

Abstract
Although multivalent binding to surfaces is an important tool in nanotechnology, quantitative information about the residual valency and orientation of surface-bound molecules is missing. To address these questions, we study streptavidin (SAv) binding to commonly used biotinylated surfaces such as supported lipid bilayers (SLBs) and self-assembled monolayers (SAMs). Stability and kinetics of SAv binding are characterized by quartz crystal microbalance with dissipation monitoring, while the residual valency of immobilized SAv is quantified using spectroscopic ellipsometry by monitoring binding of biotinylated probes. Purpose-designed SAv constructs having controlled valencies (mono-, di-, trivalent in terms of biotin-binding sites) are studied to rationalize the results obtained on regular (tetravalent) SAv. We find that divalent interaction of SAv with biotinylated surfaces is a strict requirement for stable immobilization, while monovalent attachment is reversible and, in the case of SLBs, leads to the extraction of biotinylated lipids from the bilayer. The surface density and lateral mobility of biotin, and the SAv surface coverage are all found to influence the average orientation and residual valency of SAv on a biotinylated surface. We demonstrate how the residual valency can be adjusted to one or two biotin binding sites per immobilized SAv by choosing appropriate surface chemistry. The obtained results provide means for the rational design of surface-confined supramolecular architectures involving specific biointeractions at tunable valency. This knowledge can be used for the development of well-defined bioactive coatings, biosensors and biomimetic model systems.
Introduction
Multivalent binding, through multiple interactions between specific ligand/receptor pairs, is involved in a diversity of biological interactions (e.g., antigen/antibody, cell/cell, cell/virus) and cellular processes (e.g., adhesion, chemotaxis, inflammation, secretion).1 Multivalent interactions are also an attractive tool for the design of supramolecular architectures, because they enable strong but reversible attachment, while drastically increasing binding selectivity as compared to monovalent bonds.2,3 Defining the factors governing multivalent binding at interfaces is therefore important for understanding biological systems and for the design of supramolecular nanomaterials.1−3 In particular, the dependence of multivalent self-assembly on surface properties (chemistry, packing, lateral mobility) remains difficult to assess.
Among numerous surface-binding approaches, the interaction between biotin and tetravalent SAv stands out by its high affinity (Kd ≈ 10–14 M4) and specificity. In addition, the high stability of SAv compared to most other proteins along with the availability of efficient methods for the biotinylation of biological and synthetic molecules and nano-objects render the SAv-biotin technology versatile and enable various experimental designs. These features have made the SAv-biotin pair widely used in biotechnology for labeling, detection and purification.5,6 In particular, SAv-mediated assembly at interfaces (surface coatings, nanoparticles, membranes, etc.) provided the basis for various supramolecular architectures intended for biosensors,7−10 biospecific targeting,11 drug delivery systems,12 bioactive coatings13,14 and biomimetic model systems.15,16
In the majority of these architectures, SAv acts as a multivalent linker that uses the same type of interaction to bind to the surface and to attach desired molecules (proteins,7,9,17 nucleic acids,7,8,17,18 sugars,15,16 drugs13) or nano-objects (vesicles,19,20 viruses,21 nanoparticles22) from the solution phase. Composite films, in which SAv functions as a supramolecular glue to assemble multiple layers of nano-objects, have also been realized.19,21 The efficiency of surface functionalization and the stability of the whole surface-confined architecture depend in these systems on the residual valency, i.e., the amount of biotin-binding sites that remain accessible after SAv attachment to the surface.
Among the different surface chemistry approaches, supported lipid bilayers (SLBs)23 and self-assembled monolayers (SAMs)8,24−27 are the most commonly used to study23−27 and to realize8,28 multivalent binding to surfaces. They can be formed on various solid supports (metal/metal oxide, transparent/opaque, conducting/insulating, planar macroscopic surface/curved particle surface, etc.), which provides versatile platforms to study multivalency effects by different surface sensitive techniques and in a wide range of experimental conditions.23,26,27 Combining SLBs and SAMs with established conjugation chemistries (e.g., click alkyne/azide,26−28 His-tag/NTA23) allows tuning of the nature and density of surface binding sites, while keeping lateral mobility on (fluid-phase SLBs) or off (SAMs) depending on the experimental design.
In the case of SAv, many experiments have been performed on SLBs containing laterally mobile biotinylated lipids (b-SLBs).29−33 These revealed that SAv forms dense and stable monolayers,29,31−33 which may evolve into two-dimensional crystals,29,32,33 and divalent binding was proposed based on the analysis of the crystal structure.29 Studies with SAMs containing effectively immobile biotinylated compounds (b-SAMs), formed through thiol/metal34,35 or silane/metal oxide chemistries36 have also been reported.
Despite the common use, and the growing interest in the SAv/biotin technology, our understanding of the parameters that define the stability, the orientation and the residual valency of SAv on biotinylated surfaces is still very limited. On the basis of the symmetric structure of SAv, it is often assumed that SAv binds with two binding sites to the surface and thus has a residual valency of two.8,15,29,30,37 This assumption, however, and the corresponding orientation of SAv on the surface are rarely confirmed experimentally. It has also been reported that the strength of SAv-biotin binding on SAMs can be lower than in the solution phase and depends on the nature of the SAM and the surface density of biotin,34 illustrating that the SAv binding depends sensitively on how biotin is presented by the surface. One can expect the residual valency of SAv to depend on the biotin surface density, and the flexibility and/or lateral mobility of binding sites may also influence the residual valency.9 Moreover, solution studies revealed that the SAv-biotin binding strength depends on the number and position of the occupied binding pockets,38−40 and this may additionally affect SAv attachment to biotinylated surfaces. In particular, substantial negative cooperativity due to steric hindrance was found for large biotinylated ligands (e.g., DNA or proteins) in the case of divalent binding with the occupied binding pockets arranged in cis- but not when arranged in trans-orientation.38 Recognizing this complexity raises the question of the interplay between surface chemistry and the residual valency of SAv, and multivalent molecules in general.
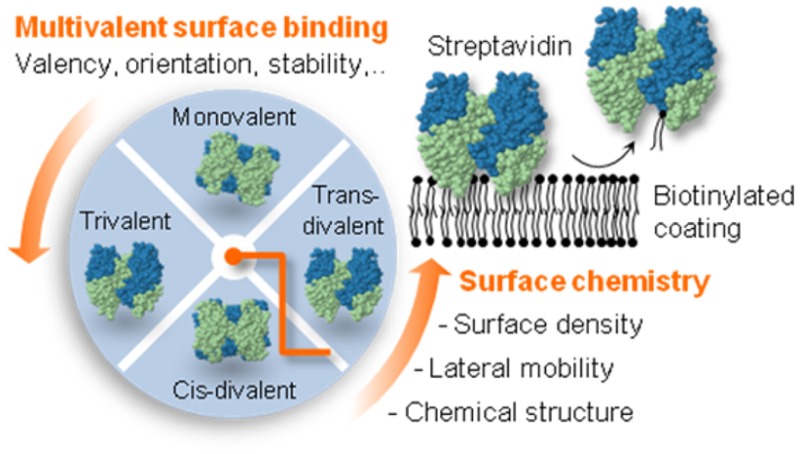
Here, we address this question with a systematic and quantitative analysis of SAv binding to both b-SAMs and b-SLBs. The lateral mobility and surface density of biotin, and the SAv surface coverage, are tuned and their influence on SAv-biotin interactions is studied. The results obtained on regular SAv are rationalized using purpose-designed synthetic SAv constructs having lower valencies but no change in binding affinity (Figure 1). We uncover that SAv can bind in several distinct orientations and valencies to b-SAMs and b-SLBs, and that the monovalent attachment of SAv on b-SLBs and b-SAMs is generally unstable. We thus show that the spectrum of SAv properties at surfaces is much richer than previously appreciated, and demonstrate how this can be exploited to tune the residual valency of SAv between one and two by choosing appropriate surface chemistry and/or SAv immobilization conditions.
Figure 1.

Tunable model system to study SAv binding to biotinylated surfaces. (A) Table of tunable parameters. (B) SAv constructs having different valencies. On the left, the structure of the SAv tetramer (ribbon diagram with each monomer in distinct color) with biotins attached to its binding pockets (ball-and-stick model) is shown; on the right, the SAv constructs used are listed schematically. (C–E) Fluorescence measurements in the presence of biotin in solution reveal the number of binding sites in SAv constructs. (C) Relative fluorescence emission intensity of tryptophan located in the binding pockets of rSAv, tSAv, dSAv-trans, mSAv (filled squares, each data point represents a single measurement) and dSAv-cis (empty triangle, mean of 2 measurements with standard error) upon biotin binding to saturation. All data fall onto a straight line (linear fit) that crosses the y axis at 100%. (D) Example of tryptophan relative intensity change upon biotin binding to SAv (the moment of biotin injection is indicated by an arrow). This data set corresponds to the last point in (C). (E) Examples of fluorescence spectra. Here, tryptophan fluorescence emission spectra of rSAv solution in the absence of biotin (blue) and 45 min after biotin injection (red) are shown, and the maxima in these spectra correspond to the first and last points, respectively, in (D). A negative control (i.e., biotin without SAv) is also shown (black).
Experimental Section
Materials
The SAv constructs used are schematically shown in Figure 1B. Regular tetravalent streptavidin (rSAv, Mw ≈ 60 kDa) was purchased from Sigma-Aldrich. Trivalent SAv (tSAv, 55.4 kDa), divalent SAv with cis- (dSAv-cis, 55.2 kDa) and trans- (dSAv-trans, 54.6 kDa) orientations of biotin-binding sites and monovalent SAv (mSAv, 53.8 kDa) were expressed in E. coli, refolded from inclusion bodies, and purified by ion-exchange chromatography as previously described.38 Dioleoylphosphatidylcholine (DOPC; 1 in Figure 2A) and dioleoylphosphatidylethanolamine-CAP-biotin (DOPE-CAP-biotin; 2 in Figure 2A) were purchased from Avanti Polar Lipids (Alabaster, AL). HS-(CH2)11-EG4-OH (EG: ethylene glycol; 3 in Figure 2B) and HS-(CH2)11-EG6-biotin (4 in Figure 2B) were purchased from Prochimia (Sopot, Poland). Biotin and O-(2-aminoethyl)-O′-[2-(biotinylamino)ethyl]octaethylene glycol (b-OEG) were purchased from Sigma-Aldrich. An oligosaccharide of HA with 15 monosaccharide units and a biotin at the reducing end (b-oHA; 3.2 kDa; Figure 3) was produced through functionalization of an HA pentadecasaccharide generated by an automated solid-phase synthesis,41 purified by HPLC and characterized by MS and 1H NMR (Supporting Methods and Figure S1). A tandem repeat of the Z domain of protein A connected through a flexible spacer (12 amino acids) to an N-terminal biotin (b-ZZ, 16.2 kDa; Figure 3) was expressed in E. coli, purified and characterized by SDS-PAGE (Supporting Methods and Figure S2). 4.95 MHz QCM-D sensors coated with gold (QSX301) or silica (QSX303) were purchased from Biolin Scientific (Västra Frölunda, Sweden). Silicon wafers with a native oxide film were purchased from University Wafer (Boston, MA). Silicon wafers with an opaque gold coating were purchased from BT Electronics (Les Ulis, France). A working buffer made of 10 mM HEPES, pH 7.4 and 150 mM NaCl was used for all QCM-D, SE and fluorescence measurements. All experiments were performed at room temperature.
Figure 2.

Biotinylated surfaces with controlled lateral mobility and biotin content. (A) Schematic representation of a b-SLB, chemical structure of lipids and a characteristic QCM-D signature of b-SLB formation using SUVs made of DOPC (1) and DOPE-CAP-biotin (2) at a 19:1 molar ratio. The time of SUV exposure is indicated by an arrow. (B) Schematic representation of b-SAM, chemical structure of thiols and characteristic images of water drops placed on a bare Au surface and on Au surfaces functionalized with HS-(CH2)11-EG4-OH (3) and its mixture with HS-(CH2)11-EG6-biotin (4) at a 9:1 molar ratio with contact angles indicated.
Figure 3.
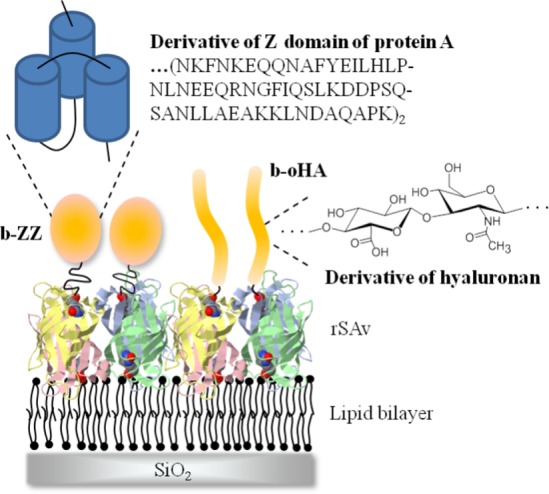
Reporters for residual SAv valency. Schematic representation of biotinylated probes, b-ZZ and b-oHA, used as reporters of the residual valency of SAv bound to a biotinylated surface (in this case b-SLB). The reporter probes are drawn approximately to scale with the rSAv and the SLB thickness.
Quartz Crystal Microbalance with Dissipation Monitoring (QCM-D) Measurements
QCM-D measurements were performed in flow mode at a flow rate of 20 μL/min using a Q-Sense E4 system equipped with four Q-Sense Flow Modules (Biolin Scientific). Silica-coated sensors (for SLBs) or gold-coated sensors (functionalized ex situ with SAMs) were used as substrates. Before injection, SAv, b-ZZ and b-oHA were dissolved in working buffer to 10, 36, and 5 μg/mL concentration, respectively. Overtones j = 3, 5, 7, 9, 11, and 13 were recorded in addition to the fundamental resonance frequency (4.95 MHz). Changes in dissipation (ΔD) and normalized frequency, Δf = Δfj/j, for j = 7 are presented; all other overtones would have provided equivalent information.
Spectroscopic Ellipsometry (SE) Measurements
SE measurements were performed on silicon wafers with a native oxide film (for SLBs) or with a gold coating (functionalized ex situ with SAMs) using a spectroscopic rotating compensator ellipsometer (M-2000 V; J. A. Woollam, Lincoln, NE) and a custom-made open cuvette (250 μL volume) equipped with a magnetic stirrer for liquid homogenization and connections to tubings for liquid flow. Sample incubations were performed in “batch mode” by injecting concentrated samples directly into the buffer-filled cuvette under stirring (no flow). SAv, b-ZZ and b-oHA were incubated at final concentrations of 10, 36, and 5 μg/mL, respectively. Rinsing in working buffer was performed in “flow mode” at a flow rate of 500 μL/min. The ellipsometric angles Δ and Ψ were acquired over a wavelength range from λ = 380 to 1000 nm, at an angle of incidence of 70°. Prior to measurements, we checked that the polarization of the light beam was not affected by the cuvette’s glass windows as previously described.42 The refractive index n(λ) and optical thickness d of the adsorbed film were determined by fitting the ellipsometric data to a multilayer model, using the software CompleteEASE (J. A. Woollam). The model relates the measured Δ and Ψ as a function of λ to the optical properties of the substrate, the adsorbed organic film(s) and the surrounding solution. The semi-infinite bulk solution was treated as a transparent Cauchy medium, with a refractive index nsol(λ) = Asol + Bsol/λ2. For the surrounding buffer solution, Asol = 1.325 and Bsol = 0.00322 μm2 were used.42 The opaque gold coating modified with SAM was treated as a homogeneous substrate. Its effective optical properties were determined from data acquired in the presence of bulk solution but in the absence of a protein film, by fitting the refractive index and extinction coefficient over the accessible λ range using a B-spline algorithm implemented in CompleteEASE. The native oxide film on Si wafers was treated as transparent and homogeneous (Cauchy medium), and its optical properties and thickness were determined from data acquired in the presence of a bulk solution, which were fitted over the accessible λ range using tabulated values for the underlying Si substrate (implemented in CompleteEASE). The solvated organic film was treated as a single layer, which we assumed to be transparent and homogeneous (Cauchy medium), with a given d and n(λ) = A + B/λ2. d and A were treated as fitting parameters, assuming B = Bsol. The χ2 value for the best fit was typically below 2, indicating a good fit. The adsorbed organic mass per unit area Γ was determined from Γ = d(A – Asol)/(dn/dc), which is equivalent to de Feijter’s equation,43 with refractive index increments dn/dc = 0.180 cm3/g for proteins43 and 0.150 cm3/g for b-oHA.44
Contact Angle Measurements
Contact angle measurements were performed on a DSA100 Drop Shape Analyzer (KRÜSS, Hamburg, Germany). Contact angles were calculated as mean and standard deviation of six measurements, with 3 μL drops of ultrapure water being positioned on different places of the modified gold surfaces.
Fluorescence Measurements
Fluorescence measurements were performed with a FluoroLog fluorometer (Horiba Jobin Yvon, Longjumeau, France). Fluorescence emission spectra of SAv solutions were recorded at λex = 280 nm in the range λem = 290 to 500 nm, corresponding to the excitation and emission wavelengths of tryptophan located in the biotin-binding pockets of SAv. Measurements were performed in working buffer at 1.5 to 3.0 μg/mL SAv, with 10 to 20-fold excess of b-SUVs (in molar concentration of biotinylated lipids per SAv tetramer) or 50-fold excess of biotin. The results are presented in the form of relative fluorescence intensity, that is (Imax/Imax0) × 100%, where Imax and Imax are the maxima in the emission spectra before and after biotin or b-SUV injection, respectively.
Formation of b-SLBs
Stock solutions at 2 mg/mL of small unilamellar vesicles (SUVs) composed of pure DOPC, and of a mixture of DOPC and DOPE-CAP-biotin (molar ratio 19:1), were prepared in working buffer as previously described.45 Silica-coated surfaces were cleaned by immersion in 2% sodium dodecyl sulfate (SDS) aqueous solution (30 min), rinsing in ultrapure water and blow drying in nitrogen gas, and activated by UV-ozone treatment (30 min). SLBs were formed by the method of vesicle spreading,46 through 10 min exposure of 50 μg/mL SUVs in working buffer supplemented with 2 mM CaCl2 to freshly activated surfaces. The biotin content in the SLBs was varied by mixing SUV stock solutions of pure DOPC and DOPC/DOPC-CAP-biotin at desired ratios (0 to 5% final molar fraction of DOPC-CAP-biotin) prior to their dilution and exposure to the substrate. Excess lipid material was removed from the bulk solution by rinsing with working buffer.
Formation of b-SAMs
Mixed SAMs were formed following the procedure developed for the HS-(CH2)11-EG4-OH/HS-(CH2)11-EG6-azide thiol pair.28 The gold-coated surfaces were first cleaned by UV-ozone treatment (5 min) and immersion in ethanol (20 min) with stirring, and then placed overnight in ethanol containing HS-(CH2)11-EG4-OH and HS-(CH2)11-EG6-biotin (1 mM total concentration). The molar fraction of HS-(CH2)11-EG6-biotin was varied from 0 to 10%. Excess thiols were removed by rinsing with ethanol, and the substrate was blow dried with nitrogen gas.
Results
Design and Characterization of SAv/Biotin Model System
Our goal was to identify the relevant parameters governing the binding stability, orientation and residual valency of surface-attached SAv. To this end, we developed a tunable model system (Figure 1A) based on a set of synthetic SAv constructs (Figure 1B), biotinylated surfaces (Figure 2) and biotinylated reporter probes (Figure 3).
Streptavidin Constructs
In addition to the regular streptavidin (rSAv) with 4 biotin binding sites, synthetic SAv constructs were designed to have 1 (mSAv), 2 (dSAv) or 3 (tSAv) binding sites, where two topologically distinct arrangements (cis, trans) are provided for dSAv (Figure 1B).38 The stoichiometry of SAv to biotin was verified by fluorescence spectroscopy, using the intrinsic fluorescence of tryptophan in SAv’s biotin-binding pockets as readout (Figure 1C–E).47,48 Four residues per pocket49 give 16 tryptophans per SAv sequence eventually involved in biotin binding. The fluorescence intensity decreased upon addition of excess biotin to the SAv solution (Figure 1D,E), as expected, and the linear dependence of the change in peak fluorescence intensity on the nominal SAv valency (Figure 1C) confirmed the expected number of biotin-binding sites in all SAv constructs.
Biotinylated Surfaces
Contrary to SAMs, where each thiol is chemically grafted to the gold surface,50 SLBs on silica allow biotinylated lipids to diffuse along the surface with a rate of a few μm2/s.51 Comparative assays with b-SLBs (“fluid surface”) and b-SAMs (“immobile surface”), therefore, enable us to evaluate the effect of biotin lateral mobility. SLBs and SAMs were designed to display biotin at tunable surface densities (Figure 2). To this end, the molar fraction of biotinylated lipids and thiols was systematically varied in the ranges 0 ≤ x ≤ 5% and 0 ≤ y ≤ 10%, respectively. The surface coatings thus formed are denoted as bx-SLBs and by-SAMs. With a typical molecular footprint of 60 Å2 per lipid49 and 28 Å2 per thiol28 one can estimate the root-mean-square (rms) distance between biotins to be lb = 3.4 nm for b5%-SLBs, and 1.7 nm for b10%-SAMs, where the latter corresponds to a surface density of 60 pmol/cm2. We have here assumed that the molar fractions of biotinylated molecules displayed on the surfaces are identical to the molar fraction in the solutions from which the SLBs and SAMs were assembled. In reality, some deviations may occur, yet the numbers should represent a reasonable order-of-magnitude estimate of the interbiotin distance at the maximal biotin densities explored.
b-SLBs were composed of DOPC (1) and DOPE-CAP-biotin (2), and their proper formation was ascertained by QCM-D (Figure 2A). The two-phase responses in the frequency shifts Δf and dissipation shifts ΔD are characteristic for the self-assembly process that involves binding, spreading and rupture of vesicles, and ultimately the coalescence of bilayer patches into a confluent SLB.46,52 b-SLBs of appropriate quality are characterized by Δf = −25 ± 1 Hz and ΔD < 0.5 × 10–6 at the end of the SLB formation process, and these were routinely obtained irrespective of the molar fraction of biotinylated lipids.
b-SAMs were composed of HS-(CH2)11-EG4-OH (3) and HS-(CH2)11-EG6-biotin (4), and their proper formation was confirmed by contact angle goniometry (Figure 2B). b-SAMs of appropriate quality were characterized by contact angles that decrease from 31 to 19° as the thiol-biotin fraction in solution is increased from 0 to 10%, whereas bare gold had a much larger contact angle (75°). Sample-to-sample reproducibility was tested for b-SAMs prepared using 10% thiol-biotin, and the relative standard deviation in the contact angle was 5.8% (19.1 ± 1.1°, n = 17).
Biotinylated Reporter Probes
Two distinct biotinylated probes were used as reporters for the mean residual valency of surface-bound SAv (Figure 3). b-ZZ is a protein construct with an N-terminal biotin followed by a flexible linker (∼5 nm contour length) and a tandem repeat of the Z domain of protein A (∼3.5 nm diameter). b-ZZ was designed to have a relatively large molecular weight (16.2 kDa) to facilitate high-resolution surface density quantification by SE while at the same time being just small enough to assess all biotin-binding sites on a SAv monolayer without appreciable steric hindrance (the flexible linker was inserted to facilitate b-ZZ binding into narrow spaces between adjacent SAv molecules and further alleviate packing constraints). b-oHA is a linear oligosaccharide of hyaluronan (15 monosaccharides, contour length 7.5 nm) connecting via a flexible linker to biotin. The persistence length of HA has been reported to lie between 4 to 8 nm,53 and oHA can thus be pictured as a slightly bent rod (∼1 nm diameter). With these dimensions, b-oHA binding should not suffer from steric constraints even on the most densely packed SAv films, although its low molecular weight (3.2 kDa) limits somewhat the surface density resolution by SE. We used b-oHA as an additional reference to confirm that b-ZZ binding to densely packed SAv monolayers is quantitative, and more generally, to validate the binding assays and quantify residual valencies with two independent probes.
Control Measurements
Before embarking on a systematic analysis, the specificity of binding to SLBs and SAMs was ascertained. None of the SAv constructs bound in detectable amounts to SLBs made from pure DOPC or to SAMs made from pure HS-(CH2)11-EG4-OH, and neither did b-ZZ or b-oHA, confirming that nonspecific interactions are negligible in our assays (Figure S3). We also verified that SAv, which typically bound very stably to b-SLBs and b-SAMs (notable exceptions are discussed below), was not released by competition with biotinylated probes in the solution phase (Figure S4). This confirmed that the intended function of b-oHA or b-ZZ to report the presence of free binding sites is not compromised by adverse secondary effects.
Next, we systematically studied SAv binding to b-SLBs and b-SAMs. QCM-D was used to assess the occurring binding events and the stability of SAv attachment (Figures 4, 5, and 8), while quantitative information about the SAv surface density and residual valency was obtained by SE (Figures 6 and 7). Among all the tunable parameters in our system (Figure 1A), the SAv concentration did not significantly affect the final SAv coverage and residual valency (Figure S5). All assays presented in the following were thus performed at a fixed SAv concentration (10 μg/mL).
Figure 4.

Binding of monovalent SAv to biotinylated surfaces. (A,B) QCM-D responses recorded during the binding of mSAv, and subsequently b-ZZ, to b10%-SAM (A) and b5%-SLB (B). Insets in A and B illustrate reversible mSAv binding and extraction of biotinylated lipids by mSAv, respectively. (C) QCM-D responses obtained upon exposure of mSAv and subsequently rSAv and b-ZZ to a b1%-SLB (squares) together with a control in which the exposure to mSAv was omitted (circles). The data indicate SAv-induced depletion of biotinylated lipids from the SLB, as illustrated schematically. (D) Representative measurement of changes in tryptophan fluorescence emission intensity upon injection of b5%-SUVs to a solution of mSAv (injection time is indicated by an arrow). The inset shows mean and standard deviation of the relative intensity decrease at equilibrium; the decrease is similar to that of free biotin in solution (Figure 1C) and indicates specific and quantitative binding of mSAv to biotinylated lipids through the biotin-binding pocket of mSAv.
Figure 5.

Binding of multivalent SAv constructs to densely biotinylated surfaces. Shown are QCM-D responses recorded during the binding of SAv constructs, and subsequently b-ZZ, to b10%-SAM (A) and b5%-SLB (B). Insets in A and B illustrate the surfaces and molecules used, and the schematic representations in C illustrate the binding features—residual valency, reported by b-ZZ, and orientation—of the different SAv constructs. Data are marked with distinct symbols for each SAv construct: rSAv (squares), tSAv (circles), dSAv-cis (upward-pointing triangles) and dSAv-trans (downward-pointing triangles).
Figure 8.
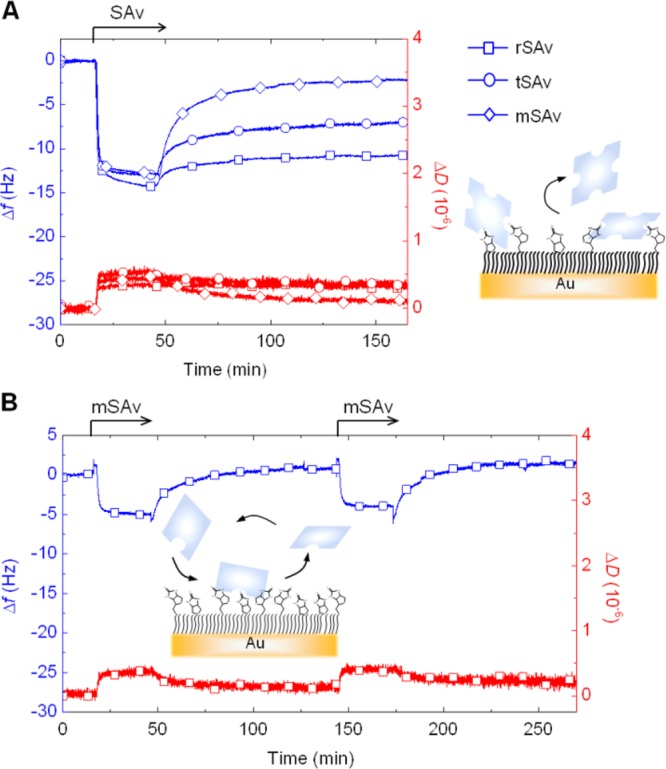
SAv binding to sparsely biotinylated surfaces. (A) QCM-D responses obtained during the binding of rSAv (squares), tSAv (circles) and mSAv (lozenges) to b0.1%-SAMs. Binding is followed by partial detachment, as illustrated schematically. (B) The QCM-D response for the exposure of mSAv to b0.05%-SAM reveals rapid unbinding upon rinsing in buffer (as illustrated schematically) and repeatable binding.
Figure 6.

SAv binding and residual valency on densely biotinylated surfaces, quantified by SE. (A) Example of binding curves obtained by SE, here for rSAv and b-oHA on a b5%-SLB. The inset illustrates the SE setup. (B) Table of surface densities of SAv, ΓSAv, and biotinylated reporter probes (b-ZZ or b-oHA), Γb, and the mean residual valency, Γb/ΓSAv. Values are presented as mean ± error, where the latter is the sum of the reproducibility error (4%; averaged from 4 independent sets of SE measurements with 2 to 4 samples in each set, cf. column 3 in Table S1) and the detection limit of the SE (1 ng/cm2). aBinding was below the detection limit of SE; bmSAv removed biotinylated lipids from b-SLB (cf. Figure 4B); cMeasured before buffer rinsing, as binding was not stable (cf. Figure 4A); db-ZZ accelerated displacement of mSAv from the surface. Conditions: SAv adsorption time = 90 min, biotinylated probe adsorption time = 15–60 min. All values were determined after adsorption and buffer rinsing once the SE response was stabilized, except for mSAv on SAMs, where the equilibrium bound amount before rinsing is given (as rinsing provokes mSAv detachment, Figure 4A).
Figure 7.

Effect of SAv coverage, and biotin surface density on SAv binding to biotinylated surfaces. The surface density of the biotinylated reporter probe vs the surface density of rSAv (squares) and tSAv (circles) was quantified by SE for immobile (A) and fluid (B) surfaces. The SAv surface density was tuned by varying either biotin surface density (incubating SAv to saturation or equilibrium; blue), or SAv adsorption time (on b10%-SAMs and b5%-SLBs, respectively; red). All values were determined after adsorption and buffer rinsing once the SE response was stabilized. The ratio Γb/ΓSAv (insets) corresponds to the mean residual valency of SAv. Lines are parabolic or linear fits; dashed lines indicate the range where SAv binding is not stable precluding quantification of residual valency. Conditions: biotin surface fraction = 10% (A, red), 5% (B, red), tuned from 1 to 10% (A, blue) or from 0.45 to 5% (B, blue); SAv adsorption time–tuned from 10 s to 90 min (A and B, red), 90 min (A, blue), 30 min (B, blue); biotinylated probe adsorption time = 15 to 60 min; see Table S1 for full details on conditions and determined values.
Binding Stability, Orientation and Residual Valency at High Surface Coverage
We first studied the binding of SAv on surfaces displaying high biotin densities, while tuning the SAv valency and the lateral mobility of biotin. Figures 4A,B and 5 compare the QCM-D signatures obtained for different SAv constructs, and subsequent b-ZZ binding, to b10%-SAMs and b5%-SLBs. Analogous measurements were performed with b-oHA, and qualitatively similar responses were obtained (Figure S6). Quantitative analysis by SE (Figure 6 and Table S1) also showed comparable levels of b-ZZ and b-oHA binding on dense SAv monolayers (e.g., on b10%-SAMs coated with rSAv until saturation) confirming that steric hindrance does not restrict b-ZZ binding, and we thus used both b-ZZ and b-oHA as reliable reporters of residual valency.
Stability of SAv Surface Attachment
We started with mSAv as this enables the quality of monovalent interactions to be directly assessed. When exposed to b10%-SAMs, mSAv rapidly bound to equilibrium (Δf = −21 Hz) but slow release occurred during rinsing in working buffer, and the release was accelerated in the presence of competing biotinylated probes in solution (Figures 4A and S6A). Upon exposure of mSAv to b5%-SLBs, Δf decreased only slightly (by less than 3 Hz), and the initial rapid decrease in Δf was followed by a slow but significant increase, indicating effective release of material, even in the presence of mSAv in the solution phase (Figure 4B). Perhaps most strikingly, rSAv binding to b-SLBs was completely impaired by preincubation of b-SLBs with mSAv (Figure 4C). We verified by intrinsic fluorescence spectroscopy that mSAv retains the capacity of rSAv to bind biotinylated lipids (Figure 4D). Collectively, these data demonstrate that mSAv is unable to bind stably to both the immobile and the fluid biotinylated surfaces, but instead has the remarkable and unexpected capacity to extract biotinylated lipids from lipid bilayers (Figure 4B, inset).
In contrast to mSAv, all multivalent forms of SAv bound stably to b10%-SAMs and b5%-SLBs (Figures 5 and S6). No significant desorption was observed upon rinsing in working buffer, indicating that multivalent binding is both necessary and sufficient for the quasi-irreversible attachment of SAv that is desired in most practical applications.
Residual Valency and Orientation of Surface-Attached SAv
Next, we used our biotinylated reporter probes (Figure 3) to characterize the residual valency of multivalent SAv constructs. Both QCM-D (Figures 5 and S6) and SE (Figure 6) data clearly showed that the reporter probes are unable to bind divalent SAv, suggesting that both available biotin-binding pockets are engaged in SAv attachment to the biotinylated surfaces. These results effectively demonstrate that two SAv binding pockets can bind simultaneously to b-SAMs or b-SLBs not only when they are arranged in cis but also when they are arranged in trans positions (Figure 5C).
The magnitudes of the frequency shift for the reporter probes were more than 2-fold smaller on tSAv than on rSAv under otherwise identical conditions (Figures 5 and S6). Equivalent measurements by SE (Figure 6B) revealed that the mean residual valency of surface-attached tSAv was below 1.0, whereas values between 1.0 and 2.0 were obtained for rSAv. These results indicate that SAv can also bind to biotinylated surfaces in a trivalent mode and that residual valencies of 0 and 1 for tSAv, and of 1 and 2 for rSAv, can coexist on the same b-SLB or b-SAM surface (Figure 5C).
Packing Density of Surface-Attached SAv
There are several subtle yet characteristic differences in the packing of the multivalent SAv constructs on b-SAMs and b-SLBs. First, |ΔfSAv| values at saturation were larger on b-SLBs than on b-SAMs for any of the SAv constructs. This reflects differences in the packing density (Figures 6B and S7A), and most likely arises from the ability of SAv to rearrange and thus pack more tightly on the fluid surface. Second, lower ΔD/−Δf values were obtained on b-SAMs compared to b-SLBs (Figure S7B). For globular proteins such as SAv, ΔD/−Δf is a measure of the softness of the linker region that connects the protein with the biotinylated surface,54 and the observed difference indicates a tighter (and thus rigid) binding interface in the case of b-SAMs. Finally, slightly higher |ΔfSAv| values were obtained for rSAv and dSAv-cis at saturation as compared to tSAv and dSAv-trans (Figure 5). SE results confirmed that this difference reflects variations in the packing densities of the cis- and trans-oriented SAv forms (Figures 6B and S7A).
Effect of Surface Coverage on Binding Stability and Residual Valency
In principle, the surface coverage of SAv can be tuned in two distinct ways: by varying the SAv incubation time or by varying the biotin surface density. We thus performed two series of measurements to study how these parameters influence the stability of SAv binding and the residual valency. The SE results are summarized in Figure 7, and additional QCM-D data are shown in Figures S8–S10.
Tuning SAv Incubation Time
First, we varied the protein incubation time while maintaining the biotin surface density high and unaltered. As expected, SAv coverage increased steadily with SAv adsorption time (Figure S8A). We tested the effect of this approach on the residual valencies of tSAv and rSAv on b5%-SLBs and b10%-SAMs.
On both fluid and immobile surfaces, both SAv constructs bound stably and the amount of bound biotinylated probes Γb increased steadily with SAv surface coverage ΓSAv (Figure S9A,B). On b-SAMs, the dependence was linear, as evidenced by SE (for rSAv; Figure 7A, red) and QCM-D (for tSAv and rSAv; Figure S10A). This implies that, for a given b-SAM, the mean residual valency is unaffected by the SAv surface density (Γb/ΓrSAv = 1.7 on b10%-SAMs; Figure 7A, inset). This was not the case on b-SLBs, where the dependence was supra-linear (Figures 7B and S10B, red). Effectively, the mean residual valency increased with the SAv surface density (inset in Figure 7B, red). At the lowest (0.8 pmol/cm2) and highest (4.5 pmol/cm2) rSAv densities tested by SE, Γb/ΓrSAv were 1.1 and 1.7, respectively, implying that attachment varied from mainly (90%) trivalent to mainly (70%) divalent. At the most extreme, purely trivalent binding is observed, as illustrated by the complete absence of b-ZZ attachment to dilute tSAv monolayers on b5%-SLBs (Figure S9B).
Tuning Biotin Surface Density
In the second set of measurements, we tested the binding of SAv on b-SLBs and b-SAMs displaying different densities of biotin with SAv being incubated to saturation (or equilibrium) throughout. Different trends were observed on immobile and fluid surfaces.
On immobile surfaces, not only the amount (Figure S8B, blue) but also the stability of SAv binding was significantly affected by biotin coverage, with partial detachment being detected at biotin fractions below 1% even for the multivalent SAv constructs (Figure 8A). The estimated rms distance between biotins on a b1%-SAM is around 5 nm, which is comparable to the spacing between binding pockets in SAv. It can thus be expected that, due to steric constraints, most SAv molecules find only one biotin to attach to at lower biotin content. We therefore attribute the reduced binding stability on diluted b-SAMs to the lack of multivalent attachment, as already revealed above for mSAv (Figure 4A). Completely reversible binding of mSAv to b0.05%-SAMs (Figure 8B) supports this interpretation. The higher apparent stability with increasing SAv valency (at constant biotin surface density; Figure 8A) and with increasing surface density (for mSAv; Figures 4A and 8) can be explained by an increased probability of rebinding to adjacent binding sites.55
Binding of the multivalent SAv constructs was essentially stable for biotin fractions of 1% and larger. Quantification revealed mean residual valencies Γb/ΓrSAv = 1.95 and Γb/ΓtSAv = 0.85 on b1%-SAMs (Figure 7A, blue), close to the values of 2.0 and 1.0, respectively, expected for purely divalent attachment. For b10%-SAMs, the values were reduced (to 1.76 and 0.74, respectively), indicating that trivalent attachment becomes increasingly important as the biotin fraction increases. Consistent with this trend, we had previously reported an even lower residual valency of Γb/ΓrSAv = 1.51 on b20%-SAMs (using different biotinylated probe and buffer).8
On fluid surfaces, SAv bound stably irrespective of the biotin content. As expected, the amount of bound SAv increased steadily with the biotin density, with a plateau being attained at about 1% (Figure S8B, red). Furthermore, SE and complementary QCM-D data (Figures 7B, S9C and S10B, blue) revealed a supra-linear relationship between the amount of bound SAv and subsequently bound biotinylated probe. One can assume that, in the absence of steric hindrance, the attachment of SAv to b-SLBs matures over time by recruiting additional biotins, thus leading to a reduced residual valency. This is similar to the case when the SAv incubation time was varied, although the effect is now less pronounced (compare blue and red data for rSAv in Figure 7B, inset).
In summary, the systematic analysis (Figure 7) shows that the mean residual valency of SAv bound to biotinylated surfaces is determined by several factors, including lateral mobility, biotin surface density and SAv coverage, and can be experimentally adjusted, between 1 and 2 for rSAv and between 0 and 1 for tSAv, by tuning one or several of these parameters.
Discussion
Stability of SAv Anchoring
Our findings on immobile biotinylated surfaces demonstrate that the details of the binding interface have an appreciable effect on the stability of SAv attachment and need careful consideration. Even though we did not quantify the affinity, it is clear from our data (Figure 8) that the lifetime of the biotin-SAv bond on b-SAMs is drastically reduced compared to its extremely high stability in solution. We propose that this effect is due to steric hindrance and/or entropic costs arising from reduced conformational freedom of flexible OEG chains (biotinylated or not) in the SAM upon SAv binding. In essence, the flexible OEG linkers of the SAM layer appear to exert a force that effectively weakens the biotin-SAv bond. This scenario is consistent with the high rigidity of the SAM interface revealed by QCM-D (Figure S7B). As a result, stable immobilization of SAv requires multivalent binding that in turn occurs at sufficiently high (≥1% in the case of HS-(CH2)11-EG4-OH and HS-(CH2)11-EG6-biotin) fraction of biotinylated thiols in b-SAMs. This suggests that earlier studies reporting weakened interactions between rSAv and b-SAMs,34,35,56 may have probed monovalent SAv/biotin binding.
Stable multivalent attachment to fluid surfaces, regardless of their biotin surface density (Figure 7B, blue), is expected and can be explained by the ability of laterally mobile binding sites to diffuse to the binding pocket over long distances. Indeed, assuming lb < 100 nm and a lipid diffusion constant Dlipid > 1 μm2/s,51 the time needed for lipids to diffuse over an area of lb2 is roughly lb2/Dlipid < 10–2 s. This implies that two biotins will get close to each other within a few milliseconds, which is much shorter than the time scale of the SAv binding process (seconds to minutes, Figure 6A).
Perhaps the most surprising finding of our study is that mSAv is able to extract biotinylated lipids from lipid bilayers. This effect is reminiscent of the action of certain lipid transfer proteins. Glycolipid transfer protein, for example, is known to recognize glycolipids with high specificity and to facilitate their extraction from lipid bilayers with the aid of a hydrophobic pocket that harbors the lipid tail and thus effectively solubilizes the lipid.57,58 We do not know what the mechanism of lipid extraction is in the case of mSAv but it appears possible that it acts in a similar way through the binding of lipid tails into the residual binding pockets. Since both mutated (inactive) and native (active) biotin binding sites in our SAv constructs are hydrophobic, it appears possible that not only mSAv but also other SAv constructs, including regular SAv, have the intrinsic ability to extract biotinylated lipids. This capacity, however, is effectively impaired when SAv binds multivalently to b-SLBs and thus difficult to observe directly for multivalent SAv constructs.
Orientation of Bound SAv
Our data provide evidence that SAv can attach stably to b-SLBs and b-SAMs in several distinct ways: divalently with the two biotin-binding sites arranged in either cis or trans positions, or trivalently (Figure 5C). This implies that the orientation of SAv on the surface also differs (Figure S11), and that the orientation corresponding to two binding sites arranged in cis positions—previously identified in two-dimensional SAv crystals on SLBs and on lipid monolayers at the air–water interface29,32,33—is only one out of several possible scenarios. Clearly, the arrangement of the biotin binding pockets on SAv allows for various ways of biotin binding, even though the linkers used for anchoring the biotin moiety to the SLB and the SAM are relatively short: 1.3 nm and 1–2 nm (depending on EG6 stretching), respectively. The access of biotins to two binding pockets in trans-orientation and formation of trivalent bonds can be facilitated by the out-of-plane mobility of lipids and the intrinsic flexibility of the EG6 linker chain, respectively. Reorganization of flexible loops on SAv, and of lipids or OEG molecules, may then bring the protein and the SLB/SAM even closer together and thereby further aid the biotins to reach their binding pockets (Figure S11). In particular, one can envisage the appearance of local indentations in the SLB or SAM induced by the tight SAv attachment (Figure S7B), where the degree of SAv insertion into the immobile coating should strongly depend on the characteristics of the OEG layer (i.e., OEG length, density and organization).
Tuning the SAv Residual Valency
Our systematic analysis demonstrates that the mean residual valency of SAv can be tuned by surface chemistry (Figure 7). On immobile surfaces, the mean residual valency is defined by the SAM’s biotin content and does not depend on the SAv surface coverage. Specifically, maximal values close to 2.0 for rSAv and 1.0 for tSAv are attained at 1% biotin, and the residual valency progressively decreases with higher biotin content as trivalent attachment increases. The present data, together with previous work8 suggest that values as low as 1.5 with rSAv and 0.5 with tSAv can be attained at 20% biotin.
Somewhat less intuitively, the mean residual valency of SAv on fluid surfaces was found to increase with SAv surface coverage. Several mechanisms can account for this behavior. First, the steric hindrance affecting dense monolayers may prevent the rearrangement of SAv to form an additional third bond with b-SLB. This may result in higher residual valency, in particular if the formation of the third bond is the rate limiting step. Second, SAv crystallization, known to occur in a divalent mode at fluid interfaces,29,32,33 may also contribute to the increase of the residual valency with SAv coverage. Furthermore, tuning SAv coverage through SAv incubation time had a stronger effect than tuning the SLB’s biotin content on the residual valency (inset in Figure 7B, blue vs red). This result suggests that the surface density of biotin does also play a role in the regulation of SAv binding. In the case of diluted (<1%) b-SLBs, the molar density of biotinylated lipids is about a few pmol/cm2, which is comparable with ΓSAv. The formation of purely trivalent complexes with diluted b-SLBs may thus be restricted by the depletion of biotins (Figure 7B, blue). When keeping the biotin coverage high (i.e., at least several times higher than ΓSAv), the residual valency can be reduced to 1.0 sharply by shortening the SAv incubation time (Figure 7B, red). The subtle dependence of SAv binding on b-SLB’s coverage thereby allows one to tune the SAv residual valency from 1.0 to 2.0 (Figure 7B).
Summary and Implications
Our findings can be summarized as follows:
-
(i)
Monovalent binding, even if strong (femtomolar affinity) in solution, is not sufficient for the stable attachment of SAv to commonly employed biotinylated surfaces.
-
(ii)
On immobile but not on fluid surfaces, stable (multivalent) SAv attachment requires a minimal surface density of biotin binding sites.
-
(iii)
The residual valency of surface-bound rSAv can vary between 1 and 2, and depends sensitively on the surface chemistry, with opposite trends on immobile and fluid surfaces.
SAv-governed self-assembly is currently used for the design of functional interfaces suitable for numerous applications, including sensors, biomaterials and nanomedicine.7−16 Our data illustrate that effects (i) and (ii) require serious consideration to ensure interface functionality. Special attention should be given to effect (iii), since it may drastically affect the efficiency of the supramolecular assembly: when “gluing” nano-objects (e.g., capsules,19,20 viruses,21 particles22) together or attaching them to the surface via SAv/biotin bonds, the SAv residual valency should be kept close to 2 to ensure stable (multivalent) attachment from both sides. Our data show immobile surfaces with moderately diluted biotin content (e.g., b1%-SAMs, lb ≈ 5 nm) as well as fluid membranes with dense biotin packing (e.g., b ≥ 5%-SLBs, lb ≤ 3.4 nm) to be optimal for such architectures. Moreover, effect (iii) also entails opportunities for the quantitative tuning of residual valency, which can be advantageously used for the development of model surfaces displaying specific functionalities either individually or in clusters of two via postmodification of SAv at residual valency 1 or 2, respectively. Such a model could be used, for instance, to probe the effect of dimeric vs monomeric presentation of cell-membrane ligands or receptors like cytokines, hormones, cadherins, hyaladherins—with dimerization being a ubiquitous signaling mechanism, whose impact on multivalent binding and resulting biological functions (cell development, adhesion, immune response, etc.) is not well studied.59,60
Conclusions
We have developed a well-defined, highly specific and tunable model system to identify the parameters governing the orientation and residual valency of SAv at biotinylated interfaces. Using the SAv/biotin pair as a model system, we have demonstrated that the stoichiometry of multivalent binding is essentially governed, and can thus be tuned, by the surface chemistry. In addition, we found that monovalent anchoring to the surface is not stable, even in the case of high affinity interactions, like between SAv and biotin, and may lead to the extraction of functional molecules from laterally mobile coatings. Our study should extend the practical applications of the SAv/biotin technology, in particular in the design of robust biomimetic and bioactive supramolecular assemblies at interfaces. The obtained knowledge about the interplay between surface chemistry, binding stability and residual valency should also be useful to control surface binding of other rigid multivalent scaffolds (i.e., proteins, nanoparticles, and dendrimers),3 thus contributing to the general understanding of multivalent interactions and their progressive use in nanobiotechnology.
Acknowledgments
This work was supported by the Marie Curie Career Integration Grant “CELLMULTIVINT” (PCIG09-GA-2011-293803) to G.V.D., the Spanish Ministry of Competitiveness and Innovation (project MAT2014-54867-R), and the European Research Council Starting Grant “JELLY” (306435) to R.P.R. M.F. and M.H. were funded by the Biotechnology and Biological Sciences Research Council (BBSRC). L. Yate (CIC biomaGUNE) is acknowledged for providing metal surface coatings.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b00540.
Supporting Methods (synthesis of b-oHA, expression and purification of b-ZZ); Supporting Figures S1–S11; Supporting Table S1; Supporting References (PDF)
The authors declare the following competing financial interest(s): M.H. is an author on a patent for monovalent streptavidin (US8586708 B2).
Supplementary Material
References
- Mammen M.; Choi S.-K.; Whitesides G. M. Angew. Chem., Int. Ed. 1998, 37 (20), 2754.. [DOI] [PubMed] [Google Scholar]
- Fasting C.; Schalley C. A.; Weber M.; Seitz O.; Hecht S.; Koksch B.; Dernedde J.; Graf C.; Knapp E.-W.; Haag R. Angew. Chem., Int. Ed. 2012, 51 (42), 10472. 10.1002/anie.201201114. [DOI] [PubMed] [Google Scholar]
- Varner C. T.; Rosen T.; Martin J. T.; Kane R. S. Biomacromolecules 2015, 16 (1), 43. 10.1021/bm5014469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green N. M. In Methods in Enzymology; Bayer M. W., Ed.; Academic Press, 1990; Avidin-Biotin Technology, Vol. 184, pp 51–67. [Google Scholar]
- Wilchek M.; Bayer E. A. Anal. Biochem. 1988, 171 (1), 1. 10.1016/0003-2697(88)90120-0. [DOI] [PubMed] [Google Scholar]
- Diamandis E. P.; Christopoulos T. K. Clin. Chem. 1991, 37 (5), 625. [PubMed] [Google Scholar]
- Tang Q.; Su X.; Loh K. P. J. Colloid Interface Sci. 2007, 315 (1), 99. 10.1016/j.jcis.2007.06.040. [DOI] [PubMed] [Google Scholar]
- Osypova A.; Thakar D.; Dejeu J.; Bonnet H.; Van der Heyden A.; Dubacheva G. V.; Richter R. P.; Defrancq E.; Spinelli N.; Coche-Guérente L.; Labbé P. Anal. Chem. 2015, 87 (15), 7566. 10.1021/acs.analchem.5b01736. [DOI] [PubMed] [Google Scholar]
- Huang N.-P.; Vörös J.; De Paul S. M.; Textor M.; Spencer N. D. Langmuir 2002, 18 (1), 220. 10.1021/la010913m. [DOI] [Google Scholar]
- Wang J.; Zhu X.; Tu Q.; Guo Q.; Zarui C. S.; Momand J.; Sun X. Z.; Zhou F. Anal. Chem. 2008, 80 (3), 769. 10.1021/ac0714112. [DOI] [PubMed] [Google Scholar]
- Howarth M.; Takao K.; Hayashi Y.; Ting A. Y. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (21), 7583. 10.1073/pnas.0503125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.-H.; Soda Y.; Izawa K.; Kobayashi S.; Tani K.; Maruyama K.; Tojo A.; Asano S. Int. J. Pharm. 2013, 454 (1), 478. 10.1016/j.ijpharm.2013.06.031. [DOI] [PubMed] [Google Scholar]
- Aumsuwan N.; Pearson H. A.; Urban M. W. Biomater. Sci. 2013, 1 (7), 711. 10.1039/c3bm00187c. [DOI] [PubMed] [Google Scholar]
- Wilson R.; Mehrabi M.; Prior I. A.; Beckett A.; Hutchinson A. Chem. Commun. 2009, (18), 2487. 10.1039/b822688a. [DOI] [PubMed] [Google Scholar]
- Migliorini E.; Thakar D.; Sadir R.; Pleiner T.; Baleux F.; Lortat-Jacob H.; Coche-Guerente L.; Richter R. P. Biomaterials 2014, 35 (32), 8903. 10.1016/j.biomaterials.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Richter R. P.; Hock K. K.; Burkhartsmeyer J.; Boehm H.; Bingen P.; Wang G.; Steinmetz N. F.; Evans D. J.; Spatz J. P. J. Am. Chem. Soc. 2007, 129 (17), 5306. 10.1021/ja068768s. [DOI] [PubMed] [Google Scholar]
- Su X.; Lin C.-Y.; O’Shea S. J.; Teh H. F.; Peh W. Y. X.; Thomsen J. S. Anal. Chem. 2006, 78 (15), 5552. 10.1021/ac0606103. [DOI] [PubMed] [Google Scholar]
- Su X.; Wu Y.-J.; Robelek R.; Knoll W. Langmuir 2005, 21 (1), 348. 10.1021/la047997u. [DOI] [PubMed] [Google Scholar]
- Roling O.; Wendeln C.; Kauscher U.; Seelheim P.; Galla H.-J.; Ravoo B. J. Langmuir 2013, 29 (32), 10174. 10.1021/la4011218. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Zhang H.; Gruzdys V.; Sun X.-L. Langmuir 2011, 27 (21), 13097. 10.1021/la2032434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz N. F.; Calder G.; Lomonossoff G. P.; Evans D. J. Langmuir 2006, 22 (24), 10032. 10.1021/la0621362. [DOI] [PubMed] [Google Scholar]
- Aslan K.; Luhrs C. C.; Pérez-Luna V. H. J. Phys. Chem. B 2004, 108 (40), 15631. 10.1021/jp036089n. [DOI] [Google Scholar]
- Wolny P. M.; Banerji S.; Gounou C.; Brisson A. R.; Day A. J.; Jackson D. G.; Richter R. P. J. Biol. Chem. 2010, 285 (39), 30170. 10.1074/jbc.M110.137562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huskens J.; Mulder A.; Auletta T.; Nijhuis C. A.; Ludden M. J. W.; Reinhoudt D. N. J. Am. Chem. Soc. 2004, 126 (21), 6784. 10.1021/ja049085k. [DOI] [PubMed] [Google Scholar]
- Bacharouche J.; Degardin M.; Jierry L.; Carteret C.; Lavalle P.; Hemmerlé J.; Senger B.; Auzély-Velty R.; Boulmedais F.; Boturyn D.; Coche-Guérente L.; Schaaf P.; Francius G. J. Mater. Chem. B 2015, 3 (9), 1801. 10.1039/C4TB01261E. [DOI] [PubMed] [Google Scholar]
- Dubacheva G. V.; Curk T.; Mognetti B. M.; Auzély-Velty R.; Frenkel D.; Richter R. P. J. Am. Chem. Soc. 2014, 136 (5), 1722. 10.1021/ja411138s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubacheva G. V.; Curk T.; Auzély-Velty R.; Frenkel D.; Richter R. P. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (18), 5579. 10.1073/pnas.1500622112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubacheva G. V.; Van der Heyden A.; Dumy P.; Kaftan O.; Auzély-Velty R.; Coche-Guerente L.; Labbé P. Langmuir 2010, 26 (17), 13976. 10.1021/la102026h. [DOI] [PubMed] [Google Scholar]
- Darst S. A.; Ahlers M.; Meller P. H.; Kubalek E. W.; Blankenburg R.; Ribi H. O.; Ringsdorf H.; Kornberg R. D. Biophys. J. 1991, 59 (2), 387. 10.1016/S0006-3495(91)82232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto D.; Uchihashi T.; Kodera N.; Ando T. Nanotechnology 2008, 19 (38), 384009. 10.1088/0957-4484/19/38/384009. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Spinke J.; Bayerl T.; Sackmann E.; Knoll W. Biophys. J. 1992, 63 (5), 1385. 10.1016/S0006-3495(92)81715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert T. L.; Leckband D. Langmuir 1997, 13 (25), 6737. 10.1021/la970590n. [DOI] [Google Scholar]
- Reviakine I.; Brisson A. Langmuir 2001, 17 (26), 8293. 10.1021/la010626i. [DOI] [Google Scholar]
- Pérez-Luna V. H.; O’Brien M. J.; Opperman K. A.; Hampton P. D.; López G. P.; Klumb L. A.; Stayton P. S. J. Am. Chem. Soc. 1999, 121 (27), 6469. 10.1021/ja983984p. [DOI] [Google Scholar]
- Bacharouche J.; Badique F.; Fahs A.; Spanedda M. V.; Geissler A.; Malval J.-P.; Vallat M.-F.; Anselme K.; Francius G.; Frisch B.; Hemmerlé J.; Schaaf P.; Roucoules V. ACS Nano 2013, 7 (4), 3457. 10.1021/nn400356p. [DOI] [PubMed] [Google Scholar]
- Singh S.; Lapin N.; Singh P. K.; Khan M. A.; Chabal Y. J. In AIP Conference Proceedings; AIP Publishing, 2009; Vol. 1147, pp 443–449. [Google Scholar]
- Wong J.; Chilkoti A.; Moy V. T. Biomol. Eng. 1999, 16 (1–4), 45. 10.1016/S1050-3862(99)00035-2. [DOI] [PubMed] [Google Scholar]
- Fairhead M.; Krndija D.; Lowe E. D.; Howarth M. J. Mol. Biol. 2014, 426 (1), 199. 10.1016/j.jmb.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stayton P. S.; Freitag S.; Klumb L. A.; Chilkoti A.; Chu V.; Penzotti J. E.; To R.; Hyre D.; Le Trong I.; Lybrand T. P.; Stenkamp R. E. Biomol. Eng. 1999, 16 (1–4), 39. 10.1016/S1050-3862(99)00042-X. [DOI] [PubMed] [Google Scholar]
- Loosli A.; Rusbandi U. E.; Gradinaru J.; Bernauer K.; Schlaepfer C. W.; Meyer M.; Mazurek S.; Novic M.; Ward T. R. Inorg. Chem. 2006, 45 (2), 660. 10.1021/ic051405t. [DOI] [PubMed] [Google Scholar]
- Walvoort M. T. C.; Volbeda A. G.; Reintjens N. R. M.; van den Elst H.; Plante O. J.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Org. Lett. 2012, 14 (14), 3776. 10.1021/ol301666n. [DOI] [PubMed] [Google Scholar]
- Carton I.; Brisson A. R.; Richter R. P. Anal. Chem. 2010, 82 (22), 9275. 10.1021/ac102495q. [DOI] [PubMed] [Google Scholar]
- Richter R. P.; Rodenhausen K. B.; Eisele N. B.; Schubert M. In Ellipsometry of Functional Organic Surfaces and Films; Hinrichts K., Eichhorn K.-J., Eds.; Springer: Berlin, 2014; Springer Series in Surface Sciences, Vol. 52, pp 223–238. [Google Scholar]
- Takahashi R.; Kubota K.; Kawada M.; Okamoto A. Biopolymers 1999, 50 (1), 87.. [DOI] [Google Scholar]
- van der Meulen S. A. J.; Dubacheva G. V.; Dogterom M.; Richter R. P.; Leunissen M. E. Langmuir 2014, 30 (22), 6525. 10.1021/la500940a. [DOI] [PubMed] [Google Scholar]
- Richter R. P.; Bérat R.; Brisson A. R. Langmuir 2006, 22 (8), 3497. 10.1021/la052687c. [DOI] [PubMed] [Google Scholar]
- Chilkoti A.; Tan P. H.; Stayton P. S. Proc. Natl. Acad. Sci. U. S. A. 1995, 92 (5), 1754. 10.1073/pnas.92.5.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panek J. J.; Ward T. R.; Jezierska A.; Novič M. J. Mol. Model. 2009, 15 (3), 257. 10.1007/s00894-008-0382-0. [DOI] [PubMed] [Google Scholar]
- Yeagle P.The Membranes of Cells; Academic Press Inc.: Orlando, 1987. [Google Scholar]
- Häkkinen H. Nat. Chem. 2012, 4 (6), 443. 10.1038/nchem.1352. [DOI] [PubMed] [Google Scholar]
- Migliorini E.; Thakar D.; Kühnle J.; Sadir R.; Dyer D. P.; Li Y.; Sun C.; Volkman B. F.; Handel T. M.; Coche-Guerente L.; Fernig D. G.; Lortat-Jacob H.; Richter R. P. Open Biol. 2015, 5 (8), 150046. 10.1098/rsob.150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C. A.; Kasemo B. Biophys. J. 1998, 75 (3), 1397. 10.1016/S0006-3495(98)74057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attili S.; Borisov O. V.; Richter R. P. Biomacromolecules 2012, 13 (5), 1466. 10.1021/bm3001759. [DOI] [PubMed] [Google Scholar]
- Johannsmann D.; Reviakine I.; Richter R. P. Anal. Chem. 2009, 81 (19), 8167. 10.1021/ac901381z. [DOI] [PubMed] [Google Scholar]
- Martinez-Veracoechea F. J.; Leunissen M. E. Soft Matter 2013, 9 (12), 3213. 10.1039/c3sm27766f. [DOI] [Google Scholar]
- Buranda T.; Jones G. M.; Nolan J. P.; Keij J.; Lopez G. P.; Sklar L. A. J. Phys. Chem. B 1999, 103 (17), 3399. 10.1021/jp983842h. [DOI] [Google Scholar]
- Brown R. E.; Mattjus P. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2007, 1771 (6), 746. 10.1016/j.bbalip.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carton I.; Malinina L.; Richter R. P. Biophys. J. 2010, 99 (9), 2947. 10.1016/j.bpj.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plopper G. In Cells; Jones & Bartlett Learning, 2007; pp 831–889. [Google Scholar]
- Matthews J. M.; Sunde M. In Protein Dimerization and Oligomerization in Biology; Matthews J. M., Ed.; Springer: New York, 2012; Advances in Experimental Medicine and Biology, pp 1–18. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.