

Summary
Protein aggregation is associated with age-related neurodegenerative disorders, such as Alzheimer’s and polyglutamine diseases. As a causal relationship between protein aggregation and neurodegeneration remains elusive, understanding the cellular mechanisms regulating protein aggregation will help develop future treatments. To identify such mechanisms, we conducted a forward genetic screen in a C. elegans model of polyglutamine aggregation and identified the protein MOAG-2/LIR-3 as a driver of protein aggregation. In the absence of polyglutamine, MOAG-2/LIR-3 regulates the RNA polymerase III-associated transcription of small non-coding RNAs. This regulation is lost in the presence of polyglutamine, which mislocalizes MOAG-2/LIR-3 from the nucleus to the cytosol. We then show biochemically that MOAG-2/LIR-3 can also catalyze the aggregation of polyglutamine-expanded huntingtin. These results suggest that polyglutamine can induce an aggregation-promoting activity of MOAG-2/LIR-3 in the cytosol. The concept that certain aggregation-prone proteins can convert other endogenous proteins into drivers of aggregation and toxicity adds to the understanding of how cellular homeostasis can be deteriorated in protein misfolding diseases.
Keywords: MOAG-2/LIR-3, protein aggregation, polyglutamine, C. elegans, non-coding RNA, tRNA, snoRNA, RNA polymerase III, protein homeostasis, protein quality control
Graphical Abstract
Highlights
-
•
Inactivation of MOAG-2/LIR-3 reduces polyglutamine aggregation
-
•
MOAG-2/LIR-3 regulates Pol III-mediated transcription of small non-coding RNAs
-
•
Polyglutamine mislocalizes MOAG-2/LIR-3 from the nucleus to the cytosol
-
•
Polyglutamine converts MOAG-2/LIR-3 into an aggregation-promoting factor
The cellular mechanisms that drive polyglutamine aggregation are poorly understood. Sin et al. show that polyglutamine relocates MOAG-2/LIR-3 from the nucleus to the cytosol, thereby converting this protein into an aggregation-promoting factor to drive protein aggregation and toxicity.
Introduction
Neurodegenerative disorders such as Alzheimer’s, Parkinson’s, and Huntington’s diseases represent a major health problem and demand a better understanding of the molecular mechanisms of pathogenesis in order to develop disease-modifying treatments. The hallmark of many neurodegenerative disorders is the presence of protein aggregates in different brain areas of affected patients (Soto, 2003). These insoluble macromolecular structures are enriched in aggregation-prone proteins that—by exposing certain regions of their amino acid sequences—associate in an aberrant manner with other proteins, thereby hampering normal cellular function (Eisenberg and Jucker, 2012, Olzscha et al., 2011; also reviewed in Chiti and Dobson, 2009, Sin and Nollen, 2015). It is not yet clear, however, whether these protein aggregates are actually a cause or a consequence of the disease. The current view is that the soluble precursors of these aggregates—in particular some oligomeric forms—are the main cytotoxic species, and that at least in some cases the aggregation process can represent a protective measure to sequester these smaller harmful species (Arrasate et al., 2004, Bolognesi et al., 2010, Kayed et al., 2003, Miller et al., 2011).
These observations focus the attention on the cellular factors that can drive protein aggregation, which are currently poorly understood. In some cases, direct aggregation-promoting factors have been identified, both in humans and in animal models, and include SH3GL3 (Davranche et al., 2011, Sittler et al., 1998), MOAG-4/SERF (Falsone et al., 2012, van Ham et al., 2010), and UNC-30 (Garcia et al., 2007). More studies, however, are needed to acquire a more comprehensive understanding of this phenomenon.
In this context, the roundworm C. elegans is a much-used animal model for neurodegenerative diseases and is proving very useful in providing a basic understanding of protein aggregation. If we can use this model organism to identify genetic modifiers of protein aggregation, then we can also obtain insight into the cellular pathways that are dysregulated in the pathogenesis of human protein misfolding diseases and target them for pharmacological intervention. Indeed, a wide variety of genetic screens have already been performed in C. elegans to find genes that regulate protein aggregation and its associated toxicity (Hamamichi et al., 2008, Kuwahara et al., 2008, Lejeune et al., 2012, Nollen et al., 2004, Silva et al., 2011, van der Goot et al., 2012, van Ham et al., 2010). The aim of the current study is to identify genes that drive protein aggregation, to discover the function of such genes, and to understand how they are related to protein aggregation. We identified MOAG-2/LIR-3 as a modifier of aggregation, since this protein in the presence of polyglutamine shifts its role from a transcriptional regulator of small non-coding RNAs (ncRNAs) to an aggregation and toxicity promoting factor.
Results
Inactivation of MOAG-2/LIR-3 Reduces Polyglutamine Aggregation
To identify genes whose products are capable of driving protein aggregation, we performed a forward genetic screen in a C. elegans model in which the body-wall muscle cells express a transgene consisting of an aggregation-prone polyglutamine stretch of 40 residues fused to YFP (Q40-YFP) (van Ham et al., 2010). We screened for mutants with reduced polyglutamine aggregation, and we named the resulting genes “modifiers of aggregation” (moag) (van Ham et al., 2010). At the fourth larval stage (L4), moag-2(pk2183) (hereafter designated as moag-2/lir-3(pk2183)) showed a reduction of about 50% in the number of aggregates relative to the wild-type Q40 worms (Figures 1A, 1B, and S1A). SNP mapping and genome sequencing allowed us to fine-map the causal mutation and revealed six genes as putative candidates for moag-2 (Figure S1B). For one of these, which mapped to lir-3 (lin-26 related; sequence: F37H8.1; GenBank: NM_063994), the causative mutation was in the start codon, replacing the first methionine with an isoleucine (Met1Ile) (Figures 1C and S1B).
Figure 1.

Identification of moag-2/lir-3 as an Aggregation-Promoting Factor
(A) Number of aggregates in Q40 worms and Q40;moag-2(pk2183) worms.
(B) Representative images of Q40 worms and Q40;moag-2(pk2183) worms. Scale bar, 75 μm. See also Figure S1A.
(C) Chromosomal location of lir-3 (F37H8.1, chromosome II, reverse strand of assembly; http://www.wormbase.org, WS248), showing the point mutation in the start codon (red arrow), the partial deletion (red bar), and the rescue fragment (green). See also Figures S1B and S1C.
(D) Number of aggregates in Q40 worms with either wild-type alleles, or heterozygous (het) or homozygous (tm813) deletion for the lir-3(tm813) allele.
(E) Number of aggregates in Q40 worms and Q40;lir-3(tm813) worms, with and without transgenic overexpression of an injected lir-3 rescue fragment, including its endogenous promoter. See also Figure S1D.
(F) Filter retardation assay with 5-fold serial dilutions of crude protein extract from Q40 worms and Q40;moag-2/lir-3 mutant worms. The results shown are from a representative experiment of three biological replicates. Q40-YFP and α-tubulin expression were included as controls. See also Figure S1E.
(G) Q40-YFP transcript expression detected by RNA sequencing in L4-stage worms and protein expression detected in urea-treated L4-stage worms and day 1 adult worms.
In all panels, aggregate counting, representative images, and filter retardation assay were performed at the L4 stage and the average of three biological replicates is represented. Data are represented as mean ± SEM and significance was calculated using a one-tailed unpaired Student’s t test. ∗∗p < 0.01; ∗∗∗p < 0.001.
The lir-3 gene encodes a LIN-26-like zinc-finger protein of unknown function (http://www.wormbase.org/, June 2016). It is predicted to have two zinc-finger domains of the C2H2 type (residues 191–214 and 224–247) at the carboxyl terminus and a nuclear localization signal spanning amino acid residues 132–141 (http://nls-mapper.iab.keio.ac.jp/, June 2016) (Figure S1C). LIR-3 shares two non-canonical C2H2 zinc-finger motifs with three other C. elegans proteins: LIN-26 (31%–35% similarity), LIR-1 (20%–31% similarity), and LIR-2 (25% similarity) (Dufourcq et al., 1999).
To assess whether moag-2 was lir-3, we used an lir-3(tm813) deletion mutant (hereafter designated as “moag-2/lir-3(tm813)”) and crossed it with the polyglutamine (Q40) worm model. This strain has a 795 bp deletion spanning residues 276–1,070 of the F37H8.1 sequence that also causes a premature stop codon (Figures 1C and S1C). This partial deletion of lir-3 caused a 35% reduction of aggregates relative to the numbers seen in wild-type worms (Figure 1D). Worms heterozygous for lir-3 deletion allele had aggregate numbers similar to those seen in the wild-type, suggesting that the reduction of aggregation was recessive and due to the loss of function of lir-3 (Figure 1D). We next asked whether overexpression of lir-3 could restore the aggregation phenotype. To this end, we injected worms with a rescue fragment, consisting of full-length lir-3, including a 1.5 kb sequence upstream of the start codon to include its endogenous promoter as well as 330 bp downstream to include the 3′ UTR (Figure 1C). Expression of the lir-3 rescue fragment in Q40; lir-3(tm813) worms was able to restore the aggregation phenotype by 2-fold (p < 0.001), confirming lir-3 as a gene responsible for driving aggregation in the polyglutamine model (Figures 1E and S1D). Taken together, these results demonstrate that lir-3 is moag-2.
A specific property of protein aggregates in the brains of neurodegenerative disease patients that is also captured by the C. elegans polyglutamine model used here is that they are typically insoluble in strong detergents such as sodium dodecyl sulfate (SDS) (Lee et al., 1999, Shankar et al., 2008, Yanamandra et al., 2015). To establish whether MOAG-2/LIR-3 promoted aggregation of SDS-insoluble polyglutamine aggregates, we performed a filter retardation assay on lysates of wild-type and moag-2/lir-3 mutant polyglutamine worms (Scherzinger et al., 1997, Wanker et al., 1999). This assay enables the detection of SDS-insoluble protein aggregates, while soluble species are not captured. Both Q40;moag-2/lir-3(pk2183) and Q40;moag-2/lir-3(tm813) mutants had fewer SDS-insoluble aggregates than their corresponding Q40 controls (Figure 1F). The reduction of SDS-insoluble aggregates was more pronounced in the point mutant (43%; p = 0.05) than in the deletion mutant (26%; p = 0.18) (Figure S1E). Mutation or partial deletion of moag-2/lir-3 did not cause a discernible reduction in the transcription or the protein expression level of Q40-YFP, indicating that moag-2/lir-3 does not reduce aggregation by reducing expression levels of the Q40-YFP protein (Figures 1F and 1G). Together, these results indicate that moag-2/lir-3 drives the formation of SDS-insoluble polyglutamine aggregates.
MOAG-2/LIR-3 Is a C2H2-Domain Protein Associated with RNA Polymerase III Promoters
Having established that mutation of moag-2/lir-3 reduces polyglutamine aggregation, we next determined the endogenous function of the corresponding protein. C2H2 zinc-finger domains are predominantly associated with DNA-binding transcription factors but may also have other functions such as mediating protein-protein interactions or binding to RNA (Brown, 2005, Hall, 2005, Krishna et al., 2003). Bioinformatics analysis combined with manual curation predicted MOAG-2/LIR-3 to be a transcription factor (Reece-Hoyes et al., 2005, Reece-Hoyes et al., 2007). To explore this possibility, we determined the subcellular localization of FLAG-tagged MOAG-2/LIR-3 protein in wild-type N2 worms. Indirect immunofluorescence using an anti-FLAG antibody revealed that MOAG/LIR-3 is localized in the nucleus (Figures 2A and S2A).
Figure 2.

MOAG-2/LIR-3 Preferentially Binds to Promoters of Small ncRNAs
(A) Subcellular localization of MOAG-2/LIR-3 in wild-type N2 worms. Scale bar, 50 μm. Arrowheads point at nuclei; asterisk is non-specific staining. See also Figure S2A.
(B) Enrichment of binding of MOAG-2/LIR-3 to different gene biotypes (1 kb upstream/downstream of TSS) relative to genes distributed genome-wide. See also Figure S2B.
(C) MOAG-2/LIR-3 binding sites within −1,000 and +1,000 bp of TSS for protein-coding, ncRNA, tRNA, and snoRNA genes.
(D) Enriched consensus DNA motifs for MOAG-2/LIR-3 with p value.
(E) Number of MOAG-2/LIR-3 binding sites containing Box A and Box B.
(F) Heatmap showing the binding of different transcription factors to C. elegans promoters of protein-coding, snoRNA, and tRNA genes. The hierarchical clustering was generated using the average linkage cluster method with a binary metric distance.
Next, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) in L4-staged worms that expressed an integrated construct of lir-3 fused to FLAG and GFP (Sarov et al., 2012, Zhong et al., 2010). This analysis yielded a total of 678 unique MOAG-2/LIR-3 binding sites, 404 of which overlapped with 813 C. elegans genes. Further analyses of these genes revealed that MOAG-2/LIR-3 binding was enriched in the transcription start sites (TSSs) of tRNA genes (35.7%, p < 0.001), small nucleolar RNA (snoRNA) genes (6.3%, p < 0.001), rRNA genes (2.5%, p < 0.001), and small nuclear RNA (snRNA) genes (2.2%, p < 0.002) (Figures 2B and S2B). While MOAG-2/LIR-3 was also found in the vicinity of protein-coding and other ncRNA genes, this binding was not significantly enriched (Figures 2B, 2C, and S2B).
We then asked whether the binding sites were enriched in any consensus sequence motif that could be recognized by MOAG-2/LIR-3. Of the 678 binding sites initially identified in our ChIP-seq data, more than half of the sites contained Box A and Box B sequence motifs, 301 of which contained both motifs (Figures 2D and 2E). Box A and Box B constitute the canonical type 2 promoter site recognized by the RNA polymerase (Pol) III complex (Ikegami and Lieb, 2013, Schramm and Hernandez, 2002). Pol III is responsible for the transcription of structural or catalytic snRNAs, of tRNAs, and of snoRNAs, which mediate chemical modifications of other RNA molecules (Bratkovič and Rogelj, 2014, Guthrie and Patterson, 1988, Schramm and Hernandez, 2002, White, 2011). These findings led us to hypothesize that MOAG-2/LIR-3 may bind to the same target promoters as Pol III. Binding in the proximity of non-coding genes has been shown on occasion for several C. elegans transcription factors, including PHA-4, PQM-1, and GEI-11 (Niu et al., 2011). These observations prompted us to ask whether the association between MOAG-2/LIR-3 and the Pol III complex resembled the binding of these transcription factors to the promoters of non-coding genes. To answer this question, we collected publicly available ChIP-seq data for known C. elegans transcription factors (http://www.modencode.org, September 2014) and analyzed binding to the promoters of protein-coding genes, snoRNA genes, and tRNA genes (Figure 2F). While there is little association between MOAG-2/LIR-3 and the promoters of protein-coding genes, the MOAG-2/LIR-3 binding profile is very similar to that of a group of clustered factors that contain representative components of the Pol III complex. These factors include Pol III, TATA binding protein (TBP-1), two subunits of the transcription factor for Pol III C (TFC-1 and TFC-4), and the nuclear pore proteins NPP-3 and NPP-13, which have recently been shown to associate with the Pol III complex to regulate tRNA and snoRNA splicing (Ikegami and Lieb, 2013). In contrast to the majority of the other transcription factors, the binding of these factors is very abundant in the promoters of snoRNAs and of tRNAs (Figure 2F). Therefore, the binding of MOAG-2/LIR-3 to the promoters of small ncRNA genes suggests that MOAG-2/LIR-3 is associated with Pol III transcription.
MOAG-2/LIR-3 Is a Positive Regulator of Pol III-Mediated Transcription of Small ncRNAs
Because we found MOAG-2/LIR-3 to bind to the promoters of small ncRNA genes, we next asked what consequences this binding had for the transcription of Pol III downstream targets. We therefore used transcriptome profiling to compare RNA expression in wild-type worms with that in moag-2/lir-3 mutant worms. In line with the absence of MOAG-2/LIR-3 at RNA Pol II promoter sites, we did not find any protein-coding genes that were differentially expressed between the mutants and wild-type N2 worms, thereby excluding MOAG-2/LIR-3 as a transcriptional regulator of protein-coding genes (Figures 3A and S3A). Mutations in moag-2/lir-3, however, did result in the downregulation of snRNAs (p < 0.001), snoRNAs (p < 0.001), and tRNAs (p < 0.001) in both mutants, demonstrating that MOAG-2/LIR-3 regulates Pol III-mediated transcription of these small ncRNAs (Figures 3A and S3A).
Figure 3.

MOAG-2/LIR-3 Regulates Transcription of Small ncRNAs
(A) Boxplot showing the relative expression of different gene biotypes in moag-2/lir-3(pk2183) worms relative to the wild-type N2 background. TPM, tags per kilobase million; Coding, protein-coding genes; ncRNA, non-coding RNA; Pseudo, pseudogenes; snRNA, small nuclear RNA; snlRNA, snRNA-like RNA; snoRNA, small nucleolar RNA; tRNA, transfer RNA. The average of three biological replicates is represented and significance was calculated using a two-tailed unpaired Student’s t test. ∗∗∗p < 0.001. See also Figure S3A.
(B) Positions of ChIP-seq signal maxima relative to TSS (right y axis) with maximum normalized read count (left y axis) for the 51 snoRNA genes and the 290 tRNA genes picked in this study. Bottom box represents the motif position of Box A and Box B relative to snoRNA and tRNA genes. See also Figures S3B and S3C.
(C) Diagram showing the positions of the Pol III factors and that of MOAG-2/LIR-3 as estimated from the data presented in (B).
(D) Co-immunoprecipitation of FLAG-tagged MOAG-2/LIR-3 protein by α-Pol II and α-Pol III protein antibodies. IP, immunoprecipitation; WT, wild-type; OE, MOAG-2/LIR-3 overexpression.
We next asked where in the TSS region MOAG-2/LIR-3 was positioned relative to the Pol III complex. To this end, we compared the positions of the ChIP-seq signals of MOAG-2/LIR-3 with those of the different components of the Pol III complex. For both the tRNA and snoRNA genes, all factors localized to the Box A- and Box B-containing promoter region, consistent with previous reports (Figures 3B, 3C, and S3B) (Ikegami and Lieb, 2013). MOAG-2/LIR-3 was also positioned at these same sites (Figures 3B and 3C).
Next, we tested whether MOAG-2/LIR-3 interacts physically with the Pol III complex by means of immunoprecipitation experiments. We found that FLAG-tagged MOAG-2/LIR-3 protein co-immunoprecipitated with Pol III, but not detectably with Pol II, confirming that MOAG-2/LIR-3 cooperates with the Pol III machinery to drive transcription of small ncRNAs (Figures 3D and S3C). The fact that MOAG-2/LIR-3 did not detectably co-immunoprecipitate with Pol II further supports the notion that MOAG-2/LIR-3 is not directly involved in the transcription of protein-coding genes (Figure 3D).
Together these results indicate that MOAG-2/LIR-3 functions as a positive regulator of the Pol III-mediated transcription of small ncRNAs in C. elegans.
Regulation of Protein Aggregation by MOAG-2/LIR-3 Is Independent of Its Role as a Transcriptional Regulator
Next, we asked whether MOAG-2/LIR-3 regulated protein aggregation via the Pol III-mediated transcription of small ncRNAs. We therefore used RNAi to knock down, one by one, the individual components of the Pol III complex in both wild-type worms and moag-2/lir-3(pk2183) mutants. To confirm RNAi knockdown, we also looked for RNAi-associated phenotypes other than aggregation. If Pol III-mediated transcription were involved in promoting protein aggregation, this would result in a reduction in the amount of aggregates in the wild-type Q40 worms, but not in the moag-2/lir-3 mutants. However, knockdown of Pol III, TBP-1, or TFC-1 did not alter aggregation in the Q40 worms or in the Q40;moag-2/lir-3 mutant strains, indicating that in the absence of moag-2/lir-3 it is not the resulting lack of Pol III-mediated transcription that is responsible for the reduction in aggregation (Figures 4A, S4A, and S4B).
Figure 4.

Polyglutamine Reduces the Transcription of Small ncRNAs
(A) Number of aggregates measured in Q40 and Q40;moag-2/lir-3(pk2183) worms after RNAi knockdown of individual components of the Pol III complex. As an internal quality control for RNAi, squares indicate penetrance (50% [half-open] and 0% [open]) of all associated visible RNAi phenotypes other than aggregation. See also Figures S4A and S4B.
(B) Number of aggregates measured in Q40 and Q40;moag-2/lir-3(pk2183) worms upon RNAi knockdown of tRNA-processing enzymes. See also Figure S4C.
(C and D) Boxplot showing the relative expression of different gene biotypes in Q40 and Q40;moag-2/lir-3(pk2183) worms (C) and in N2 and Q40 wild-type worms (Q40 wild-type outcrossed from pk2183) (D). TPM, tags per kilobase million; Coding, protein-coding genes; ncRNA, non-coding RNA; Pseudo, pseudogenes; snRNA, small nuclear RNA; snlRNA, snRNA-like RNA; snoRNA, small nucleolar RNA; tRNA, transfer RNA. See also Figures S4D and S4E.
In (A) and (B), aggregate counting was performed at the L4 stage and the average of three biological replicates is represented. Data are represented as mean ± SEM. In (C) and (D), the average of three biological replicates is represented and significance was calculated using a two-tailed unpaired Student’s t test. ∗∗∗p < 0.001; ns is not significant. See also Figures S4D and S4E.
The nuclear pore protein npp-13 has been shown to interact with the Pol III complex and regulate the processing of small ncRNAs (Ikegami and Lieb, 2013). The aggregation phenotype was not altered by knockdown of npp-13 or of tRNA-processing enzymes, indicating that neither small RNA processing nor the availability of mature tRNAs is involved in reducing aggregation (Figures 4A, 4B, and S4A–S4C). These results indicate that the regulation of protein aggregation by MOAG-2/LIR-3 is separate from its involvement in RNA transcription together with the Pol III complex.
Polyglutamine Suppresses Transcription of Small ncRNAs
We next asked why the involvement of MOAG-2/LIR-3 in driving protein aggregation is independent of its role as a transcriptional regulator. We therefore first compared the RNA expression profiles of wild-type Q40 worms with those of moag-2/lir-3 mutant Q40 worms. In contrast to wild-type N2 worms, there was no longer a reduction in the relative expression levels of the small ncRNAs in the presence of polyglutamine (Figures 4C and S4D). When we then measured the absolute levels of all RNAs, we found that in contrast to the protein-coding RNAs, pseudogenes, and other ncRNAs, the expression of snRNAs, snoRNAs, and tRNAs was already strongly reduced in wild-type Q40 worms (p < 0.001; Figures 4D and S4E). These findings explained why mutations in moag-2/lir-3 could no longer reduce expression in these Q40 worms (Figures 4C and S4D) and indicated that presence of polyglutamine results in downregulation of small ncRNA expression.
MOAG-2/LIR-3 Increases Polyglutamine Toxicity
Having identified MOAG-2/LIR-3 as a regulator of polyglutamine aggregation, we asked whether it regulates the toxicity of polyglutamine as well. To answer to this question, we compared the motilities of worms with or without overexpression of FLAG-tagged MOAG-2/LIR-3 in absence or presence of polyglutamine over the course of 12 days. In absence of polyglutamine, FLAG-tagged MOAG-2/LIR-3 had no effect on the motility of young adults but improved the motility of old adult animals (Figure 5A). In contrast, in worms expressing polyglutamine, overexpression of MOAG-2/LIR-3 accelerated the age-dependent decline in motility (Figure 5B). To note, a deletion in moag-2/lir-3 had no significant effect on the motility of polyglutamine worms up to 13 days of adulthood (Figure S5A). Our results with the MOAG-2/LIR-3-overexpressing worms imply that the effect of a deletion may not have been strong enough to be detected in our assays because they indicate that increasing the levels of MOAG-2/LIR-3 enhances the toxicity of polyglutamine.
Figure 5.

Polyglutamine Moves MOAG-2/LIR-3 Cytosol Where It Turns into a Positive Regulator of Protein Aggregation
(A and B) Number of body bends in N2 wild-type worms and worms overexpressing MOAG-2/LIR-3 (OE) (A) and Q40 wild-type and Q40 worms overexpressing MOAG-2/LIR-3 (Q40;OE) (B). The average of three biological replicates is represented. Data are represented as mean ± SEM and significance was calculated using a two-tailed unpaired Student’s t test. ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figure S5A.
(C) Thioflavin T reaction profiles of 4 μM HttQ48 or HttQ23 solutions in the absence or presence of equimolar concentrations of MOAG-2/LIR-3 (black) or the modifier of aggregation MOAG-4 (blue). The concentration of MOAG-2/LIR-3 in whole-organism lysates has been determined to be between 0.3 and 8.5 ppm (http://pax-db.org). The average of four replicates is represented and error bars indicate mean ± SD.
(D) Western blot of subcellular fractionation of MOAG-3/LIR-3, which was detected using α-FLAG antibody. Q40-YFP, LMN-1 (nuclear marker), and α-tubulin (cytosolic marker) were used as controls. See Figure S5C.
(E) Western blot analysis of MOAG-2/LIR-3 SDS solubility from wild-type (WT) and MOAG-2/LIR-3-overexpressing (OE) worms in N2 and polyglutamine worms (Q40). Q40-YFP and β-actin expression were included as controls.
In (D) and (E), the results were generated from L4-stage animals and a representative experiment of three biological replicates is shown.
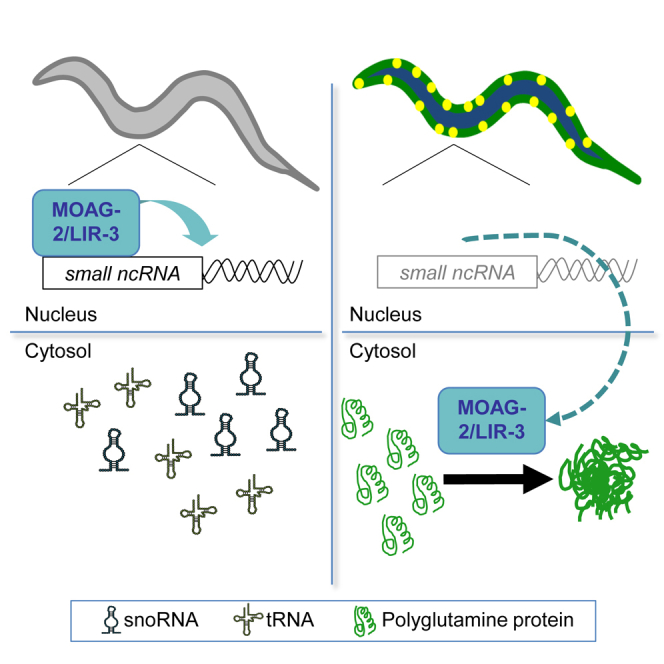
(F) Model proposing how MOAG-2/LIR-3 drives polyglutamine aggregation. MOAG-2/LIR-3 normally regulates the transcription of small ncRNAs in the nucleus. In the presence of polyglutamine, MOAG-2/LIR-3 is moved to the cytosol, where it exposes an ectopic aggregation-promoting activity.
MOAG-2/LIR-3 Can Directly Drive Polyglutamine Aggregation
Our results indicate that MOAG-2/LIR-3 regulates protein aggregation independently of its role in small ncRNA transcription and processing. A possible mechanism by which loss of MOAG-2/LIR-3 could suppress aggregation indirectly could be by increasing the cellular folding capacity. The transcription of protein-coding genes was not affected, however, indicating that increased transcription of folding genes was not involved (Table S3). Alternatively, MOAG-2/LIR-3 may normally sequester folding factors, which then may be released in the Q40;moag-2/lir-3 mutants, a mechanism that has been previously proposed for an increase in protein aggregation by temperature-sensitive mutant proteins (Gidalevitz et al., 2006). If such a mechanism would be responsible for the suppression of aggregation in the Q40;moag-2/lir-3 mutants, knockdown of folding factors in these animals would increase aggregation again. To test this possibility, we knocked down by RNAi several folding factors that have been previously shown to regulate polyglutamine aggregation (Hsu et al., 2003, Nillegoda et al., 2015, Nollen et al., 2004, Rampelt et al., 2012). The depletion of these factors could not revert the aggregation phenotype in the Q40;moag-2/lir-3 mutants (Figure S5B). These results suggest that suppression of protein aggregation by inactivation of MOAG-2/LIR-3 is not caused by an increase in the expression or a release of folding factors.
Another possibility is that MOAG-2/LIR-3 drives aggregation directly. Precedents for aggregation-promoting factors include MOAG-4/SERF, which can drive aggregation of a variety of disease proteins via a transient direct interaction with early aggregation intermediates (Davranche et al., 2011, Falsone et al., 2012, Sittler et al., 1998, van Ham et al., 2010). To determine whether MOAG-2/LIR-3 can drive aggregation directly, we incubated GST-tagged huntingtin exon 1 containing 48 CAG repeats (HttQ48) with purified MOAG-2/LIR-3 protein and monitored amyloid formation by measuring thioflavin T (ThT) fluorescence in vitro. Our kinetic data revealed that the presence of an equimolar concentration of MOAG-2/LIR-3 accelerated the aggregation of HttQ48, but not of the non-pathogenic form HttQ23, similarly to what was observed in the case of MOAG-4, the C. elegans ortholog of SERF1A (Figure 5C). These results indicate that MOAG-2/LIR-3 can catalyze the deposition of aggregation-prone proteins directly.
Polyglutamine Mislocalizes MOAG-2/LIR-3 from the Nucleus to the Cytosol
For a lack of an aggregation-catalyzing effect to explain the reduction of protein aggregation in Q40;moag-2/lir-3 mutants, MOAG-2/LIR-3 and polyglutamine would, in the wild-type animals, have to localize in the same cellular compartment. Thus, to determine their localization, we performed subcellular fractionations with wild-type and Q40 worms with or without the expression of FLAG-tagged MOAG-2/LIR-3. We found that in the absence of polyglutamine, MOAG-2/LIR-3::FLAG was primarily located in the nucleus (Figure 5D, OE; Figure S5C). Polyglutamine was mostly localized in the cytosol, which was independent of the presence of MOAG-2/LIR-3::FLAG (Figure 5D, Q40 and Q40;OE; Figure S5C). In Q40 worms, however, MOAG-2/LIR-3::FLAG was enriched in the cytosol (Figure 5D, OE and Q40;OE; Figure S5C), which suggests that the presence of polyglutamine altered the localization of MOAG-2/LIR-3. Their joint presence in the cytosol supports a direct interaction between the proteins to drive protein aggregation.
Proteins involved in RNA metabolism have been described to co-aggregate with polyglutamine (Doi et al., 2010, Doi et al., 2008, Schwab et al., 2008), which could explain the retention of MOAG-2/LIR-3 in the cytosol. We therefore fractionated SDS-soluble and insoluble material to determine whether MOAG-2/LIR-3 co-aggregated with polyglutamine. Q40-YFP was detected as low molecular weight monomers and as SDS-resistant species retained in the stacking gel (Figure 5D). The solubility of MOAG-2/LIR-3::FLAG was not altered in the presence of polyglutamine (Figure 5D), suggesting that MOAG-2/LIR-3 is not recruited to SDS-resistant aggregates, but may be associated with the soluble species.
Together, these data suggest that the presence of polyglutamine moves MOAG-2/LIR-3 from the nucleus to the cytosol, where it turns into a positive regulator of protein aggregation, which is at the expense of its function as a transcriptional regulator (Figure 5F).
Discussion
From a genetic screen in a C. elegans model of protein aggregation disease, we identified MOAG-2/LIR-3 as a regulator of Pol III transcription in the nucleus and showed that—in the presence of polyglutamine—this protein switches into a positive regulator of polyglutamine aggregation in the cytosol.
Our finding that MOAG-2/LIR-3 is a regulator of transcription confirms predictions about the function of the lir-3 gene. Its role as a transcriptional regulator has been suggested on the basis of its structural similarity to the C2H2 zinc fingers of LIN-26, a fate regulator responsible for the differentiation of non-neuronal ectodermal cells and somatic gonad epithelium (den Boer et al., 1998, Dufourcq et al., 1999, Labouesse et al., 1996). We showed that MOAG-2/LIR-3 is required for the transcription of snRNA, snoRNA, and tRNA genes. In addition, we also showed that unlike other transcription factors that have on occasion been found to bind to the promoters of small ncRNAs, MOAG-2/LIR-3 is associated with the same target genes as the Pol III complex (Niu et al., 2011).
MOAG-2/LIR-3 is known to be expressed in the nuclei of a subset of cell types of C. elegans, namely body wall muscle cells, the vulval muscles, the spermatheca, the head and tail ganglia, and the ventral nerve cord, from embryogenesis throughout adulthood (https://transgeneome.mpi-cbg.de/transgeneomics/index.html, March 2014; Reece-Hoyes et al., 2005, Reece-Hoyes et al., 2007). A microarray performed in two distinct mechanosensory neurons—the touch receptor neurons and the FLP neurons—revealed that moag-2/lir-3 is upregulated in the FLP sensory neurons, suggesting that moag-2/lir-3 is required for FLP differentiation (Topalidou and Chalfie, 2011). The observation that the expression of MOAG-2/LIR-3 is limited to a subset of cell types suggests that this protein could be a tissue-specific regulator of transcription rather than a core component of the Pol III machinery. Such cell-type-specific regulators of Pol III have been described for human cells and proposed to accommodate cell-specific needs for small ncRNAs (Alla and Cairns, 2014, Oler et al., 2010; also reviewed in Marshall and White, 2008, White, 2011). Whether MOAG-2/LIR-3 has a similar role remains to be established, but it could explain why mutations in the moag-2/lir-3 gene do not result in any obvious abnormalities in terms of growth or viability.
In C. elegans, the nuclear pore protein NPP-13 has been described to associate with the Pol III complex, which regulates the efficient processing of snoRNA and tRNA transcripts (Ikegami and Lieb, 2013). Knockdown of NPP-13 results in abnormally long snoRNA and tRNA transcripts that cannot be processed into their mature form (Ikegami and Lieb, 2013). In moag-2/lir-3 mutant worms, we did not find unprocessed transcripts for snoRNA or for tRNAs (data not shown), which excludes the possibility that MOAG-2/LIR-3 is required for Pol III transcript processing. Moreover, knockdown of NPP-13 did not alter aggregation, which suggests that mutations in moag-2/lir-3 do not alter aggregation by interfering with the nuclear pore complex.
Polyglutamine proteins can undergo post-translational modifications, including acetylation, phosphorylation, ubiquitation, and sumoylation (reviewed in Ehrnhoefer et al., 2011, Pennuto et al., 2009). These post-translational modifications have been shown to modulate polyglutamine toxicity and aggregation (Gu et al., 2009, Jana et al., 2005, Matsumoto et al., 2004, Steffan et al., 2004, Thomas et al., 2004). One possibility that could explain the reduction of aggregation observed in the polyglutamine model is that mutations in moag-2/lir-3 alter the post-translational status of polyglutamine. Although with our present results we cannot exclude this possibility, our in vitro data (Figure 5C) show that MOAG-2/LIR-3 is able to drive polyglutamine aggregation directly in conditions where post-translational modifications do not occur, supporting a functional interaction between MOAG-2/LIR-3 and polyglutamine.
In this study, expression of polyglutamine downregulated the levels of snRNAs, snoRNAs, and tRNAs, demonstrating that aggregation-prone proteins can affect small ncRNA homeostasis. A possible explanation for this downregulation is that the aggregation-prone proteins blocked the nuclear localization of MOAG-2/LIR-3 and perhaps also components of the Pol III complex. Indeed, our data suggest that polyglutamine retains MOAG-2/LIR-3 in the cytosol, which is consistent with the recent finding that aggregation-prone proteins block trafficking in and out of the nucleus (Woerner et al., 2016). Several intrinsically disordered proteins—which include proteins involved in transcriptional regulation—are known to be sequestered by aggregation-prone proteins (Iakoucheva et al., 2002, Liu et al., 2006, Minezaki et al., 2006, Olzscha et al., 2011, Walther et al., 2015). One example is Sp1, which can no longer bind to its DNA targets due to sequestration by mutant huntingtin (Dunah et al., 2002, Li et al., 2002). We found that MOAG-2/LIR-3 is not sequestered in the polyglutamine aggregates. The concentration of MOAG-2/LIR-3 in whole-worm lysates has been determined to be between 0.3 and 8.5 ppm (http://pax-db.org). Although these concentrations do not take into account cell- or stage-specific differences in protein expression, its relatively low abundance would support a model in which MOAG-2/LIR-3 drives aggregation via transient interactions with rare SDS-soluble polyglutamine species.
In summary, this work has revealed that protein aggregation can affect the transcription of the non-coding genome by altering the expression of ncRNA genes. Our findings also open up another potential perspective on how aggregation-prone proteins can impair cellular homeostasis: these proteins can convert from their normal functions into aggregation-promoting factors by relocation. These results imply a cellular mechanism whereby aggregation-prone disease-associated proteins inactivate, recruit, and use endogenous proteins that promote their own aggregation and toxicity, which would resemble viral self-catalysis of pathogenesis. We anticipate that interfering with this class of gene products that promote aggregation-prone interactions can be explored for the development of therapies to slow down progression of age-related protein aggregation diseases.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-GFP for ChIP-seq experiment | gift from Kevin White | N/A |
| Mouse anti-FLAG M2 | Sigma-Aldrich | Cat# F3165, RRID: AB_259529 |
| Mouse anti-GFP | Clontech Laboratories | Cat# 632381, RRID: AB_2313808 |
| Mouse α-tubulin | Sigma-Aldrich | Cat# T6074, RRID: AB_477582 |
| Goat anti-mouse Cyanine 5 | Thermo Fisher | Cat# A10524, RRID: AB_2534033 |
| Rabbit anti-Pol II (ama-1 subunit) | Novus Biologicals | Cat# 38520002, RRID: AB_10709680 |
| Rabbit anti-Pol III (rpc-1 subunit) | Novus Biologicals | Cat# 5333.00.02, RRID: AB_2616364 |
| Rabbit anti-LMN-1 | Novus Biologicals | Cat# 38530002, RRID: AB_10005072 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trizol | Life Technologies | Cat#15596-018 |
| Vectashield Mounting Medium (DAPI) | Vector Labs | Cat#H-1200 |
| Critical Commercial Assays | ||
| ECL Prime Western Blotting Detection Reagent | Amersham | Cat#RPN2232 |
| RevertAid H Minus First Strand cDNA Synthesis kit | Life Technologies | Cat#K1632 |
| SYBR Green Dye | Bio-Rad | Cat#172-5125 |
| Qubit RNA HS Assay Kit | Thermo Fisher | Cat#Q32852 |
| TruSeq Sample Preparation V2 Kit | Illumina | RS-122-2001, RS-122-2002 |
| Ribominus Eukaryote Kit | Invitrogen | Cat#A10837-08 |
| Fragmentation Buffer | Ambion | Cat#AM8740 |
| DNase I | Fermentas | Cat#EN0521 |
| Deposited Data | ||
| ChIP-seq data | ENCODE | ENCODE: ENCSR408FDZ |
| ChIP-seq data | ArrayExpress | ArrayExpress: E-MTAB-4174 |
| RNA-seq data | ArrayExpress | ArrayExpress: E-MTAB-4172 |
| Mendeley Data | This paper | http://dx.doi.org/10.17632/knzkvgbf6x.2 |
| Experimental Models: Organisms/Strains | ||
| C. elegans strains are listed in Table S1. | N/A | N/A |
| Oligonucleotides | ||
| Primer F1: CGCTCACAGTCAACGTCG | This paper | N/A |
| Primer R1: CCATGCGATTTGACACATTTCG | This paper | N/A |
| Primer F2: CGGCATTGCTCTTGTCGTGC | This paper | N/A |
| Primer R2: GCATCTCATGAAACCAGACGC | This paper | N/A |
| cdc-42_F3: TGAAAGCAGTGAAATACGTTGAA | This paper | N/A |
| cdc-42_R3: TGTTGTGGTGGGTCGAGAG | This paper | N/A |
| lir-3_F4: TTCTCCATATCCAGTGCATGA | This paper | N/A |
| lir-3_R4: TGAAGCTTCCTGTCGGATG | This paper | N/A |
| rpc-1_F1: GGAAGCCTATAAAACATCACTTC | This paper | N/A |
| rpc1_R1: GAGTCGATGGTTCTCCAATACTAG | This paper | N/A |
| rps-21_F1: CGTTCCACGCAAGTGCTCTTCG | This paper | N/A |
| rps-21_R1: CTTTCCTGGGATCATGCGGCC | This paper | N/A |
| Recombinant DNA | ||
| pGEM-T Easy Vector | Promega | Cat#A1380 |
| pPD136.61 [P(unc-54)::CFP] | Addgene | Cat#1682 |
| pENG603 [P(lir-3)::lir-3] | This paper | N/A |
| Software and Algorithms | ||
| CLC Bio | www.clcbio.com | N/A |
| MAQGene | Bigelow et al., 2009 | http://maqweb.sourceforge.net/ |
| Leica Application Suite X | Leica | N/A |
| TopHat 2.0.9 | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/index.shtml |
| EdgeR | Robinson et al., 2010, McCarthy et al., 2012 | http://bioconductor.org |
| ImageJ | Open source | N/A |
| SMART | Letunic et al., 2012, Schultz et al., 1998 | http://smart.embl-heidelberg.de/ |
| NLS Mapper | Kosugi et al., 2009 | http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi |
| FoldIndex | Prilusky et al., 2005 | http://bioportal.weizmann.ac.il/fldbin/findex |
| MEME Suite | Bailey et al., 2009 | http://meme-suite.org/ |
| BLAST | Open source | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| T-Coffee | Notredame et al., 2000 | http://www.ebi.ac.uk/Tools/msa/tcoffee/ |
| ORF Finder | Open source | https://www.ncbi.nlm.nih.gov/gorf/orfig.cgi |
| Other | ||
| Confocal laser scanning microscope: TCS SP8 | Leica | N/A |
| FastPrep 24 Instrument | MP Biomedicals | Cat#116004500 |
| 48-well Bio-Dot microfiltration system | Bio-Rad | Cat#1703938 |
| HiSeq 2500 Instrument | Illumina | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ellen A.A. Nollen (e.a.a.nollen@umcg.nl). Published research materials and reagents from the ERIBA are shared with the academic community under a Material Transfer Agreement (MTA).
Experimental Model and Subject Details
C. elegans
The strains used in this study are listed in Table S1. Standard methods were used for culturing C. elegans at 20°C (Brenner, 1974). The LIR-3 overexpression strain (OP312) was generated by biolistic transformation to produce an integrated, low-copy transgene of the WRM0637aB05 fosmid, recombined with GFP::3xFLAG in frame at the carboxy terminus of the lir-3 locus (Sarov et al., 2012). To synchronize animals, eggs were collected from gravid hermaphrodites by hypochlorite bleaching and hatched overnight in M9 buffer. The desired numbers of L1 animals were subsequently cultured on nematode growth medium (NGM) agar plates seeded with OP50 bacteria.
Method Details
EMS mutagenesis and mapping
Mutagenesis was performed using standard C. elegans ethyl methanesulfonate (EMS) methodology (Jorgensen and Mango, 2002). 8000 mutagenized genomes were screened for suppressors of aggregation. moag-2(pk2183) was identified by single-nucleotide polymorphism mapping to a region between base 9,400,743 and 11,827,697 on linkage group II (Wicks et al., 2001). Next generation sequencing was performed in that region to identify candidate genes for moag-2. CLC Bio and MAQGene software were utilized for mapping the mutation in F37H8.1 (listed in Key Resources Table).
RNA interference
RNAi experiments were performed on NGM agar plates containing 1 mM IPTG and 50 mg/ml ampicillin and seeded with RNAi bacteria induced with IPTG to produce dsRNA. Worms were synchronized by hypochlorite bleaching; L1 worms were grown on RNAi plates and used for the experiments at L4 stage, unless stated otherwise. Plates were coded so that the experimenter was blind to the genotype of the animal.
Motility assay
At day 1, 4, 8 and 12 of adulthood, animals were placed in a drop of M9 and were allowed to recover for 30 s after which the number of body bends was counted for 30 s. Fifteen animals were counted per experiment and the data from three biological replicates was combined. Plates were coded so that the experimenter was blind to the genotype of the animal. The experiments with the N2 worms and the Q40 animals were performed independently due to growth differences between the strains. In our laboratory conditions, N2 worms take approximately 72 hr to reach L4 stage after hypochlorite treatment, whereas Q40 take approximately 96 hr.
Generation of transgenic strains
For the rescue experiment, a genomic construct of lir-3 spanning 1500 bp upstream to 330 bp downstream of F37H8.1 was amplified from N2 genomic DNA by nested PCR using primers F1; R1; F2 and R2 (primer sequences listed in the Key Resources Table). The resulting PCR fragment was cloned into the pGEM-T Easy Vector (#A1380, Promega) and sequenced. Transgenic lines were made by injecting ∼20 ng/μl of construct along with ∼10 ng/μl of pPD136.61 [P(unc-54::CFP)] into N2 animals. Two independent lines were obtained for each transgene of interest.
Quantitative PCR
Total RNA was extracted from synchronized populations in L4 stage using Trizol (#15596-018, Life Technologies) according to the manufacturer’s description. Total RNA quality and concentration were assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific). cDNA was made from 2 μg total C. elegans RNA with a RevertAid H Minus First Strand cDNA Synthesis kit (#K1632, Life Technologies) using random hexamer primers. Quantitative real-time PCR was performed using a Roche LightCycler 480 Instrument II (Roche Diagnostics) with SYBR green dye (#172-5125, Bio-Rad) to detect DNA amplification. The following cycle conditions were used: 50°C for 10 min, 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 57°C for 30 s. Relative transcript levels were quantitated using a standard curve of pooled cDNA solutions. Expression levels were normalized against the endogenous reference gene cdc-42. The following primers were used: cdc-42_F3; cdc-42_R3; lir-3_F4 and lir-3_R4 (primer sequences listed in the Key Resources Table).
To measure the efficiency of Pol III knockdown by RNAi, total RNA from 20 L4 animals was extracted using Trizol (Life Technologies). As a quality control step, total RNA was measured using Qubit RNA HS Assay Kit (#Q32852, Thermo Fisher). Following DNase I digestion (#EN0521, Fermentas), total RNA was purified with Trizol (Life Technologies). First strand synthesis was carried out using Superscript II (Invitrogen) using random hexamer primers. Expression levels were normalized to the expression of rps-21 following the 2−ΔΔCT method (Livak and Schmittgen, 2001). The primers used were rpc-1_F1; rpc1_R1; rps-21_F1 and rps-21_R1 (primer sequences listed in the Key Resources Table).
Chromatin immunoprecipitation sequencing
ChIP assays were conducted as previously described (Niu et al., 2011, Zhong et al., 2010). Worm staging was achieved by bleaching and L1 starvation. Arrested L1 worms were plated on peptone-enriched NGM plates seeded with OP50 bacteria and grown for 48 hr for L4 collection at 20°C. Samples were cross-linked with 2% formaldehyde for 30 min at room temperature and then quenched with 1 M Tris pH 7.5. The pelleted worms were subsequently flash frozen in liquid nitrogen and stored at −80°C. Samples were sonicated using a microtip to obtain DNA fragments mostly 200 to 800 bp in length. For each sample, 2.2 or 4.4 mg of cell extract was immunoprecipitated using a goat α-GFP antibody, GoatV (gift from Kevin White).
For library preparation and sequencing we used the enriched DNA fragments and input control (genomic DNA from the same sample) for two biological replicates as previously described (Kasper et al., 2014). Briefly, samples were multiplexed using the Ovation Ultralow DR Multiplex Systems 1-8 and 9-16 (NuGEN Technologies) following the manufacturer’s protocol, with the exception that QIAGEN MinElute PCR purification kits were used to isolate the DNA. Library size selection in the 200-800 bp range was achieved using the SPRIselect reagent kit (Beckman Coulter) and sequencing was performed on the Illumina HiSeq 2000 platform. To search for MOAG-2/LIR-3-specific binding sites, we only considered binding sites consistent across both replicates and within a range of −400 bp to +100 bp relative to the TSS. In addition, to avoid false positives in our analysis we excluded highly occupied target regions (Gerstein et al., 2010). The ChIP seq data used was obtained from modENCODE DCC (http://www.modencode.org, September 2014). All ChIP seq data were deposited in the ENCODE database, under ID code ENCSR408FDZ and in ArrayExpress, under accession number E-MTAB-4174.
RNA sequencing
Worms were grown to L4 stage and total RNA was extracted using Trizol (Life Technologies) according to the manufacturer’s description. For polyA RNA sequencing, the TruSeq Sample Preparation V2 Kit was used (Illumina). For RNA sequencing (mRNA, snoRNA, tRNA and large ncRNA), a protocol has been described previously (Ikegami and Lieb, 2013). Briefly, total RNA was treated with DNaseI and depleted from rRNA with the Ribominus Eukaryote Kit (#A10837-08, Invitrogen). Fragmentation of RNA was performed using Fragmentation Buffer (#AM8740, Ambion) and cDNA was generated using the Superscript II Kit (Invitrogen). cDNA libraries were subjected to high-throughput single-end sequencing (50 bp) in an Illumina HiSeq 2500 instrument. RNA sequencing data was mapped to the WS220 genome reference using the TopHat 2.0.9 program (Kim et al., 2013) and gene annotation was derived from Ensembl release 66. All RNA sequencing has been submitted to ArrayExpress, under accession number E-MTAB-4172.
Immunofluorescence
Indirect immunofluorescence was performed using the “freeze-cracking” method (Duerr, 2006). Briefly, synchronized worms at L4 stage were fixed in 4% paraformaldehyde in 100 mM sodium phosphate buffer for 1 hr. Samples were washed three times with TBS-T for 10 min and blocked in 10% goat serum in antibody buffer (PBS, 0.5% Triton X-100, 1mM EDTA, 0.1% BSA, 0.05% sodium azide, pH 7.2) for 1 hr. Anti-FLAG primary antibody (#F3165, Sigma-Aldrich) was applied at a 1:100 dilution overnight at 4°C. The next day, samples were washed three times in antibody buffer for 20 min and incubated with the secondary antibody Cy5-labeled goat anti-mouse IgG (#A10524, Thermo Fisher) at a 1:500 dilution for 4 hr at room temperature. After washing with antibody buffer three times, samples were mounted in Vectashield mounting medium with DAPI (#H-1200, Vector Labs) and analyzed on a TCS SP8 laser scanning confocal microscope with a 40x/1.30 NA objective lens (Leica).
Filter retardation assay
The protocol was adapted from Wanker et al. (Scherzinger et al., 1997, Wanker et al., 1999). Briefly, crude worm lysates from synchronized L4 animals were resuspended in FTA sample buffer (10 mM Tris-Cl pH 8.0, 150 mM NaCl, 2% SDS) with protease inhibitors (Complete, Roche) and disrupted using a bead beater (FastPrep 24, MP Biomedicals) for 7 cycles of 20 s with 5 min rest in between cycles. Supernatants were transferred to new 1.5 mL tubes, and protein concentration was determined. To detect SDS-insoluble aggregates, 100 μg of total protein was mixed with 1 M DTT and FTA sample buffer (final concentration 40 μg/100 μl) and heated at 98°C for 5 min. Samples were filtered through a 0.22 micron cellulose acetate membrane using a 48-well Bio-Dot microfiltration system (Bio-Rad) and 100 μg of total protein was used for the assay plus two five-fold serial dilutions. Proteins were blocked for 30 min with 5% milk in TBS-T. Membranes were incubated with the primary antibodies α-GFP (#632381, Clontech Laboratories) or α-tubulin (#T6074, Sigma-Aldrich) at a 1:5000 dilution overnight at 4°C. Incubation with the secondary α-mouse antibody was at a 1:10 000 dilution for 1 hr at room temperature. Antibody binding was visualized with an ECL kit (#RPN2232, Amersham).
Co-immunoprecipitation
The protocol was adapted from (Ikegami et al., 2010). Briefly, crude worm lysates from synchronized L4 animals were resuspended in FA buffer (50 mM HEPES/KOH pH 7.5, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 150 mM NaCl) supplemented with protease inhibitors (Complete, Roche) and 1% sodium lauroyl sarcosinate (sarkosyl). Samples were cross-linked with formaldehyde for 30 min and 2.5 M glycine for 5 min. Worms were disrupted using a bead beater (FastPrep 24, MP Biomedicals) for 7 cycles of 20 s with 5 min rest in between cycles. Supernatants were transferred to new 1.5 mL tubes and protein concentration was determined. Lysates were pre-cleared for 30 min with Protein G-Sepharose beads (Amersham) and incubated with primary antibodies α-Pol II (#38520002, Novus Biologicals) or α-Pol III (#53330002, Novus Biologicals) at a dilution of 1:100 overnight at 4°C. Lysates were coupled to Protein G-Sepharose beads (Amersham) for 1 hr and washed five times for 5 min with FA buffer supplemented with 1 mM PMSF and protease inhibitor cocktail (Complete, Roche). Proteins were eluted by boiling the beads in SDS sample buffer and analyzed by western blotting.
Protein insolubility assay
Synchronized worms at L4 stage animals were resuspended in FA buffer and disrupted using a bead beater (FastPrep 24, MP Biomedicals) for 7 cycles of 20 s with 5 min rest in between cycles, followed by 15 sonication steps of 30 s with 30 s rest between cycles. Protein concentration was determined and equal amounts of lysates were incubated with 2% SDS for 1 hr at room temperature. The lysates were centrifuged at 13 300 rpm for 30 min and the supernatant (soluble fraction) was collected. The pellet (insoluble fraction) was washed two times with FA buffer and resuspended in urea buffer (8M urea, 2% SDS, 50 mM DTT, 50 mM Tris pH 8) for 1 hr. The samples were subsequently analyzed by western blotting.
Protein purification
Recombinant moag-2/lir-3 was expressed and purified fused to the glutathione S-transferase (GST) from the pGEX-6-P1 vector in E. coli BL21 (DE) gold strain (Stratagene). Cells were grown in Overnight Express Instant TB Medium (Merck Millipore) supplemented with ampicillin (100 μg/ml) overnight at 30°C under constant shaking at 250 rpm. The cells were harvested by centrifugation, resuspended in lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA and EDTA-Free protease inhibitor cocktail (Complete, Roche) and lysed by sonication. The cell debris was removed by centrifugation at 18 000 rpm (JA-25.50 rotor, Beckman Coulter). The supernatant was loaded onto a column containing Glutathione Sepharose 4 Fast Flow resin (GE Healthcare LifeSciences) and equilibrated with lysis buffer. The column was washed with 40 CV of lysis buffer and the protein was eluted in 50 mM Tris pH 8, 10 mM reduced glutathione and dialyzed in 50 mM Tris pH 7.4, 150 mM NaCl for the aggregation experiments.
Aggregation kinetics
4 μM solutions of GST-HttQ48 or GST-HttQ23 alone or with an equimolar concentration of moag-2/lir-3 or moag-4 were incubated in the presence of 7U of PreScission protease (GE Healthcare LifeSciences) per nmol of Htt protein in 50 mM Tris pH 7.4, 150 mM NaCl, 20 μM Thioflavin T at 37°C. The aggregation reactions were performed under constant linear shaking at 500 rpm and the ThT fluorescence was monitored in low-binding, clear-bottomed half-area 96-well plates. Emissions at 480 nm were recorded every 300 s with excitation at 440 nm, using a CLARIOstar plate reader (BMG Labtech). Fluorescence values were analyzed with the following sigmoidal equation:
where A0 and A are the values at the beginning and the end of the aggregation, mA0 and mA are the slopes of the lag phase and the plateau, assuming for them a linear dependency of normalized fluorescence values with the incubation time, t50% is the midpoint of aggregation and kagg is the apparent aggregation rate constant.
Subcellular fractionation
The protocol was adapted from (Chen et al., 2000). Briefly, crude worms lysates from synchronized L4 animals were resuspended in hypotonic buffer (15 mM HEPES KOH pH 7.6, 10 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 350 mM Sucrose, 1 mM PMSF, 1 mM DTT, supplemented with protease inhibitors cocktail (Complete, Roche). Animals were homogenized using microtube pestle rods and motor (Kontes) by applying 10 strokes of 1 min followed by 1 min rest in between strokes. Worm debris was removed by centrifugation at 500 g for 5 min at 4°C. The pellet was discarded and the supernatant was used as input of the cell fractionation. Nuclei were pelleted at 4000 g for 5 min at 4°C. The supernatant containing the cytosolic fraction was centrifuged at 17 000 g for 30 min at 4°C to remove membrane fraction and contaminants. The nuclear fraction was washed two times with hypotonic buffer and resuspended in hypertonic buffer (15 mM HEPES KOH pH 7.6, 400 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 0.1% Tween 20, 10% Glycerol, 1 mM PMSF, 1 mM DTT, supplemented with protease inhibitors cocktail (Complete, Roche)). The nuclear fraction was homogenized using 29G ± inch needle insulin syringe (Terumo) and treated with 25 U/μl Benzonase (Millipore) for 30 min at 4°C. Ten μg of each fraction was loaded on a 12% SDS-PAGE. Gels were transferred to nitrocellulose membranes (Brunschwig Chemie) and incubated with the primary antibodies α-FLAG (#F3165, Sigma-Aldrich) at a 1:1000 dilution; α-LMN-1 (#38530002, Novus Biologicals) at a 1:1000 dilution; α-GFP (#632381, Clontech Laboratories) and α-tubulin (#T6074-200UL, Sigma-Aldrich) at a 1:5000 dilution overnight at 4°C. Incubation with the secondary antibody was at a 1:10 000 dilution for 1 hr at room temperature. Antibody binding was visualized with an ECL kit (#RPN2232, Amersham).
Bioinformatic analysis
Conserved domains were identified using SMART (Simple Modular Architecture Research Tool) (Letunic et al., 2012, Schultz et al., 1998). Nuclear signal localization was predicted using NLS Mapper (Kosugi et al., 2009). Protein folding was predicted using FoldIndex (Prilusky et al., 2005). The algorithm used for motif discovery was the MEME Suite (Bailey et al., 2009). Orthologs were identified using protein BLAST search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and aligned with T-Coffee multiple sequence alignment tool (Notredame et al., 2000). Amino acid predictions were performed using ORF Finder (https://www.ncbi.nlm.nih.gov/gorf/orfig.cgi).
Quantification and Statistical Analyses
Quantification of aggregates and motility assay
The number of aggregates present in whole worms was counted using a fluorescence dissection microscope (Leica). A minimum of 20 worms was counted and the data from three or four biological replicates were combined. For the motility assay, 15 animals were counted per experiment and the data from three biological replicates was combined.
A biological replicate was defined as an independently grown worm population, before extraction of embryos from gravid adult hermaphrodites to obtain synchronized populations (see Experimental Model and Subject Details). The exact number of biological replicates is indicated in the figure legends. Data are represented as mean ± SEM and significance was calculated using a one-tailed or two-tailed unpaired Student’s t test. The exact type of Student’s t test is indicated in the figure legend. ∗p < 0.05 ∗∗p < 0.01; ∗∗∗p < 0.001.
RNA sequencing data analysis
All RNA seq are represented as the mean of n = 3 biological replicates, with error bars representing mean ± SEM. A generalized linear model was used to identify differential gene expression between the Q40 wild-type and the Q40 mutants with EdgeR (Robinson et al., 2010, McCarthy et al., 2012). Replicates 1-3 were introduced as a technical factor to correct for batch effect. Genes with average expression level below 1 fragment per million (FPM) were excluded from the analysis. The library normalization was left at the standard setting (trimmed mean of M-values, TMM). The resulting p values were corrected for multiple testing using the Benjamini-Hochberg procedure. Per gene expression data was normalized as FPM. Data visualization and statistical tests were conducted using R scripts (available upon request).
Quantitative PCR analysis
All quantitative PCR are represented as the mean of n = 3 biological replicates, with error bars representing mean ± SEM. To measure the relative lir-3 mRNA expression, lir-3 transcript levels were quantitated using a standard curve of pooled cDNA solutions and expression levels were normalized against the endogenous reference gene cdc-42. To measure the relative Pol II or Pol III mRNA expression, the corresponding transcript levels were normalized to the expression of rps-21 following the 2-ΔΔCT method (Livak and Schmittgen, 2001) (see Method Details).
Filter retardation assay
Immunoblots were quantified by densitometry using ImageJ (listed in Key Resources Table). To quantify the relative amount of SDS-insoluble protein, a ratio (fold change) was calculated by dividing the values of Q40;moag-2/lir-3 mutants by their corresponding wild-types (corrected to α-tubulin as a loading control).
Data and Software Availability
The accession numbers for the ChIP-seq data reported in this paper are ENCODE: ENCSR408FDZ and ArrayExpress: E-MTAB-4174. The accession number for the RNA-seq data reported in this paper is ArrayExpress: E-MTAB-4172. Full images used in this study have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/knzkvgbf6x.2.
Author Contributions
O.S. and E.A.A.N. designed the research; O.S. performed most experiments; A.M.-C., M.K., M.A.Z., F.A.A., R.I.S., E.S., C.N.M., H.H.W., W.H., and A.S. participated in the experiments; O.S., T.d.J., R.W.P., E.E.W., M.V., C.F.C., V.R., and V.G. analyzed the data; and O.S. and E.A.A.N. wrote the manuscript with input from all authors.
Acknowledgments
We thank the Caenorhabditis Genetics Centre (funded by the NIH National Centre for Research Resources and the NIH Office of Research Infrastructure Programs [P40 OD010440]) and the Mitani laboratory for the C. elegans strains (funded by the Japan National BioResource Project). We thank Pieter van der Vlies and Jelkje Bergsma for practical assistance with whole-genome sequencing. We thank Pieter Neerincx for help with installing MAQGene. We thank Marianna Bevova and Diana Spierings for assistance with RNA sequencing. We thank Kohta Ikegami for the npp3 RNAi vector and for the protocols on RNA sequencing and co-immunoprecipitation. We thank the modERN project (NIH U41HG007355) for sharing the MOAG-2/LIR-3 ChIP-seq data prior to publication. We thank Klaas Sjollema for help with C. elegans imaging (UMIC, UMCG). We thank Sally Hill for editing this manuscript. This study was supported by funding from the Berlin Institute of Health Collaborative Research Grant no. 1.1.2.a.3 funded by the German Federal Ministry for Education and Research (BMBF) (to E.E.W). This project was funded by a Meervoud Grant from NWO (836.09.001) (to E.A.A.N.), a European Research Council (ERC) starting grant (281622 PDControl) (to E.A.A.N.), the Fundação para a Ciência e Tecnologia fellowship (SFRH/BD/51009/2010) (to O.S.), the Alumni chapter Gooische Groningers facilitated by the Ubbo Emmius Fonds, and a Groningen University Institute for Drug Exploration (GUIDE) fellowship (O.S.).
Published: March 16, 2017
Footnotes
Supplemental Information includes five figures and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.02.022.
Contributor Information
Victor Guryev, Email: v.guryev@umcg.nl.
Ellen A.A. Nollen, Email: e.a.a.nollen@umcg.nl.
Supplemental Information
Related to Figures 1, 4, S4, and S5. This table includes raw data showing the average number of aggregates and p values for the panel(s) in Figures 1, 4, S4, and S5.
Related to Figures 4 and S4. This table includes raw data showing the changes in transcription between Q40 and Q40;moag-2/lir-3 mutants in Figures 4 and S4.
References
- Alla R.K., Cairns B.R. RNA polymerase III transcriptomes in human embryonic stem cells and induced pluripotent stem cells, and relationships with pluripotency transcription factors. PLoS ONE. 2014;9:e85648. doi: 10.1371/journal.pone.0085648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M., Mitra S., Schweitzer E.S., Segal M.R., Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Bailey T.L., Bodén M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow H., Doitsidou M., Sarin S., Hobert O. MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat. Methods. 2009;6:549. doi: 10.1038/nmeth.f.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B., Kumita J.R., Barros T.P., Esbjorner E.K., Luheshi L.M., Crowther D.C., Wilson M.R., Dobson C.M., Favrin G., Yerbury J.J. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 2010;5:735–740. doi: 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- Bratkovič T., Rogelj B. The many faces of small nucleolar RNAs. Biochim. Biophys. Acta. 2014;1839:438–443. doi: 10.1016/j.bbagrm.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R.S. Zinc finger proteins: getting a grip on RNA. Curr. Opin. Struct. Biol. 2005;15:94–98. doi: 10.1016/j.sbi.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Chen F., Hersh B.M., Conradt B., Zhou Z., Riemer D., Gruenbaum Y., Horvitz H.R. Translocation of C. elegans CED-4 to nuclear membranes during programmed cell death. Science. 2000;287:1485–1489. doi: 10.1126/science.287.5457.1485. [DOI] [PubMed] [Google Scholar]
- Chiti F., Dobson C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- Davranche A., Aviolat H., Zeder-Lutz G., Busso D., Altschuh D., Trottier Y., Klein F.A.C. Huntingtin affinity for partners is not changed by polyglutamine length: aggregation itself triggers aberrant interactions. Hum. Mol. Genet. 2011;20:2795–2806. doi: 10.1093/hmg/ddr178. [DOI] [PubMed] [Google Scholar]
- den Boer B.G., Sookhareea S., Dufourcq P., Labouesse M. A tissue-specific knock-out strategy reveals that lin-26 is required for the formation of the somatic gonad epithelium in Caenorhabditis elegans. Development. 1998;125:3213–3224. doi: 10.1242/dev.125.16.3213. [DOI] [PubMed] [Google Scholar]
- Doi H., Okamura K., Bauer P.O., Furukawa Y., Shimizu H., Kurosawa M., Machida Y., Miyazaki H., Mitsui K., Kuroiwa Y., Nukina N. RNA-binding protein TLS is a major nuclear aggregate-interacting protein in huntingtin exon 1 with expanded polyglutamine-expressing cells. J. Biol. Chem. 2008;283:6489–6500. doi: 10.1074/jbc.M705306200. [DOI] [PubMed] [Google Scholar]
- Doi H., Koyano S., Suzuki Y., Nukina N., Kuroiwa Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci. Res. 2010;66:131–133. doi: 10.1016/j.neures.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Duerr J.S. WormBook. WormBook; 2006. Immunohistochemistry; pp. 1–61. [Google Scholar]
- Dufourcq P., Chanal P., Vicaire S., Camut E., Quintin S., den Boer B.G., Bosher J.M., Labouesse M. lir-2, lir-1 and lin-26 encode a new class of zinc-finger proteins and are organized in two overlapping operons both in Caenorhabditis elegans and in Caenorhabditis briggsae. Genetics. 1999;152:221–235. doi: 10.1093/genetics/152.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah A.W., Jeong H., Griffin A., Kim Y.-M., Standaert D.G., Hersch S.M., Mouradian M.M., Young A.B., Tanese N., Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D.E., Sutton L., Hayden M.R. Small changes, big impact: posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist. 2011;17:475–492. doi: 10.1177/1073858410390378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D., Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsone S.F., Meyer N.H., Schrank E., Leitinger G., Pham C.L.L., Fodero-Tavoletti M.T., Holmberg M., Dulle M., Scicluna B., Gesslbauer B. SERF protein is a direct modifier of amyloid fiber assembly. Cell Rep. 2012;2:358–371. doi: 10.1016/j.celrep.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia S.M., Casanueva M.O., Silva M.C., Amaral M.D., Morimoto R.I. Neuronal signaling modulates protein homeostasis in Caenorhabditis elegans post-synaptic muscle cells. Genes Dev. 2007;21:3006–3016. doi: 10.1101/gad.1575307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M.B., Lu Z.J., Van Nostrand E.L., Cheng C., Arshinoff B.I., Liu T., Yip K.Y., Robilotto R., Rechtsteiner A., Ikegami K., modENCODE Consortium Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidalevitz T., Ben-Zvi A., Ho K.H., Brignull H.R., Morimoto R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Gu X., Greiner E.R., Mishra R., Kodali R., Osmand A., Finkbeiner S., Steffan J.S., Thompson L.M., Wetzel R., Yang X.W. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828–840. doi: 10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C., Patterson B. Spliceosomal snRNAs. Annu. Rev. Genet. 1988;22:387–419. doi: 10.1146/annurev.ge.22.120188.002131. [DOI] [PubMed] [Google Scholar]
- Hall T.M.T. Multiple modes of RNA recognition by zinc finger proteins. Curr. Opin. Struct. Biol. 2005;15:367–373. doi: 10.1016/j.sbi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Hamamichi S., Rivas R.N., Knight A.L., Cao S., Caldwell K.A., Caldwell G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA. 2008;105:728–733. doi: 10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu A.-L., Murphy C.T., Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Iakoucheva L.M., Brown C.J., Lawson J.D., Obradović Z., Dunker A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- Ikegami K., Lieb J.D. Integral nuclear pore proteins bind to Pol III-transcribed genes and are required for Pol III transcript processing in C. elegans. Mol. Cell. 2013;51:840–849. doi: 10.1016/j.molcel.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami K., Egelhofer T.A., Strome S., Lieb J.D. Caenorhabditis elegans chromosome arms are anchored to the nuclear membrane via discontinuous association with LEM-2. Genome Biol. 2010;11:R120. doi: 10.1186/gb-2010-11-12-r120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana N.R., Dikshit P., Goswami A., Kotliarova S., Murata S., Tanaka K., Nukina N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 2005;280:11635–11640. doi: 10.1074/jbc.M412042200. [DOI] [PubMed] [Google Scholar]
- Jorgensen E.M., Mango S.E. The art and design of genetic screens: caenorhabditis elegans. Nat. Rev. Genet. 2002;3:356–369. doi: 10.1038/nrg794. [DOI] [PubMed] [Google Scholar]
- Kasper D.M., Wang G., Gardner K.E., Johnstone T.G., Reinke V. The C. elegans SNAPc component SNPC-4 coats piRNA domains and is globally required for piRNA abundance. Dev. Cell. 2014;31:145–158. doi: 10.1016/j.devcel.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R., Head E., Thompson J.L., McIntire T.M., Milton S.C., Cotman C.W., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S., Hasebe M., Tomita M., Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S.S., Majumdar I., Grishin N.V. Structural classification of zinc fingers: survey and summary. Nucleic Acids Res. 2003;31:532–550. doi: 10.1093/nar/gkg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T., Koyama A., Koyama S., Yoshina S., Ren C.-H., Kato T., Mitani S., Iwatsubo T. A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum. Mol. Genet. 2008;17:2997–3009. doi: 10.1093/hmg/ddn198. [DOI] [PubMed] [Google Scholar]
- Labouesse M., Hartwieg E., Horvitz H.R. The Caenorhabditis elegans LIN-26 protein is required to specify and/or maintain all non-neuronal ectodermal cell fates. Development. 1996;122:2579–2588. doi: 10.1242/dev.122.9.2579. [DOI] [PubMed] [Google Scholar]
- Lee V.M., Wang J., Trojanowski J.Q. Purification of paired helical filament tau and normal tau from human brain tissue. Methods Enzymol. 1999;309:81–89. doi: 10.1016/s0076-6879(99)09008-4. [DOI] [PubMed] [Google Scholar]
- Lejeune F.-X., Mesrob L., Parmentier F., Bicep C., Vazquez-Manrique R.P., Parker J.A., Vert J.-P., Tourette C., Néri C. Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC Genomics. 2012;13:91. doi: 10.1186/1471-2164-13-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Doerks T., Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40:D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.-H., Cheng A.L., Zhou H., Lam S., Rao M., Li H., Li X.-J. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 2002;22:1277–1287. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Perumal N.B., Oldfield C.J., Su E.W., Uversky V.N., Dunker A.K. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Marshall L., White R.J. Non-coding RNA production by RNA polymerase III is implicated in cancer. Nat. Rev. Cancer. 2008;8:911–914. doi: 10.1038/nrc2539. [DOI] [PubMed] [Google Scholar]
- Matsumoto M., Yada M., Hatakeyama S., Ishimoto H., Tanimura T., Tsuji S., Kakizuka A., Kitagawa M., Nakayama K.I. Molecular clearance of ataxin-3 is regulated by a mammalian E4. EMBO J. 2004;23:659–669. doi: 10.1038/sj.emboj.7600081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J., Arrasate M., Brooks E., Libeu C.P., Legleiter J., Hatters D., Curtis J., Cheung K., Krishnan P., Mitra S. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 2011;7:925–934. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minezaki Y., Homma K., Kinjo A.R., Nishikawa K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 2006;359:1137–1149. doi: 10.1016/j.jmb.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Nillegoda N.B., Kirstein J., Szlachcic A., Berynskyy M., Stank A., Stengel F., Arnsburg K., Gao X., Scior A., Aebersold R. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature. 2015;524:247–251. doi: 10.1038/nature14884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W., Lu Z.J., Zhong M., Sarov M., Murray J.I., Brdlik C.M., Janette J., Chen C., Alves P., Preston E. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res. 2011;21:245–254. doi: 10.1101/gr.114587.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollen E.A.A., Garcia S.M., van Haaften G., Kim S., Chavez A., Morimoto R.I., Plasterk R.H.A. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C., Higgins D.G., Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Oler A.J., Alla R.K., Roberts D.N., Wong A., Hollenhorst P.C., Chandler K.J., Cassiday P.A., Nelson C.A., Hagedorn C.H., Graves B.J., Cairns B.R. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat. Struct. Mol. Biol. 2010;17:620–628. doi: 10.1038/nsmb.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzscha H., Schermann S.M., Woerner A.C., Pinkert S., Hecht M.H., Tartaglia G.G., Vendruscolo M., Hayer-Hartl M., Hartl F.U., Vabulas R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- Pennuto M., Palazzolo I., Poletti A. Post-translational modifications of expanded polyglutamine proteins: impact on neurotoxicity. Hum. Mol. Genet. 2009;18(R1):R40–R47. doi: 10.1093/hmg/ddn412. [DOI] [PubMed] [Google Scholar]
- Prilusky J., Felder C.E., Zeev-Ben-Mordehai T., Rydberg E.H., Man O., Beckmann J.S., Silman I., Sussman J.L. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- Rampelt H., Kirstein-Miles J., Nillegoda N.B., Chi K., Scholz S.R., Morimoto R.I., Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012;31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes J.S., Deplancke B., Shingles J., Grove C.A., Hope I.A., Walhout A.J.M. A compendium of Caenorhabditis elegans regulatory transcription factors: a resource for mapping transcription regulatory networks. Genome Biol. 2005;6:R110. doi: 10.1186/gb-2005-6-13-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes J.S., Shingles J., Dupuy D., Grove C.A., Walhout A.J.M., Vidal M., Hope I.A. Insight into transcription factor gene duplication from Caenorhabditis elegans Promoterome-driven expression patterns. BMC Genomics. 2007;8:27. doi: 10.1186/1471-2164-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarov M., Murray J.I., Schanze K., Pozniakovski A., Niu W., Angermann K., Hasse S., Rupprecht M., Vinis E., Tinney M. A genome-scale resource for in vivo tag-based protein function exploration in C. elegans. Cell. 2012;150:855–866. doi: 10.1016/j.cell.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G.P., Davies S.W., Lehrach H., Wanker E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Schramm L., Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002;16:2593–2620. doi: 10.1101/gad.1018902. [DOI] [PubMed] [Google Scholar]
- Schultz J., Milpetz F., Bork P., Ponting C.P. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C., Arai T., Hasegawa M., Yu S., McGeer P.L. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 2008;67:1159–1165. doi: 10.1097/NEN.0b013e31818e8951. [DOI] [PubMed] [Google Scholar]
- Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M.C., Fox S., Beam M., Thakkar H., Amaral M.D., Morimoto R.I. A genetic screening strategy identifies novel regulators of the proteostasis network. PLoS Genet. 2011;7:e1002438. doi: 10.1371/journal.pgen.1002438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin O., Nollen E.A.A. Regulation of protein homeostasis in neurodegenerative diseases: the role of coding and non-coding genes. Cell. Mol. Life Sci. 2015;72:4027–4047. doi: 10.1007/s00018-015-1985-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittler A., Wälter S., Wedemeyer N., Hasenbank R., Scherzinger E., Eickhoff H., Bates G.P., Lehrach H., Wanker E.E. SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates. Mol. Cell. 1998;2:427–436. doi: 10.1016/s1097-2765(00)80142-2. [DOI] [PubMed] [Google Scholar]
- Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- Steffan J.S., Agrawal N., Pallos J., Rockabrand E., Trotman L.C., Slepko N., Illes K., Lukacsovich T., Zhu Y.-Z., Cattaneo E. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Thomas M., Dadgar N., Aphale A., Harrell J.M., Kunkel R., Pratt W.B., Lieberman A.P. Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J. Biol. Chem. 2004;279:8389–8395. doi: 10.1074/jbc.M311761200. [DOI] [PubMed] [Google Scholar]
- Topalidou I., Chalfie M. Shared gene expression in distinct neurons expressing common selector genes. Proc. Natl. Acad. Sci. USA. 2011;108:19258–19263. doi: 10.1073/pnas.1111684108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Goot A.T., Zhu W., Vázquez-Manrique R.P., Seinstra R.I., Dettmer K., Michels H., Farina F., Krijnen J., Melki R., Buijsman R.C. Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl. Acad. Sci. USA. 2012;109:14912–14917. doi: 10.1073/pnas.1203083109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham T.J., Holmberg M.A., van der Goot A.T., Teuling E., Garcia-Arencibia M., Kim H.-E., Du D., Thijssen K.L., Wiersma M., Burggraaff R. Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell. 2010;142:601–612. doi: 10.1016/j.cell.2010.07.020. [DOI] [PubMed] [Google Scholar]
- Walther D.M., Kasturi P., Zheng M., Pinkert S., Vecchi G., Ciryam P., Morimoto R.I., Dobson C.M., Vendruscolo M., Mann M., Hartl F.U. Widespread proteome remodeling and aggregation in aging C. elegans. Cell. 2015;161:919–932. doi: 10.1016/j.cell.2015.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker E.E., Scherzinger E., Heiser V., Sittler A., Eickhoff H., Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 1999;309:375–386. doi: 10.1016/s0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- White R.J. Transcription by RNA polymerase III: more complex than we thought. Nat. Rev. Genet. 2011;12:459–463. doi: 10.1038/nrg3001. [DOI] [PubMed] [Google Scholar]
- Wicks S.R., Yeh R.T., Gish W.R., Waterston R.H., Plasterk R.H. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 2001;28:160–164. doi: 10.1038/88878. [DOI] [PubMed] [Google Scholar]
- Woerner A.C., Frottin F., Hornburg D., Feng L.R., Meissner F., Patra M., Tatzelt J., Mann M., Winklhofer K.F., Hartl F.U., Hipp M.S. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351:173–176. doi: 10.1126/science.aad2033. [DOI] [PubMed] [Google Scholar]
- Yanamandra K., Jiang H., Mahan T.E., Maloney S.E., Wozniak D.F., Diamond M.I., Holtzman D.M. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann. Clin. Transl. Neurol. 2015;2:278–288. doi: 10.1002/acn3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M., Niu W., Lu Z.J., Sarov M., Murray J.I., Janette J., Raha D., Sheaffer K.L., Lam H.Y.K., Preston E. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet. 2010;6:e1000848. doi: 10.1371/journal.pgen.1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figures 1, 4, S4, and S5. This table includes raw data showing the average number of aggregates and p values for the panel(s) in Figures 1, 4, S4, and S5.
Related to Figures 4 and S4. This table includes raw data showing the changes in transcription between Q40 and Q40;moag-2/lir-3 mutants in Figures 4 and S4.