkeson GS, Wakeland EK, Li QZ, Langefeld CD, Marion MC, Williams AH, Divers J, Alarcon GS, Brown EE, Kimberly RP, Edberg JC, Ramsey-Goldman R, Reveille JD, McGwin G, Jr, Vila LM, Petri MA, Vyse TJ, Merrill JT, James JA, Nath SK, Harley JB, Guthridge JM. Replication of the BANK1 genetic association with systemic lupus erythematosus in a European-derived population. Genes Immun. 2009;10:531–538. doi: 10.1038/gene.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann MJ, Walter A, Schreppel T, Ehlis AC, Pauli P, Lesch KP, Fallgatter AJ. D4 receptor gene variation modulates activation of prefrontal cortex during working memory. Eur J Neurosci. 2007;26:2713–2718. doi: 10.1111/j.1460-9568.2007.05921.x. [DOI] [PubMed] [Google Scholar]
- Hibar DP, Stein JL, Kohannim O, Jahanshad N, Saykin AJ, Shen L, Kim S, Pankratz N, Foroud T, Huentelman MJ, Potkin SG, Jack CR, Jr, Weiner MW, Toga AW, Thompson PM. Voxelwise gene-wide association study (vGeneWAS): Multivariate gene-based association testing in 731 elderly subjects. Neuroimage. 2011;56:1875–1891. doi: 10.1016/j.neuroimage.2011.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, Guennel T, Shin Y, Johnson MB, Krsnik Z, Mayer S, Fertuzinhos S, Umlauf S, Lisgo SN, Vortmeyer A, Weinberger DR, Mane S, Hyde TM, Huttner A, Reimers M, Kleinman JE, Sestan N. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, Markov V, Eggermann T, Krach S, Zerres K, Stocker T, Shah NJ, Schneider F, Nothen MM, Treutlein J, Rietschel M, Kircher T. Genetic variation in the schizophrenia-risk gene neuregulin1 correlates with differences in frontal brain activation in a working memory task in healthy individuals. Neuroimage. 2008;42:1569–1576. doi: 10.1016/j.neuroimage.2008.05.058. [DOI] [PubMed] [Google Scholar]
- Lai WS, Xu B, Westphal KG, Paterlini M, Olivier B, Pavlidis P, Karayiorgou M, Gogos JA. Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc Natl Acad Sci U S A. 2006;103:16906–16911. doi: 10.1073/pnas.0604994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte V, Ait-Ghezala G, Volmar CH, Ganey C, Ganey N, Wood M, Mullan M. CD40 ligation mediates plaque-associated tau phosphorylation in beta-amyloid overproducing mice. Brain Res. 2008;1231:132–142. doi: 10.1016/j.brainres.2008.06.032. [DOI] [PubMed] [Google Scholar]
- LeBlanc M, Kulle B, Sundet K, Agartz I, Melle I, Djurovic S, Frigessi A, Andreassen OA. Genome-wide study identifies PTPRO and WDR72 and FOXQ1-SUMO1P1 interaction associated with neurocognitive function. J Psychiatr Res. 2012;46:271–278. doi: 10.1016/j.jpsychires.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Li D, Collier DA, He L. Meta-analysis shows strong positive association of the neuregulin 1 (NRG1) gene with schizophrenia. Hum Mol Genet. 2006;15:1995–2002. doi: 10.1093/hmg/ddl122. [DOI] [PubMed] [Google Scholar]
- Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity (Edinb) 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- Macey PM, Macey KE, Kumar R, Harper RM. A method for removal of global effects from fMRI time series. Neuroimage. 2004;22:360–366. doi: 10.1016/j.neuroimage.2003.12.042. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K, Glahn DC, Peluso MA, Hatch JP, Monkul ES, Najt P, Sanches M, Zamarripa F, Li J, Lancaster JL, Fox PT, Gao JH, Soares JC. Prefrontal hyperactivation during working memory task in untreated individuals with major depressive disorder. Mol Psychiatry. 2007;12:158–166. doi: 10.1038/sj.mp.4001894. [DOI] [PubMed] [Google Scholar]
- Matthews SC, Simmons AN, Strigo I, Jang K, Stein MB, Paulus MP. Heritability of anterior cingulate response to conflict: an fMRI study in female twins. Neuroimage. 2007;38:223–227. doi: 10.1016/j.neuroimage.2007.07.015. [DOI] [PubMed] [Google Scholar]
- McEvoy BP, Montgomery GW, McRae AF, Ripatti S, Perola M, Spector TD, Cherkas L, Ahmadi KR, Boomsma D, Willemsen G, Hottenga JJ, Pedersen NL, Magnusson PK, Kyvik KO, Christensen K, Kaprio J, Heikkila K, Palotie A, Widen E, Muilu J, Syvanen AC, Liljedahl U, Hardiman O, Cronin S, Peltonen L, Martin NG, Visscher PM. Geographical structure and differential natural selection among North European populations. Genome Res. 2009;19:804–814. doi: 10.1101/gr.083394.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medland SE, Nyholt DR, Painter JN, McEvoy BP, McRae AF, Zhu G, Gordon SD, Ferreira MA, Wright MJ, Henders AK, Campbell MJ, Duffy DL, Hansell NK, Macgregor S, Slutske WS, Heath AC, Montgomery GW, Martin NG. Common variants in the trichohyalin gene are associated with straight hair in Europeans. Am J Hum Genet. 2009;85:750–755. doi: 10.1016/j.ajhg.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci. 2006;26:2862–2870. doi: 10.1523/JNEUROSCI.4048-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafò MR, Brown SM, Hariri AR. Serotonin transporter (5-HTTLPR) genotype and amygdala activation: a meta-analysis. Biol Psychiatry. 2008;63:852–857. doi: 10.1016/j.biopsych.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MC, Baker SM, Xie G, Maes HH. Mx: Statistical Modeling. 6th. Department of Psychiatry, University of Virginia; VCU Box 900126, Richmond, VA 23298: 2002. [Google Scholar]
- Nichols LM, Masdeu JC, Mattay VS, Kohn P, Emery M, Sambataro F, Kolachana B, Elvevag B, Kippenhan S, Weinberger DR, Berman KF. Interactive effect of apolipoprotein e genotype and age on hippocampal activation during memory processing in healthy adults. Arch Gen Psychiatry. 2012;69:804–813. doi: 10.1001/archgenpsychiatry.2011.1893. [DOI] [PubMed] [Google Scholar]
- Nicodemus KK, Callicott JH, Higier RG, Luna A, Nixon DC, Lipska BK, Vakkalanka R, Giegling I, Rujescu D, St Clair D, Muglia P, Shugart YY, Weinberger DR. Evidence of statistical epistasis between DISC1, CIT and NDEL1 impacting risk for schizophrenia: biological validation with functional neuroimaging. Hum Genet. 2010a;127:441–452. doi: 10.1007/s00439-009-0782-y. [DOI] [PubMed] [Google Scholar]
- Nicodemus KK, Law AJ, Radulescu E, Luna A, Kolachana B, Vakkalanka R, Rujescu D, Giegling I, Straub RE, McGee K, Gold B, Dean M, Muglia P, Callicott JH, Tan HY, Weinberger DR. Biological validation of increased schizophrenia risk with NRG1, ERBB4, and AKT1 epistasis via functional neuroimaging in healthy controls. Arch Gen Psychiatry. 2010b;67:991–1001. doi: 10.1001/archgenpsychiatry.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon DC, Prust MJ, Sambataro F, Tan HY, Mattay VS, Weinberger DR, Callicott JH. Interactive effects of DAOA (G72) and catechol-O-methyltransferase on neurophysiology in prefrontal cortex. Biol Psychiatry. 2011;69:1006–1008. doi: 10.1016/j.biopsych.2010.10.031. [DOI] [PubMed] [Google Scholar]
- Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, Nikolov I, Hamshere M, Carroll L, Georgieva L, Dwyer S, Holmans P, Marchini JL, Spencer CC, Howie B, Leung HT, Hartmann AM, Moller HJ, Morris DW, Shi Y, Feng G, Hoffmann P, Propping P, Vasilescu C, Maier W, Rietschel M, Zammit S, Schumacher J, Quinn EM, Schulze TG, Williams NM, Giegling I, Iwata N, Ikeda M, Darvasi A, Shifman S, He L, Duan J, Sanders AR, Levinson DF, Gejman PV, Cichon S, Nothen MM, Gill M, Corvin A, Rujescu D, Kirov G, Owen MJ, Buccola NG, Mowry BJ, Freedman R, Amin F, Black DW, Silverman JM, Byerley WF, Cloninger CR. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- Orozco G, Abelson AK, Gonzalez-Gay MA, Balsa A, Pascual-Salcedo D, Garcia A, Fernandez-Gutierrez B, Petersson I, Pons-Estel B, Eimon A, Paira S, Scherbarth HR, Alarcon-Riquelme M, Martin J. Study of functional variants of the BANK1 gene in rheumatoid arthritis. Arthritis Rheum. 2009;60:372–379. doi: 10.1002/art.24244. [DOI] [PubMed] [Google Scholar]
- Papassotiropoulos A, Henke K, Stefanova E, Aerni A, Muller A, Demougin P, Vogler C, Sigmund JC, Gschwind L, Huynh KD, Coluccia D, Mondadori CR, Hanggi J, Buchmann A, Kostic V, Novakovic I, van den Bussche H, Kaduszkiewicz H, Weyerer S, Bickel H, RiedelHeller S, Pentzek M, Wiese B, Dichgans M, Wagner M, Jessen F, Maier W, de Quervain DJ. A genome-wide survey of human short-term memory. Mol Psychiatry. 2011;16:184–192. doi: 10.1038/mp.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus FM, Bedenbender J, Krach S, Pyka M, Krug A, Sommer J, Mette M, Nothen MM, Witt SH, Rietschel M, Kircher T, Jansen A. Association of rs1006737 in CACNA1C with alterations in prefrontal activation and fronto-hippocampal connectivity. Hum Brain Mapp. 2014;35:1190–1200. doi: 10.1002/hbm.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus FM, Krach S, Bedenbender J, Pyka M, Sommer J, Krug A, Knake S, Nothen MM, Witt SH, Rietschel M, Kircher T, Jansen A. Partial support for ZNF804A genotype-dependent alterations in prefrontal connectivity. Hum Brain Mapp. 2013;34:304–313. doi: 10.1002/hbm.21434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietilainen OP, Paunio T, Loukola A, Tuulio-Henriksson A, Kieseppa T, Thompson P, Toga AW, van Erp TG, Silventoinen K, Soronen P, Hennah W, Turunen JA, Wedenoja J, Palo OM, Silander K, Lonnqvist J, Kaprio J, Cannon TD, Peltonen L. Association of AKT1 with verbal learning, verbal memory, and regional cortical gray matter density in twins. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:683–692. doi: 10.1002/ajmg.b.30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletikos M, Sousa AM, Sedmak G, Meyer KA, Zhu Y, Cheng F, Li M, Kawasawa YI, Sestan N. Temporal specification and bilaterality of human neocortical topographic gene expression. Neuron. 2014;81:321–332. doi: 10.1016/j.neuron.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomarol-Clotet E, Fatjo-Vilas M, McKenna PJ, Monte GC, Sarro S, Ortiz-Gil J, Aguirre C, Gomar JJ, Guerrero A, Landin R, Capdevila A, Fananas L, Salvador R. COMT Val158Met polymorphism in relation to activation and de-activation in the prefrontal cortex: A study in patients with schizophrenia and healthy subjects. Neuroimage. 2010;53:899–907. doi: 10.1016/j.neuroimage.2010.04.018. [DOI] [PubMed] [Google Scholar]
- Potkin SG, Turner JA, Fallon JA, Lakatos A, Keator DB, Guffanti G, Macciardi F. Gene discovery through imaging genetics: Identification of two novel genes associated with schizophrenia. Mol Psychiatry. 2009a;14:416–428. doi: 10.1038/mp.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potkin SG, Turner JA, Guffanti G, Lakatos A, Fallon JH, Nguyen DD, Mathalon D, Ford J, Lauriello J, Macciardi F. A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophr Bull. 2009b;35:96–108. doi: 10.1093/schbul/sbn155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda B, Gourh P, Broen J, Agarwal SK, Simeon C, Ortego-Centeno N, Vonk MC, Coenen M, Riemekasten G, Hunzelmann N, Hesselstrand R, Tan FK, Reveille JD, Assassi S, Garcia-Hernandez FJ, Carreira P, Camps M, Fernandez-Nebro A, Garcia de la Pena P, Nearney T, Hilda D, Gonzalez-Gay MA, Airo P, Beretta L, Scorza R, Radstake TR, Mayes MD, Arnett FC, Martin J. BANK1 functional variants are associated with susceptibility to diffuse systemic sclerosis in Caucasians. Ann Rheum Dis. 2010;69:700–705. doi: 10.1136/ard.2009.118174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon E, Van der Linden M, Collette F, Delfiore G, Maquet P, Degueldre C, Luxen A, Franck G. Regional brain activity during working memory tasks. Brain. 1996;119(Pt 5):1617–1625. doi: 10.1093/brain/119.5.1617. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Stevens B. Microglia Function in Central Nervous System Development and Plasticity. Cold Spring Harbor perspectives in biology. 2015;7:a020545. doi: 10.1101/cshperspect.a020545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrout PE, Fleiss JL. Intraclass correlations: Uses in assessing rater reliability. Psychol Bull. 1979;86:420–428. doi: 10.1037//0033-2909.86.2.420. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Sarginson J, Kong A, Yates P, Steinthorsdottir V, Gudfinnsson E, Gunnarsdottir S, Walker N, Petursson H, Crombie C, Ingason A, Gulcher JR, Stefansson K, St Clair D. Association of neuregulin 1 with schizophrenia confirmed in a Scottish population. Am J Hum Genet. 2003;72:83–87. doi: 10.1086/345442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, Chou TT, Hjaltason O, Birgisdottir B, Jonsson H, Gudnadottir VG, Gudmundsdottir E, Bjornsson A, Ingvarsson B, Ingason A, Sigfusson S, Hardardottir H, Harvey RP, Lai D, Zhou M, Brunner D, Mutel V, Gonzalo A, Lemke G, Sainz J, Johannesson G, Andresson T, Gudbjartsson D, Manolescu A, Frigge ML, Gurney ME, Kong A, Gulcher JR, Petursson H, Stefansson K. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JL, Hua X, Lee S, Ho AJ, Leow AD, Toga AW, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen AN, Corneveaux JJ, Dechairo BM, Potkin SG, Weiner MW, Thompson P. Voxelwise genome-wide association study (vGWAS) Neuroimage. 2010;53:1160–1174. doi: 10.1016/j.neuroimage.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl JC, Esslinger C, Tost H, Bilek E, Kirsch P, Ohmle B, Viviani R, Walter H, Rietschel M, Meyer-Lindenberg A. Genetic variation in CYP2D6 impacts neural activation during cognitive tasks in humans. Neuroimage. 2012;59:2818–2823. doi: 10.1016/j.neuroimage.2011.07.052. [DOI] [PubMed] [Google Scholar]
- Stollstorff M, Foss-Feig J, Cook EH, Jr, Stein MA, Gaillard WD, Vaidya CJ. Neural response to working memory load varies by dopamine transporter genotype in children. Neuroimage. 2010;53:970–977. doi: 10.1016/j.neuroimage.2009.12.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, Hawrylycz M, Dang C. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013;41:D996–D1008. doi: 10.1093/nar/gks1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HY, Chen AG, Kolachana B, Apud JA, Mattay VS, Callicott JH, Chen Q, Weinberger DR. Effective connectivity of AKT1-mediated dopaminergic working memory networks and pharmacogenetics of anti-dopaminergic treatment. Brain. 2012;135:1436–1445. doi: 10.1093/brain/aws068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HY, Chen Q, Sust S, Buckholtz JW, Meyers JD, Egan MF, Mattay VS, Meyer-Lindenberg A, Weinberger DR, Callicott JH. Epistasis between catechol-Omethyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. Proc Natl Acad Sci U S A. 2007;104:12536–12541. doi: 10.1073/pnas.0610125104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HY, Nicodemus KK, Chen Q, Li Z, Brooke JK, Honea R, Kolachana BS, Straub RE, Meyer-Lindenberg A, Sei Y, Mattay VS, Callicott JH, Weinberger DR. Genetic variation in AKT1 is linked to dopamine-associated prefrontal cortical structure and function in humans. J Clin Invest. 2008;118:2200–2208. doi: 10.1172/JCI34725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tura E, Turner JA, Fallon JH, Kennedy JL, Potkin SG. Multivariate analyses suggest genetic impacts on neurocircuitry in schizophrenia. Neuroreport. 2008;19:603–607. doi: 10.1097/WNR.0b013e3282fa6d8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan J. Radiofrequency coils for imaging and spectroscopy. U S Patent. 1999;5:886. 596. [Google Scholar]
- Wei Y, Williams JM, Dipace C, Sung U, Javitch JA, Galli A, Saunders C. Dopamine transporter activity mediates amphetamine-induced inhibition of Akt through a Ca2+/calmodulin-dependent kinase II-dependent mechanism. Mol Pharmacol. 2007;71:835–842. doi: 10.1124/mol.106.026351. [DOI] [PubMed] [Google Scholar]
- Winterer G, Coppola R, Egan MF, Goldberg TE, Weinberger DR. Functional and effective frontotemporal connectivity and genetic risk for schizophrenia. Biol Psychiatry. 2003;54:1181–1192. doi: 10.1016/s0006-3223(03)00532-8. [DOI] [PubMed] [Google Scholar]
- Wright MJ, Martin NG. The Brisbane Adolescent Twin Study: Outline of study methods and research projects. Aust J Psychol. 2004;56:65–78. [Google Scholar]
- Zeng H, Constable RT. Image distortion correction in EPI: Comparison of field mapping with point spread function mapping. Magn Reson Med. 2002;48:137–146. doi: 10.1002/mrm.10200. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17:400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- Zhang F, Sarginson J, Crombie C, Walker N, St Clair D, Shaw D. Genetic association between schizophrenia and the DISC1 gene in the Scottish population. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:155–159. doi: 10.1002/ajmg.b.30274. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Shi Y, Tang J, Tang R, Yu L, Gu N, Feng G, Zhu S, Liu H, Xing Y, Zhao S, Sang H, Guan Y, St Clair D, He L. A case control and family based association study of the neuregulin1 gene and schizophrenia. J Med Genet. 2004;41:31–34. doi: 10.1136/jmg.2003.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.