

ABSTRACT
Insights into the host-microbial virulence factor interaction, especially the immune signaling mechanisms, could provide novel prevention and treatment options for pneumococcal diseases. Streptococcus pneumoniae endopeptidase O (PepO) is a newly discovered and ubiquitously expressed pneumococcal virulence protein. A PepO-mutant strain showed impaired adherence to and invasion of host cells compared with the isogenic wild-type strain. It is still unknown whether PepO is involved in the host defense response to pneumococcal infection. Here, we demonstrated that PepO could enhance phagocytosis of Streptococcus pneumoniae and Staphylococcus aureus by peritoneal exudate macrophages (PEMs). Further studies showed that PepO stimulation upregulated the expression of microRNA-155 (miR-155) in PEMs in a time- and dose-dependent manner. PepO-induced enhanced phagocytosis was decreased in cells transfected with an inhibitor of miR-155, while it was increased in cells transfected with a mimic of miR-155. We also revealed that PepO-induced upregulation of miR-155 in PEMs was mediated by Toll-like receptor 2 (TLR2)–NF-κB signaling and that the increased expression of miR-155 downregulated expression of SHIP1. Taken together, these results indicate that PepO induces upregulation of miR-155 in PEMs, contributing to enhanced phagocytosis and host defense response to pneumococci and Staphylococcus aureus.
KEYWORDS: PepO, TLR2, macrophages, miR-155, phagocytosis
INTRODUCTION
Streptococcus pneumoniae endopeptidase O (PepO) is a newly discovered and ubiquitously expressed virulence protein. Through binding to plasminogen, fibronectin, and complement component C1q, PepO helps pneumococci adhere to host cells (1). Furthermore, PepO modulates the complement attack by binding to C1q and the classical complement pathway inhibitor C4BP (2). It is still unknown whether PepO is involved in the host defense response to pneumococcal infection. Further research is needed to clarify the interaction between PepO and host immune cells.
Alveolar macrophages are important players in the pulmonary immune system (3–8). During pneumococcal infection, clearance of pneumococci is initiated by macrophages, not neutrophils. Recruited macrophages utilize a variety of mechanisms to control invading pneumococci. Decreased macrophage trafficking in infants results in delayed clearance of pneumococcal colonization (9). Reducing the bactericidal function of alveolar macrophages with glucocorticoid leads to inhibition of pulmonary pneumococcal clearance in mice (10), while promoting CCL2-mediated macrophage recruitment with macrolides accelerates clearance of nasopharyngeal pneumococcal colonization in mice (11). Although the significance of macrophages in clearance of pneumococci has been well established, the related molecular mechanisms remain poorly understood.
MicroRNAs are a class of noncoding small RNAs complementary to the noncoding region of mRNA transcripts. Through promoting the degradation or preventing the translation of mRNAs (12–16), microRNAs regulate many aspects of physiology and pathology processes of mammals (17, 18). Increasing evidence shows that a number of microRNAs participate in modulation of host defense response against Gram-positive and Gram-negative bacterial infection. As a canonical multifunctional microRNA, miR-155 plays diverse roles in the host defense response to microbial infection (17). Francisella novicida-induced increased expression of miR-155 results in decreased SH2 domain-containing inositol phosphatase (SHIP) expression and enhanced proinflammatory cytokine production (19). Mycobacterium tuberculosis virulence-associated secreted protein ESAT-6-induced upregulation of miR-155 inhibits production of cyclooxygenase 2 (Cox-2) and interleukin-6 (IL-6), which promotes Mycobacterium tuberculosis survival (20). A recent study on Streptococcus pneumoniae shows that miR-155 is also required for clearance of pneumococci from the nasopharynx (21). However, the related molecular mechanism is still unclear.
It is well reported that macrophages recognize microbial components to modulate expression of miR-155. Lipopolysaccharide (LPS) from Gram-negative bacteria and lipoprotein from Gram-positive bacteria upregulate expression of miR-155 via binding to Toll-like receptor 4 (TLR4) and TLR2, respectively (17, 22). Borna disease virus-encoded phosphoprotein inhibits expression of miR-155 (23). What kind of pneumococcal components are involved in the regulation of miR-155 is still unclear. We demonstrated that PepO promoted phagocytosis of pneumococci and Staphylococcus aureus by macrophages. Further research revealed that PepO induced increased expression of miR-155 in a TLR2–NF-κB-dependent manner and that the upregulation of miR-155 downregulated SHIP1, which suggests that PepO may be a novel ligand of TLR2.
RESULTS
PepO enhanced phagocytosis of Staphylococcus aureus and Streptococcus pneumoniae by PEMs.
Phagocytosis assays were performed to investigate the effect of PepO on macrophages. As shown in Fig. 1A and C, phagocytosis of Staphylococcus aureus was increased in PepO-treated peritoneal exudate macrophages (PEMs) in a time-dependent manner, with the maximum bacterial count occurring in cells stimulated with PepO for 24 h. A protease K-digested or heat-killed PepO preparation failed to enhance phagocytosis of Staphylococcus aureus by PEMs (Fig. 1A). Recombinant ΔA146Ply, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and MarR, which were also purified on a Ni2+-charged column, had no effect on the phagocytosis of bacteria by PEMs (Fig. 1B). These results suggested a role for PepO in modulating macrophage phagocytosis. Figure 1D shows that fluorescein isothiocyanate (FITC)-labeled pneumococci were increased in PepO-treated PEMs in a time-dependent manner. We also performed experiments to discriminate between extracellular and intracellular bacteria. As shown in Fig. 1E, there was no significant difference in bacterial counts between antibiotic-incubated and non-antibiotic-treated macrophages. Most bacteria cannot be reached by anti-Streptococcus pneumoniae antibody without permeabilization of macrophages (Fig. 1F), which suggested that at 30 min of incubation most bacteria have been ingested by macrophages in our model. With the same amount of protein stimulation, attached and ingested bacteria increased as incubation time was prolonged, which excluded the possibility that protein stimulation enhanced phagocytosis of bacteria by macrophages through binding to them.
FIG 1.

PepO enhanced phagocytosis of Staphylococcus aureus and Streptococcus pneumoniae by PEMs. (A) After pretreatment with PepO (1 μg/ml) or heat-killed (HK) or protease K (PK)-digested PepO for 24 h, PEMs were incubated with Staphylococcus aureus at a multiplicity of infection of 100 and the phagocytic bacteria were enumerated. Protease K was inactivated after treatment. (B and C) Staphylococcus aureus bacteria phagocytosed by cells stimulated with PepO (1 μg/ml), ΔA146Ply (1 μg/ml), GAPDH (Gap, 1 μg/ml), or MarR (1 μg/ml) for 24 h (B) or with PepO for indicated times (C) were counted. (D) After stimulation with PepO for indicated times, PEMs were incubated with FITC-labeled Streptococcus pneumoniae at a multiplicity of infection of 100 and phagocytosis was assessed by fluorescence microscopy. Representative pictures are shown, and the phagocytosis percentages are from three independent experiments. (E) Staphylococcus aureus in antibiotic-incubated or non-antibiotic-treated PepO-stimulated macrophages was assessed by dilution plating. Amp, ampicillin; Gm, gentamicin. (F) PepO-stimulated macrophages were incubated with unlabeled D39, and bacteria were stained using specific anti-Streptococcus pneumoniae antibody following Alexa Fluor 488-conjugated secondary antibody with or without permeabilization of macrophages. The attached or phagocytic bacteria were assessed by fluorescence microscopy. The data in panels A to E are shown as the mean ± SD (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
PepO-induced enhanced phagocytosis relied on the upregulation of miR-155 in PEMs.
We speculated that PepO might influence phagocytosis through the alteration of macrophage signaling pathways. miR-155 plays an important role in host defense response, and bacterial components can mediate the expression of miR-155. As shown in Fig. 2A, miR-155 was upregulated in PepO-stimulated PEMs, while miR-146a and miR-27a were not. PepO mediated the upregulation of miR-155 in a time- and dose-dependent manner (Fig. 2D and E). The final concentration of LPS in PepO preparation was under 0.1 endotoxin units (EU)/ml. As shown in Fig. 2B, expression of miR-155 induced by protease K-digested PepO was significantly less than that induced by untreated PepO, while there was no significant difference between protease K-digested LPS- and LPS-induced expression of miR-155, which suggested that the effect of protein preparation on expression of miR-155 was dependent on PepO, not the contaminated LPS. Furthermore, recombinant ΔA146Ply, GAPDH, and MarR had no effect on the expression of miR-155 (Fig. 2C), which excludes the contamination of proteins from Escherichia coli. Expression of miR-155 increased in a time- and dose-dependent manner, which suggested that PepO stimulation changed signal transduction leading to enhanced phagocytosis of bacteria by macrophages. To investigate whether miR-155 participates in the regulation of phagocytosis by PEMs, mimics or inhibitors of miR-155 were transfected into cells. The efficacy of miR-155 mimics and inhibitors was quantified by showing the fold change of miR-155 with subsequent stimulation with PepO (Fig. 3A). PepO-induced phagocytosis of Staphylococcus aureus was significantly augmented in cells transfected with a mimic of miR-155, while it was decreased in cells transfected with an inhibitor of miR-155. Phagocytosis of Staphylococcus aureus was also enhanced in cells merely transfected with a mimic of miR-155, indicating that miR-155 can enhance phagocytosis by macrophages (Fig. 3B). In addition, PepO-induced phagocytosis of FITC-labeled pneumococci was also dramatically enhanced in cells transfected with a mimic of miR-155, while it was decreased in cells transfected with an inhibitor of miR-155 (Fig. 3C and D). Taken together, these results demonstrate that PepO-induced expression of miR-155 regulated the phagocytosis of bacteria by PEMs.
FIG 2.

PepO induced increased expression of miR-155 in PEMs. (A) Quantitative PCR analysis was used to determine the expression of miR-155, miR-146a, and miR-27a in PEMs treated with medium, LPS (100 ng/ml), peptidoglycan (PGN) (5 μg/ml), or PepO (1 μg/ml) for 6 h. (B) Cells were incubated with LPS (100 ng/ml), PepO (1 μg/ml), or protease K (PK)-digested LPS or PepO for 6 h, and the expression of miR-155 was measured by quantitative PCR analysis. Protease K was inactivated after treatment. (C to E) The expression of miR-155 in cells stimulated with PepO (1 μg/ml), ΔA146Ply (1 μg/ml), GAPDH (Gap, 1 μg/ml), or MarR (1 μg/ml) for 6 h (C) or with different doses of PepO for 6 h (D) or with PepO (1 μg/ml) for indicated times (E) was evaluated by PCR analysis. The data are shown as the mean ± SD (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
FIG 3.

PepO-induced enhanced phagocytosis relied on the upregulation of miR-155 in PEMs. (A) The fold change of miR-155 in macrophages transfected with inhibitors or mimics of miR-155 for 24 h with subsequent stimulation with PepO for another 24 h was assessed by PCR analysis. (B to D) Macrophages were transfected with inhibitors or mimics of miR-155 for 24 h and then stimulated with PepO or medium for another 24 h, after which they were incubated with Staphylococcus aureus (B) or FITC-labeled Streptococcus pneumoniae (C and D) at a multiplicity of infection of 100, and the phagocytic bacteria were enumerated. Panels C and D show representative pictures, and the phagocytosis percentages are from three independent experiments. The data are shown as the mean ± SD (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
PepO-induced upregulation of miR-155 downregulated expression of SHIP1.
It is well demonstrated that SHIP1 regulates phagocytosis by macrophages and that miR-155 can target a SHIP1 gene. In our model, SHIP1 transcript was decreased in PepO-stimulated PEMs in a time- and dose-dependent manner (Fig. 4A and B). Figure 4C shows that SHIP1 transcript was decreased in cells transfected with a mimic of miR-155 but was increased in cells transfected with an inhibitor of miR-155. To verify the interaction between miR-155 and the 3′ untranscribed region (UTR) of SHIP1, luciferase reporter gene analysis was performed. Luciferase activity of SHIP1 reporter plasmid was inhibited by miR-155 (Fig. 4D). These results suggested that PepO-induced upregulation of miR-155 inhibited expression of SHIP1.
FIG 4.

PepO-induced upregulation of miR-155 downregulated expression of SHIP1. (A and B) The SHIP1 transcripts in PEMs stimulated by different doses of PepO for 6 h (A) or by PepO (1 μg/ml) for different times (B) were analyzed by quantitative PCR. (C) PEMs were pretreated with or without an miR-155 mimic or inhibitor for 24 h and then incubated with medium or PepO (1 μg/ml) for another 6 h. PCR analysis was used to determine the expression of SHIP1. (D) PEMs were transfected with basic reporter plasmid or SHIP1 reporter plasmid for 6 h and then transfected with miR-155 or scramble microRNA (miRNA) for another 36 h. The luciferase activity of transfected cells was measured. The data are shown as the mean ± SD (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
PepO-induced upregulation of miR-155 was mediated by the TLR2 signaling pathway.
It is widely accepted that LPS from Gram-negative bacteria can be recognized by TLR4 and that peptidoglycan, teichoic acid, and lipoprotein from Gram-positive bacteria can be recognized by TLR2. Whether TLRs mediate PepO-induced enhanced phagocytosis by PEMs is still unknown. Our results showed that in TLR2-deficient PEMs, PepO failed to enhance phagocytosis of Staphylococcus aureus (Fig. 5A). Consistently, in TLR2-deficient cells PepO failed to stimulate the upregulation of miR-155 or the downregulation of SHIP1 (Fig. 5B and C), which implied that the effect of PepO on macrophages was mediated by TLR2. Complement receptor 3 (CR3), one of the most important phagocytosis-associated receptors on macrophages, is also negatively regulated by SHIP1 (24). As shown in Fig. 5D, PepO induced increased expression of CR3 in PEMs in a time-dependent manner, while it failed to upregulate the expression of CR3 in TLR2-deficient cells.
FIG 5.

PepO-induced upregulation of miR-155 was mediated by the TLR2 signaling pathway. (A) Wild-type (WT) or TLR2-deficient PEMs were stimulated with medium, LPS (100 ng/ml), or PepO (1 μg/ml) for 24 h and then incubated with Staphylococcus aureus at a multiplicity of infection of 100, and the phagocytic bacteria were enumerated. (B) WT or TLR2-deficient PEMs were stimulated with medium, LPS (100 ng/ml), peptidoglycan (PGN) (5 μg/ml), or PepO (1 μg/ml) for 6 h, after which miR-155 transcripts were determined by PCR analysis. (C and D) After treatment with PepO for indicated times, SHIP1 (C) and CR3 (D) expression in WT or TLR2-deficient cells was analyzed by Western blotting and immunofluorescence assays, respectively. (E) PEMs were pretreated with LY294002 (20 μM), BAY7085 (20 μM), SB203580 (20 μM), or U0126 (20 μM) for 1 h and then incubated with PepO for another 6 h. miR-155 transcripts were determined by quantitative PCR. (F) WT or TLR2-deficient cells were treated with PepO for indicated times. The activation of Akt was analyzed by Western blotting. The data are shown as the mean ± SD (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Other researchers have proved that phosphatidylinositol 3-kinase (PI3K)/Akt and NF-κB signaling mediate the expression of miR-155. Our results showed that PepO failed to upregulate the expression of miR-155 in PEMs pretreated with LY294002 (a PI3K inhibitor) or BAY7058 (an IκB-α phosphorylation inhibitor). PepO-induced upregulation of miR-155 was not suppressed by SB203580 (a p38 inhibitor) and U0126 (an extracellular signal-regulated kinase [ERK] inhibitor) (Fig. 5E). Western blot analysis also showed the rapid phosphorylation of p65 and Akt (Fig. 5F and 6B), which suggests that in our model PepO-induced upregulation of miR-155 is mediated by PI3K/Akt and NF-κB signaling. Figure 6A shows the translocation of p65 to the nucleus of PepO-treated wild-type (WT) cells. p65 was primarily present in the cytoplasm of unstimulated cells. In TLR2-deficient cells, PepO failed to induce the nuclear translocation of p65. Western blot analysis also showed that the phosphorylation of p65 occurred at 15 min of stimulation and was sustained until 24 h of stimulation, while in TLR2-deficient cells, PepO failed to induce activation of p65 (Fig. 6B).
FIG 6.

PepO induces nuclear translocation and phosphorylation of p65 in a TLR2-dependent manner. Immunofluorescence (A) and Western blot (B) assays were performed to determine the translocation and phosphorylation of NF-κB p65 in WT or TLR2-deficient PEMs. Representative blots of three independent experiments with consistent outcomes are shown. For panel A, arrows indicate cells where nuclear translocation has occurred. Additionally, 100 cells were examined and percentages of cells that showed evidence of nuclear translocation are indicated. The data are shown as the mean ± SD (n = 3). ***, P < 0.001; ns, not significant.
Interestingly, there was no significant difference in activation of Akt between WT and TLR2-deficient cells (Fig. 5F), which suggested that other receptors may mediate PepO-induced Akt activation. To verify it, direct binding experiments were performed. Figure 7A and B show that PepO could bind to the surface of WT and TLR2- and TLR4-deficient macrophages but that the PepO level on TLR2- or TLR4-deficient cells is less than that on WT cells. These results indicated that both TLR2 and TLR4 mediated recognition of PepO. Our previous work also has shown that PepO elicits cytokine production and cellular infiltration in mice partially through TLR2 and TLR4 signaling pathways (25). As shown in Fig. 5B, in this model PepO-induced overexpression of miR-155 was mediated only by TLR2 signaling. These results suggested that different responses to PepO stimuli were mediated by different signaling pathways.
FIG 7.
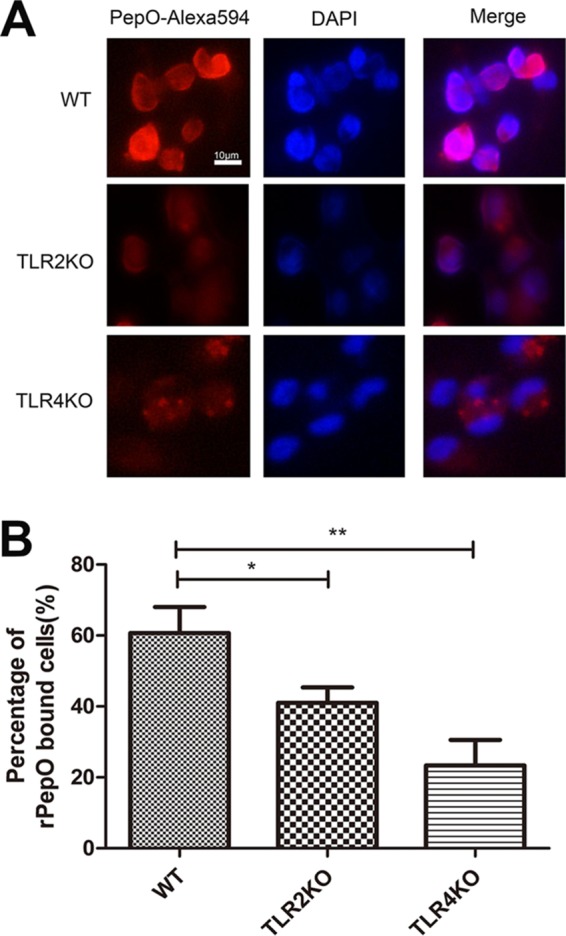
PepO binds to WT or TLR2- or TLR4-deficient macrophages. (A) WT or TLR2- or TLR4-deficient macrophages were treated with PepO (1 μg/ml) for 1 h, fixed, and stained with Alexa 594-conjugated anti-His antibody and DAPI. Representative fluorescence pictures are shown. (B) Recombinant PepO (rPepO)-bound cells in 10 high-power fields were counted, and the percentages of rPepO-bound cells were calculated. The data are shown as the mean ± SD (n = 3). *, P < 0.05; **, P < 0.01.
DISCUSSION
The significance of macrophages in clearance of nasopharyngeal pneumococcal colonization has been well established, but the related regulatory mechanisms remain poorly understood. miR-155 plays diverse roles in the host defense response to microbial infection. In Pseudomonas aeruginosa-induced keratitis, miR-155 inhibits macrophage-mediated phagocytosis and intracellular killing to suppress bacterial clearance by targeting Rheb (26), while in tuberculosis miR-155 promotes autophagy to clear intracellular mycobacteria through targeting Rheb (27). Pathways for Pseudomonas aeruginosa and mycobacteria are different, maybe because Pseudomonas aeruginosa is an extracellular bacterium while mycobacteria are intracellular bacteria. Our results showed that PepO enhanced phagocytosis and expression of miR-155 in macrophages in a time-dependent manner. In our model, miR-155 promoted PepO-induced clearance of bacteria by macrophages. PepO-induced phagocytosis was increased in cells transfected with a mimic of miR-155, while it was decreased in cells transfected with an inhibitor of miR-155. This is the first demonstration that the pneumococcal component PepO contributes to phagocytosis by macrophages via modulating expression of miR-155. Further research is still needed to investigate whether Rheb is involved in this process.
Other researchers have proved that miR-155 participates in regulation of phagocytosis (28–30), but the related mechanism is still unclear. Maybe different signaling mechanisms are utilized to respond to different stimuli. In our model, we detected the activation of the PI3K/Akt signaling pathway. It is well known that SHIP1 is a negative regulator of PI3K/Akt signaling (31–34), and SHIP1 plays an important role in modulation of phagocytosis by macrophages (35, 36). Our results demonstrated that SHIP1 was downregulated in PepO-stimulated macrophages in an miR-155-dependent manner. In our research, it is still unknown how SHIP1 downregulation results in enhanced phagocytosis by macrophages. Researchers have proved that the effect of SHIP on regulation of phagocytosis is mediated by Fc gamma receptors and complement receptor 3 (CR3) (36). Opsonophagocytosis mediated by Fc gamma receptors is IgG dependent (37–39), and phagocytosis mediated by CR3 is complement dependent. There is no IgG or complement involved in PepO-induced phagocytosis by macrophages. Increasing evidence shows that CR3 is also a multifunctional pattern recognition receptor of the innate immune system (40–43). Once activated by its ligand, CR3 mediates a series of signaling transductions, including rearrangement of actin, which promotes phagocytosis of bacteria by macrophages and contributes to the host defense response to bacterial infection. In our model, we detected the upregulation of CR3 in PepO-stimulated macrophages in a time-dependent manner. We hypothesize that PepO-induced upregulation of miR-155 inhibits expression of SHIP1 and SHIP1 downregulation leads to augmented activity of CR3, which promotes phagocytosis of bacteria by macrophages (Fig. 8). The upregulation of CR3 in some way supports our hypothesis. Further research is still needed to investigate the direct regulatory role of SHIP1 in activity of CR3, as well as the regulatory role of CR3 in PepO-induced enhanced phagocytosis.
FIG 8.

Schematic model demonstrating PepO-induced signaling transduction in macrophages. PepO induces upregulation of miR-155 in a TLR2-, NF-κB-, and Akt-dependent manner. miR-155 inhibits expression of SHIP1, and SHIP1 downregulation leads to augmented activity of CR3, which may promote phagocytosis of bacteria by macrophages.
TLRs play an important role in the host innate immune system. A variety of pathogen-associated molecular patterns (PAMPs) can be recognized by TLRs. It is widely accepted that LPS from Gram-negative bacteria is recognized by TLR4 (44) and that peptidoglycan, teichoic acid, and lipoprotein are recognized by TLR2 (45–47). In the context of Streptococcus pneumoniae, some virulence proteins (RrgA pneumococcal pilus type 1 protein, GHIP, et al.) bind to TLR2 (48, 49) and Ply binds to TLR4 (50). Our results demonstrated that PepO-induced miR-155 upregulation relied on TLR2. In TLR2-deficient PEMs, PepO failed to modulate the expression of miR-155, SHIP1, and CR3, as well as phagocytosis. These results imply that PepO enhances phagocytosis by macrophages in a TLR2- and miR-155-dependent manner. PepO-induced upregulation of miR-155 was blocked by a PI3K inhibitor and an IκB-α phosphorylation inhibitor. PepO failed to stimulate the phosphorylation of p65 in TLR2-deficient cells, which proves that PepO induces the expression of miR-155 via a TLR2–NF-κB signaling pathway. We are still exploring whether PepO is a direct ligand of TLR2.
Agarwal et al. have proved that PepO is highly conserved within pneumococci and other Streptococcus species (1). To date, the enzyme has not been identified in any high-throughput screens or vaccine studies. The present report is the first demonstration that PepO enhances phagocytosis of pneumococci and Staphylococcus aureus by macrophages in a TLR2- and miR-155-dependent manner. As a virulence protein, PepO also plays an important role in the host defense response to pneumococcal infection, which may contribute to the prevention and treatment of pneumococcal diseases.
MATERIALS AND METHODS
Reagents.
The Ni2+-charged column chromatograph was bought from GE Healthcare (Buckinghamshire, United Kingdom). Polymyxin B agarose used for removing lipopolysaccharide (LPS) in the protein preparation was purchased from Sigma Corp. (Santa Clara, CA). Mouse antiactin monoclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and rabbit anti-phospho-Akt, anti-phospho-p65, and anti-Akt monoclonal antibodies were from Cell Signaling Technology (Beverly, MA). Mouse anti-SHIP1 polyclonal antibody was bought from Millipore Corp. (Bedford, MA). IκB-α phosphorylation inhibitor BAY7085, phosphatidylinositol 3-OH kinase (PI3K) inhibitor LY294002, p38 mitogen-activated protein kinase (MAPK) inhibitor SB203580, and extracellular signal-regulated kinase inhibitor U0126 were bought from Cell Signaling Technology.
Preparation of recombinant pneumococcal proteins.
In brief, the amplified full-length genes for PepO, ΔA146Ply, GAPDH, and MarR were cloned in pET28a (Novagen) for protein expression, after which N-terminal His6-tagged pneumococcal proteins (PepO, ΔA146Ply [51], GAPDH, and MarR) were expressed in E. coli BL21(DE3) (Stratagene) and purified with the use of a Ni2+-charged column chromatograph according to the manufacturer's instructions. Polymyxin B agarose was used to remove LPS in the protein preparation as much as possible. Residual LPS in the protein preparation was measured by Limulus amoebocyte lysate (LAL) assay (Lonza), according to the manufacturer's instructions.
Bacterial strains.
Streptococcus pneumoniae strain D39 was purchased from the American Type Culture Collection (ATCC) and cultured in C+Y medium in 5% CO2 at 37°C for 5 h. Staphylococcus aureus was obtained from the ATCC and seeded on Columbia sheep blood agar in 5% CO2 at 37°C overnight.
Mice.
TLR2-deficient (TLR2-knockout [TLRKO]) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All mice used are on a C57BL/6 background. All experimental protocols involving animals were approved by the Animal Ethics and Research Committee of China University Graduate School of Medicine.
Cell culture.
PEMs were isolated from 6- to 8-week-old C57 or TLR2KO mice with intraperitoneal injection of 1 ml paroline 4 days prior to the experiment. Cells were washed and seeded on a 24-well plate (5 × 105/well) or a 6-well plate (1 × 106/well). After removal of nonadherent cells, adherent cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% penicillin-streptomycin and 10% fetal bovine serum (FBS) (HyClone, Barrington, IL, USA) and stimulated as described below.
Quantitative PCR (Q-PCR) analysis.
Total RNA was isolated from PEMs using RNAiso Plus reagent (TaKaRa) according to the manufacturer's instructions. The sequences of primers are listed in Table 1. For quantitative analysis, a Primer PCR SYBR green assay kit (TaKaRa) was used in a Bio-Rad real-time PCR machine. The annealing temperature and extension time for PCR analysis were 60°C and 10 s, respectively. The gene for GAPDH, U6, was amplified as an endogenous reference. Both relative threshold cycle (ΔΔCT) and a standard curve were used for determining quantification.
TABLE 1.
Primers used in this study
| Primer name | Nucleotide sequence (5′–3′) |
|---|---|
| U6 forward | CTCGCTTCGGCAGCACA |
| U6 reverse | AACGCTTCACGAATTTGCGT |
| miR-155 forward | GGCGTTAATGCTAATTGTGAT |
| miR-155 reverse | GTGCAGGGTCCGAGGT |
| miR-155 mimic | UUAAUGCUAAUUGUGAUAGGGGU |
| CCCUAUCACAAUUAGCAUUAAUU | |
| miR-155 inhibitor | ACCCCUAUCACAAUUAGCAUUAA |
| Mus GAPDH forward | AGGTCGGTGTGAACGGATTTG |
| Mus GAPDH reverse | GGGGTCGTTGATGGCAACA |
| SHIP1 forward | TCCAAGAATGGTCCTGGCAC |
| SHIP1 reverse | CAAACCGTACCACCAGCTCT |
| miR-146a forward | GGCCTGAGAACTGAATTCCA |
| miR-146a reverse | GTGCAGGGTCCGAGGT |
| miR-27a forward | GCAGGGCTTAGCTGCTTG |
| miR-27a reverse | GTGCAGGGTCCGAGGT |
Cell transfection and reporter gene assay.
The miR-155 mimic, miR-155 inhibitor, control mimic, and control inhibitor were purchased from GenePharma (Shanghai, China). These RNA oligomers and plasmid were transfected into PEMs using Lipofectamine 2000 (Invitrogen). Methods for transfection and luciferase reporter assays followed the manufacturer's instructions. For transfection of PEMs, liposome-DNA complexes were formed by incubating plasmid DNA with Lipofectamine 2000 with appropriate proportions and fixed volumes for 20 min. The Lipofectamine-DNA mixture was added to cells in antibiotic-free medium later. After 24 h of incubation, cells were then exposed to PepO. Luciferase activities of cell lysates were normalized to protein content.
Western blot analysis.
After washing with prechilled phosphate-buffered saline (PBS), PEMs in a 24-well plate were lysed with 0.2 ml lysis buffer (radioimmunoprecipitation assay [RIPA] buffer containing phosphorylase inhibitor and protease inhibitor mixed with SDS loading buffer). After cell debris was removed, protein extract was boiled for 5 min and then centrifuged at 12,000 × g for 5 min. Before transfer to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA), an equal volume of protein (10 μg) was subjected to 10% SDS-PAGE. The membrane was blocked with 5% defatted milk for 2 h at 37°C and incubated with the indicated antibody at 4°C overnight. After being washed three times, the membrane was incubated with corresponding horseradish peroxidase-marked secondary goat anti-rabbit or goat anti-mouse antibodies for 1 h at 37°C. An enhanced chemiluminescence (ECL) detection system was used to detect antibody-antigen complexes.
Immunofluorescence assays.
PEMs cultured on glass coverslips were stimulated with PepO for indicated times. After fixation with 4% paraformaldehyde, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min, washed with PBS, blocked with 2% bovine serum albumin (BSA) in PBS for 2 h, and incubated with rabbit anti-phospho-p65 or anti-CR3 antibody (1:100 dilution) at 4°C overnight, followed by 3 washing procedures with PBS and incubation with Alexa Fluor 488- or 594-conjugated secondary antibody. Cells were then stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. A Nikon Eclipse 80i microscope equipped with a Nikon Intensilight C-HGFI was used to observe cell morphology and fluorescence intensity.
Phagocytosis assays.
Phagocytosis assays were performed in the presence of heat-inactivated (56°C, 30 min) serum. PepO-treated PEMs were infected with Staphylococcus aureus for 30 min at 37°C. After washing with prechilled PBS, cells were lysed with 0.2% Triton X-100. Cell lysates were diluted and seeded on Columbia sheep blood agar in 5% CO2 at 37°C overnight. Phagocytosis of Streptococcus pneumoniae was performed as described earlier (52–54). In brief, PepO-treated PEMs were infected with FITC-labeled D39 for 30 min at 37°C. After washing with prechilled PBS, macrophages were fixed with 4% paraformaldehyde and stained with DAPI. Phagocytosis was assessed by a Nikon Eclipse 80i microscope equipped with a Nikon Intensilight C-HGFI. Phagocytosis percentage was quantified by counting FITC-positive cells in 100 macrophages. To discriminate between extracellular and intracellular bacteria, attached Staphylococcus aureus cells were killed with the use of ampicillin (10 μg/ml) and gentamicin (200 μg/ml) for 20 min (Fig. 1E). In Fig. 1F, PepO-stimulated macrophages were incubated with unlabeled Streptococcus pneumoniae D39 and phagocytosed bacteria were stained using specific anti-Streptococcus pneumoniae antibody following Alexa Fluor 488-conjugated secondary antibody with or without permeabilization of macrophages.
Statistical analysis.
All data are shown as means ± standard deviations (SDs) from triple independent experiments. Differences between groups were determined by unpaired t test. Difference with P values of <0.05 were deemed significant. All analyses were implemented using the Statistical Package for Prism 5 statistical software (GraphPad Prism, La Jolla, CA, USA).
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (grant no. 81371778), the Project of Science and Technology of Chinese Ministry of Education (grant no. V201308), and the Project of Medical Scientific Research of Chongqing (grant no. 20142004).
We declare that no competing interests exist.
REFERENCES
- 1.Agarwal V, Kuchipudi A, Fulde M, Riesbeck K, Bergmann S, Blom AM. 2013. Streptococcus pneumoniae endopeptidase O (PepO) is a multifunctional plasminogen- and fibronectin-binding protein, facilitating evasion of innate immunity and invasion of host cells. J Biol Chem 288:6849–6863. doi: 10.1074/jbc.M112.405530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agarwal V, Sroka M, Fulde M, Bergmann S, Riesbeck K, Blom AM. 2014. Binding of Streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. J Biol Chem 289:15833–15844. doi: 10.1074/jbc.M113.530212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eichinger KM, Egana L, Orend JG, Resetar E, Anderson KB, Patel R, Empey KM. 2015. Alveolar macrophages support interferon gamma-mediated viral clearance in RSV-infected neonatal mice. Respir Res 16:122. doi: 10.1186/s12931-015-0282-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soltysiak KA, van Schaik EJ, Samuel JE. 2015. Surfactant protein D binds to Coxiella burnetii and results in a decrease in interactions with murine alveolar macrophages. PLoS One 10:e0136699. doi: 10.1371/journal.pone.0136699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis KM, Nakamura S, Weiser JN. 2011. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Invest 121:3666–3676. doi: 10.1172/JCI57761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorrington MG, Roche AM, Chauvin SE, Tu Z, Mossman KL, Weiser JN, Bowdish DM. 2013. MARCO is required for TLR2- and Nod2-mediated responses to Streptococcus pneumoniae and clearance of pneumococcal colonization in the murine nasopharynx. J Immunol 190:250–258. doi: 10.4049/jimmunol.1202113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemon JK, Miller MR, Weiser JN. 2015. Sensing of interleukin-1 cytokines during Streptococcus pneumoniae colonization contributes to macrophage recruitment and bacterial clearance. Infect Immun 83:3204–3212. doi: 10.1128/IAI.00224-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z, Clarke TB, Weiser JN. 2009. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest 119:1899–1909. doi: 10.1172/JCI36731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siegel SJ, Tamashiro E, Weiser JN. 2015. Clearance of pneumococcal colonization in infants is delayed through altered macrophage trafficking. PLoS Pathog 11:e1005004. doi: 10.1371/journal.ppat.1005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stolberg VR, McCubbrey AL, Freeman CM, Brown JP, Crudgington SW, Taitano SH, Saxton BL, Mancuso P, Curtis JL. 2015. Glucocorticoid-augmented efferocytosis inhibits pulmonary pneumococcal clearance in mice by reducing alveolar macrophage bactericidal function. J Immunol 195:174–184. doi: 10.4049/jimmunol.1402217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwanaga N, Nakamura S, Oshima K, Kajihara T, Takazono T, Miyazaki T, Izumikawa K, Yanagihara K, Sugawara A, Sunazuka T, Omura S, Kohno S. 2015. Macrolides promote CCL2-mediated macrophage recruitment and clearance of nasopharyngeal pneumococcal colonization in mice. J Infect Dis 212:1150–1159. doi: 10.1093/infdis/jiv157. [DOI] [PubMed] [Google Scholar]
- 12.Slezak-Prochazka I, Durmus S, Kroesen BJ, van den Berg A. 2010. MicroRNAs, macrocontrol: regulation of miRNA processing. RNA 16:1087–1095. doi: 10.1261/rna.1804410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He L, Hannon GJ. 2004. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 14.Huntzinger E, Izaurralde E. 2011. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 15.Friedman RC, Farh KK, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghildiyal M, Zamore PD. 2009. Small silencing RNAs: an expanding universe. Nat Rev Genet 10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staedel C, Darfeuille F. 2013. MicroRNAs and bacterial infection. Cell Microbiol 15:1496–1507. doi: 10.1111/cmi.12159. [DOI] [PubMed] [Google Scholar]
- 18.Eulalio A, Schulte L, Vogel J. 2012. The mammalian microRNA response to bacterial infections. RNA Biol 9:742–750. doi: 10.4161/rna.20018. [DOI] [PubMed] [Google Scholar]
- 19.Cremer TJ, Ravneberg DH, Clay CD, Piper-Hunter MG, Marsh CB, Elton TS, Gunn JS, Amer A, Kanneganti TD, Schlesinger LS, Butchar JP, Tridandapani S. 2009. MiR-155 induction by F. novicida but not the virulent F. tularensis results in SHIP down-regulation and enhanced pro-inflammatory cytokine response. PLoS One 4:e8508. doi: 10.1371/journal.pone.0008508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar R, Halder P, Sahu SK, Kumar M, Kumari M, Jana K, Ghosh Z, Sharma P, Kundu M, Basu J. 2012. Identification of a novel role of ESAT-6-dependent miR-155 induction during infection of macrophages with Mycobacterium tuberculosis. Cell Microbiol 14:1620–1631. doi: 10.1111/j.1462-5822.2012.01827.x. [DOI] [PubMed] [Google Scholar]
- 21.Verschoor CP, Dorrington MG, Novakowski KE, Kaiser J, Radford K, Nair P, Anipindi V, Kaushic C, Surette MG, Bowdish DM. 2014. MicroRNA-155 is required for clearance of Streptococcus pneumoniae from the nasopharynx. Infect Immun 82:4824–4833. doi: 10.1128/IAI.02251-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch M, Mollenkopf HJ, Klemm U, Meyer TF. 2012. Induction of microRNA-155 is TLR- and type IV secretion system-dependent in macrophages and inhibits DNA-damage induced apoptosis. Proc Natl Acad Sci U S A 109:E1153–E1162. doi: 10.1073/pnas.1116125109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhai A, Qian J, Kao W, Li A, Li Y, He J, Zhang Q, Song W, Fu Y, Wu J, Chen X, Li H, Zhong Z, Ling H, Zhang F. 2013. Borna disease virus encoded phosphoprotein inhibits host innate immunity by regulating miR-155. Antiviral Res 98:66–75. doi: 10.1016/j.antiviral.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Horan KA, Watanabe K, Kong AM, Bailey CG, Rasko JE, Sasaki T, Mitchell CA. 2007. Regulation of FcgammaR-stimulated phagocytosis by the 72-kDa inositol polyphosphate 5-phosphatase: SHIP1, but not the 72-kDa 5-phosphatase, regulates complement receptor 3 mediated phagocytosis by differential recruitment of these 5-phosphatases to the phagocytic cup. Blood 110:4480–4491. doi: 10.1182/blood-2007-02-073874. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Kang L, Yao H, He Y, Wang X, Xu W, Song Z, Yin Y, Zhang X. 2016. Streptococcus pneumoniae endopeptidase O (PepO) elicits a strong innate immune response in mice via TLR2 and TLR4 signaling pathways. Front Cell Infect Microbiol 6:23. doi: 10.3389/fcimb.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang K, Wu M, Li M, Li D, Peng A, Nie X, Sun M, Wang J, Wu Y, Deng Q, Zhu M, Chen K, Yuan J, Huang X. 2014. miR-155 suppresses bacterial clearance in Pseudomonas aeruginosa-induced keratitis by targeting Rheb. J Infect Dis 210:89–98. doi: 10.1093/infdis/jiu002. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Yang K, Zhou L, Wu M, Wu Y, Zhu M, Lai X, Chen T, Feng L, Li M, Huang C, Zhong Q, Huang X. 2013. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog 9:e1003697. doi: 10.1371/journal.ppat.1003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domingo-Gonzalez R, Katz S, Serezani CH, Moore TA, Levine AM, Moore BB. 2013. Prostaglandin E2-induced changes in alveolar macrophage scavenger receptor profiles differentially alter phagocytosis of Pseudomonas aeruginosa and Staphylococcus aureus post-bone marrow transplant. J Immunol 190:5809–5817. doi: 10.4049/jimmunol.1203274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R, Meinl E. 2009. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain 132:3342–3352. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 30.Kim SM, Lim SM, Yoo JA, Woo MJ, Cho KH. 2015. Consumption of high-dose vitamin C (1250 mg per day) enhances functional and structural properties of serum lipoprotein to improve anti-oxidant, anti-atherosclerotic, and anti-aging effects via regulation of anti-inflammatory microRNA. Food Funct 6:3604–3612. doi: 10.1039/C5FO00738K. [DOI] [PubMed] [Google Scholar]
- 31.Rajaram MV, Ganesan LP, Parsa KV, Butchar JP, Gunn JS, Tridandapani S. 2006. Akt/protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J Immunol 177:6317–6324. doi: 10.4049/jimmunol.177.9.6317. [DOI] [PubMed] [Google Scholar]
- 32.Parsa KV, Ganesan LP, Rajaram MV, Gavrilin MA, Balagopal A, Mohapatra NP, Wewers MD, Schlesinger LS, Gunn JS, Tridandapani S. 2006. Macrophage pro-inflammatory response to Francisella novicida infection is regulated by SHIP. PLoS Pathog 2:e71. doi: 10.1371/journal.ppat.0020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajaram MV, Butchar JP, Parsa KV, Cremer TJ, Amer A, Schlesinger LS, Tridandapani S. 2009. Akt and SHIP modulate Francisella escape from the phagosome and induction of the Fas-mediated death pathway. PLoS One 4:e7919. doi: 10.1371/journal.pone.0007919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santic M, Al-Khodor S, Abu Kwaik Y. 2010. Cell biology and molecular ecology of Francisella tularensis. Cell Microbiol 12:129–139. doi: 10.1111/j.1462-5822.2009.01400.x. [DOI] [PubMed] [Google Scholar]
- 35.Ai J, Maturu A, Johnson W, Wang Y, Marsh CB, Tridandapani S. 2006. The inositol phosphatase SHIP-2 down-regulates FcgammaR-mediated phagocytosis in murine macrophages independently of SHIP-1. Blood 107:813–820. doi: 10.1182/blood-2005-05-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox D, Dale BM, Kashiwada M, Helgason CD, Greenberg S. 2001. A regulatory role for Src homology 2 domain-containing inositol 5′-phosphatase (SHIP) in phagocytosis mediated by Fc gamma receptors and complement receptor 3 (alpha(M)beta(2); CD11b/CD18). J Exp Med 193:61–71. doi: 10.1084/jem.193.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dewitt S, Tian W, Hallett MB. 2006. Localised PtdIns(3,4,5)P3 or PtdIns(3,4)P2 at the phagocytic cup is required for both phagosome closure and Ca2+ signalling in HL60 neutrophils. J Cell Sci 119:443–451. doi: 10.1242/jcs.02756. [DOI] [PubMed] [Google Scholar]
- 38.Marshall JG, Booth JW, Stambolic V, Mak T, Balla T, Schreiber AD, Meyer T, Grinstein S. 2001. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fc gamma receptor-mediated phagocytosis. J Cell Biol 153:1369–1380. doi: 10.1083/jcb.153.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ninomiya N, Hazeki K, Fukui Y, Seya T, Okada T, Hazeki O, Ui M. 1994. Involvement of phosphatidylinositol 3-kinase in Fc gamma receptor signaling. J Biol Chem 269:22732–22737. [PubMed] [Google Scholar]
- 40.Gasque P. 2004. Complement: a unique innate immune sensor for danger signals. Mol Immunol 41:1089–1098. doi: 10.1016/j.molimm.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 41.Le Cabec V, Carreno S, Moisand A, Bordier C, Maridonneau-Parini I. 2002. Complement receptor 3 (CD11b/CD18) mediates type I and type II phagocytosis during nonopsonic and opsonic phagocytosis, respectively. J Immunol 169:2003–2009. doi: 10.4049/jimmunol.169.4.2003. [DOI] [PubMed] [Google Scholar]
- 42.Hajishengallis G, Harokopakis E. 2007. Porphyromonas gingivalis interactions with complement receptor 3 (CR3): innate immunity or immune evasion? Front Biosci 12:4547–4557. doi: 10.2741/2409. [DOI] [PubMed] [Google Scholar]
- 43.Hajishengallis G, Wang M, Liang S, Shakhatreh MA, James D, Nishiyama S, Yoshimura F, Demuth DR. 2008. Subversion of innate immunity by periodontopathic bacteria via exploitation of complement receptor-3. Adv Exp Med Biol 632:203–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 45.Dziarski R, Gupta D. 2005. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: a reevaluation. Infect Immun 73:5212–5216. doi: 10.1128/IAI.73.8.5212-5216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimbrell MR, Warshakoon H, Cromer JR, Malladi S, Hood JD, Balakrishna R, Scholdberg TA, David SA. 2008. Comparison of the immunostimulatory and proinflammatory activities of candidate Gram-positive endotoxins, lipoteichoic acid, peptidoglycan, and lipopeptides, in murine and human cells. Immunol Lett 118:132–141. doi: 10.1016/j.imlet.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451. doi: 10.1016/S1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 48.Orrskog S, Rounioja S, Spadafina T, Gallotta M, Norman M, Hentrich K, Falker S, Ygberg-Eriksson S, Hasenberg M, Johansson B, Uotila LM, Gahmberg CG, Barocchi M, Gunzer M, Normark S, Henriques-Normark B. 2012. Pilus adhesin RrgA interacts with complement receptor 3, thereby affecting macrophage function and systemic pneumococcal disease. mBio 4:e00535-12. doi: 10.1128/mBio.00535-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basset A, Zhang F, Benes C, Sayeed S, Herd M, Thompson C, Golenbock DT, Camilli A, Malley R. 2013. Toll-like receptor (TLR) 2 mediates inflammatory responses to oligomerized RrgA pneumococcal pilus type 1 protein. J Biol Chem 288:2665–2675. doi: 10.1074/jbc.M112.398875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernatoniene J, Zhang Q, Dogan S, Mitchell TJ, Paton JC, Finn A. 2008. Induction of CC and CXC chemokines in human antigen-presenting dendritic cells by the pneumococcal proteins pneumolysin and CbpA, and the role played by toll-like receptor 4, NF-kappaB, and mitogen-activated protein kinases. J Infect Dis 198:1823–1833. doi: 10.1086/593177. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Wang H, Zhang S, Zeng L, Xu X, Wu K, Wang W, Yin N, Song Z, Zhang X, Yin Y. 2014. Mucosal immunization with recombinant fusion protein DnaJ-DeltaA146Ply enhances cross-protective immunity against Streptococcus pneumoniae infection in mice via interleukin 17A. Infect Immun 82:1666–1675. doi: 10.1128/IAI.01391-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin CC, Yu CS, Yang JS, Lu CC, Chiang JH, Lin JP, Kuo CL, Chung JG. 2012. Chrysin, a natural and biologically active flavonoid, influences a murine leukemia model in vivo through enhancing populations of T- and B-cells, and promoting macrophage phagocytosis and NK cell cytotoxicity. In Vivo 26:665–670. [PubMed] [Google Scholar]
- 53.Ninkovic J, Roy S. 2014. High throughput fluorometric technique for assessment of macrophage phagocytosis and actin polymerization. J Vis Exp 2014(93):e52195. doi: 10.3791/52195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song Z, Zhang J, Zhang X, Li D, Wang H, Xu X, Xu W, Yin Y, Cao J. 2015. Interleukin 4 deficiency reverses development of secondary Pseudomonas aeruginosa pneumonia during sepsis-associated immunosuppression. J Infect Dis 211:1616–1627. doi: 10.1093/infdis/jiu668. [DOI] [PubMed] [Google Scholar]