

ABSTRACT
Clostridium botulinum C2 toxin consists of an enzyme component (C2I) and a binding component (C2II). Activated C2II (C2IIa) binds to a cell receptor, giving rise to lipid raft-dependent oligomerization, and it then assembles with C2I. The whole toxin complex is then endocytosed into the cytosol, resulting in the destruction of the actin cytoskeleton and cell rounding. Here, we showed that C2 toxin requires acid sphingomyelinase (ASMase) activity during internalization. In this study, inhibitors of ASMase and lysosomal exocytosis blocked C2 toxin-induced cell rounding. C2IIa induced Ca2+ influx from the extracellular medium to cells. C2 toxin-induced cell rounding was enhanced in the presence of Ca2+. ASMase was released extracellularly when cells were incubated with C2IIa in the presence of Ca2+. Small interfering RNA (siRNA) knockdown of ASMase reduced C2 toxin-induced cell rounding. ASMase hydrolyzes sphingomyelin to ceramide on the outer leaflet of the membrane at acidic pH. Ceramide was detected in cytoplasmic vesicles containing C2IIa. These results indicated that ASMase activity is necessary for the efficient internalization of C2 toxin into cells. Inhibitors of ASMase may confer protection against infection.
KEYWORDS: C. botulinum C2 toxin, acid sphingomyelinase, endocytosis
INTRODUCTION
Clostridium botulinum produces a binary toxin, C2, which consists of two nonlinked proteins called C2I (active enzymatic component) and C2II (binding component) (1–4). The two components represent separate proteins, which are devoid of toxic activity when injected alone (4). Only the combination of the two components shows a cytopathic effect on cells. C2I mono-ADP-ribosylates G-actin at arginine-177, which converts G-actin into a capping molecule and interferes with the additional polymerization of actin filaments. This leads to actin filament depolymerization and cell rounding (1, 3). C2 toxin is a member of the binary actin-ADP-ribosylating toxin family. Members of this toxin family include Clostridium perfringens iota-toxin, Clostridium spiroforme iota-like toxin, Clostridium difficile ADP-ribosyltransferase, and vegetative insecticidal protein from Bacillus cereus (1–4).
The binding and internalization of C2I depend on C2IIa, which binds asparagine-linked carbohydrates on target cells and forms heptamers that bind C2I (5). Binding of C2I to membrane-bound C2IIa oligomers in lipid rafts induced the activation of phosphatidylinositol 3-kinase (PI3K), which in turn caused C2 toxin internalization via a lipid raft-mediated process (3, 6). After endocytosis of C2 toxin, it was trafficked to early endosomes (1, 3, 7). The oligomer of C2IIa entered the endosomal membrane under acidic conditions and formed pores. Thereafter, C2I was translocated in an unfolded conformation through the endosomal membranes into the cytosol via the C2IIa pore (1, 3). The membrane translocation of C2I was reported to be facilitated by the host cell factors Hsp90 (8), cyclophilin A (9), and FK506-binding protein (10). After translocation to the cytosol, C2I ADP-ribosylates G-actin in the cytosol. Some amounts of both components were returned to the plasma membranes through recycling endosomes. On the other hand, the rest of C2IIa was transported to late endosomes and lysosomes for degradation (7).
We previously reported that the binding component (Ib) of iota-toxin causes cell necrosis in A431 and A549 cells (11). These cytotoxic effects are responsible for the pore formation of Ib in the plasma membrane. Our previous study also reported that Ib causes an elevation of intracellular Ca2+ levels from the extracellular space during endocytosis (11). On the other hand, bacterial pore-forming protein-induced Ca2+ influx and the binding of pathogenic microorganisms lead to the lysosomal exocytosis and release of the lysosomal acid sphingomyelinase (ASMase) extracellularly (12, 13). Subsequently, in both cases, ASMase hydrolyzes sphingomyelin (SM) into ceramide (14, 15). Ceramide self-associates in plasma membrane microdomains that bud into cells, generating endosomes that induce endocytosis (16). These results indicated that the cleavage of sphingomyelin by secreted ASMase generates ceramide-enriched microdomains in the plasma membrane, promoting endocytosis (14–16). It is as yet unknown whether the activation of ASMase is related to the uptake of C2 toxin in target cells. Madin-Darby canine kidney (MDCK) cells provide a good model system to study the binding and internalization of C2 toxin (6, 7). Here, we investigated whether ceramide-generating lysosomal ASMase plays a role in the endocytosis process of C2 toxin. We found that the internalization of C2 toxin into host cells was dependent on ASMase activity.
RESULTS
Effect of Ca2+ on the cytotoxicity of C2 toxin.
To determine the effect of Ca2+ on the host cell internalization of C2 toxin, we examined the cell-rounding activity of C2 toxin in the presence or absence of Ca2+ in the extracellular medium. As shown in Fig. 1A, MDCK cells incubated with C2 toxin in the presence of Ca2+ underwent rapid rounding. In contrast, the cell-rounding activity of C2 toxin under Ca2+-free conditions was weak over time. On the other hand, the ADP-ribosylating activity of C2 toxin in the presence or absence of Ca2+ was not altered (data not shown). To investigate the effect of Ca2+ on the internalization of C2IIa, C2IIa was incubated with MDCK cells in the presence or absence of Ca2+ in the extracellular medium. As shown in Fig. 1B, C2IIa was efficiently internalized into the cytosol of MDCK cells at 37°C in the presence of Ca2+. In contrast, without Ca2+, the immunofluorescent signal for C2IIa was largely localized to the plasma membrane. Next, we examined the effect of Ca2+ on the binding of C2IIa to MDCK cells (Fig. 1C). When MDCK cells were incubated with C2IIa in the presence or absence of Ca2+ at 37°C for 30 min, the binding of the C2IIa oligomer and monomer was not affected. These results suggested that the binding of C2IIa to cells and the formation of the C2IIa oligomer do not require extracellular Ca2+. Idone et al. (14) revealed previously that Ca2+ influx triggers a rapid form of endocytosis. Therefore, we investigated whether C2IIa causes Ca2+ influx into MDCK cells (Fig. 1D). Intracellular Ca2+ levels ([Ca2+]i) were measured by using Fura-2, a fluorescent calcium probe. MDCK cells were incubated with Fura-2–pentaacetotoxymethyl ester (Fura-2/AM) and treated with C2IIa. [Ca2+]i was then estimated from the fluorescence intensity. In normal Ca2+ medium, C2IIa evoked a transient increase in [Ca2+]i, which showed a rapid rise and a gradual decay phase, in a dose-dependent manner (Fig. 1D). No increase in [Ca2+]i was caused by heat-inactivated C2IIa, and the increase in [Ca2+]i evoked by C2IIa was completely inhibited by an anti-C2IIa antibody (data not shown). In Ca2+-free medium, C2IIa did not cause Ca2+ uptake (data not shown). These data show that C2IIa induces Ca2+ uptake from the extracellular medium during endocytosis.
FIG 1.

Effect of Ca2+ on the action of C2 toxin. (A) MDCK cells were incubated in either Ca2+ (1.8 mM) medium or Ca2+-free medium with C2I (100 ng/ml) and C2IIa (200 ng/ml) at 37°C for the periods indicated. Pictures were taken. About 100 cells were counted per picture, and the number of rounded cells was determined as a percentage. Values are given as means ± standard deviations (n = 3). (B) MDCK cells were incubated with C2IIa (1 μg/ml) at 37°C for the periods indicated. Cells were fixed, permeabilized, and stained with an anti-C2IIa antibody and DAPI. C2IIa (red) and the nucleus (blue) were viewed by using a confocal microscope. The experiments were repeated three times, and a representative result is shown. Bar, 7.5 μm. (C) MDCK cells were incubated in either Ca2+ (1.8 mM) medium or Ca2+-free medium with C2IIa (1 μg/ml) at 37°C for 30 min. The cells were rinsed and subjected to Western blot analyses of C2IIa and β-actin (control). A typical result from one of three experiments is shown. (D) MDCK cells were loaded with the intracellular Ca2+ indicator Fura-2/AM. [Ca2+]i was calculated as described in Materials and Methods. Changes in [Ca2+]i induced by C2IIa were measured in cells in extracellular buffer containing 1.8 mM CaCl2. C2IIa was added at the time indicated by the arrow. A typical result from three independent experiments is shown.
C2 toxin activates acid but not neutral sphingomyelinase.
We investigated whether the lysosomal enzyme ASMase, which was previously shown to promote endocytosis (15), was required for the internalization of C2 toxin. As shown in Fig. 2A and B, ASMase activity levels in the culture supernatant increased in a dose- and time-dependent manner when cells were treated with C2I plus C2IIa and C2IIa alone in the presence of Ca2+. Consistent with the Ca2+ requirement for lysosomal exocytosis, the increase in secreted ASMase activity by C2 toxin was not observed in the absence of Ca2+ (Fig. 2A). Furthermore, C2IIa plus C2I and C2IIa alone dose-dependently increased extracellular ASMase activity (Fig. 2B). Next, we determined whether the secretion of ASMase into the culture supernatant was increased by treatment with C2IIa. We found that control cells secreted very low levels of ASMase, whereas incubation with C2IIa showed increases in the secretion of this enzyme in a time-dependent manner (Fig. 2C). To determine if C2 toxin induces lysosomal exocytosis, the culture supernatants of toxin-treated cells were analyzed for the presence of the lysosomal enzyme β-hexosaminidase (βHex) as a marker for the secretion lysosomal contents. As shown in Fig. 2C, there was an increase in βHex activity in the culture supernatants of toxin-treated cells compared to that in untreated cells. For ASMase to access sphingomyelin on the cell surface, lysosomal exocytosis must occur. Next, we investigated whether C2IIa also enhanced neutral sphingomyelinase (NSMase). As shown in Fig. 2D, cells treated with C2IIa showed an increase in the activity of ASMase in the culture supernatant. In contrast, no increase in the activity of NSMase was observed. Moreover, NSMase was not detected in the culture supernatant of C2 toxin-treated cells (data not shown).
FIG 2.

Activity of acid sphingomyelinase in culture supernatants of C2 toxin-treated MDCK cells. (A) MDCK cells were incubated in either Ca2+ (1.8 mM) medium or Ca2+-free medium with C2I (200 ng/ml) and C2IIa (500 ng/ml) or C2IIa (500 ng/ml) only at 37°C for the periods indicated. (B) MDCK cells were incubated in either Ca2+ (1.8 mM) medium with C2I (200 ng/ml) and various amounts of C2IIa (500 ng/ml) or various amounts of C2IIa (500 ng/ml) only at 37°C for 30 min. The activity of ASMase in the culture supernatant was determined as described in Materials and Methods. Untreated cells, used as controls, were set at a basal activity level of 1.0. Data are reported as percentages of the values obtained with untreated controls (means ± standard deviations for four independent experiments). (C) MDCK cells were incubated with C2I (200 ng/ml) and C2IIa (500 ng/ml) at 37°C for the indicated times. ASMase in the culture supernatant was detected by Western blotting using a specific antibody. A typical result from one of three experiments is shown. For the βHex assay, MDCK cells were incubated with C2I (200 ng/ml) and C2IIa (500 ng/ml) at 37°C. At the indicated time points, the culture supernatants were assayed for βHex activity. All enzyme activities are expressed as a percentage of the total enzyme activity found in the supernatant and cells (means ± standard deviations for four independent experiments). (D) Activity of NSMase and ASMase in C2 toxin-treated cells. Cells were stimulated with C2I (200 ng/ml) and C2IIa (500 ng/ml) at 37°C for 60 min. The activity of ASMase in the culture supernatant and the activity of NSMase in cell lysates were determined as described in Materials and Methods. Data are reported as percentages of the values obtained with untreated controls (means ± standard deviations for four independent experiments).
Acid sphingomyelinase activity is required for the endocytosis of C2 toxin.
We examined whether ASMase played a role in the endocytosis of C2 toxin. Specific ASMase inhibitors, such as amitriptyline (Ami) and imipramine (Imi), are water-soluble compounds that block the function of ASMase both at the cell surface and within lysosomes. As shown in Fig. 3A, Ami and Imi inhibited cell rounding induced by C2 toxin. Bromoenol lactone (BEL) inhibited lysosomal exocytosis and also suppressed toxin-induced cell rounding. In contrast, the NSMase inhibitor GW4869 (GW) did not affect toxin-induced cell rounding. We then examine the effect of inhibitors on the increase in extracellular ASMase activity induced by C2IIa. Amitriptyrine, imipramine, and BEL but not GW4869 inhibited the enhanced activity of ASMase induced by C2IIa (Fig. 3B). Next, we determined the influence of amitriptyrine on the internalization of C2IIa. When MDCK cells were incubated with C2IIa in the presence of a vehicle at 37°C for 30 min, C2IIa was present in cytoplasmic vesicles (Fig. 3C). On the other hand, after treatment of MDCK cells with C2IIa in the presence of amitriptyrine, the internalization of C2IIa decreased, and C2IIa was detected in the plasma membrane. The initial process of C2 toxin internalization is not attributed to the calcium-driven activation of ASMase inside cells. These findings indicate that the extracellular release of ASMase is necessary for the internalization of C2IIa into sensitive cells.
FIG 3.

Inhibition of acid sphingomyelinase prevents C2 toxin-induced cytotoxic effects in MDCK cells. (A) MDCK cells were pretreated with various inhibitors (25 μM) at 37°C for 1 h. The cells were incubated with C2I (200 ng/ml) and C2IIa (500 ng/ml) at 37°C for 4 h. About 100 cells were counted per picture, and the number of rounded cells was determined as a percentage. Values are given as means ± standard deviations (n = 4). One-way analysis of variance was employed to assess statistical significance. *, P < 0.01 (significantly different from dimethyl sulfoxide [DMSO] plus C2 toxin). Abbreviations: Ami, amitriptyline; Imi, imipramine; BEL, bromoenol lactone. (B) MDCK cells were pretreated with various inhibitors (25 μM) at 37°C for 1 h. The cells were incubated with C2I (200 ng/ml) and C2IIa (500 ng/ml) at 37°C for 1 h. The activity of ASMase in the culture supernatant was determined as described in Materials and Methods. Data are reported as percentages of the values obtained with untreated controls (means ± standard deviations for four independent experiments). One-way analysis of variance was employed to assess statistical significance. *, P < 0.01 (significantly different from DMSO plus C2 toxin). (C) MDCK cells were pretreated with amitriptyline (25 μM) at 37°C for 1 h and then rinsed. Cells were incubated with C2I (500 ng/ml) and C2IIa (1000 ng/ml) at 37°C for 30 min. Cells were fixed, permeabilized, and stained with the anti-C2IIa antibody, Alexa Fluor 488-phallodin, and DAPI. C2IIa (red), actin (green), and the nucleus (blue) were viewed by using a confocal microscope. The experiments were repeated three times, and a representative result is shown. Bar, 7.5 μm.
Effects of acid sphingomyelinase siRNA and treatment of B. cereus sphingomyelinase on C2 toxin-induced cytotoxicity.
To investigate the role of ASMase in C2 toxin-induced cytotoxicity, an RNA interference (RNAi) assay was performed to knock down the expression of ASMase. When cells were transfected with ASMase small interfering RNA (siRNA), the protein levels of the ASMase were significantly lower than those in cells that were intact or transfected with negative-control siRNA (NC-siRNA) (Fig. 4A). As shown in Fig. 4B, C2 toxin induced cell rounding of intact and NC-siRNA-transfected cells. On the other hand, the knockdown of the ASMase by siRNA inhibited toxin-induced cell rounding. To determine whether sphingomyelinase plays a role in the internalization of C2 toxin, MDCK cells were pretreated with different concentrations of B. cereus sphingomyelinase (Bc-SMase) at 37°C for 60 min, washed to remove excess Bc-SMase, and incubated with C2 toxin at 37°C. As shown in Fig. 4C, cells treated with Bc-SMase were more sensitive to C2 toxin than were those that received mock treatment. No cell rounding was evoked by Bc-SMase only under our experimental conditions.
FIG 4.
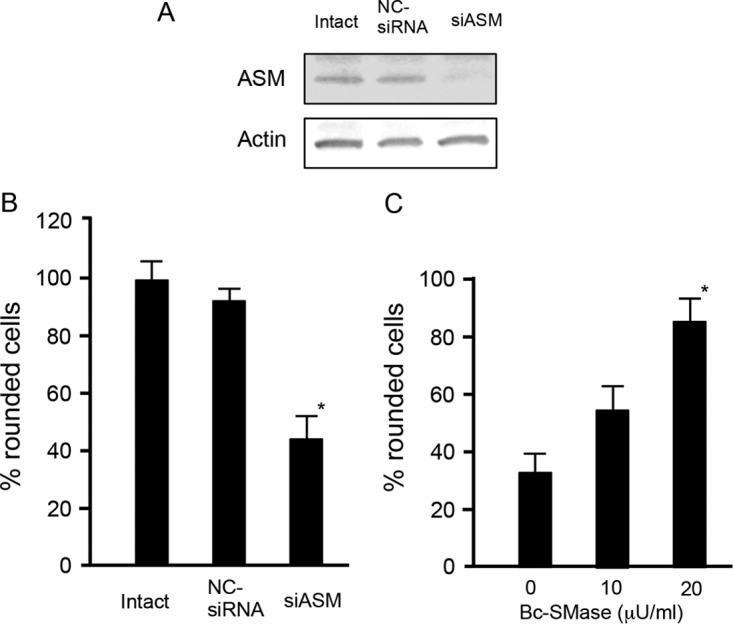
Role of acid sphingomyelinase in C2 toxin-induced cytotoxic effects on Madin-Darby canine kidney cells. siRNAs were used to reduce cellular ASMase levels (siASM) with a nonsilencing siRNA used as a control (NC-siRNA). (A) Western blots were used to determine the reduction in ASMase levels. A typical result from one of three experiments is shown. (B) After siRNA treatment, cells were incubated with C2I (200 ng/ml) and C2IIa (500 ng/ml) at 37°C for 4 h. About 100 cells were counted per picture, and the number of rounded cells was determined as a percentage. Values are given as means ± standard deviations (n = 4). Two-tailed Student's t test was employed to assess statistical significance. *, P < 0.01 (significantly different from NC-siRNA-treated cells plus C2 toxin). (C) MDCK cells were treated with or without 10 or 20 μU/ml of sphingomyelinase (SMase) from Bacillus cereus for 30 min and then rinsed. Cells were incubated with C2I (100 ng/ml) and C2IIa (200 ng/ml) at 37°C for 4 h. About 100 cells were counted per picture, and the number of rounded cells was determined as a percentage. Values are given as means ± standard deviations (n = 3). Two-tailed Student's t test was employed to assess statistical significance. *, P < 0.01 (significantly different from Bc-SMase-untreated cells plus C2 toxin).
C2 toxin induces the recruitment of acid sphingomyelinase and an increase in ceramide levels.
ASMase converts sphingomyelin to ceramide both in lysosomes and on the outer leaflet of the plasma membrane (15, 17). Therefore, we tested if the toxin induced the recruitment of ASMase to the plasma membrane. When the cells were incubated with C2IIa at 37°C for 30 min, ASMase was found to be localized in plasma membranes, indicating that ASMase is recruited to the membrane (Fig. 5A). To substantiate the induction of ceramide levels due to C2IIa, C2IIa-treated or untreated MDCK cells were labeled with an antibody specific for ceramide, as described in Materials and Methods. As shown in Fig. 5A, untreated cells showed very slight ceramide labeling. On the other hand, treatment of cells with C2IIa for 30 min induced a clear enhancement of ceramide labeling.
FIG 5.
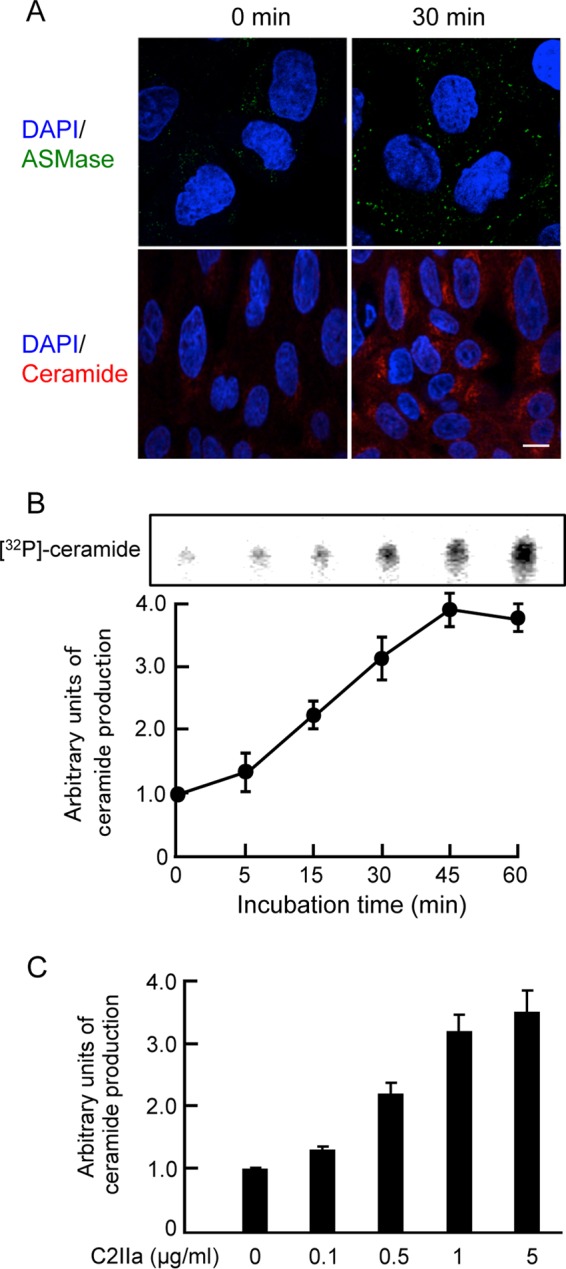
Ceramide generation and localization of acid sphingomyelinase in C2 toxin-treated cells. (A) MDCK cells were incubated with C2I (200 ng/ml) and C2IIa (500 ng/ml) at 37°C for 30 min, washed, fixed, and permeabilized. ASMase was labeled by using an anti-ASMase antibody (green), and the cell nuclei were stained with DAPI (blue). Ceramide was stained with an anticeramide antibody and viewed by using a confocal microscope. These results are representative of data from three independent experiments. Bar, 7.5 μm. (B) MDCK cells were treated with C2IIa (1 μg/ml) for the indicated periods at 37°C. Ceramide levels were determined as described in Materials and Methods. Data from one representative experiment of three are shown. (C) MDCK cells were treated with various doses of C2IIa for 45 min at 37°C. Ceramide levels were determined as described in Materials and Methods. For quantitative analysis, the level of ceramide was measured by densitography. The control level for untreated cells was set to 1. Values represent means ± standard deviations (n = 3).
Next, we evaluated whether C2IIa induces ceramide production in MDCK cells. When C2IIa was incubated with the cells at 37°C, the toxin caused an increase in the production of ceramide in a dose- and time-dependent manner (Fig. 5B and C). Heat-inactivated C2IIa did not induce the production of ceramide, and ceramide production induced by C2IIa was blocked by an antibody against C2IIa (data not shown).
DISCUSSION
In the present study, the C2IIa oligomer triggered the release of ASMase and the subsequent production of ceramide in the outer plasma membrane. The generation of ceramide in the membrane promotes the endocytosis of C2 toxin. Our studies showed that ASMase activity is critical for the internalization of C2 toxin into host cells.
The binding of several microbial pathogens to target cells can give rise to the exocytosis of lysosomal ASMase to the extracellular leaflet of the cell membrane, where it hydrolyzes outer surface SM (18–21). Subsequently, the cleavage of sphingomyelin to ceramide results in membrane invagination and endocytosis, leading to the uptake of particles, including pathogens, into cells (20). Bacterial pore-forming protein-induced Ca2+ influx from the extracellular space promotes the exocytosis of lysosomal ASMase (12, 13). ASMase subsequently induces endocytosis as well as mediating the uptake of pathogens into cells. C2IIa forms ion-permeable channels in planar phospholipid bilayer membranes (4). In the present study, C2IIa was easily internalized in the presence of Ca2+ but not in the absence of Ca2+. Moreover, C2IIa caused Ca2+ influx into cells. It is therefore likely that the internalization process of C2IIa is similar to that of microbial pathogens and bacterial pore-forming proteins. These results led us to hypothesize that the Ca2+-dependent exocytosis of lysosomal ASMase might indicate an initial step in the endocytosis of C2IIa to sensitive cells. In this study, we provide several lines of evidence in support of this view. First, C2IIa was internalized into the host cells in a Ca2+-dependent manner. Second, C2IIa caused the release of ASMase from lysosomes. Third, an ASMase inhibitor, a lysosome exocytosis blocker, or ASMase knockdown reduced the susceptibility of host cells to C2 toxin. Fourth, C2 toxin caused the generation of ceramide. Fifth, exogenously added sphingomyelinase increased the susceptibility of untreated cells to C2 toxin. Previous reports as to ASMase activity at neutral pH showed that despite its optimal activity at acidic pH, recombinant ASMase can act extracellularly, facilitating endocytosis and plasma membrane repair under physiological conditions (15). On the other hand, ASMase possesses sufficient activity at neutral pH to hydrolyze sphingomyelin in the outer leaflet of the plasma membrane (18). Mechanisms accelerating ASMase activity in the extracellular environment may include acidic microenvironments generated at the cell surface during lysosomal exocytosis (22) or enzyme activation by lysosomal lipids such as lysobisphosphatidic acid (23). On the basis of those previous reports, we think that ASMase can act in the culture medium. Collectively, our results indicate that C2 toxin is internalized into cells by endocytosis using Ca2+-dependent exocytosis of lysosomal ASMase.
Secreted ASMase catalyzes ceramide production in the outer leaflet of the plasma membrane. Ceramide-enriched plasma membrane domains fuse into enlarged ceramide-enriched domains (24). In addition to changing membrane rigidity and fluidity, ceramide-enriched domains contribute to the sorting and, ultimately, condensing of membrane receptors (24). CIIa monomers that are bound to their receptor move in confined spaces equivalent to lipid rafts. This confinement results in an enrichment of toxin molecules and promotes their interactions and subsequent oligomerization.
Ceramide is an essential messenger signaling molecule capable of evoking signaling cascades for either proapoptotic or antiapoptotic cases (25). A predominant metabolite of ceramide is ceramide-1-phosphate (C1P), which can be formed via the phosphorylation of ceramide by ceramide kinase (25, 26). The C1P receptor is coupled to inhibitory guanine nucleotide-binding regulatory (Gi) proteins and causes the activation of the PI3K/Akt signaling pathway to facilitate cell survival (25, 27, 28). We reported previously that C2 toxin in lipid rafts activates the PI3K/Akt signaling pathway (6). As C2 toxin stimulates the generation of ceramide via ASMase activity, the activation of the PI3K/Akt signaling pathway induced by the toxin may be responsible for the generation of C1P. The generation of ceramide and its metabolite induced by C2 toxin resulted in cell survival via the activation of the PI3K/Akt signaling pathway. Therefore, the mechanisms to antagonize the cytotoxic effect may be built into the host cell defense response to the toxin.
The pore-forming activity of C2IIa has been analyzed with lipid bilayers. The addition of C2IIa to diphytanoyl phosphatidylcholine membranes results in the formation of ion-permeable channels that are formed by C2IIa heptamers (29–31). The C2IIa heptamer inserts were oriented in the membrane in the absence of ceramide or sphingomyelin (31). However, C2IIa has not been shown to have any lipid preference. By using planar lipid bilayers, previous studies showed pore formation of C2IIa but not internalization of C2IIa. However, in the present study, we addressed the initial step of C2IIa internalization and found that via C2IIa-induced Ca2+-dependent lysosome exocytosis, ASMase is released to the outer leaflet of the plasma membrane. The hydrolysis of sphingomyelin in lipid rafts to ceramide resulted in membrane invagination and endosome formation. Namely, ceramide production by ASMase plays an important role in the internalization of C2IIa.
Iota-toxin of C. perfringens is highly related to C2 toxin and shares a binding domain that forms a heptamer and an enzyme domain that modifies actin. C2IIa and a binding component (Ib) of iota-toxin were shown previously to induce the endocytosis of their receptors via a lipid raft-mediated process (6, 7, 32, 33). Based on our present results with C2 toxin, we speculate that iota-toxin is endocytosed by the same ASMase-dependent process.
We recently reported that oligomer formation of the epsilon-toxin monomer is facilitated by the production of ceramide through the activation of NSMase caused by the toxin in MDCK cells (34). In the present study, the NSMase inhibitor GW4869 did not affect C2 toxin-induced cell rounding in MDCK cells. Moreover, C2IIa did not show an increase in the activity of NSMase. Namely, NSMase is not responsible for the intoxication of C2 toxin. In the case of C2 toxin, cleavage of sphingomyelin by ASMase secreted by C2IIa generates a ceramide-enriched microdomain on the plasma membrane, triggering the endocytosis of C2 toxin. ASMase is involved in the internalization of C2 toxin into cells. In contrast, NSMase is important for oligomer formation of epsilon-toxin.
The actin cytoskeleton is one of the main targets of clostridial binary toxins and thus is of major importance for the host-pathogen interaction. C2 toxin is an enterotoxin correlated with serious animal enteric diseases (4). In the present study, ASMase inhibitors blocked the internalization of C2 toxin; therefore, the blocking of ASMase may provide new avenues for drug treatment.
In conclusion, we provide evidence of a critical function of ASMase in C2 toxin internalization into target cells. The elevation of the intracellular Ca2+ concentration induced by C2 toxin triggers the exocytosis of lysosomes. Lysosomal ASMase is delivered to the outer leaflets of the plasma membrane, where it converts sphingomyelin into ceramide. Ceramide-rich microdomains bud into the cell, generating endosomes that promote the internalization of C2 toxin into the cytosol. Thus, C2 toxin utilizes Ca2+-dependent exocytosis of lysosomal ASMase to enter the target cell.
MATERIALS AND METHODS
Materials.
Recombinant C2I and C2II were expressed and fused with glutathione S-transferase (GST) in Escherichia coli BL21 cells (6). To obtain C2IIa, C2II was activated by incubation with trypsin (6). Rabbit anti-C2I and anti-C2II antibodies were prepared as described previously (6). The expression and purification of sphingomyelinase from Bacillus cereus were performed according to a method used previously (35). Amitriptyline hydrochloride and imipramine hydrochloride were obtained from Wako Pure Chemical (Osaka, Japan). BEL, GW4869 hydrate, p-nitrophenyl N-acetyl-β-d-glucosaminide, and monoclonal anticeramide IgM antibody (clone 15B4) produced in mouse were obtained from Sigma-Aldrich (Tokyo, Japan). Rabbit anti-acid sphingomyelinase (H-181) antibody and anti-β-actin antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and an Amplex Red sphingomyelinase assay kit was obtained from Invitrogen (Tokyo, Japan). Dulbecco's modified Eagle's medium (DMEM) and Hanks' balanced salt solution (HBSS) were obtained from Gibco BRL (New York, NY, USA). Alexa Fluor 568-conjugated goat anti-rabbit IgG, Alexa Fluor 564-conjugated goat anti-mouse IgM (μ chain), Alexa Fluor 488 phalloidin conjugate, and 4′,6′-diamidino-2-phenylindole (DAPI) were obtained from Life Technologies (Tokyo, Japan). An anti-neutral sphingomyelinase antibody was obtained from Abcam (Tokyo, Japan). Horseradish peroxidase-labeled anti-rabbit IgG and enhanced chemiluminescence kits were purchased from GE Healthcare (Tokyo, Japan). All other chemicals were of the highest grade available from commercial sources.
Cell culture and assay of cytotoxicity.
MDCK cells were obtained from the Riken Cell Bank (Tsukuba, Japan). Cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS), 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 2 mM glutamine (FCS-DMEM). All incubation steps were carried out at 37°C in a 5% CO2 atmosphere. Cells for cytotoxicity assays were inoculated in FCS-DMEM in 48-well tissue culture plates (Falcon, Oxnard, CA, USA). Various concentrations of C2I and C2IIa were mixed in FCS-DMEM and inoculated onto cell monolayers. Cells were observed for morphological alterations 4 h after inoculation, as described previously (7). For the study of the role of Ca2+ in the cytotoxicity of the C2 toxin, cells were incubated in HBSS with or without 1.8 mM Ca2+.
Inhibitor treatments and heat inactivation.
Cells were treated with 25 μM amitriptyrine, 25 μM imipramine, 25 μM BEL, or 25 μM GW4869 at 37°C for 1 h before experiments. In some experimental studies, heat-inactivated C2IIa was prepared by heating for 5 min at 95°C.
Determination of acidic or neutral sphingomyelinase activities.
Following treatment with C2 toxin, the supernatants of the cells were mixed in neutral lysis buffer (20 mM Tris-HCl buffer [pH 7.5], 1% Triton X-100, 1 mM EDTA) or were mixed with acid lysis buffer (50 mM sodium acetate buffer [pH 5.0], 1% Triton X-100, 1 mM EDTA) with a freshly added protease inhibitor cocktail (Nacalai, Kyoto, Japan). Activities of NSMase (at pH 7.5) and ASMase (at pH 5.0) were analyzed by using an Amplex Red sphingomyelinase assay kit according to the manufacturer's protocol.
Western blotting.
Methods for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and standard Western blotting, including the use of antibodies against C2II, β-actin, and ASMase, were performed as described previously (36).
Measurement of intracellular Ca2+ concentrations.
The intracellular Ca2+ concentration was determined by using Fura-2/AM (4.5 μM) (Dojindo, Kumamoto, Japan) as described previously (36). Cells were loaded with Fura-2/AM (4.5 μM) at 37°C for 30 min in dye-loading solution (Hanks' HEPES buffer containing 2.5 mM probenecid, 1% fetal bovine serum [FBS], and 0.05% pluronic F-127). After washing, cells were incubated in an assay solution (Hanks' HEPES buffer containing 2.5 mM probenecid and 1% bovine serum albumin [BSA]) at 37°C for an additional 15 min. Fura-2 fluorescence from these cells was monitored at 37°C by the ratio technique (excitation at 340 and 380 nm and emission at 500 nm) using an inverted microscope (TMD-300; Nikon, Tokyo, Japan) with fluorescence software (Aqua-cosmos; Hamamatsu Photonics, Hamamatsu, Japan). The imaging system was standardized with a Fura-2 calcium-imaging calibration kit (Invitrogen) as described previously (36).
β-Hexosaminidase assays.
MDCK cells in a 24-well plate were incubated with phosphate-buffered saline (PBS) or C2 toxin at 37°C. At the indicated time points, the β-hexosaminidase activity in 200-μl supernatant samples was measured by incubation with 200 μl of 1 mM p-nitrophenyl N-acetyl-β-d-glucosaminide in 0.1 M citrate buffer (0.05 M citric acid, 0.05 M sodium citrate [pH 4.5]) for 1 h at 37°C. Reactions were terminated by the addition of 400 μl 0.1 M sodium carbonate buffer (0.1 M Na2CO3 and 0.1 M NaHCO3 [pH 9.8]) to the mixture. The absorbance was read at 405 nm. Enzyme activities are expressed as a percentage of the total enzyme activity found in the supernatant and lysate prepared by using 1% Triton X-100.
Immunofluorescence analysis.
For immunofluorescence staining of the cells with antibodies, MDCK cells were fixed with 4% paraformaldehyde for 15 min at room temperature after incubation with C2 toxin. Fixed cells were quenched with 15 mM NH4Cl for 15 min and incubated with PBS containing 0.1% Triton X-100 for 20 min at room temperature. Cells were then blocked with PBS containing 4% BSA for 1 h. For C2IIa staining, cells were incubated with rabbit anti-C2II antibody at 4°C overnight. The cells were rinsed again and incubated with Alexa Fluor 568-conjugated anti-rabbit IgG in PBS containing 4% BSA at room temperature for 1 h. The cell nuclei were stained with 0.4 μg/ml DAPI for 30 min (7). For ceramide staining, cells were incubated with mouse anticeramide IgM monoclonal antibody for 1 h. Cells were rinsed and then incubated with anti-mouse IgM Alexa Fluor 594 secondary antibody for 1 h. For ASMase staining, cells were incubated with rabbit anti-acid sphingomyelinase (H-181) antibody for 1 h. Cells were rinsed and then incubated with Alexa Fluor 568-conjugated anti-rabbit IgG antibody for 1 h. The cells were visualized by using a Nikon A1 laser scanning confocal microscope (Nikon, Tokyo, Japan).
Level of ceramides.
MDCK cells (2 × 107 cells/ml) were treated with C2IIa at 37°C. After incubation, the reaction was terminated by the addition of chloroform-methanol-acetic acid-water (25:15:4:2, vol/vol/vol/vol) to the mixture, and ceramide was extracted (34). Ceramide was phosphorylated by 1,2-diacylglycerol kinase (Merck Millipore, Tokyo, Japan) in the presence of [γ-32P]ATP (PerkinElmer, Tokyo, Japan), separated by thin-layer chromatography (TLC), and analyzed by autoradiography according to a previously described method (34).
Small interfering RNA transfection.
siRNAs for sphingomyelin phosphodiesterase 1 (SMPD1) and siRNA negative controls were obtained from Qiagen (Tokyo, Japan). In the knockdown procedure, MDCK cells were mixed with siRNA (5 × 106 cells plus 500 pmol siRNA) and then electroporated by using a Neon transfection system (Life Technologies) in accordance with the manufacturer's instructions. After electroporation, cells were transferred to 24-well plates containing prewarmed antibiotic-free medium and incubated at 37°C. Experiments were carried out 48 h after transfection with siRNAs (36).
Statistical analysis.
All statistical analyses were performed with Easy R (Saitama Medical Center, Jichi Medical University) (37). Differences between two groups were evaluated by using two-tailed Student's t test. One-way analysis of variance (ANOVA) followed by the Tukey test was used to evaluate differences among three or more groups. Differences were considered to be significant at P values of <0.01.
ACKNOWLEDGMENTS
We thank K. Kashiwazaki and H. Segawa for technical assistance.
This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, MEXT.SENRYAKU, 2015.
We declare no competing financial interests.
REFERENCES
- 1.Aktories K, Lang AE, Schwan C, Mannherz HG. 2011. Actin as target for modification by bacterial protein toxins. FEBS J 278:4526–4543. doi: 10.1111/j.1742-4658.2011.08113.x. [DOI] [PubMed] [Google Scholar]
- 2.Aktories K, Schwan C, Papatheodorou P, Lang AE. 2012. Bidirectional attack on the actin cytoskeleton. Bacterial protein toxins causing polymerization or depolymerization of actin. Toxicon 60:572–581. doi: 10.1016/j.toxicon.2012.04.338. [DOI] [PubMed] [Google Scholar]
- 3.Stiles BG, Pradhan K, Fleming JM, Samy RP, Barth H, Popoff MR. 2014. Clostridium and Bacillus binary enterotoxins: bad for the bowels, and eukaryotic being. Toxins (Basel) 6:2626–2656. doi: 10.3390/toxins6092626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knapp O, Benz R, Popoff MR. 2016. Pore-forming activity of clostridial binary toxins. Biochim Biophys Acta 1858:512–525. doi: 10.1016/j.bbamem.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Eckhardt M, Barth H, Blöcker D, Aktories K. 2000. Binding of Clostridium botulinum C2 toxin to asparagine-linked complex and hybrid carbohydrates. J Biol Chem 275:2328–2334. doi: 10.1074/jbc.275.4.2328. [DOI] [PubMed] [Google Scholar]
- 6.Nagahama M, Hagiyama T, Kojima T, Aoyanagi K, Takahashi C, Oda M, Sakaguchi Y, Oguma K, Sakurai J. 2009. Binding and internalization of Clostridium botulinum C2 toxin. Infect Immun 77:5139–5148. doi: 10.1128/IAI.00638-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagahama M, Takahashi C, Aoyanagi K, Tashiro R, Kobayashi K, Sakaguchi Y, Ishidoh K, Sakurai J. 2014. Intracellular trafficking of Clostridium botulinum C2 toxin. Toxicon 82:76–82. doi: 10.1016/j.toxicon.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Haug G, Leemhuis J, Tiemann D, Meyer DK, Aktories K, Barth H. 2003. The host cell chaperone Hsp90 is essential for translocation of the binary Clostridium botulinum C2 toxin into the cytosol. J Biol Chem 278:32266–32274. doi: 10.1074/jbc.M303980200. [DOI] [PubMed] [Google Scholar]
- 9.Kaiser E, Pust S, Kroll C, Barth H. 2009. Cyclophilin A facilitates translocation of the Clostridium botulinum C2 toxin across membranes of acidified endosomes into the cytosol of mammalian cells. Cell Microbiol 11:780–795. doi: 10.1111/j.1462-5822.2009.01291.x. [DOI] [PubMed] [Google Scholar]
- 10.Kaiser E, Böhm N, Ernst K, Langer S, Schwan C, Aktories K, Popoff M, Fischer G, Barth H. 2012. FK506-binding protein 51 interacts with Clostridium botulinum C2 toxin and FK506 inhibits membrane translocation of the toxin in mammalian cells. Cell Microbiol 14:1193–1205. doi: 10.1111/j.1462-5822.2012.01788.x. [DOI] [PubMed] [Google Scholar]
- 11.Nagahama M, Umezaki M, Oda M, Kobayashi K, Tone S, Suda T, Ishidoh K, Sakurai J. 2011. Clostridium perfringens iota-toxin b induces rapid cell necrosis. Infect Immun 79:4353–4360. doi: 10.1128/IAI.05677-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Idone V, Tam C, Andrews NW. 2008. Two-way traffic on the road to plasma membrane repair. Trends Cell Biol 18:552–559. doi: 10.1016/j.tcb.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Los FCO, Randis TM, Aroian RV, Ratner AJ. 2013. Role of pore-forming toxins in bacterial infectious diseases. Microbiol Mol Biol Rev 77:173–207. doi: 10.1128/MMBR.00052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. 2008. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol 180:905–914. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, Schuchman E, Tabas I, Andrews NW. 2010. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol 189:1027–1038. doi: 10.1083/jcb.201003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corrotte M, Fernandes MC, Tam C, Andrews NW. 2012. Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic 13:483–494. doi: 10.1111/j.1600-0854.2011.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews NW, Almeida PE, Corrotte M. 2014. Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol 24:734–742. doi: 10.1016/j.tcb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grassmé H, Jendrossek V, Riehle A, von Kürthy G, Berger J, Schwarz H, Weller M, Kolesnick R, Gulbins E. 2003. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat Med 9:322–330. doi: 10.1038/nm823. [DOI] [PubMed] [Google Scholar]
- 19.Fernandes MC, Cortez M, Flannery AR, Tam C, Mortara RA, Andrews NW. 2011. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J Exp Med 208:909–921. doi: 10.1084/jem.20102518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. 2012. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol 86:7473–7483. doi: 10.1128/JVI.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simonis A, Hebling S, Gulbins E, Schneider-Schaulies S, Schubert-Unkmeir A. 2014. Differential activation of acid sphingomyelinase and ceramide release determines invasiveness of Neisseria meningitidis into brain endothelial cells. PLoS Pathog 10:e1004160. doi: 10.1371/journal.ppat.1004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baron R, Neff L, Louvard D, Courtoy PJ. 1985. Cell-mediated extracellular acidification and bone resorption: evidence for a low pH in resorbing lacunae and localization of a 100-kD lysosomal membrane protein at the osteoclast ruffled border. J Cell Biol 101:2210–2222. doi: 10.1083/jcb.101.6.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linke T, Wilkening G, Lansmann S, Moczall H, Bartelsen O, Weisgerber J, Sandhoff K. 2001. Stimulation of acid sphingomyelinase activity by lysosomal lipids and sphingolipid activator proteins. Biol Chem 382:283–290. [DOI] [PubMed] [Google Scholar]
- 24.Stancevic B, Kolesnick R. 2010. Ceramide-rich platforms in transmembrane signaling. FEBS Lett 584:1728–1740. doi: 10.1016/j.febslet.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gómez-Muñoz A. 2006. Ceramide 1-phosphate/ceramide, a switch between life and death. Biochim Biophys Acta 1758:2049–2056. doi: 10.1016/j.bbamem.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Muñoz A, Gangoiti P, Arana L, Ouro A, Rivera IG, Ordoñez M, Trueba M. 2013. New insights on the role of ceramide 1-phosphate in inflammation. Biochim Biophys Acta 1831:1060–1066. doi: 10.1016/j.bbalip.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Gómez-Muñoz A, Kong JY, Parhar K, Wang SW, Gangoiti P, González M, Eivemark S, Salh B, Duronio V, Steinbrecher UP. 2005. Ceramide-1-phosphate promotes cell survival through activation of the phosphatidylinositol 3-kinase/protein kinase B pathway. FEBS Lett 579:3744–3750. doi: 10.1016/j.febslet.2005.05.067. [DOI] [PubMed] [Google Scholar]
- 28.Rivera IG, Ordoñez M, Presa N, Gomez-Larrauri A, Simón J, Trueba M, Gomez-Muñoz A. 2015. Sphingomyelinase D/ceramide 1-phosphate in cell survival and inflammation. Toxins (Basel) 7:1457–1466. doi: 10.3390/toxins7051457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmid A, Benz R, Just I, Aktories K. 1991. Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes. Formation of cation-selective channels and inhibition of channel function by chloroquine. J Biol Chem 269:16706–16711. [PubMed] [Google Scholar]
- 30.Barth H, Blocker D, Behlke J, Bergsma-Schutter W, Brisson A, Benz R, Aktories K. 2000. Cellular uptake of Clostridium botulinum C2 toxin requires oligomerization and acidification. J Biol Chem 275:18704–18711. doi: 10.1074/jbc.M000596200. [DOI] [PubMed] [Google Scholar]
- 31.Bachmeyer C, Benz R, Barth H, Aktories K, Gilbert M, Popoff MR. 2001. Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes and Vero cells: inhibition of channel function by chloroquine and related compounds in vitro and intoxification in vivo. FASEB J 15:1658–1660. [DOI] [PubMed] [Google Scholar]
- 32.Hale ML, Marvaud JC, Popoff MR, Stiles BG. 2004. Detergent-resistant membrane microdomains facilitate Ib oligomer formation and biological activity of Clostridium perfringens iota-toxin. Infect Immun 72:2186–2193. doi: 10.1128/IAI.72.4.2186-2193.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagahama M, Yamaguchi A, Hagiyama T, Ohkubo N, Kobayashi K, Sakurai J. 2004. Binding and internalization of Clostridium perfringens iota-toxin in lipid rafts. Infect Immun 72:3267–3275. doi: 10.1128/IAI.72.6.3267-3275.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takagishi T, Oda M, Takehara M, Kobayashi K, Nagahama M. 2016. Oligomer formation of Clostridium perfringens epsilon-toxin is induced by activation of neutral sphingomyelinase. Biochim Biophys Acta 1858:2681–2688. doi: 10.1016/j.bbamem.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Oda M, Fujita A, Okui K, Miyamoto K, Shibutani M, Takagishi T, Nagahama M. 2013. Bacillus cereus sphingomyelinase recognizes ganglioside GM3. Biochem Biophys Res Commun 431:164–168. doi: 10.1016/j.bbrc.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Nagahama M, Seike S, Shirai H, Takagishi T, Kobayashi K, Takehara M, Sakurai J. 2015. Role of P2X7 receptor in Clostridium perfringens beta-toxin-mediated cellular injury. Biochim Biophys Acta 1850:2159–2167. doi: 10.1016/j.bbagen.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Kanda Y. 2013. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant 48:452–458. doi: 10.1038/bmt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]