

Abstract

We have recently reported on several Fe catalysts for N2-to-NH3 conversion that operate at low temperature (−78 °C) and atmospheric pressure while relying on a very strong reductant (KC8) and acid ([H(OEt2)2][BArF4]). Here we show that our original catalyst system, P3BFe, achieves both significantly improved efficiency for NH3 formation (up to 72% for e– delivery) and a comparatively high turnover number for a synthetic molecular Fe catalyst (84 equiv of NH3 per Fe site), when employing a significantly weaker combination of reductant (Cp*2Co) and acid ([Ph2NH2][OTf] or [PhNH3][OTf]). Relative to the previously reported catalysis, freeze-quench Mössbauer spectroscopy under turnover conditions suggests a change in the rate of key elementary steps; formation of a previously characterized off-path borohydrido–hydrido resting state is also suppressed. Theoretical and experimental studies are presented that highlight the possibility of protonated metallocenes as discrete PCET reagents under the present (and related) catalytic conditions, offering a plausible rationale for the increased efficiency at reduced driving force of this Fe catalyst system.
Short abstract
A synthetic Fe complex catalyzes nitrogen fixation at lower driving force increasing its relevance as a functional model of nitrogenase. Theory and experiment lead to the proposal that protonated cobaltocenes may play a role as PCET reagents.
The reduction of N2 to NH3 is critical for life and is performed on a massive scale both industrially and biologically.1 The high stability of the N≡N triple bond necessitates catalysts and high-energy reagents/conditions to achieve the desired transformation.2 Synthetic studies of catalytic N2-to-NH3 conversion by model complexes are of interest to constrain hypotheses concerning the mechanism/s of biological (or industrial) N2-fixation and to map fundamental catalyst design principles for multielectron reductive transformations.3,4 Interest in Fe model systems that catalyze N2-to-NH3 conversion has grown in part due to the postulate that one or more Fe centers in the FeMo-cofactor of FeMo-nitrogenase may serve as the site of N2 binding and activation during key bond-breaking and -making steps.5 Previous examples of synthetic molecular Fe catalysts that mediate N2-to-NH3 conversion operate with high driving force, relying on a very strong acid (pKa ca. 0) and reductant (E° ≤ −3.0 V vs Fc+/0).6−9 In contrast, several Mo catalysts have been shown to facilitate N2-to-NH3 conversion with significantly lower driving force.10−13 There is thus interest in exploring the viability of Fe-mediated catalytic N2-to-NH3 conversion under less forcing conditions from a practical perspective, and to continue assessing these systems as functional models of biological nitrogenases, in which 8 ATP are consumed per NH3 formed providing a total driving force of 58 kcal/mol.2
Herein we demonstrate that catalytic conversion of N2 to NH3 by P3BFe+ (P3B = tris(o-diisopropylphosphinophenyl)borane) can be achieved with a significantly lower driving force by coupling Cp*2Co with [Ph2NH2]+ or [PhNH3]+ (Figure 1). Such conditions additionally afford unusually high selectivity and catalytic turnover for NH3.20 Moreover, we note that the use of milder reagents as reductant (E0; eq 1) and acid (pKa; eq 1) engenders a higher effective bond dissociation enthalpy (BDEeffective; eq 1).15,21 This may in turn afford access to proton-coupled electron transfer (PCET) pathways (e.g., FeN2 + H• → FeN2H) in addition to electron transfer (ET)/proton transfer (PT) pathways, thus enhancing overall catalytic efficiency. Theoretical considerations, including DFT calculations, and experimental details are discussed that suggest the viability of a decamethylcobaltocene-mediated PCET pathway in this system; by extension we suggest that metallocene-mediated (e.g., Cp*2Cr) PCET pathways may be operative in previously studied Mo and Fe N2-fixing systems that use metallocene reductants.10−13,20
| 1 |
Figure 1.

Summary of conditions used for catalytic N2-to-NH3 conversion by P3BFe+ highlighting the estimated enthalpic driving force (ΔΔHf).14−19
Various observations of P3BFe complexes in the presence of acids and reductants suggested that this system might be capable of N2-to-NH3 conversion with lower driving force than that originally reported. Accordingly, we had observed that the treatment of P3BFeN2– with KC8 and weaker acids (pKa > 0) led to greater than stoichiometric NH3 formation (e.g., under unoptimized conditions [2,6-dimethylanilinium][OTf] afforded 2.1 equiv of NH3 per Fe).22 Similarly, the treatment of P3BFeN2– with [H(OEt2)2][BArF4] (HBArF4, BArF4 = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate) and weaker reductants led to modest yields of NH3. For example, under unoptimized conditions we had observed that decamethylcobaltocene (Cp*2Co) and HBArF4 afforded 0.6 equiv of NH3 per Fe.22,23 Most recently, an apparent catalytic response was observed during a cyclic voltammetry experiment at the P3BFeN20/– couple (−2.1 V vs Fc+/0) upon addition of excess HBArF4 under an N2 atmosphere. Electrolytic NH3 generation by P3BFe+ was observed at −2.4 V vs Fc+/0 in Et2O,23 and Na/Hg (−2.4 V vs Fc+/0 in THF)16 could instead be used for N2-to-NH3 conversion catalysis (albeit less selectively and with low turnover). Finally, mixing P3BFe+ with Cp*2Co in Et2O at −78 °C under N2 generates some P3BFeN2– as observed by X-band EPR and Mössbauer spectroscopy (see the Supporting Information), suggesting that Cp*2Co is in principle a sufficiently strong reductant to trigger catalysis by P3BFe+.
Treatment of P3BFe+ with Cp*2Co and [Ph2NH2][OTf], [Ph2NH2][BArF4], or [PhNH3][OTf] in Et2O at −78 °C under an N2 atmosphere affords catalytic yields of NH3 (Table 1). Notably, the highest selectivity for NH3 obtained among this series (72% at standard substrate loading; entry 1) is significantly improved compared to all previously described (molecular) Fe catalysts for N2-to-NH3 conversion.20,24 Tripling the initial substrate loading (entry 2) nearly triples the NH3 production with only modest loss in efficiency for NH3 (63%). Preliminary attempts to further increase the initial substrate loading led to substantially decreased efficiency (entry 3). However, substrate reloading experiments (entries 4 and 5) maintained greater than 50% efficiency for NH3 overall; a turnover number for NH3 generation via two reloadings has been achieved as high as 89 in a single run (84 ± 8; entry 5). This is a high turnover number for a molecular Fe N2-to-NH3 conversion catalyst under any conditions.20,25
Table 1. N2-to-NH3 Conversion with P3EM Complexes (M = Fe, Co)a.
| catalyst | Cp*2Co (equiv) | acid (equiv) | equiv of NH3/Fe | % yield of NH3/e– | |
|---|---|---|---|---|---|
| 1 | P3BFe+ | 54 | 108c | 12.8 ± 0.5 | 72 ± 3 |
| 2 | P3BFe+ | 162 | 322c | 34 ± 1 | 63 ± 2 |
| 3 | P3BFe+ | 322 | 638c | 26.7 ± 0.9 | 25 ± 1 |
| 4b | P3BFe+ | [162] × 2 | [322] × 2c | 56 ± 9 | 52 ± 9 |
| 5b | P3BFe+ | [162] × 3 | [322] × 3c | 84 ± 8 | 52 ± 5 |
| 6 | P3BFe+ | 54 | 108d | 8 ± 1 | 42 ± 6 |
| 7 | P3BFe+ | 54 | 108e | 7 ± 1 | 38 ± 7 |
| 8 | P3BFe+ | 162 | 322e | 16 ± 3 | 29 ± 4 |
| 9 | P3SiFeN2 | 54 | 108c | 1.2 ± 0.1 | 6 ± 1 |
| 10 | P3BCoN2– | 54 | 108c | 1.1 ± 0.4 | 6 ± 2 |
| 11 | P3SiCoN2 | 54 | 108c | 0 ± 0 | 0 ± 0 |
The catalyst, acid, Cp*2Co, and Et2O were sealed in a vessel at −196 °C under an N2 atmosphere followed by warming to −78 °C and stirring. Yields are reported as an average of at least 2 runs; for individual experiments see the Supporting Information.
For these experiments the reaction was allowed to proceed for 3 h at −78 °C before cooling to −196 °C and furnishing with additional substrate and solvent.
[Ph2NH2][OTf].
[Ph2NH2][BArF4].
[PhNH3][OTf].
The use of the more soluble acid [Ph2NH2][BArF4] (entry 6) provides significantly lower, but still catalytic, yields of NH3. This more soluble acid presumably increases background reactivity with Cp*2Co (see the Supporting Information). Perhaps more significantly, [PhNH3][OTf] is a considerably weaker acid than [Ph2NH2][OTf] (Figure 1), but still provides substantial catalytic yields of NH3 (entries 7 and 8) and at efficiencies that compare well with those obtained previously using HBArF4 and KC8 despite a difference in driving force of nearly 100 kcal/mol.23
We also screened several related phosphine-ligated Fe–N2 and Co–N2 complexes26,27 under the new standard reaction conditions with [Ph2NH2][OTf] and Cp*2Co (entries 9–11) but found that none of these other systems were competent catalysts. While we anticipate that other catalyst systems for N2-to-NH3 conversion may yet be found that function under the conditions described herein,20 certain features of the P3BFe system correlate with unusually productive catalysis.27
Also significant is that when P3BFe+ is loaded with 322 equiv of [Ph2NH2][OTf] and 162 equiv of Cp*2Co in Et2O at −78 °C, very modest levels of N2H4 are detected (<1 equiv per Fe; see the Supporting Information).9,20 We had previously reported that catalytic N2 reduction with KC8 and HBArF4 yielded no detectable hydrazine, but observed that if hydrazine was added at the outset of a catalytic run, it was consumed.6 When 5 equiv of N2H4 were added at the beginning of a catalytic run (again with 322 equiv of [Ph2NH2][OTf] and 162 equiv of Cp*2Co), only 0.22 equiv of N2H4 (4.4% recovery) remained after workup. This result indicates that liberated hydrazine can also be reduced or disproportionated under the present conditions. That N2H4 is detected to any extent in the absence of initially added N2H4 under these conditions indicates that a late N–N cleavage mechanism to produce NH3 (e.g., alternating or hybrid crossover) is accessible.4,28 A recent report by Ashley and co-workers describes a phosphine-supported Fe system for which catalytic hydrazine formation is kinetically dominant.20 Whether such a pathway is kinetically dominant in this system is as yet unclear.23,29
The P3BFe speciation under turnover conditions was probed via freeze-quench Mössbauer spectroscopy.23 The Mössbauer spectrum of a catalytic reaction mixture after 5 min of reaction time (Figure 2) reveals the presence of multiple species featuring well-resolved sets of quadrupole doublets. The spectrum is satisfactorily simulated with P3BFeN2 (δ = 0.55 mm/s, ΔEQ = 3.24 mm/s, 32%; Figure 2, green), P3BFeN2– (δ = 0.40 mm/s, ΔEQ = 0.98 mm/s, 26%; Figure 2, blue),23,30 an unknown, likely P3B metalated Fe species (δ = 0.42 mm/s, ΔEQ = 1.84 mm/s, 18%; Figure 2, yellow), and a final species that is modeled with δ = 0.96 mm/s and ΔEQ = 3.10 mm/s (24%; Figure 2, orange). The broad nature of this last signal and its overlap with other features in the spectrum prevents its precise assignment, but its high isomer shift and large quadrupole splitting are suggestive of a tetrahedral, S = 2 Fe(II) complex.31,32 The Mössbauer spectrum of a catalytic reaction mixture after 30 min was also analyzed (see the Supporting Information). The spectrum still shows P3BFeN2 (53%), the same unknown P3BFe species (18%), and again a tetrahedral, high-spin Fe(II) component (22%). However, P3BFe+ is now present (δ = 0.75 mm/s, ΔEQ = 2.55 mm/s, 8%) and P3BFeN2– is no longer observed. The reloading experiments described above provide strong evidence that “P3BFe” species represent an “active catalyst” population; interpretation of the relative speciation via spectroscopy should hence bear on the mechanism of the overall catalysis.
Figure 2.
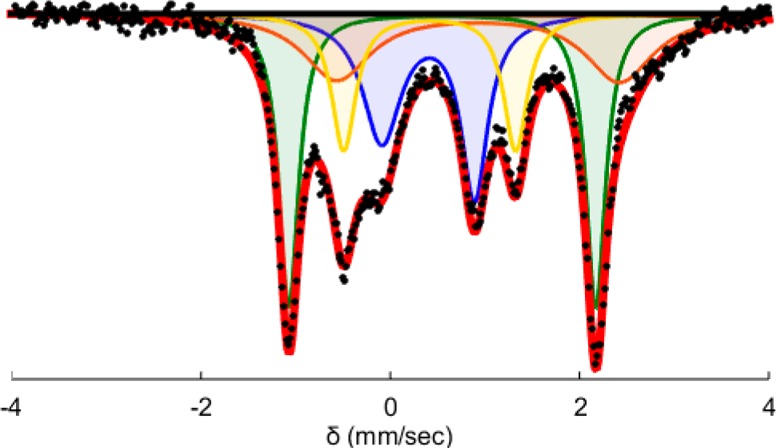
Mössbauer spectrum at 80 K with 50 mT applied parallel field of a freeze-quenched catalytic reaction (54 equiv of Cp*2Co, 108 equiv of [Ph2NH2][OTf], 1 equiv of P3B[57Fe]+) after 5 min of reaction time.
The appearance of a presumed high-spin (S = 2), tetrahedral Fe(II) species during catalysis (ca. 25%) might arise via dechelation of a phosphine arm. This species could represent an off-path state or a downstream deactivation product. Interestingly, under the present catalytic conditions we do not observe the borohydrido–hydrido species P3B(μ-H)Fe(H)(L) (L = N2 or H2); this species was postulated to be an off-path state during N2-to-NH3 conversion catalysis using HBArF4 and KC8 and was the major component observed at early times (ca. 60% at 5 min).23 It therefore appears that a larger fraction of the “P3BFe” species are in a catalytically on-path state at early reaction times under these new catalytic conditions.
Additionally, the presence of a significant degree of P3BFeN2– (Figure 2) at an early time point is distinct from conditions with HBArF4 and KC8.23 This observation is consistent with the notion that protonation of P3BFeN2– is slowed under the present conditions, likely as a result of the insolubility of the triflate salt [Ph2NH2][OTf] and its attenuated acidity relative to HBArF4.17,18,33 Clearly, differences in the rates of key elementary steps under the new conditions described here may lead to new mechanistic scenarios for N2-to-NH3 conversion.
The improved catalytic efficiency at significantly lower driving force warrants additional consideration. When using HBArF4 and KC8 we have previously suggested that protonation of P3BFeN2–, which itself can be generated by reduction of P3BFeN2, to produce P3BFe–N=NH is a critical first step; P3BFe–N=NH can then be trapped by acid to produce spectroscopically observable P3BFe=N–NH2+.29 These steps, shown in eqs 2a and 2b, represent an ET–PT pathway. A PT–ET pathway, where P3BFeN2 is sufficiently basic to be protonated to generate P3BFe–N=NH+ as a first step, followed by ET, is also worth considering (eqs 3a and 3b). A direct PCET pathway (eq 4), where H atom delivery to P3BFeN2 occurs, thus obviating the need to access either P3BFeN2– or P3BFe–N=NH+, needs also to be considered.
| 2a |
| 2b |
| 3a |
| 3b |
| 4 |
Initial PT to P3BFeN2 to generate P3BFe–N=NH+ (eq 3a) is unlikely under the present conditions due to the high predicted acidity of P3BFe–N=NH+ (pKa = −3.7; estimated via DFT; see the Supporting Information); efficient generation of such a species seems implausible for acids whose pKa’s are calculated at 1.4 (Ph2NH2+) and 6.8 (PhNH3+) in Et2O (Table 2). We note that [Ph2NH2][OTf] does not react productively with P3BFeN2 at −78 °C in Et2O, as analyzed by Mössbauer spectroscopy.
Table 2. Calculated pKa Values and BDEs of Selected Speciesa.
| species | pKa | BDEb |
|---|---|---|
| Ph2NH2+ | 1.4c | |
| PhNH3+ | 6.8 | |
| lutidinium | 14.5 | |
| endo-Cp*Co(η4-C5Me5H)+ | 16.8 | 31 |
| exo-Cp*Co(η4-C5Me5H)+ | 16.8 | 31 |
| endo-Cp*Cr(η4-C5Me5H)+ | 17.3 | 37 |
| exo-Cp*Cr(η4-C5Me5H)+ | 12.1 | 30 |
| P3BFe–N=NH+ | –3.7 | |
| P3BFe–N=NH | 38.7 | 35 |
| P3BFe=N–NH2+ | 14.4 | 51 |
| P3BFe=N–NH2 | 47 | |
| [HIPTN3N]Mo–N=NH | 51 |
Calculations were performed using the M06-L34 functional with a def2-TZVP basis set on Fe and Mo and a def2-SVP basis set on all other atoms35 (see the Supporting Information).
In kcal/mol.
pKa values were calculated in Et2O and reported relative to (Et2O)2H+.
Focusing instead on the PCET pathway (eq 4), the DFT-calculated BDEN–H for P3BFe–N=NH (35 kcal/mol; Table 2; see the Supporting Information for details)36 is larger than the effective BDE21 of either Cp*2Co/Ph2NH2+ or Cp*2Co/PhNH3+ (25 and 31 kcal/mol, respectively). This suggests that PCET (eq 4) is plausible on thermodynamic grounds. Given that we have employed Cp*2Co in this study, and that this and also Cp2Co and Cp*2Cr have been effective in other N2-fixing molecular catalyst systems,10−13,20 we have explored via DFT several putative metallocene-derived PCET reagents. Independent studies of H2 evolution from cobaltocene have invoked a protonated cobaltocene intermediate.37−39 The observation of a background H2 evolution reaction (HER) when employing metallocene reductants, but in the absence of an N2-to-NH3 conversion catalyst, suggests that metallocene protonation is kinetically competent.13,40 Based on the analysis we describe below, we propose that protonated metallocenes may serve as discrete and highly active H• sources for PCET.
We find that the formation of endo- and exo-Cp*Co(η4-C5Me5H)+ is predicted to be thermodynamically favorable via protonation of Cp*2Co by either Ph2NH2+ or PhNH3+ (−21 and −13 kcal/mol, respectively; Figure 3A).41,42 We have calculated the BDEC–H’s for both endo- and exo-Cp*Co(η4-C5Me5H)+ as 31 kcal/mol (Figure 3B; Table 2), indicating that they should be among the strongest PCET reagents accessible in this catalyst cocktail. Indeed, they would be among the strongest PCET reagents known.21
Figure 3.

(A) Calculated free-energy changes for the protonation of Cp*2Co. (B) DFT optimized structure of endo-Cp*Co(η4-C5Me5H)+ (methyl protons omitted for clarity). (C) The unfavorable reduction of 2,6-lutidinium by Cp*2Cr with the calculated free energy change. (D) The favorable protonation of Cp*2Cr by lutidinium with the calculated free energy change.
We anticipate that these species would be extremely unstable in solution and hence difficult to detect in situ, but via trapping in the solid state by rapid precipitation from toluene we have isolated a species whose EPR data and chemical behavior are consistent with {Cp*Co(η4-C5Me5H)}{OTf}. Accordingly, slow addition of a toluene solution of Cp*2Co at −78 °C to triflic acid (HOTf) leads to the instantaneous precipitation of a purple solid that can be handled only at low temperature. The purple solid is characterized at 77 K by powder EPR spectroscopy via its highly structured signal. By contrast, at this temperature S = 1/2 Cp*2Co does not display a discernible EPR signal (see the Supporting Information). The new signal shows strong Co hyperfine coupling and significant g-anisotropy, consistent with a new S = 1/2 cobalt species (Figure 4). Furthermore, the resulting EPR signal is slightly perturbed when this purple solid is instead generated from the reaction between deuterated triflic acid (DOTf) and Cp*2Co (see the Supporting Information), suggesting that the acidic proton is directly associated with the new Co species and consistent with its assignment as a protonated decamethylcobaltocene species. Close inspection of these spectra indicates that they likely represent a mixture of two signals arising from similar Co-containing complexes. This observation is consistent with the presence of both endo- and exo-Cp*Co(η4-C5Me5H)+, as is to be expected given that they are predicted to be nearly isoenergetic. Allowing the purple precipitate to warm to room temperature either as a solid or as a stirred suspension in toluene leads to the formation of H2 and Cp*2Co+ (see the Supporting Information).
Figure 4.
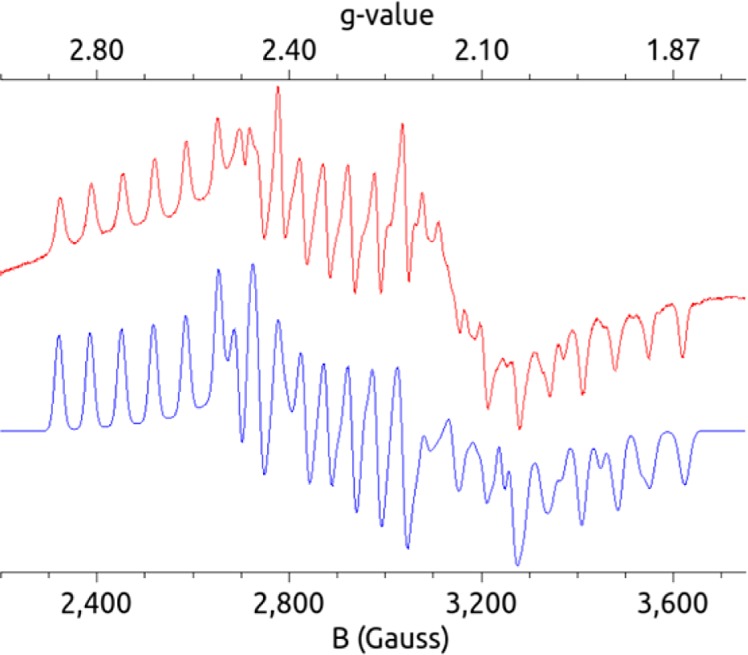
X-band 77 K powder EPR spectrum (red) and simulation (blue) of the isolated purple precipitate (assigned as endo- and exo-Cp*Co(η4-C5Me5H)+) from reaction between Cp*2Co and HOTf at −78 °C (see the Supporting Information for simulation parameters).
To better understand the potential role of PCET in N2-to-NH3 conversion catalysis by P3BFe, we have additionally calculated the N–H bond strengths (Table 2) of several early stage candidate intermediates, including the aforementioned P3BFe–N=NH (35 kcal/mol), P3BFe=N–NH2+ (51 kcal/mol), and P3BFe=N–NH2 (47 kcal/mol). We conclude that PCET from Cp*Co(η4-C5Me5H)+ to generate intermediates of these types is thermodynamically favorable in each case.43 To generate the first and most challenging intermediate (eq 5), the enthalpic driving force for PCET is estimated at ∼4 kcal/mol (ΔGcalc = −9 kcal/mol). This driving force and, hence, the plausibility of PCET steps, increase sharply as further downstream Fe–NxHy intermediates are considered.44−47
| 5 |
Given the prevalence of metallocene reductants in N2-to-NH3 (or -N2H4) conversion,10−13,20 especially for the well-studied Mo catalyst systems, it is worth considering metallocene-mediated PCET more generally. For instance, a role for ET/PT steps (or conversely PT/ET) in N2-to-NH3 conversion catalyzed by [HIPTN3N]Mo (HIPTN3N = [(3,5-(2,4,6- iPr3C6H2)2C6H3NCH2CH2)3N]3–, a bulky triamidoamine ligand) has been frequently posited.48−52 But PCET steps may play a critical role, too. In the latter context, we note reports from Schrock and co-workers that have shown that both acid and reductant are required to observe productive reactivity with [HIPTN3N]MoN2. These observations are consistent with PCET to generate [HIPTN3N]Mo–N=NH.52 A PCET scenario has been discussed in the general context of N2-to-NH3 conversion, where a lutidinyl radical intermediate formed via ET from Cp*2Cr was suggested as a PCET reagent that can be generated in situ.40,53 However, our own calculations predict that the lutidinyl radical should not be accessible with Cp*2Cr as the reductant (ΔGcalc = +10 kcal/mol; Figure 3C).54−56 We instead propose protonation of Cp*2Cr by the lutidinium acid as more plausible (ΔGcalc = −5.3 kcal/mol; Figure 3D) to generate a highly reactive decamethylchromocene-derived PCET reagent.
While N–H bond strengths have not been experimentally determined for the [HIPTN3N]Mo system, using available published data we deduce the N–H bond of [HIPTN3N]Mo–N=NH to be ca. 49 kcal/mol and we calculate it via DFT (truncated HIPTN3N; see the Supporting Information) as 51 kcal/mol.57 The BDEN–H for this Mo diazenido species is hence much larger than we predict for P3BFe–N=NH (35 kcal/mol), perhaps accounting for its higher stability.52 A PCET reaction between endo-Cp*Cr(η4-C5Me5H)+ (BDEcalc = 37 kcal/mol) and [HIPTN3N]MoN2 to generate [HIPTN3N]Mo–N=NH and Cp*2Cr+ would be highly exergonic. Furthermore, we predict a similarly weak BDEC–H for Cp-protonated cobaltocene, CpCo(η4-C5H6)+ (BDEcalc = 35 kcal/mol). These considerations are consistent with the reported rapid formation of [HIPTN3N]Mo–N=NH using either Cp*2Cr or Cp2Co in the presence of lutidinium acid.58
To close, we have demonstrated catalytic N2-to-NH3 conversion by P3BFe+ at a much lower driving force (nearly 100 kcal/mol) than originally reported via combination of a weaker reductant (Cp*2Co) and acid ([Ph2NH2][OTf] or [PhNH3][OTf]). Significantly improved efficiency for NH3 formation is observed (up to 72% at standard substrate loading), and by reloading additional substrate at low temperature a turnover number that is comparatively high for a synthetic molecular Fe catalyst (84 ± 8 equiv of NH3 per Fe) has been achieved. Freeze-quench Mössbauer spectroscopy under turnover conditions reveals differences in the speciation of P3BFe compared to previous studies with HBArF4 and KC8, suggesting changes in the rates of key elementary steps. Using DFT calculations we have considered the viability of a decamethylcobaltocene-mediated PCET pathway as an additional contributor to the previously formulated ET–PT and PT–ET pathways. Based on our calculations, we propose that protonated metallocenes should serve as discrete, very reactive PCET reagents in N2-to-NH3 conversion catalysis. Furthermore, we present preliminary experimental data that suggests that protonated decamethylcobaltocene can be accessed synthetically and that such a species may be a potent PCET reagent. Indeed, the achievement of high efficiency for N2-to-NH3 conversion by both P3BFe and various Mo catalysts that benefit from metallocene reductants raises the intriguing possibility that metallocene-based PCET reactivity is a potentially widespread and overlooked mechanism. Efforts are underway to experimentally probe such pathways.
Acknowledgments
This work was supported by the NIH (GM 070757) and the Gordon and Betty Moore Foundation. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant No. ACI-1053575. M.J.C., T.J.D.C., and B.D.M. acknowledge the support of the NSF for Graduate Fellowships (GRFP).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00014.
Author Contributions
† M.J.C., T.J.D.C., and B.D.M. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Smil V.Enriching the Earth: MIT Press: Cambridge, 2001. [Google Scholar]
- van der Ham C. J. M.; Koper M. T. M.; Hetterscheid D. G. H. Challenges in Reduction of Dinitrogen by Proton and Electron Transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. 10.1039/C4CS00085D. [DOI] [PubMed] [Google Scholar]
- Shaver M. P.; Fryzuk M. D. Activation of Molecular Nitrogen: Coordination, Cleavage and Functionalization of N2 Mediated By Metal Complexes. Adv. Synth. Catal. 2003, 345, 1061–1076. 10.1002/adsc.200303081. [DOI] [Google Scholar]
- MacLeod K. C.; Holland P. L. Recent Developments in the Homogeneous Reduction of Dinitrogen by Molybdenum and Iron. Nat. Chem. 2013, 5, 559–565. 10.1038/nchem.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev. 2014, 114, 4041–4062. 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. S.; Rittle J.; Peters J. C. Catalytic Conversion of Nitrogen to Ammonia by an Iron Model Complex. Nature 2013, 501, 84–87. 10.1038/nature12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creutz S. E.; Peters J. C. Catalytic Reduction of N2 to NH3 by an Fe–N2 Complex Featuring a C-Atom Anchor. J. Am. Chem. Soc. 2014, 136, 1105–1115. 10.1021/ja4114962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ung G.; Peters J. C. Low-Temperature N2 Binding to Two-Coordinate L2Fe0 Enables Reductive Trapping of L2FeN2– and NH3 Generation. Angew. Chem., Int. Ed. 2015, 54, 532–535. 10.1002/anie.201409454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama S.; Arashiba K.; Nakajima K.; Matsuo Y.; Tanaka H.; Ishii K.; Yoshizawa K.; Nishibayashi Y. Catalytic Transformation of Dinitrogen into Ammonia and Hydrazine by Iron-Dinitrogen Complexes Bearing Pincer Ligand. Nat. Commun. 2016, 7, 12181. 10.1038/ncomms12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandulov D. V.; Schrock R. R. Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]
- Arashiba K.; Miyake Y.; Nishibayashi Y. A Molybdenum Complex Bearing PNP-Type Pincer Ligands Leads to the Catalytic Reduction of Dinitrogen into Ammonia. Nat. Chem. 2011, 3, 120–125. 10.1038/nchem.906. [DOI] [PubMed] [Google Scholar]
- Kuriyama S.; Arashiba K.; Nakajima K.; Tanaka H.; Kamaru N.; Yoshizawa K.; Nishibayashi Y. Catalytic Formation of Ammonia from Molecular Dinitrogen by Use of Dinitrogen-Bridged Dimolybdenum–Dinitrogen Complexes Bearing PNP-Pincer Ligands: Remarkable Effect of Substituent at PNP-Pincer Ligand. J. Am. Chem. Soc. 2014, 136, 9719–9731. 10.1021/ja5044243. [DOI] [PubMed] [Google Scholar]
- Arashiba K.; Kinoshita E.; Kuriyama S.; Eizawa A.; Nakajima K.; Tanaka H.; Yoshizawa K.; Nishibayashi Y. Catalytic Reduction of Dinitrogen to Ammonia by Use of Molybdenum–Nitride Complexes Bearing a Tridentate Triphosphine as Catalysts. J. Am. Chem. Soc. 2015, 137, 5666–5669. 10.1021/jacs.5b02579. [DOI] [PubMed] [Google Scholar]
- The enthalpic driving force (ΔΔHf) has been estimated here by taking 3(BDEH• – BDEeffective), where BDE is bond dissociation enthalpy. This allows for an evaluation of the driving force for a given reaction with respect to that for a hypothetical N2-to-NH3 conversion catalyst that uses H2 as the proton and electron source. This is achieved by using Bordwell’s equation (with the assumption that S(X•) = S(XH); see the Supporting Information) and literature values (see refs (15−19) and (21)) for pKa, redox potential, the enthalpy of reaction for H+ + e– → H• (CH = 66 kcal/mol in THF), and the energy of H• in THF (52 kcal/mol).
- Bordwell F. G.; Cheng J. P.; Harrelson J. A. Homolytic Bond Dissociation Energies in Solution from Equilibrium Acidity and Electrochemical Data. J. Am. Chem. Soc. 1988, 110, 1229–1231. 10.1021/ja00212a035. [DOI] [Google Scholar]
- Connelly N. G.; Geiger W. E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- Garrido G.; Rosés M.; Ràfols C.; Bosch E. Acidity of Several Anilinium Derivatives in Pure Tetrahydrofuran. J. Solution Chem. 2008, 37, 689–700. 10.1007/s10953-008-9262-6. [DOI] [Google Scholar]
- Kaljurand I.; Kütt A.; Sooväli L.; Rodima T.; Mäemets V.; Leito I.; Koppel I. A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- Cappellani E. P.; Drouin S. D.; Jia G.; Maltby P. A.; Morris R. H.; Schweitzer C. T. Effect of the Ligand and Metal on the pKa Values of the Dihydrogen Ligand in the Series of Complexes [M(H2)H(L)2]+, M = Fe, Ru, Os, Containing Isosteric Ditertiaryphosphine Ligands, L. J. Am. Chem. Soc. 1994, 116, 3375–3388. 10.1021/ja00087a024. [DOI] [Google Scholar]
- While initiating our studies we became aware of a phosphine-supported Fe system that catalyzes N2-to-N2H4 conversion using Cp*2Co and [Ph2NH2][OTf] with efficiency as high as 72% for e– delivery to N2:Hill P. J.; Doyle L. R.; Crawford A. D.; Myers W. K.; Ashley A. E. Selective Catalytic Reduction of N2 to N2H4 by a Simple Fe Complex. J. Am. Chem. Soc. 2016, 138, 13521–13524. 10.1021/jacs.6b08802. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Tronic T. A.; Mayer J. M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010, 110, 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. S.Catalytic conversion of nitrogen to ammonia by an iron model complex; Ph.D. Thesis, California Institute of Technology: September 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo T. J.; Thompson N. B.; Peters J. C. A Synthetic Single-Site Fe Nitrogenase: High Turnover, Freeze-Quench 57Fe Mössbauer Data, and a Hydride Resting State. J. Am. Chem. Soc. 2016, 138, 5341–5350. 10.1021/jacs.6b01706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previously reported molecular Fe catalysts for N2-to-NH3 conversion utilize KC8 and HBArF4 and achieve NH3 selectivities ≤45% with respect to their limiting reagent (see refs (6−9)) at a similar reductant loading. Lower selectivities are observed with higher loading (see refs (6) and (23)).
- In catalytic runs performed with labeled [Ph215NH2][OTf] under an atmosphere of natural abundance 14N2 the production of exclusively 14NH3 is observed, demonstrating that the NH3 formed during catalysis is derived from N2 and not degradation of the acid (see the Supporting Information).
- Whited M. T.; Mankad N. P.; Lee Y.; Oblad P. F.; Peters J. C. Dinitrogen Complexes Supported by Tris(phosphino)silyl Ligands. Inorg. Chem. 2009, 48, 2507–2517. 10.1021/ic801855y. [DOI] [PubMed] [Google Scholar]
- Del Castillo T. J.; Thompson N. B.; Suess D. L. M.; Ung G.; Peters J. C. Evaluating Molecular Cobalt Complexes for the Conversion of N2 to NH3. Inorg. Chem. 2015, 54, 9256–9262. 10.1021/acs.inorgchem.5b00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittle J.; Peters J. C. An Fe-N2 Complex That Generates Hydrazine and Ammonia via Fe=NNH2: Demonstrating a Hybrid Distal-to-Alternating Pathway for N2 Reduction. J. Am. Chem. Soc. 2016, 138, 4243–4248. 10.1021/jacs.6b01230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. S.; Cutsail G. E.; Rittle J.; Connor B. A.; Gunderson W. A.; Zhang L.; Hoffman B. M.; Peters J. C. Characterization of an Fe≡N–NH2 Intermediate Relevant to Catalytic N2 Reduction to NH3. J. Am. Chem. Soc. 2015, 137, 7803–7809. 10.1021/jacs.5b03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The presence of P3BFeN2– was confirmed by freeze-quench EPR spectroscopy experiments (see the Supporting Information). The asymmetry observed in the Mössbauer lineshapes is characteristic of this species. A redox equilibrium between P3BFeN20/– and Cp*2Co+/0 is also observed in the reaction of P3BFe+ with excess Cp*2Co in the absence of acid (see the Supporting Information).
- The distinct properties of tetrahedral, high spin Fe(II) lead to high isomer shifts (0.9–1.3) and large quadrupole splittings (>2.5) that are characteristic of these types of species:Münck E. In Physical Methods in Bioinorganic Chemistry: Spectroscopy and Magnetism; Que L., Jr., Ed.; University Science Books: Sausalito, CA, 2000; pp 287–320. [Google Scholar]
- Daifuku S. L.; Kneebone J. L.; Snyder B. E. R.; Neidig M. L. Iron(II) Active Species in Iron–Bisphosphine Catalyzed Kumada and Suzuki–Miyaura Cross-Couplings of Phenyl Nucleophiles and Secondary Alkyl Halides. J. Am. Chem. Soc. 2015, 137, 11432–11444. 10.1021/jacs.5b06648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamashima Y.; Somei H.; Shimura Y.; Tamura T.; Sodeoka M. Amine-Salt-Controlled, Catalytic Asymmetric Conjugate Addition of Various Amines and Asymmetric Protonation. Org. Lett. 2004, 6, 1861–1864. 10.1021/ol0493711. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2006, 125, 194101. 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Experimental BDEN-H’s for related species (P3SiFe–C=NH+, P3SiFe–C=NH, P3SiFe–C=N(Me)H+, P3SiFe–C=N(Me)H, and P3SiFe–N=N(Me)H+) have been measured and are in good agreement with the BDEN–H values calculated using the DFT methods described in this work (see the Supporting Information for full details):Rittle J.; Peters J. C.. N−H Bond Dissociation Enthalpies and Facile H Atom Transfers for Early Intermediates of Fe−N2 and Fe−CN Reductions. J. Am. Chem. Soc. 2017, in press, DOI: 10.1021/jacs.6b12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelle U.; Infelta P. P.; Graetzel M. Kinetics and Mechanism of the Reduction of Protons to Hydrogen by Cobaltocene. Inorg. Chem. 1988, 27, 879–883. 10.1021/ic00278a026. [DOI] [Google Scholar]
- Pitman C. L.; Finster O. N. L.; Miller A. J. M. Cyclopentadiene-Mediated Hydride Transfer from Rhodium Complexes. Chem. Commun. 2016, 52, 9105–9108. 10.1039/C6CC00575F. [DOI] [PubMed] [Google Scholar]
- Quintana L. M. A.; Johnson S. I.; Corona S. L.; Villatoro W.; Goddard W. A.; Takase M. K.; VanderVelde D. G.; Winkler J. R.; Gray H. B.; Blakemore J. D. Proton–hydride Tautomerism in Hydrogen Evolution Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 6409–6414. 10.1073/pnas.1606018113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munisamy T.; Schrock R. R. An Electrochemical Investigation of Intermediates and Processes Involved in the Catalytic Reduction of Dinitrogen by [HIPTN3N]Mo (HIPTN3N = (3,5-(2,4,6-I-Pr3C6H2)2C6H3NCH2CH2)3N). Dalton Trans. 2012, 41, 130–137. 10.1039/C1DT11287B. [DOI] [PubMed] [Google Scholar]
- Efforts to instead optimize a metal hydride species, [Cp*2Co–H]+, led to hydride transfer to the ring system. Nevertheless, reactive transition metal hydride radical cations are also known to exhibit PCET behavior.
- Hu Y.; Shaw A. P.; Estes D. P.; Norton J. R. Transition-Metal Hydride Radical Cations. Chem. Rev. 2016, 116, 8427–8462. 10.1021/acs.chemrev.5b00532. [DOI] [PubMed] [Google Scholar]
- The dissolution equilibria and kinetics of the insoluble reagents used complicate analysis of the kinetics of individual ET, PT, and PCET steps. However, the low activation barriers (G⧧ < 9 kcal/mol) calculated for all proposed PCET reactions are consistent with these reactions being kinetically accessible (see the Supporting Information for full details).
- Studies (see refs (45−47)) have shown that the Marcus cross-relation holds quite well for many PCET reactions. This is indicative of a substantial correlation between thermodynamic driving force and reaction kinetics; it is, however, unclear whether the proposed reactivity would demonstrate such behavior.
- Roth J. P.; Yoder J. C.; Won T.-J.; Mayer J. M. Application of the Marcus Cross Relation to Hydrogen Atom Transfer Reactions. Science 2001, 294, 2524–2526. 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- Mayer J. M.; Rhile I. J. Thermodynamics and Kinetics of Proton-Coupled Electron Transfer: Stepwise vs. Concerted Pathways. Biochim. Biophys. Acta, Bioenerg. 2004, 1655, 51–58. 10.1016/j.bbabio.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Theoretical Perspectives on Proton-Coupled Electron Transfer Reactions. Acc. Chem. Res. 2001, 34, 273–281. 10.1021/ar9901117. [DOI] [PubMed] [Google Scholar]
- Studt F.; Tuczek F. Energetics and Mechanism of a Room-Temperature Catalytic Process for Ammonia Synthesis (Schrock Cycle): Comparison with Biological Nitrogen Fixation. Angew. Chem., Int. Ed. 2005, 44, 5639–5642. 10.1002/anie.200501485. [DOI] [PubMed] [Google Scholar]
- Reiher M.; Le Guennic B.; Kirchner B. Theoretical Study of Catalytic Dinitrogen Reduction under Mild Conditions. Inorg. Chem. 2005, 44, 9640–9642. 10.1021/ic0517568. [DOI] [PubMed] [Google Scholar]
- Studt F.; Tuczek F. Theoretical, Spectroscopic, and Mechanistic Studies on Transition-Metal Dinitrogen Complexes: Implications to Reactivity and Relevance to the Nitrogenase Problem. J. Comput. Chem. 2006, 27, 1278–1291. 10.1002/jcc.20413. [DOI] [PubMed] [Google Scholar]
- Thimm W.; Gradert C.; Broda H.; Wennmohs F.; Neese F.; Tuczek F. Free Reaction Enthalpy Profile of the Schrock Cycle Derived from Density Functional Theory Calculations on the Full [MoHIPTN3N] Catalyst. Inorg. Chem. 2015, 54, 9248–9255. 10.1021/acs.inorgchem.5b00787. [DOI] [PubMed] [Google Scholar]
- Yandulov D. V.; Schrock R. R. Studies Relevant to Catalytic Reduction of Dinitrogen to Ammonia by Molybdenum Triamidoamine Complexes. Inorg. Chem. 2005, 44, 1103–1117. 10.1021/ic040095w. [DOI] [PubMed] [Google Scholar]
- Pappas I.; Chirik P. J. Catalytic Proton Coupled Electron Transfer from Metal Hydrides to Titanocene Amides, Hydrazides and Imides: Determination of Thermodynamic Parameters Relevant to Nitrogen Fixation. J. Am. Chem. Soc. 2016, 138, 13379–13389. 10.1021/jacs.6b08009. [DOI] [PubMed] [Google Scholar]
- Although our calculations for a hypothetical lutidinyl radical predict a weak N–H bond (BDEN–H ∼ 35 kcal/mol), the oxidation potential of this species is calculated to be −1.89 V vs Fc+/0 in THF (see the Supporting Information). Experimental determination of this reduction potential for calibration has been contentious; however, our calculated reduction potential is similar to that previously calculated for pyridinium in aqueous media (−1.37 V vs SCE; see refs (55) and (56)).
- Yan Y.; Zeitler E. L.; Gu J.; Hu Y.; Bocarsly A. B. Electrochemistry of Aqueous Pyridinium: Exploration of a Key Aspect of Electrocatalytic Reduction of CO2 to Methanol. J. Am. Chem. Soc. 2013, 135, 14020–14023. 10.1021/ja4064052. [DOI] [PubMed] [Google Scholar]
- Keith J. A.; Carter E. A. Theoretical Insights into Pyridinium-Based Photoelectrocatalytic Reduction of CO2. J. Am. Chem. Soc. 2012, 134, 7580–7583. 10.1021/ja300128e. [DOI] [PubMed] [Google Scholar]
- It has been reported that [HIPTN3N]MoN2–/[HIPTN3N]Mo–N=NH is in equilibrium with DBU/DBUH+ (DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; pKa = 18.5 in THF; see refs (17) and (18)). Taken with the reported reduction potential of [HIPTN3N]MoN2 (E1/2 = – 1.81 V vs Fc+/0 in THF, see ref (52)), the experimental BDE can be approximated with the Bordwell equation and the enthalpy of reaction for H+ + e– → H• (see refs (15) and (19)).
- In addition to lutidinium salts, [Et3NH][OTf] has been shown to affect the formation of [HIPTN3N]Mo–N=NH from [HIPTN3N]MoN2 in the presence of metallocene reductants (see ref (52)).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.