

Abstract
Bacterial fatty acid synthesis is essential for many pathogens and different from the mammalian counterpart. These features make bacterial fatty acid synthesis a desirable target for antibiotic discovery. The structural divergence of the conserved enzymes and the presence of different isozymes catalyzing the same reactions in the pathway make bacterial fatty acid synthesis a narrow spectrum target rather than the traditional broad spectrum target. Furthermore, bacterial fatty acid synthesis inhibitors are single-targeting, rather than multi-targeting like traditional monotherapeutic, broad-spectrum antibiotics. The single-targeting nature of bacterial fatty acid synthesis inhibitors makes overcoming fast-developing, target-based resistance a necessary consideration for antibiotic development. Target-based resistance can be overcome through multi-targeting inhibitors, a cocktail of single-targeting inhibitors, or by making the single targeting inhibitor sufficiently high affinity through a pathogen selective approach such that target-based mutants are still susceptible to therapeutic concentrations of drug. Many of the pathogens requiring new antibiotic treatment options encode for essential bacterial fatty acid synthesis enzymes. This review will evaluate the most promising targets in bacterial fatty acid metabolism for antibiotic therapeutics development and review the potential and challenges in advancing each of these targets to the clinic and circumventing target-based resistance.
Keywords: antibiotic discovery, bacterial fatty acid synthesis, resistance
INTRODUCTION
Phospholipid synthesis is one of the major biosynthetic pathways in all living organisms, and fatty acids form the acyl chains found on all phospholipids. Fatty acid synthesis is essential in many bacterial pathogens, and significant differences exists between the structures of mammalian type I fatty acid synthesis and bacterial type II fatty acid synthesis (FASII). The essential nature of bacterial fatty acid synthesis coupled with the ability to target bacterial fatty acid synthesis selectively over the mammalian counterpart has made FASII a focus for developing novel antibiotics. In this review, we will discuss the potential of the various molecular targets in fatty acid synthesis to be targeted for antibiotic discovery. We will start with a brief review of antibiotic discovery to understand how history has shaped thinking in the field. Next, we will discuss the antibiotic discovery targeting bacterial fatty acid metabolism and the potential of enzymes in bacterial fatty acid metabolism as antibiotic targets. Finally, we will discuss current thinking in antibiotic discovery and how to develop novel therapeutics against the targets in bacterial lipid metabolism.
A BRIEF HISTORY OF ANTIBIOTIC DISCOVERY
Activity Based Synthetic Compound Screening – The First Step
The history of antibiotic discovery started from the crossroads of microbiology and organic chemistry in the early 1900s. Having recently shown microorganisms as the cause of infectious diseases, early researchers screened for compounds that kill the infectious agent with reduced toxicity to the human patient (as measured in animal models). The German Nobel laureate Paul Ehrlich was the first person to formalize the idea of a hypothetical agent (“magic bullet”) that kills the specific microbe responsible for the diseases without harming the body [1]. Through synthesizing and screening analogs of the organoarsenic compound Atoxyl which had antisyphilitic activity but also severe side effects, Ehrlich’s team discovered arsphenamine (Salvarsan) and neoarsphenamine (Neosalvarsan), the first modern chemotherapeutic agents. The first antibiotic to reach commercial success was sulfonamide drugs discovered by scientists in Bayer AG [2]. A red aniline dye was found to effectively inhibit bacterial infections in mice after screening hundreds of dyes. The compound was named Prontosil and was highly effective against Gram-positive streptococci. Further research found that Prontosil was a prodrug of the active sulfanilamide. Sulfanilamide was the first broad-spectrum antibiotic, and was used on the battlefields of World War II for treating wound infections.
Activity Based Natural Product Screening – The Golden Age
The Golden Age of antibiotic discovery occurred from the activity screening of natural products. Alexander Fleming was the first to show that the pure Penicillium mold was able to produce an agent with antimicrobial activity in 1928. Fleming was able to isolate the active compound and demonstrated its antimicrobial activity. Penicillin had greater clinical efficacy than sulfanilamide and reduced toxic side effects, but Fleming was unable to garner interest for research into the industrial production of penicillin due to the success of the recently commercialized sulfanilamide antibiotics. The mass scale production of penicillin in the United States was a triumph of the allied war effort during World War II. Penicillin was so successful that it defined the characteristics of the ideal antibiotic – broad-spectrum, monotherapeutic, and low toxicity.
The success of penicillin stimulated natural product screening efforts and spawned the “Golden Age” of antibiotic discovery from the 1940s to the 1970s [3]. Advances in structural biology and medicinal chemistry allowed researchers to chemically modify the natural products to produce semi-synthetic antibiotics with improved clinical properties. The majority of the broad-spectrum antibiotic classes we use even today including the β-lactams, tetracycline, and macrolides were discovered through this process. Unfortunately, resistance quickly caught up with these newly discovered antibiotics, as the discovery of new antibiotic classes through natural product screening decreased over time [4,5]. Additional screens largely rediscovered previously found chemical entities and failed to find promising new chemical entities. The decreasing return caused many pharmaceutical companies to leave antibiotic research.
Target-based Discovery – The Draught
Major advances in genetics and molecular biology by the 1990s allowed the identification of the molecular targets of the antibiotics [6,7]. The first sequenced bacterial genomes were also released at this time, with the various proteins encoded by the genomes rapidly characterized [8]. Several hundred proteins essential for bacterial growth were identified, and thought to be potential antibiotic targets [9]. The success of fluoroquinolone, a synthetic antibiotic rationally designed against DNA topoisomerase suggested that other essential molecular targets could be exploited as novel antibiotic targets as well [10]. Antibiotic discovery entered a new phase. Rather than screen for active compounds and determine their molecular target, hits against essential molecular targets were identified and chemically modified to become successful antibiotics [11]. However, the end goal for target-based antibiotic discovery remained the same - to discover another broad-spectrum, monotherapeutic, and low toxicity penicillin-like antibiotic. To rationally design such an antibiotic, researchers started with an essential molecular target found in a broad spectrum of bacteria [9]. Furthermore, this target would be nonexistent or significantly different in humans to decrease the probability of toxicity. High-throughput screening technology was used to find the lead compounds against the molecular target, and the lead compound was modified via medicinal chemistry to penetrate the bacterial cell membrane, possess the ideal broad-spectrum, and have drug like pharmacokinetic properties. This compound would then be tested in mouse models, and would be brought to clinical trials in humans if successful.
Several large pharmaceutical companies conducted systemic target-based discovery campaigns in the late 1990s. GlaxoSmithKline carried out the best documented target-based antibiotic discovery campaign [9]. The goal of the campaign was to discover novel antibacterial inhibitors with either Gram-positive or broad-spectrum activity. Over 350 conserved gene targets were identified through comparing the genome sequences of Haemophilus influenzae, Streptococcus pneumoniae, and Staphylococcus aureus. These conserved genes were genetically verified for essentiality, and 127 genes were essential in at least one of the three bacterial species. Several genes encoding for fatty acid synthesis were validated as conserved and essential in these screens. A total of 67 high throughput screening campaigns against the SmithKline Beecham compound collection were conducted against the subset of essential and conserved targets with known function and amenable assays. Only 5 leads resulted from the HTS screens, and two of the leads targeted fatty acid synthesis enzymes FabI and FabH. Nearly a decade later, none of the leads have passed phase III trials, although some are still under clinical development. The target-based antibiotic discovery efforts by the other pharmaceutical companies also did not yield clinical products despite significant investment [12]. The systematic failure of target-based antibiotic discovery suggests that some underlying assumptions about the discovery process must be incomplete or incorrect.
ANTIBIOTIC TARGETS IN FATTY ACID METABOLISM
Fatty acids make up the acyl chains of glycerophospholipids, the major and essential component of bacterial membrane [13,14]. Bacteria synthesize fatty acids using the type II fatty acid synthesis system, where fatty acids are synthesized from acetyl-CoA precursors through rounds of 2 carbon elongations using discrete, monofunctional enzymes [15] (Fig. 1). In contrast, fatty acids are synthesized by the multifunctional type I fatty acid synthase in mammals, allowing for selective targeting of FASII [16]. FASII is a validated target, with two of the five leads discovered in the GlaxoSmithKline HTS campaign targeting FASII (FabH and FabI) [9]. Targeting fatty acid synthesis for novel antibiotic therapies via target-based discovery is also an active area of research, with the FabI inhibitor AFN-1252 having passed phase II clinical trials and currently undergoing further clinical development [17-22]. This section summarizes the antibiotic targets in bacterial fatty acid metabolism.
Fig. 1.

The conserved, core enzymes in bacterial FASII. The first committed step of fatty acid synthesis is the irreversible carboxylation of acetyl-CoA by the acetyl-CoA carboxylase enzyme complex to make malonyl-CoA, the building block of fatty acids. Next, malonyl-CoA is converted into malonyl-ACP by FabD. FabH initiates fatty acid synthesis by catalyzing the Claisen condensation of acetyl-CoA with malonyl-ACP to make acetoacetyl-ACP. From here, the acyl-ACP is elongated 2 carbons per cycle by the elongation enzymes. First, the β-ketoacyl-ACP (including acetoacetyl-ACP) is reduced by FabG to make β-hydroxyacyl-ACP. Next, β-hydroxyacyl-ACP is dehydrated by FabZ to make trans-2-enoyl-ACP. trans-2-Enoyl-ACP is reduced by enoyl-ACP reductase to make acyl-ACP. Acyl-ACP is elongated by the Claisen condensation reaction with a malonyl-ACP by FabF to increase the acyl chain by another 2 carbons and start another round of elongation. Acyl-ACP of sufficient length is used by the acyltransferase system to make phosphatidic acid from glycerol-3-phosphate. Certain bacteria make branched-chain anteiso fatty acids. The FabH of these bacteria use 2-methylbutyryl-CoA instead of acetyl-CoA in the first Claisen condensation reaction. The rate-limiting enzymatic steps that would make good drug targets are in a green box. The equilibrium enzymatic steps that would make poor drug targets are in a red box.
Bacterial Fatty Acid Metabolism Is a Narrow Spectrum Antibiotic Target
The initiation and elongation steps of FASII are shown in Fig. 1 [13,15]. Some of the validated inhibitors of FASII are shown in Fig. 2. Two features of bacterial fatty acid synthesis reduce the potential spectrum of antibiotics targeting fatty acid synthesis [23]. First, fatty acid synthesis is not essential in certain bacteria, such as the key pathogen Streptococcus pneumoniae, if they are provided an extracellular source of fatty acids [24,25]. These bacteria can potentially bypass the inhibition of endogenous fatty acid synthesis by using host fatty acids to make their phospholipids, making them refractory to FASII inhibition. Second, there are different enzyme isoforms performing the same enzymatic reactions in several key steps of FASII in different bacteria. For example, there are four structurally distinct enoyl-ACP reductase enzyme forms [26-29] (Fig. 1). This feature further reduces the spectrum of potential inhibitors of bacterial fatty acid synthesis. Inhibitors targeting bacterial fatty acid metabolism will have at its inception narrower spectrum than the broad-spectrum, monotherapeutic antibiotics commonly deployed in clinics today. This factor must be considered when designing inhibitors of bacterial fatty acid synthesis.
Fig. 2.

Structures of validated FASII inhibitors. The validated molecular targets in FASII are the acetyl-CoA carboxylase, the condensing enzymes (FabH and FabF), and the enoyl-ACP reductase FabI.
Enzymes catalyzing rate-limiting reactions in key regulatory steps make for the best chemotherapeutic targets. In contrast, fast turnover enzymes catalyzing equilibrium reactions make for bad chemotherapeutic targets because a large amount of inhibition is required to cause relatively little phenotypic effect. There are three fast turnover enzymes catalyzing equilibrium reactions – FabD, FabZ, and FabA – which make for poor drug targets. In contrast, the condensation enzymes FabF and FabH, enoyl-ACP reductases, and the acetyl-CoA carboxylase enzyme complex are rate-determining reactions at key regulatory steps, making them desirable drug targets [30]. A number of natural products and synthetic inhibitors are known for these enzymes, validating them as antibacterial targets [31,32].
Acetyl-CoA Carboxylase
The acetyl-CoA carboxylase complex catalyzes the irreversible carboxylation of acetyl-CoA to make malonyl-CoA using the energy from hydrolyzing ATP to ADP [33]. This reaction is the first committed, regulated, and rate-limiting step in fatty acid synthesis, making it a good chemotherapeutic target. The acetyl-CoA carboxylase complex is composed of 4 protein subunits catalyzing two half reactions in bacteria. Biotin carboxylase (AccC) catalyzes the carboxylation of the biotin prosthetic group attached to biotin-carboxyl carrier protein (AccB) in the first half reaction. The carboxyltransferase complex (AccA and AccD) transfers the carboxyl group from the biotin to acetyl-CoA to form malonyl-CoA. The bacterial acetyl-CoA carboxylase complex is highly conserved [31,34] and structurally different from the mammalian acetyl-CoA carboxylase complex which is organized as a single polypeptide rather than separate protein subunits [35], making it a potential broad-spectrum antibiotic target.
The effective spectrum of acetyl-CoA carboxylase inhibition is still being investigated [25]. Several natural products such as moiramide B and andrimid block the growth of bacteria through on-target inhibition of fatty acid synthesis in standard laboratory culture media without fatty acids [36]. Plants also encode for a bacteria-like acetyl-CoA carboxylase complex, and the plant enzyme is targeted by commercial herbicides [37]. Extensive effort at designing ACC inhibitors for antibacterial therapy has been attempted [38,39], including designing dual-ligand inhibitors that simultaneously inhibit two subunits of the acetyl-CoA carboxylase complex [40]. Exogenous fatty acids can overcome Andrimid inhibition in both Staphylococcus aureus and Streptococcus pneumoniae in planktonic growth, and a Staphylococcus aureus strain encoding an inactive acetyl-CoA carboxylase can grow in laboratory culture media supplemented with exogenous fatty acids [25]. However, this same strain of Staphylococcus aureus cannot proliferate in a mouse sepsis model illustrating the importance of in vivo testing [41]. Acetyl-CoA carboxylase inhibition is expected to be effective against the model Gram-negative bacterium Escherichia coli because the essential lipopolysaccharide synthesis requires β-hydroxyacyl-ACP made from endogenous fatty acid synthesis [42,43]. Acetyl-CoA carboxylase is also essential for Pseudomonas aeruginosa [44]. Whether acetyl-CoA carboxylase is essential for Gram-negative bacteria with nonessential lipopolysaccharides, such as Neisseria [45,46], remains to be validated.
Condensation Enzymes
The condensation enzymes, FabH, FabF, and FabB, catalyze a Claisen condensation using malonyl-ACP as the nucleophile to elongate the acyl chain by two carbons at a time [30,47]. FabH initiates fatty acid synthesis by condensing a malonyl-ACP with acetyl-CoA to acetoacetyl-ACP [15]. Branched-chain acyl-CoA precursors such as 2-methylbutyryl-CoA are used in place of acetyl-CoA for the synthesis of branched chain fatty acids [48]. FabF initiates each round of 2 carbon elongation through the condensation of malonyl-ACP with acyl-ACP [15]. FabB has similar function as FabF, but is essential for the elongation of unsaturated fatty acids in bacteria expressing a FabA [49,50]. FabF and FabB have a Cys-His-His catalytic triad, and FabH has a Cys-His-Asn triad [51]. Condensing enzymes proceed via a Ping-Pong mechanism where the active site cysteine attacks the thioester of the acetyl-CoA to make the acyl-cysteine thioester intermediate [52-54]. After the release of the CoA, decarboxylation of the carboxyl group of malonyl-ACP makes an enolate intermediate. Nucleophilic attack from the enolate group on the acyl-cysteine thioester elongates the acyl group by two carbons.
The condensing enzymes have been an active area of antibacterial research. FabH was genetically essential in the GlaxoSmithKline antibacterial screening efforts and FabH inhibitors have demonstrated in vivo activity [9]. A variety of natural product and synthetic inhibitors also target the condensing enzymes [13,32]. Due to the similar enzymatic reactions catalyzed by the condensing enzymes, inhibitors against the condensing enzymes have the potential to be dual targeting. Several natural product inhibitors have been demonstrated to target both classes of condensing enzymes [55,55-57], including cerulenin which is a covalent inhibitor of FabB and FabF [58,59].
Reductases
There are two reduction reactions in each elongation cycle of fatty acid synthesis [15]. Enoyl-ACP reductase catalyzes the reduction of trans-2-enoyl-ACP into acyl-ACP. Because the dehydration step preceding enoyl-ACP reductase in the elongation cycle is an equilibrium reaction, enoyl-ACP reductase is one of the rate-determining steps in fatty acid synthesis [60,61]. There are four characterized isoforms of enoyl-ACP reductase. The FabI enoyl-ACP reductase isoform was first discovered in E. coli and a target of the biocide triclosan [26,62,63]. The FabL isoform is distantly related to FabI and found in addition to FabI in Bacillus species [27]. The FabK isoform is a flavoprotein with no structural relationship to FabI [28]. FabK is the primary enoyl-ACP reductase in Streptococcus species and a significant number of other firmicutes [64]. The FabV isoform is the least characterized isoform and is found predominately in γ-Proteobacteria clades such as Vibrio, Yersinia, and Pseudomonas species [29,65]. The FabV isoform may have originated from Pseudomonas fluorescens encoding for the synthesis of natural product inhibitors of FabI [66].
FabI is a validated antibiotic target [63,67], and FabI inhibitors are currently being evaluated in clinical trials [17-22]. Although FabI is not the most widely distributed enoyl-ACP reductase isoform, FabI is the essential enoyl-ACP reductase in a number of key pathogens including Staphylococcus, Neisseria, Enterococcus, Acinetobacter, and Enterobacter species [25,68,69]. The biocide triclosan targets the FabI in Staphylococcus aureus and E. coli [63,67] in addition to acting as a nonspecific biocide [28,70]. The anti-tuberculosis drug isoniazid also targets InhA in mycolic acid synthesis, which is the homologue of FabI in Mycobacterium tuberculosis [71]. Although concerns have been raised that exogenous fatty acids could bypass FabI inhibition [24], experiments in Staphylococcus and Neisseria have conclusively shown that exogenous fatty acids cannot bypass FabI inhibition in these organisms [25,68]. Because FabI is not broadly distributed in bacteria, most FabI antibiotic discovery has focused on Staphylococcus.
The β-ketoacyl-ACP reductase FabG catalyzes the reduction of β-ketoacyl-ACP into β-hydroxyacyl-ACP [15]. In contrast to enoyl-ACP reductase, there is only one highly conserved β-ketoacyl-ACP reductase isoform that has been characterized, suggesting that inhibitors targeting this enzyme would have broad-spectrum activity. However, FabG does not appear to have a rate-controlling role in fatty acid synthesis, making it less desirable as a target. While several natural product FabG inhibitors have been reported [72,73], there has been very little follow through research to validate the selectivity of these inhibitors or develop the compounds further. FabG remains to be validated as a suitable target for drug discovery.
Acyltransferases
Acyl-ACP of the appropriate length becomes substrate for the acyltransferases and the synthesis of phosphatidic acid, the precursor to all phospholipids in bacteria [48]. The structures of the enzymes involved in bacterial phospholipid synthesis are poorly defined because they are typically membrane bound proteins. Phosphatidic acid is made by two successive acylations of glycerol-3-phosphate using acyl-ACP. The PlsC enzyme involved in the second acylation step is conserved in both bacteria and mammals and make a poor antibiotic target. Three enzymes, PlsB, PlsE, and PlsY, are involved in the first acylation step. The PlsX/PlsY acyltransferase system is the most widely distributed in bacteria [74]. All characterized bacteria use the PlsX/PlsY acyltransferase system except for γ-Proteobacteria which uses PlsB and Chlamydia which uses PlsE [75]. PlsB and PlsE share the same HX4D catalytic motif and substrate as PlsC and may not be expected to allow for selective antibacterial targeting [76]. In contrast, PlsY belongs to another protein family and uses acylphosphate made by PlsX for the acylation reaction [77]. Analogues of acyl-phosphate show that the on-target inhibition of PlsY inhibits bacterial growth [78,79]. However, current acylphosphate analogues are too hydrophobic to be “drug like”, and the lack of PlsY structure hampers improved compound design [78,79]. Because PlsX catalyzes the reversible conversion between acyl-ACP and acyl-phosphate [80,81], acyl-phosphate analogues could be dual targeting inhibitors of PlsX and PlsY.
ANTIBIOTIC DISCOVERY IN BACTERIAL FATTY ACID METABOLISM
Resistance in Antibiotic Discovery
In addition to targeting an essential molecular target, resistance must also be considered in antibiotic discovery. The mechanism of resistance against an inhibitor determines the time frame that the inhibitor is therapeutically relevant. If high level resistance to the inhibitor is able to develop during the course of treatment, then the inhibitor stands a significant chance of failure during therapeutic treatment and will likely fail clinical trials. Resistance development to the inhibitor must be considered during target selection, inhibitor design, and clinical development. There are four mechanisms for bacterial resistance to antibiotics [82]. First, bacteria can modify the cell wall/membrane to decrease the permeability of the antibiotic to render the intracellular concentration of the antibiotic ineffective. Second, bacteria can increase the efflux of the antibiotic to again decrease the intracellular concentration of the antibiotic. Third, bacteria can modify the antibiotic to render it ineffective such as hydrolyzing the β-lactam ring or methylation of aminoglycosides. Finally, bacteria can modify the molecular target of the antibiotic to decrease the affinity of the antibiotic to the molecular target.
These four resistance mechanisms arise and spread by lateral gene transfer and spontaneous mutation. The mode that the resistance mechanism occurs affects the rate of resistance development [82]. Of the four resistance mechanisms, modification of cell wall, increase in efflux, and molecular target modification occur through spontaneous mutations. Spontaneous mutation arises from errors in DNA replication leading to changes in gene function or the expression level of the gene function. Missense mutations occur at approximately one in 109 bacterial cells, approximating the error rate in DNA replication [83]. Environmental stresses can further accelerate the mutation rate. This feature of bacterial physiology means that a strain encoding for a mutant gene product resistant to the antibiotic might already be present at low populations in nature, and could quickly develop if it is not already present. While resistance through modification of cell wall or increased efflux lead to low level of antibiotic resistance, single amino acid changes via a missense mutation in the drug binding pocket of the molecular target can increase resistance against the antibiotic by several hundred fold. This makes the modification of the molecular target resistance mechanism a key consideration during antibiotic discovery because this mechanism gives rise to fast-developing, high level resistance. One of the key reasons for the failure of target based antibiotic discovery (which targets a single molecular target) is due to high level resistance quickly developing via modification of the molecular target [84]. Therefore, overcoming resistance development, particularly missense mutations in the gene target, must be considered in the early phases of antibiotic discovery.
In contrast, chemical modification of antibiotics causes high level resistance by spreading through lateral gene transfer. Spontaneous mutations to an existing gene that give it the ability to modify a xenobiotic are possible in theory, but evolving such new gene functions requires long periods of selection in practice [85]. Therefore, resistance through chemical modification of antibiotics develops slowly at first, but spreads with frightening speed once a certain threshold has been reached. The development of this mode of resistance is largely unpredictable, and the appropriate focus is to slow the spread of this resistance mechanism through proper use of the antibiotic [82].
Multi-Targeting Monotherapeutic Antibiotics - FabH/FabF/FabB Inhibitors
The well-reasoned and provocative “multi-target” hypothesis posits that all broad-spectrum, monotherapeutic antibiotics in clinical use today target multiple gene targets, and stipulates that the multi-targeting feature of these antibiotics limit the fast development of high level resistance through modifying the molecular target [84]. Antibiotics targeting two or more gene targets require simultaneous resistance causing missense mutations in all the gene targets to cause high level resistance. The probability of developing a resistant missense mutation in a gene target is approximately one in 109 bacteria [83,86] meaning that a resistant bacteria can be found in an approximately 1 ml OD600 = 1 culture. However, the probability of developing two resistant missense mutations simultaneously is approximately one in 1018 bacteria, meaning a 1,000,000 liter OD600 = 1 culture would be required to find the resistance mutant. The multiplicative probability of developing resistant missense mutations means that targeting two or more essential targets simultaneously is an effective way of slowing the emergence of resistance through modifying the molecular target. A summary of the design principles of a broad-spectrum antibiotic is summarized in Table 1. Resistance against these antibiotics occurs via a combination of decreasing cell permeability against the antibiotic, efflux of the antibiotic, modification of the antibiotic, and stepwise accumulation of multiple target-based resistance mutations [84,87].
Table 1.
Summary of the design principles of penicillin (broad-spectrum, monotherapeutic antibiotic) versus AFN-1252 (pathogen-selective antibiotic).
| Penicillin | AFN-1252 | |
|---|---|---|
| Spectrum: | Broad-spectrum | Pathogen-selective |
| Targeting: | Multi-target (cell wall synthesis) | Single-target (FabI) |
| Affinity: | Micromolar affinity | Nanomolar affinity |
| Resistance: | Multi-targeting prevents target- based resistance |
Therapeutic dose still effective against resistant mutants |
| Screening: | Natural product phenotype screen | Target-based molecular screen |
| Development: | Modification of natural product | Structure-based design |
| Microbiome: | Disturbs microbiome | Minimize effect on microbiome |
A monotherapeutic antibiotic under this model must inhibit molecular target(s) encoded for by 2 or more genes in the bacterial genome. This means that the active site and function of these molecular targets must be sufficiently similar to be inhibited by a single pharmacophore. These molecular target(s) must also be highly conserved in a wide spectrum of bacteria if broad-spectrum activity is desired. Despite having nearly a dozen different classes of antibiotics, all broad-spectrum monotherapeutic antibiotics target 3 molecular target classes - penicillin binding proteins, ribosomes, and topoisomerases [84]. Each of these targets represents a family of essential and related protein functions encoded for by multiple genes. These three targets are also highly conserved in a wide range of bacteria. With the genomes of many bacterial pathogens sequenced and the protein functions of model organisms like E. coli largely characterized, an educated guess can be ventured on whether there are more molecular target(s) that are highly conserved, widely distributed, and encoded for 2 or more times in the bacterial genome. There appear to be few enzyme classes that fulfill all three criteria.
There are three dual targeting inhibitors of bacterial fatty acid metabolism. The natural product inhibitor platencin is a dual inhibitor of the condensing enzymes FabH and FabF in Staphylococcus aureus [55,88]. Platencin has relatively balanced activity against the two enzymes (IC50 of 4.6 μM vs FabF and 9.2 μM vs FabH). Unfortunately, platencin has poor pharmacokinetic properties and a continuous-infusion is required to effectively treat Staphylococcus aureus in the mouse model [88]. Little progress has been made in modifying platencin to have better pharmacokinetic properties due to the complex chemical nature of platencin that makes generating chemical analog libraries difficult [89].
The natural product inhibitor thiolactomycin is a dual inhibitor of FabF and FabB in bacteria encoding both enzymes such as E. coli [56,57,90-92]. Staphylococcus aureus only encodes for FabF, so thiolactomycin is only a single-target inhibitor of FabF in Staphylococcus aureus. Thiolactomycin is a structural mimic of malonyl-ACP and binds to the acyl-enzyme (Pong form) of the condensing enzyme [53,93]. Thiolactomycin has an antibacterial spectrum against both Gram-positive and Gram-negative bacteria, low cytotoxicity, effectiveness in a mouse model, and favorable pharmacokinetic properties [94,95]. However, despite considerable effort to structurally improve thiolactomycin [96-98], successful therapeutic products have not emerged from this lead. Optimizing thiolactomycin remains an active area of continuing research [99].
The natural product inhibitor cerulenin is a covalent dual inhibitor of FabF and FabB [58,59]. Like thiolactomycin, cerulenin is only single-targeting in bacteria with only FabF. The catalytic cysteine in the condensing enzymes attacks the epoxide group in cerulenin to form a covalent intermediate that inhibits the enzyme reaction [100]. The covalent natural of cerulenin inhibition means that selectivity is for the active site catalytic cysteine, and the inhibitor could tolerate some differences in the rest of the active site architecture (similar to β-lactam antibiotics). Unfortunately, cerulenin reacts with eukaryotic fatty acid synthase and sterol synthesis enzymes using similar enzymatic mechanisms as the condensing enzymes [101-103], so it lacks bacterial selectivity. The covalent mode of inhibition and the lack of bacterial selectivity suggest that designing a bacterial selective cerulenin derivative will be challenging and little progress has been made to improve its antibacterial properties.
Pathogen-selective Antibiotic Discovery - FabI
Three companies continue to be involved in developing FabI inhibitors for antibiotic therapy. Mutabilis [104], CrystalGenomics [105-107], and Affinium [17,20,21] are all focused on developing FabI inhibitors with Staphylococcus selective activity rather than broad-spectrum activity. These inhibitors are the first reported single-target, pathogen-selective inhibitors to enter into clinical evaluation. CG400549 from CrystalGenomics has successfully passed phase 1 trials and recently completely phase 2a trials (the results are currently under review). AFN-1252 from Affinium has successfully completed phase 2 trials and is undergoing further clinical development by Debiopharm [19]. The extensive research surrounding AFN-1252 provides an informative case study in the potential benefits and challenges of designing single-target pathogen-selective inhibitors, Fig. 3.
Fig. 3.
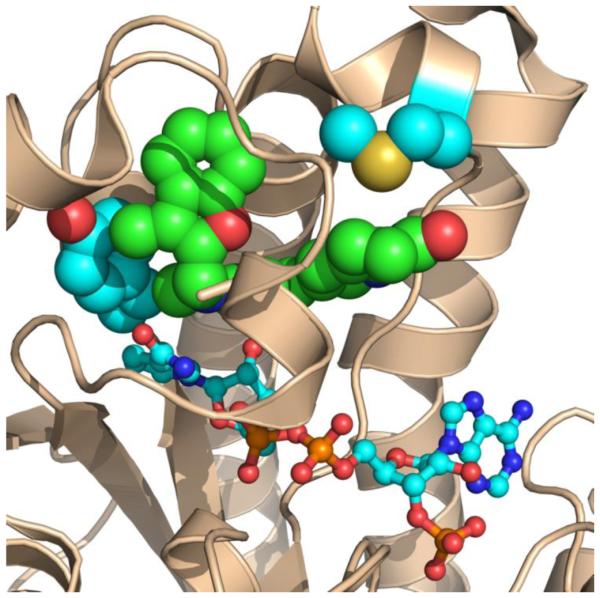
The advantage and disadvantage of side-chain interactions in single-target antibiotic design. In this example of a single-target antibiotic, AFN-1252 and NADPH bind to the active site of Staphylococcus aureus FabI (PDBID: 4FS3). The FabI protein structure is depicted as a wheat colored cartoon diagram. NADPH is depicted as sticks, and the Met-99 and Tyr-147 side-chains (light blue) contact the AFN-1252 (green). Tyr-147 interacts with the 3-methylbenzofuran portion of AFN-1252 and is involved in catalysis. Although the mutation of Tyr-147 residue to histidine imparts resistance, it also results in a severe catalytic defect and a slower growth rate. The variable Met-99 interacts with the oxotetrahydronaphthyridine portion of AFN-1252. Mutation of the Met-99 to threonine caused a 128 fold increase in resistance with minimal effects on the strain fitness, meaning that this missense mutation could lead to rapid development of clinical resistance. The Met-99 is only found in Staphylococcus FabI and is responsible for the high-affinity, pathogen-selective property of AFN-1252.
Pathogen-selective inhibitors could potentially overcome target-based resistance by being so potent that therapeutic doses of antibiotics are still curative against the missense resistant mutant. Rather than compromising affinity to target a broad spectrum of bacteria, pathogen-selective inhibitors can be designed to have extremely high affinity and activity against the particular pathogen. AFN-1252 has a 4 nM apparent Ki against the Staphylococcus FabI and 4 ng/ml minimal inhibitory concentration against Staphylococcus aureus in planktonic growth [18,20,108]. The only mutations causing high level resistance to AFN-1252 are missense mutations to two residues (M99T and Y147H) in FabI. The M99T mutation causes a 64 fold increase in AFN-1252 resistance (MIC of 250 ng/ml) with minimal change in growth fitness. The Y147H mutation causes a 128 fold increase (MIC of 500 ng/ml) in AFN-1252 resistance along with severe growth defects [25,108]. Mutants with higher levels of resistance were not found by selecting the single missense mutants, while a mutant enzyme with both the M99T and Y147H mutations is catalytically inactive [108]. This result suggests that there is a ceiling in short term resistance development (at least in Staphylococcus aureus), and that a sufficiently potent inhibitor can overcome target-based resistance. The key question for AFN-1252 is whether the drug can be dosed at the levels required to eradicate the M99T and Y147H mutants.
Pathogen-selective inhibitors also have the potential to minimize the collateral damage to the microbiome. Recent advances in microbiome research show that broad-spectrum antibiotics damage the beneficial microbiome of the human body, leading to a variety of adverse short and long term consequences [109,110]. In particular, broad-spectrum antibiotics are known to cause antibiotic associated secondary infections. These diseases arise from broad-spectrum antibiotic treatment wiping out the beneficial host microbiome and allowing drug resistant pathogens that can’t normally compete with the microflora to proliferate [111,112]. The effect of AFN-1252 on the mouse gut microbiome was examined as a case study [64]. AFN-1252 caused no significant change in gut bacterial abundance and minimal disturbance in bacterial composition in contrast to broad-spectrum antibiotics that reduced the gut bacterial abundance several thousand fold along with severe disturbance in bacterial composition. Similar results are expected in humans, but this conclusion requires verification. One of the major demands for new antibiotics comes from patients with weakened immune systems [112,113]. A pathogen-selective approach would benefit these patients by minimizing the disturbance to the beneficial microbiome.
A summary of the design principles of a pathogen-selective antibiotic are summarized in Table 1. This pathogen-selective paradigm faces two major hurdles. First, how resistance will develop against an inhibitor must be taken into account during the course of development. The inhibitor must be designed to be high-affinity while minimizing interactions with the side chains of non-conserved residues that can undergo mutations that decrease the affinity of the inhibitor [114]. Resistance against the inhibitor must be evaluated early in the discovery phase, and minimizing the impact of resistance must be an additional criterion that guides the structure optimization process. The inhibitor must also be tested against the resistant mutant in animal models to verify that the inhibitor is still effective against the missense resistant mutants. Second, the specific nature of a pathogen-selective inhibitor requires accurate pathogen diagnostic technology. Because the antibiotic must be matched with the correct pathogen, rapid and accurate identification of the pathogen responsible for the disease must precede antibiotic prescription. One major challenge in pathogen detection is determining which bacterial species is responsible for diseases because many diseases causing pathogens are also permanent residents of the human microflora [115]. Fortunately, rapid pathogen detection is an area of major research and advances [116].
Combinations of Single-target Antibiotics
The success of the β-lactam antibiotics has made combinations of antibiotics a less favorable choice due to the difficulty in dosing multiple compounds. However, combinational antibiotic therapy has long been the standard for treating Mycobacterium tuberculosis. Because Mycobacterium tuberculosis encodes for and produces extended spectrum β-lactamases, β-lactam drugs are largely ineffective [117]. Mycobacterium tuberculosis also encodes only a single rRNA gene so ribosome inhibitors must be used in combination therapy with other antibiotics to prevent fast-developing, target-based resistance [118]. Fluoroquinolones are effective against tuberculosis, but has been largely held in reserve for treatment of drug resistant tuberculosis. Instead, the front line treatment against tuberculosis is a combination of isoniazid, rifampin, ethambutol, and pyrazinamide. Each of these drugs target a single gene target and experiences high rates of resistance development when used individually [119], but the drug cocktail is effective at preventing resistant mutants. The large scale use of this effective drug cocktail against M. tuberculosis suggests that the combination of single antibiotics can be an effective therapeutic strategy to fight target-based resistance. Another example of this strategy is the sulfamethoxazole-trimethoprim combination [120]. Sulfamethoxazole is a sulfanilamide analogue that inhibits dihydropteroate synthetase in tetrahydrofolic acid synthesis. Trimethoprim inhibits dihydrofolate reductase, another enzyme in tetrahydrofolic acid synthesis. Target-based resistance arises quickly for sulfamethoxazole or trimethoprim treatment alone, which limits the clinical efficiency of these drugs. However, combining these two drugs for treatment decreases the occurrence of spontaneous target-based mutations.
Given the difficulties of finding new monotherapeutic, broad-spectrum antibiotics, combination therapy is gaining more interest. Combining β-lactam and β-lactamase inhibitors is a significant area of recent antibiotic development [121]. Given the rather limited target options in developing broad-spectrum, monotherapeutic antibiotics, combination therapy of single-target inhibitors with orthogonal gene targets should be considered as an alternative. The rate-determining steps in bacterial fatty acid synthesis are validated targets for these single-target inhibitors. Combining two inhibitors targeting two different gene targets provides the benefit of minimizing resistance via single missense mutations. Furthermore, if resistance is the key reason why single-target, broad-spectrum antibiotics failed to reach the clinic, then combining these failed single-target antibiotics should provide an effective therapy. This hypothesis could be tested directly using the low toxicity, FASII inhibitors that have already been discovered [13,32]. The challenge to multi-drug therapy is avoiding adverse drug interactions and matching the dosage of the drug components so both drugs are at active concentrations over the course of treatment to avoid stepwise resistance. Stepwise resistance against one component of the cocktail at a time when the other components are low due to improper antibiotic use is a major concern for the development of resistance in Mycobacterium tuberculosis [122].
A variety of single-target FASII inhibitors have been described in literature [13,32]. These include both natural product inhibitors and synthetic inhibitors. The two best characterized molecular targets are FabI and FabH, which have been subject to numerous discovery campaigns by a variety of investigators. One major recurring result from these campaigns is the difficulty in getting both spectrum and potency [9,21,99,104-107,123]. For example, a recent FabH discovery campaign reported a series of 22 thiolactomycin analogues and tested these analogues against Mycobacterium tuberculosis, Francisella tularensis, Yersinia pestis, and Staphylococcus aureus [99]. While several analogues were able to achieve low μg/ml minimal inhibitory concentration against one of the bacterial species, only 1 analogue was able to achieve low μg/ml minimal inhibitory concentration against two bacterial species and none against 3 or more species. These results show that FASII enzymes are sufficiently structurally divergent that even broad-spectrum single-targeting is exceptionally difficult. Outside of cell wall synthesis, ribosome, and DNA synthesis, there simply do not appear to be more highly conserved molecular targets in bacteria. A cocktail of narrow spectrum antibiotics should be considered in future antibiotic applications.
SUMMARY
The Centers for Disease Control released the “Antibiotic resistance in the United States” report in 2013 [124]. The report identified Clostridium difficile, Neisseria gonorrhoeae, Enterobacter, Acinetobacter, Campylobacter, Psuedomonas aeruginosa, Salmonella, Shigella, Staphylococcus, Enterococcus, and Streptococcus as major pathogen threats in need of new antibiotic treatments. Bacterial fatty acid metabolism has been validated as essential in most of these pathogens. Of these pathogens, only Streptococcus has the ability to bypass FASII inhibition through using exogenous fatty acids [23-25]. FASII has been demonstrated to be essential in Staphylococcus, Enterocococcus, Neisseria, and E. coli [23,25,68,69], so FabI, FabH, FabF, and acetyl-CoA carboxylase inhibitors are predicted to be effective against these pathogens. Bioinformatic predications suggest that the organization of fatty acid, phospholipid, and lipopolysaccharide synthesis in Enterobacter, Acinetobacter, Campylobacter, Salmonella and Shigella are similar to E. coli, so inhibition of the enzymes in FASII would be expected to work in these pathogens as well. Additionally, dual FabF and FabB targeting would be possible for the Gram-negative pathogens in the above list because these Gram-negative bacteria synthesize unsaturated fatty acids using a FabB [59,125]. Psuedomonas aeruginosa encodes for multiple enoyl-ACP reductase and FabH isoforms [66,126], but is predicted via bioinformatics or validated experimentally to encode only a single, essential copy of acetyl-CoA carboxylase [44], FabF, and FabB. Finally, Clostridium difficile is an opportunistic pathogen that proliferates in the gut when the normal microflora is eliminated by broad-spectrum antibiotic treatment [112,127,128]. The narrow spectrum of a FabI inhibitor designed using a pathogen-selective paradigm preserves the gut microflora [64] and prevents Clostridium difficile colitis from occurring in the first place. The various application of FASII inhibition holds great potential for the future development of antibiotics against the most urgent pathogens.
Highlights.
Bacterial fatty acid synthesis is a narrow spectrum antibiotic target.
Antibiotics must overcome fast-developing resistance to be therapeutically successful.
A pathogen-selective discovery paradigm minimizes collateral damage to the microbiome.
Bacterial fatty acid synthesis holds promise for future antibiotic development.
ACKNOWLEDGEMENTS
This work was supported in part by National Institutes of Health Grants GM034496, Cancer Center Support Grant CA21765 and the American Lebanese Syrian Associated Charities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
No conflict of interest.
References
- [1].Strebhardt K, Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat. Rev. Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- [2].Lesch JE. Prontosil, The first miracle drugs: how the sulfa drugs transformed medicine. Oxford University Press; 2006. p. 51. [Google Scholar]
- [3].Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature (London) 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- [4].Silver LL. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Silver LL, Bostian KA. Discovery and development of new antibiotics: the problem of antibiotic resistance. Antimicrob. Agents Chemother. 1993;37:377–383. doi: 10.1128/aac.37.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kelly JA, Moews PC, Knox, Frere JM, Ghuysen JM. Penicillin target enzyme and the antibiotic binding site. Science. 1982;218:479–481. doi: 10.1126/science.7123246. [DOI] [PubMed] [Google Scholar]
- [7].Kelly JA, Dideberg O, Charlier P, Wery JP, Libert M, Moews PC, Knox, Duez C, Fraipont C, Joris B. On the origin of bacterial resistance to penicillin: comparison of a beta-lactamase and a penicillin target. Science. 1986;231:1429–1431. doi: 10.1126/science.3082007. a. et. [DOI] [PubMed] [Google Scholar]
- [8].Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. e. al. [DOI] [PubMed] [Google Scholar]
- [9].Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- [10].Hooper DC, Wolfson JS. The fluoroquinolones: pharmacology, clinical uses, and toxicities in humans. Antimicrob. Agents Chemother. 1985;28:716–721. doi: 10.1128/aac.28.5.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cohen SS. A strategy for the chemotherapy of infectious disease. Science. 1977;197:431–432. doi: 10.1126/science.195340. [DOI] [PubMed] [Google Scholar]
- [12].Fernandes P. Antibacterial discovery and development-the failure of success? Nat. Biotechnol. 2006;24:1497–1503. doi: 10.1038/nbt1206-1497. [DOI] [PubMed] [Google Scholar]
- [13].Parsons JB, Rock CO. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery? Curr. Opin. Microbiol. 2011;14:544–549. doi: 10.1016/j.mib.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Payne DJ, Warren PV, Holmes DJ, Ji Y, Lonsdale JT. Bacterial fatty-acid biosynthesis: a genomics-driven target for antibacterial drug discovery. Drug Discov. Today. 2001;6:537–544. doi: 10.1016/s1359-6446(01)01774-3. [DOI] [PubMed] [Google Scholar]
- [15].Rock CO, Jackowski S. Forty years of fatty acid biosynthesis. Biochem. Biophys. Res. Commun. 2002;292:1155–1166. doi: 10.1006/bbrc.2001.2022. [DOI] [PubMed] [Google Scholar]
- [16].Asturias FJ, Chadick JZ, Cheung IK, Stark H, Witkowski A, Joshi AK, Smith S. Structure and molecular organization of mammalian fatty acid synthase. Nat. Struct. Mol. Biol. 2005 doi: 10.1038/nsmb899. [DOI] [PubMed] [Google Scholar]
- [17].Banevicius MA, Kaplan N, Hafkin B, Nicolau DP. Pharmacokinetics, pharmacodynamics and efficacy of novel FabI inhibitor AFN-1252 against MSSA and MRSA in the murine thigh infection model. J. Chemother. 2013;25:26–31. doi: 10.1179/1973947812Y.0000000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, Dorsey M, Hafkin B, Ramnauth J, Romanov V, Schmid MB, Thalakada R, Yethon J, Pauls HW. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob. Agents Chemother. 2012;56:5865–5874. doi: 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kaplan N, Garner C, Hafkin B. AFN-1252 in vitro absorption studies and pharmacokinetics following microdosing in healthy subjects. Eur. J. Pharm. Sci. 2013;50:440–446. doi: 10.1016/j.ejps.2013.08.019. [DOI] [PubMed] [Google Scholar]
- [20].Kaplan N, Awrey D, Bardouniotis E, Berman J, Yethon J, Pauls HW, Hafkin B. In vitro activity (MICs and rate of kill) of AFN-1252, a novel FabI inhibitor, in the presence of serum and in combination with other antibiotics. J. Chemother. 2013;25:18–25. doi: 10.1179/1973947812Y.0000000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Karlowsky JA, Kaplan N, Hafkin B, Hoban DJ, Zhanel GG. AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother. 2009;53:3544–3548. doi: 10.1128/AAC.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Parsons JB, Kukula M, Jackson P, Pulse M, Simecka JW, Valtierra D, Weiss WJ, Kaplan N, Rock CO. Perturbation of Staphylococcus aureus gene expression by the enoyl-acyl carrier protein reductase inhibitor AFN-1252. Antimicrob. Agents Chemother. 2013;57:2182–2190. doi: 10.1128/AAC.02307-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yao J, Rock CO. How bacterial pathogens eat host lipids: Implications for the development of fatty acid synthesis therapeutics. J. Biol. Chem. 2015;290:5940–5946. doi: 10.1074/jbc.R114.636241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature (London) 2009;458:83–86. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- [25].Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2011;108:15378–15383. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, Aschauer H, Kollenz G, Högenauer G, Turnowsky F. Protein envM is the NADH-dependent enoyl-ACP reductase fabI of Escherichia coli. J. Biol. Chem. 1994;269:5493–5496. [PubMed] [Google Scholar]
- [27].Heath RJ, Su N, Murphy CK, Rock CO. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 2000;275:40128–40133. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]
- [28].Marrakchi H, DeWolf WE, Jr, Quinn C, West J, Polizzi BJ, So CY, Holmes DJ, Reed SL, Heath RJ, Payne DJ, Rock CO, Wallis NG. Characterization of Streptococcus pneumoniae enoyl-[acyl carrier protein] reductase (FabK) Biochem. J. 2003;370:1055–1062. doi: 10.1042/BJ20021699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Massengo-Tiasse RP, Cronan JE. Vibrio cholerae fabV defines a new class of enoyl acyl-carrier-protein reductase. J. Biol. Chem. 2008;283:1308–1316. doi: 10.1074/jbc.M708171200. [DOI] [PubMed] [Google Scholar]
- [30].Zhang Y-M, White SW, Rock CO. Inhibiting bacterial fatty acid synthesis. J. Biol. Chem. 2006;281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- [31].Heath RJ, White SW, Rock CO. Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics. Appl. Microbiol. Biotechnol. 2001;58:695–703. doi: 10.1007/s00253-001-0918-z. [DOI] [PubMed] [Google Scholar]
- [32].Wang Y, Ma S. Recent advances in inhibitors of bacterial fatty acid synthesis type II (FASII) system enzymes as potential antibacterial agents. ChemMedChem. 2013;8:1589–1608. doi: 10.1002/cmdc.201300209. [DOI] [PubMed] [Google Scholar]
- [33].Cronan JE, Jr., Waldrop GL. Multi-subunit acetyl-CoA carboxylases. Prog. Lipid Res. 2002;41:407–435. doi: 10.1016/s0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- [34].Rock CO, Cronan JE., Jr. Escherichia coli as a model for the regulation of dissociable (type II) fatty acid biosynthesis. Biochim. Biophys. Acta. 1996;1302:1–16. doi: 10.1016/0005-2760(96)00056-2. [DOI] [PubMed] [Google Scholar]
- [35].Abu-Elheiga L, Jayakumar A, Baldini A, Chirala SS, Wakil SJ. Human acetyl-CoA carboxylase: characterization, molecular cloning, and evidence for two isoforms. Proc. Nalt. Acad. Sci. U. S. A. 1995;92:4011–4015. doi: 10.1073/pnas.92.9.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jin M, Fischbach MA, Clardy J. A biosynthetic gene cluster for the acetyl-CoA carboxylase inhibitor andrimid. J. Am. Chem. Soc. 2006;128:10660–10661. doi: 10.1021/ja063194c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nikolskaya T, Zagnitko O, Tevzadze G, Haselkorn R, Gornicki P. Herbicide sensitivity determinant of wheat plastid acetyl-CoA carboxylase is located in a 400-amino acid fragment of the carboxyltransferase domain. Proc. Natl. Acad. Sci. U. S. A. 1999;96:14647–14651. doi: 10.1073/pnas.96.25.14647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Freiberg C, Pohlmann J, Nell PG, Endermann R, Schuhmacher J, Newton B, Otteneder M, Lampe T, Häbich D, Ziegelbauer K. Novel bacterial acetyl coenzyme A carboxylase inhibitors with antibiotic efficacy in vivo. Antimicrob. Agents Chemother. 2006;50:2707–2712. doi: 10.1128/AAC.00012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pohlmann J, Lampe T, Shimada M, Nell PG, Pernerstorfer J, Svenstrup N, Brunner NA, Schiffer G, Freiberg C. Pyrrolidinedione derivatives as antibacterial agents with a novel mode of action. Bioorg. Med. Chem. Lett. 2005;15:1189–1192. doi: 10.1016/j.bmcl.2004.12.002. [DOI] [PubMed] [Google Scholar]
- [40].Silvers MA, Robertson GT, Taylor CM, Waldrop GL. Design, synthesis, and antibacterial properties of dual-ligand inhibitors of acetyl-CoA carboxylase. J. Med. Chem. 2014;57:8947–8959. doi: 10.1021/jm501082n. [DOI] [PubMed] [Google Scholar]
- [41].Parsons JB, Frank MW, Rosch JW, Rock CO. Staphylococcus aureus fatty acid auxotrophs do not proliferate in mice. Antimicrob. Agents Chemother. 2013;57:5729–5732. doi: 10.1128/AAC.01038-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Raetz CRH. Biochemistry of endotoxins. Annu. Rev. Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- [43].Zhang G, Meredith TC, Kahne D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr. Opin. Microbiol. 2013;16:779–785. doi: 10.1016/j.mib.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc. Natl. Acad. Sci. U. S. A. 2015;112:4110–4115. doi: 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bos MP, Tommassen J. Viability of a capsule- and lipopolysaccharide-deficient mutant of Neisseria meningitidis. Infect. Immun. 2005;73:6194–6197. doi: 10.1128/IAI.73.9.6194-6197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Piet JR, Zariri A, Fransen F, Schipper K, van der Ley P, van de Beek D, van der Ende A. Meningitis caused by a lipopolysaccharide deficient Neisseria meningitidis. J. Infect. 2014;69:352–357. doi: 10.1016/j.jinf.2014.06.005. [DOI] [PubMed] [Google Scholar]
- [47].Campbell JW, Cronan JE., Jr. Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu. Rev. Microbiol. 2001;55:305–332. doi: 10.1146/annurev.micro.55.1.305. [DOI] [PubMed] [Google Scholar]
- [48].Yao J, Rock CO. Phosphatidic acid synthesis in bacteria. Biochim. Biophys. Acta. 2013;1831:495–502. doi: 10.1016/j.bbalip.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Feng Y, Cronan JE. Escherichia coli unsaturated fatty acid synthesis: Complex transcription of the fabA gene and in vivo identification of the essential reaction catalyzed by FabB. J. Biol. Chem. 2009;284:29526–29535. doi: 10.1074/jbc.M109.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hoang TT, Schweizer HP. Fatty acid biosynthesis in Pseudomonas aeruginosa: cloning and characterization of the fabAB operon encoding β-hydroxyacyl-acyl carrier protein dehydratase (FabA) and β-ketoacyl-acyl carrier protein synthase I (FabB) J. Bacteriol. 1997;179:5326–5332. doi: 10.1128/jb.179.17.5326-5332.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Borgaro JG, Chang A, Machutta CA, Zhang X, Tonge PJ. Substrate recognition by β-ketoacyl-ACP synthases. Biochemistry. 2011;50:10678–10686. doi: 10.1021/bi201199x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tsay J-T, Oh W, Larson TJ, Jackowski S, Rock CO. Isolation and characterization of the β-ketoacyl-acyl carrier protein synthase III gene (fabH) from Escherichia coli K-12. J. Biol. Chem. 1992;267:6807–6814. [PubMed] [Google Scholar]
- [53].Heath RJ, Rock CO. The Claisen condensation in biology. Nat. Prod. Reports. 2002;19:581–596. doi: 10.1039/b110221b. [DOI] [PubMed] [Google Scholar]
- [54].Davies C, Heath RJ, White SW, Rock CO. The 1.8 Å cystal structure and active site architecture of β-ketoacyl-[acyl carrier protein] synthase III (FabH) from Escherichia coli. Structure. 2000;8:185–195. doi: 10.1016/s0969-2126(00)00094-0. [DOI] [PubMed] [Google Scholar]
- [55].Jayasuriya H, Herath KB, Zhang C, Zink DL, Basilio A, Genilloud O, Diez MT, Vicente F, Gonzalez I, Salazar O, Pelaez F, Cummings R, Ha S, Wang J, Singh SB. Isolation and structure of platencin: a FabH and FabF dual inhibitor with potent broad-spectrum antibiotic activity. Angew. Chem. Int. Ed Engl. 2007;46:4684–4688. doi: 10.1002/anie.200701058. [DOI] [PubMed] [Google Scholar]
- [56].Hayashi T, Yamamoto O, Sasaki H, Kawaguchi A, Okazaki H. Mechanism of action of the antibiotic thiolactomycin inhibition of fatty acid synthesis of Escherichia coli. Biochem. Biophys. Res. Commun. 1983;115:1108–1113. doi: 10.1016/s0006-291x(83)80050-3. [DOI] [PubMed] [Google Scholar]
- [57].Hayashi T, Yamamoto O, Sasaki H, Okazaki H. Inhibition of fatty acid synthesis by the antibiotic thiolactomycin. J. Antibiot. (Tokyo) 1984;37:1456–1461. doi: 10.7164/antibiotics.37.1456. [DOI] [PubMed] [Google Scholar]
- [58].D'Agnolo G, Rosenfeld IS, Awaya J, Omura S, Vagelos PR. Inhibition of fatty acid biosynthesis by the antibiotic cerulenin. Specific inactivation of β-ketoacyl-acyl carrier protein synthetase. Biochim. Biophys. Acta. 1973;326:155–166. doi: 10.1016/0005-2760(73)90241-5. [DOI] [PubMed] [Google Scholar]
- [59].Kauppinen S, Siggaard-Anderson M, van Wettstein-Knowles P. β-Ketoacyl-ACP synthase I of Escherichia coli: Nucleotide sequence of the fabB gene and identification of the cerulenin binding residue. Carlsberg. Res. Commun. 1988;53:357–370. doi: 10.1007/BF02983311. [DOI] [PubMed] [Google Scholar]
- [60].Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J. Biol. Chem. 1995;270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- [61].Heath RJ, Li J, Roland GE, Rock CO. Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J. Biol. Chem. 2000;275:4654–4659. doi: 10.1074/jbc.275.7.4654. [DOI] [PubMed] [Google Scholar]
- [62].Heath RJ, Rubin JR, Holland DR, Zhang E, Snow ME, Rock CO. Mechanism of triclosan inhibition of bacterial fatty acid synthesis. J. Biol. Chem. 1999;274:11110–11114. doi: 10.1074/jbc.274.16.11110. [DOI] [PubMed] [Google Scholar]
- [63].Levy CW, Roujeinikova A, Sedelnikova S, Baker PJ, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. Molecular basis of triclosan activity. Nature (London) 1999;398:383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- [64].Yao J, Carter RA, Vuagniaux G, Barbier M, Rosch JW, Rock CO. A pathogen-selective antibiotic minimizes disturbance to the microbiome. Antimicrob. Agents Chemother. 2016;60:4264–4273. doi: 10.1128/AAC.00535-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hirschbeck MW, Kuper J, Lu H, Liu N, Neckles C, Shah S, Wagner S, Sotriffer CA, Tonge PJ, Kisker C. Structure of the Yersinia pestis FabV enoyl-ACP reductase and its interaction with two 2-pyridone inhibitors. Structure. 2012;20:89–100. doi: 10.1016/j.str.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mattheus W, Masschelein J, Gao LJ, Herdewijn P, Landuyt B, Volckaert G, Lavigne R. The Kalimantacin/Batumin biosynthesis operon encodes a self-resistance isoform of the FabI bacterial target. Chem. Biol. 2010;17:1067–1071. doi: 10.1016/j.chembiol.2010.07.015. [DOI] [PubMed] [Google Scholar]
- [67].Heath RJ, Yu Y-T, Shapiro MA, Olson E, Rock CO. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J. Biol. Chem. 1998;273:30316–30321. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- [68].Yao J, Bruhn DF, Frank MW, Lee RE, Rock CO. Activation of exogenous fatty acids to acyl-acyl carrier protein cannot bypass FabI inhibition in Neisseria. J. Biol. Chem. 2016;291:171–181. doi: 10.1074/jbc.M115.699462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bi H, Zhu L, Wang H, Cronan JE. Inefficient translation renders the Enterococcus faecalis fabK enoyl-acyl carrier protein reductase phenotypically cryptic. J. Bacteriol. 2014;196:170–179. doi: 10.1128/JB.01148-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Heath RJ, Rock CO. A triclosan-resistant bacterial enzyme. Nature (London) 2000;406:145–146. doi: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- [71].Vilcheze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, Jacobs WR. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 2006;12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- [72].Sohn MJ, Zheng CJ, Kim WG. Macrolactin S, a new antibacterial agent with FabG-inhibitory activity from Bacillus sp. AT28. J. Antibiot. (Tokyo) 2008;61:687–691. doi: 10.1038/ja.2008.98. [DOI] [PubMed] [Google Scholar]
- [73].Zhang Y-M, Rock CO. Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty acid synthase. J. Biol. Chem. 2004;279:30994–31001. doi: 10.1074/jbc.M403697200. [DOI] [PubMed] [Google Scholar]
- [74].Lu Y-J, Zhang Y-M, Grimes KD, Qi J, Lee RE, Rock CO. Acyl-phosphates initiate membrane phospholipid synthesis in gram-positive pathogens. Molec. Cell. 2006;23:765–772. doi: 10.1016/j.molcel.2006.06.030. [DOI] [PubMed] [Google Scholar]
- [75].Yao J, Cherian PT, Frank MW, Rock CO. Chlamydia trachomatis relies on autonomous phospholipid synthesis for membrane biogenesis. J. Biol. Chem. 2015;290:18874–18888. doi: 10.1074/jbc.M115.657148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Turnbull AP, Rafferty JB, Sedelnikova SE, Slabas AR, Schierer TP, Kroon JT, Simon JW, Fawcett T, Nishida I, Murata N, Rice DW. Analysis of the structure, substrate specificity, and mechanism of squash glycerol-3-phosphate (1)-acyltransferase. Structure. 2001;9:347–353. doi: 10.1016/s0969-2126(01)00595-0. [DOI] [PubMed] [Google Scholar]
- [77].Lu Y-J, Zhang F, Grimes KD, Lee RE, Rock CO. Topology and active site of PlsY: the bacterial acylphosphate:glycerol-3-phosphate acyltransferase. J. Biol. Chem. 2007;282:11339–11346. doi: 10.1074/jbc.M700374200. [DOI] [PubMed] [Google Scholar]
- [78].Cherian P, Yao J, Leonardi R, Luna VA, Rock CO, Lee RE. Acyl-sulfamates target the essential glycerol-phosphate acyltransferase (PlsY) in Gram-positive bacteria. Bioorg. Med. Chem. 2012;20:4985–4994. doi: 10.1016/j.bmc.2012.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Grimes KD, Lu Y-J, Zhang Y-M, Luna VA, Hurdle JG, Carson EI, Qi J, Kudrimoti S, Rock CO, Lee RE. Novel acylphosphate mimics target PlsY, an essential acyltransferase in gram-positive bacteria. Chem. Med. Chem. 2008;3:1936–1945. doi: 10.1002/cmdc.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Parsons JB, Frank MW, Jackson P, Subramanian C, Rock CO. Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol. Microbiol. 2014;92:234–245. doi: 10.1111/mmi.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Parsons JB, Broussard TC, Bose JL, Rosch JW, Jackson P, Subramanian C, Rock CO. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 2014;111:10532–10537. doi: 10.1073/pnas.1408797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. Molecular mechanisms of antibiotic resistance. Nat. Rev. Micro. 2015;13:42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- [83].Meyerovich M, Mamou G, Ben-Yehuda S. Visualizing high error levels during gene expression in living bacterial cells. Proc. Natl. Acad. Sci. U. S. A. 2010;107:11543–11548. doi: 10.1073/pnas.0912989107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Silver LL. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 2007;6:41–55. doi: 10.1038/nrd2202. [DOI] [PubMed] [Google Scholar]
- [85].Blount ZD, Borland CZ, Lenski RE. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7899–7906. doi: 10.1073/pnas.0803151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Martinez JL, Baquero F. Mutation frequencies and antibiotic resistance. Antimicrob. Agents Chemother. 2000;44:1771–1777. doi: 10.1128/aac.44.7.1771-1777.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wright GD. Mechanisms of resistance to antibiotics. Curr. Opin. Chem. Biol. 2003;7:563–569. doi: 10.1016/j.cbpa.2003.08.004. [DOI] [PubMed] [Google Scholar]
- [88].Wang J, Kodali S, Lee SH, Galgoci A, Painter R, Dorso K, Racine F, Motyl M, Hernandez L, Tinney E, Colletti SL, Herath K, Cummings R, Salazar O, Gonzalez I, Basilio A, Vicente F, Genilloud O, Pelaez F, Jayasuriya H, Young K, Cully DF, Singh SB. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7612–7616. doi: 10.1073/pnas.0700746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Nicolaou KC, Tria GS, Edmonds DJ, Kar M. Total syntheses of (+/−)-platencin and (−)-platencin. J Am. Chem Soc. 2009;131:15909–15917. doi: 10.1021/ja906801g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Jackowski S, Murphy CM, Cronan JE, Jr., Rock CO. Acetoacetyl-acyl carrier protein synthase: a target for the antibiotic thiolactomycin. J. Biol. Chem. 1989;264:7624–7629. [PubMed] [Google Scholar]
- [91].Jackowski S, Zhang Y-M, Price AC, White SW, Rock CO. A missense mutation in the fabB (β-ketoacyl-acyl carrier protein synthase I) gene confers thiolactomycin resistance to Escherichia coli. Antimicrob. Agents Chemother. 2002;46:1246–1252. doi: 10.1128/AAC.46.5.1246-1252.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Brown MS, Akopiants K, Resceck DM, McArthur HA, McCormick E, Reynolds KA. Biosynthetic origins of the natural product, thiolactomycin: A unique and selective inhibitor of type II dissociated fatty acid synthases. J. Am. Chem. Soc. 2003;125:10166–10167. doi: 10.1021/ja034540i. [DOI] [PubMed] [Google Scholar]
- [93].Price AC, Choi KH, Heath RJ, Li Z, Rock CO, White SW. Inhibition of β-ketoacyl-[acyl carrier protein] synthases by thiolactomycin and cerulenin: structure and mechanism. J. Biol. Chem. 2001;276:6551–6559. doi: 10.1074/jbc.M007101200. [DOI] [PubMed] [Google Scholar]
- [94].Miyakawa S, Suzuki K, Noto T, Harada Y, Okazaki H. Thiolactomycin a new antibiotic. IV. Biological properties and chemotherapeutic activity in mice. J. Antibiot. (Tokyo) 1982;35:411–419. doi: 10.7164/antibiotics.35.411. [DOI] [PubMed] [Google Scholar]
- [95].Noto T, Miyakawa S, Oishi H, Endo H, Okazaki H. Thiolactomycin, a new antibiotic. III. In vitro antibacterial activity. J. Antibiot. (Tokyo) 1982;35:401–410. doi: 10.7164/antibiotics.35.401. [DOI] [PubMed] [Google Scholar]
- [96].Sakya SM, Suarez-Contreras M, Dirlam JP, O'Connell TN, Hayashi SF, Santoro SL, Kamicker BJ, George DM, Ziegler CB. Synthesis and structure-activity relationships of thiotetronic acid analogues of thiolactomycin. Bioorg. Med. Chem. Lett. 2001;11:2751–2754. doi: 10.1016/s0960-894x(01)00567-4. [DOI] [PubMed] [Google Scholar]
- [97].Senior SJ, Illarionov PA, Gurcha SS, Campbell IB, Schaeffer ML, Minnikin DE, Besra GS. Biphenyl-based analogues of thiolactomycin, active against Mycobacterium tuberculosis mtFabH fatty acid condensing enzyme. Bioorg. Med. Chem. Lett. 2003;13:3685–3688. doi: 10.1016/j.bmcl.2003.08.015. [DOI] [PubMed] [Google Scholar]
- [98].Senior SJ, Illarionov PA, Gurcha SS, Campbell IB, Schaeffer ML, Minnikin DE, Besra GS. Acetylene-based analogues of thiolactomycin, active against Mycobacterium tuberculosis mtFabH fatty acid condensing enzyme. Bioorg. Med. Chem. Lett. 2004;14:373–376. doi: 10.1016/j.bmcl.2003.10.061. [DOI] [PubMed] [Google Scholar]
- [99].Bommineni GR, Kapilashrami K, Cummings JE, Lu Y, Knudson SE, Gu C, Walker SG, Slayden RA, Tonge PJ. Thiolactomycin-based inhibitors of bacterial β-ketoacyl-ACP synthases with in vivo activity. J. Med. Chem. 2016;59:5377–5390. doi: 10.1021/acs.jmedchem.6b00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Moche M, Schneider G, Edwards P, Dehesh K, Lindqvist Y. Structure of the complex between the antibiotic cerulenin and its target, β-ketoacyl-acyl carrier protein synthase. J. Biol. Chem. 1999;274:6031–6034. doi: 10.1074/jbc.274.10.6031. [DOI] [PubMed] [Google Scholar]
- [101].Nomura S, Horiuchi T, Omura S, Hata T. The action mechanism of cerulenin. I. Effect of cerulenin on sterol and fatty acid biosynthesis in yeast. J. Biochem. (Tokyo) 1972;71:783–796. doi: 10.1093/oxfordjournals.jbchem.a129827. [DOI] [PubMed] [Google Scholar]
- [102].Shu IW, Lindenberg DL, Mizuno TM, Roberts JL, Mobbs CV. The fatty acid synthase inhibitor cerulenin and feeding, like leptin, activate hypothalamic pro-opiomelanocortin (POMC) neurons. Brain Res. 2003;985:1–12. doi: 10.1016/s0006-8993(03)02806-3. [DOI] [PubMed] [Google Scholar]
- [103].Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, Lupu R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Nalt. Acad. Sci. U. S. A. 2004;101:10715–10720. doi: 10.1073/pnas.0403390101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Escaich S, Prouvensier L, Saccomani M, Durant L, Oxoby M, Gerusz V, Moreau F, Vongsouthi V, Maher K, Morrissey I, Soulama-Mouze C. The MUT056399 inhibitor of FabI is a new antistaphylococcal compound. Antimicrob. Agents Chemother. 2011;55:4692–4697. doi: 10.1128/AAC.01248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Bogdanovich T, Clark C, Kosowska-Shick K, Dewasse B, McGhee P, Appelbaum PC. Antistaphylococcal activity of CG400549, a new experimental FabI inhibitor, compared with that of other agents. Antimicrob. Agents Chemother. 2007;51:4191–4195. doi: 10.1128/AAC.00550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Park HS, Yoon YM, Jung SJ, Kim CM, Kim JM, Kwak JH. Antistaphylococcal activities of CG400549, a new bacterial enoyl-acyl carrier protein reductase (FabI) inhibitor. J. Antimicrob. Chemother. 2007;60:568–574. doi: 10.1093/jac/dkm236. [DOI] [PubMed] [Google Scholar]
- [107].Yum JH, Kim CK, Yong D, Lee K, Chong Y, Kim CM, Kim JM, Ro S, Cho JM. In vitro activities of CG400549, a novel FabI inhibitor, against recently isolated clinical staphylococcal strains in Korea. Antimicrob. Agents Chemother. 2007;51:2591–2593. doi: 10.1128/AAC.01562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Yao J, Maxwell JB, Rock CO. Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI) J. Biol. Chem. 2013;288:36261–36271. doi: 10.1074/jbc.M113.512905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Blaser M, Bork P, Fraser C, Knight R, Wang J. The microbiome explored: recent insights and future challenges. Nat. Rev. Micro. 2013;11:213–217. doi: 10.1038/nrmicro2973. [DOI] [PubMed] [Google Scholar]
- [110].Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat. Rev. Micro. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Bohnhoff M, Miller CP. Enhanced susceptibility to Salmonella infection in streptomycin-treated mice. J. Infect. Dis. 1962;111:117–127. doi: 10.1093/infdis/111.2.117. [DOI] [PubMed] [Google Scholar]
- [112].Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 2012;80:62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- [114].Yao J, Rock CO. Resistance mechanisms and the future of bacterial enoyl-acyl carrier protein reductase (FabI) antibiotics. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a027045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Otto M. Staphylococcus epidermidis – the "accidental" pathogen. Nat. Rev. Microbiol. 2009;7:555–567. doi: 10.1038/nrmicro2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kothari A, Morgan M, Haake DA. Emerging technologies for rapid identification of bloodstream pathogens. Clin. Infect. Dis. 2014;59:272–278. doi: 10.1093/cid/ciu292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Hackbarth CJ, Unsal I, Chambers HF. Cloning and sequence analysis of a class A beta-lactamase from Mycobacterium tuberculosis H37Ra. Antimicrob. Agents Chemother. 1997;41:1182–1185. doi: 10.1128/aac.41.5.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Bercovier H, Kafri O, Sela S. Mycobacteria possess a surprisingly small number of ribosomal RNA genes in relation to the size of their genome. Biochem. Biophys. Res. Commun. 1986;136:1136–1141. doi: 10.1016/0006-291x(86)90452-3. [DOI] [PubMed] [Google Scholar]
- [119].Gillespie SH. Evolution of drug resistance in Mycobacterium tuberculosis: clinical and molecular perspective. Antimicrob. Agents Chemother. 2002;46:267–274. doi: 10.1128/AAC.46.2.267-274.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Eliopoulos GM, Huovinen P. Resistance to trimethoprim-sulfamethoxazole. Clin. Infect. Dis. 2001;32:1608–1614. doi: 10.1086/320532. [DOI] [PubMed] [Google Scholar]
- [121].Zasowski EJ, Rybak JM, Rybak MJ. The β-lactams strike back: Ceftazidime-Avibactam. Pharmacotherapy. 2015;35:755–770. doi: 10.1002/phar.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Perdigão J, Macedo R, Silva C, Machado D, Couto I, Viveiros M, Jordao L, Portugal I. From multidrug-resistant to extensively drug-resistant tuberculosis in Lisbon, Portugal: the stepwise mode of resistance acquisition. J. Antimicrob. Chemother. 2013;68:27–33. doi: 10.1093/jac/dks371. [DOI] [PubMed] [Google Scholar]
- [123].McKinney DC, Eyermann CJ, Gu RF, Hu J, Kazmirski SL, Lahiri SD, McKenzie AR, Shapiro AB, Breault G. Antibacterial FabH inhibitors with mode of action validated in Haemophilus influenzae by in vitro resistance mutation mapping. ACS Infect. Dis. 2016;2:456–464. doi: 10.1021/acsinfecdis.6b00053. [DOI] [PubMed] [Google Scholar]
- [124].Centers for Disease Control and Prevention Antibiotic Resistance in the United States. 2016 http://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf. [Google Scholar]
- [125].Edwards P, Nelsen JS, Metz JG, Dehesh K. Cloning of the fabF gene in an expression vector and in vitro characterization of recombinant fabF and fabB encoded enzymes from Escherichia coli. FEBS Lett. 1997;402:62–66. doi: 10.1016/s0014-5793(96)01437-8. [DOI] [PubMed] [Google Scholar]
- [126].Yuan Y, Sachdeva M, Leeds JA, Meredith TC. Fatty acid biosynthesis in Pseudomonas aeruginosa is initiated by the FabY class of β-ketoacyl acyl carrier protein synthases. J. Bacteriol. 2012;194:5171–5184. doi: 10.1128/JB.00792-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Abou Chakra CN, Pepin J, Sirard S, Valiquette L. Risk factors for recurrence, complications and mortality in Clostridium difficile infection: a systematic review. PLoS ONE. 2014;9:e98400. doi: 10.1371/journal.pone.0098400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Knecht H, Neulinger SC, Heinsen FA, Knecht C, Schilhabel A, Schmitz RA, Zimmermann A, dos Santos VM, Ferrer M, Rosenstiel PC, Schreiber S, Friedrichs AK, Ott SJ. Effects of β-lactam antibiotics and fluoroquinolones on human gut microbiota in relation to Clostridium difficile associated diarrhea. PLoS ONE. 2014;9:e89417. doi: 10.1371/journal.pone.0089417. [DOI] [PMC free article] [PubMed] [Google Scholar]