

Abstract
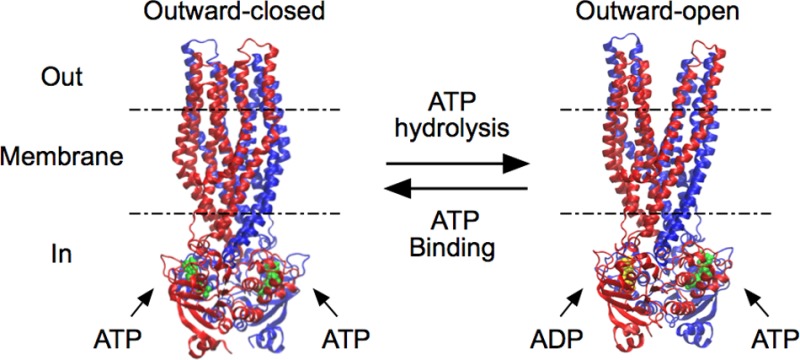
ATP-binding cassette (ABC) transporters use the energy of ATP binding and hydrolysis to move a large variety of compounds across biological membranes. P-glycoprotein, involved in multidrug resistance, is the most investigated eukaryotic family member. Although a large number of biochemical and structural approaches have provided important information, the conformational dynamics underlying the coupling between ATP binding/hydrolysis and allocrite transport remains elusive. To tackle this issue, we performed molecular dynamic simulations for different nucleotide occupancy states of Sav1866, a prokaryotic P-glycoprotein homologue. The simulations reveal an outward-closed conformation of the transmembrane domain that is stabilized by the binding of two ATP molecules. The hydrolysis of a single ATP leads the X-loop, a key motif of the ATP binding cassette, to interfere with the transmembrane domain and favor its outward-open conformation. Our findings provide a structural basis for the unidirectionality of transport in ABC exporters and suggest a ratio of one ATP hydrolyzed per transport cycle.
Short abstract
An outward-closed state of the Sav1866 efflux pump is stabilized by the binding of two ATP molecules. Hydrolysis at one ATP binding site favors the outward-open conformation. An export mechanism is proposed based on these two states.
Introduction
ATP binding cassette (ABC) transporters use the energy of ATP binding and hydrolysis to move a large variety of compounds (allocrites)1 across biological membranes as importers or exporters. The functional unit of ABC transporters consists of two highly conserved nucleotide-binding domains (NBDs) forming together two nucleotide-binding sites (NBSs, Figure 1B–E) that allow for ATP hydrolysis, and two less conserved transmembrane domains (TMDs) that allow for allocrite binding and transport. In prokaryotes, the functional unit generally consists of a homo- or heterodimer, each monomer comprising an NBD and a TMD; in eukaryotes the functional unit is often a monomer comprising two NBDs and two TMDs.
Figure 1.
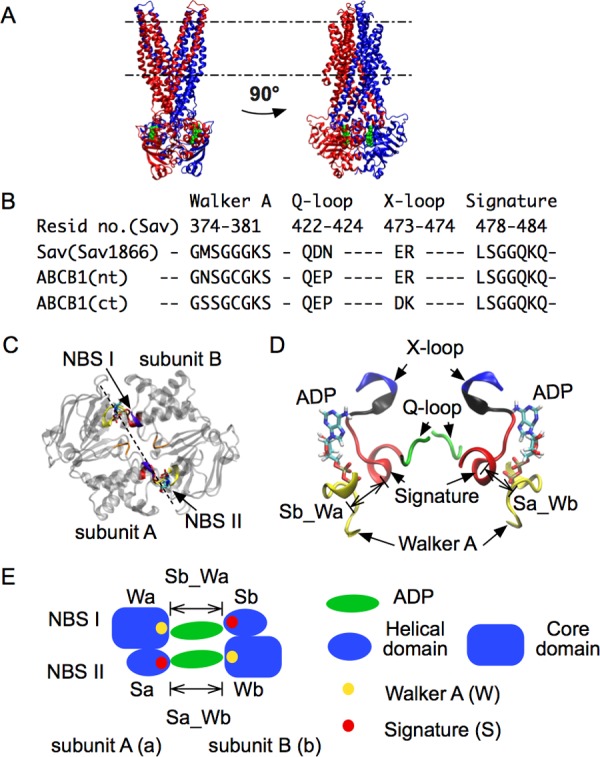
Architecture of ABC exporters. The X-ray structure of Sav1866 (PDB ID: 2HYD) is shown with bound ADP molecules. A: Side views of the whole structure. The two subunits are respectively rendered in blue and red. Membrane boundaries are indicated by dashed lines: upper, extracellular side; lower, intracellular side. ADP is shown in green. B: Sequence alignment showing the conservation of motifs in Sav1866 (Sav) and P-glycoprotein (Pgp, ABCB1). nt, ct: N-terminal and C-terminal side in the heterodimer Pgp (Sav is a homodimer). C: Top view of NBD with motifs highlighted. Two NBDs (subunits A and B) form two nucleotide binding sites, NBS I and NBS II. D: Side view of NBD motifs. The X-loop and the signature motif are connected by a short loop, colored in black. E: Scheme of the two NBSs, highlighting the two binding distances, Sb_Wa and Sa_Wb. Sa, Sb: signature motif from subunit A(a) and B(b). Wa, Wb: Walker A from the two subunits. Sb_Wa: distance between Sb and Wa, at NBS I. Sa_Wb: distance between Sa and Wb, at NBS II.
The most investigated eukaryotic exporter is the monomeric P-glycoprotein (Pgp, ABCB1). It protects cells from intrusion of toxins and drugs, and plays a major role in the acquisition of cellular resistance to antimicrobial and cancer chemotherapeutics.2,3 The protein has been crystallized in the nucleotide free form (apo-form)4,5 revealing a conformation in which the NBDs are far apart from each other. The apo-form of MsbA, a homodimer from Escherichia coli that flops lipopolysaccharides and amphiphilic cations,6 was crystallized in conformations with NBDs either separated or associated, revealing the high flexibility of these transporters.7 The prokaryotic homodimeric Sav1866 (Sav) from Staphylococcus aureus is a homologue of Pgp that has been crystallized in nucleotide-bound conformations, maintaining the NBDs together8,9 (Figure 1A). Although its physiological function is not yet known, Sav has been demonstrated to transport amphiphilic compounds across cell membranes10,11 like Pgp.12,13
The basal and drug stimulated ATP hydrolysis rates of Pgp and Sav can be assessed rather accurately by monitoring either the release of phosphate from inside-out plasma membrane vesicles or reconstituted systems,14 or the release of lactate from live cells.15 Measuring transport is more difficult because allocrites are amphiphilic (comprising a hydrophilic and a hydrophobic moiety). They partition into the lipid membrane, bind to exporters in the cytosolic membrane leaflet,16 and are transported to the extracellular membrane leaflet. As only a fraction of the transported allocrites reaches the extracellular aqueous phase, where the allocrite concentration is monitored (e.g. ref (17)), transport is generally underestimated, particularly if compounds are hydrophobic. In addition, a non-negligible fraction of allocrites escapes the transporter and diffuses across the membrane according to the concentration gradient. Passive diffusion thus tends to reduce net transport in cells and to enhance it in inside-out systems.18 The experimentally determined number of ATP molecules hydrolyzed per allocrite molecule transported therefore varied from one19 to approximately three,20 depending on the systems and the allocrites used. These stoichiometric uncertainties led to different mechanistic transport models.
Senior and co-workers showed that both NBSs of Pgp were catalytically active, and trapping of Mg·ADP with vanadate at just one site resulted in full inhibition of the transporter’s ATPase activity.21−23 It was concluded that the two NBSs cannot function as catalytic sites simultaneously, but must alternate in catalysis (i.e., from a state in which two ATP molecules are bound, only one can hydrolyze), suggesting that one ATP is hydrolyzed per drug transported (“alternating catalytic sites scheme”).23,24 This model was further supported by experiments with the purified maltose transporter complex.25 Ambudkar and co-workers also suggested that transport is coupled to the hydrolysis of one ATP, but assumed that the hydrolysis of a second ATP is required for resetting the transporter.26
With the first low-resolution structures of Pgp in the absence and presence of nucleotides,27 a different model called “alternating switch model” was proposed (for review see ref (28)). It assumes that binding of two ATP molecules at the interface of the NBDs induces an inward-closed/outward-open conformation and hydrolysis of two ATP molecules leads to an inward-open/outward-closed conformation. The model was supported by the high-resolution structure of the homodimeric Sav1866 (Sav), which shows an outward-open conformation in the presence of two AMP-PNP molecules (a poorly hydrolyzable ATP analogue),29 and the structures of apo-Pgp, which show outward-closed/inward-open conformations.4,5 The model with two ATPs hydrolyzed per transport cycle gained further support by experiments with isolated NBDs.30−32 The biochemical analysis of trapped intermediate states of NBDs in the presence of labeled ATP molecules suggested sequential hydrolysis of two ATPs per drug transported subsumed in the “processive-clamp model”.31 This model is compatible with the alternating switch model mentioned above28 but not with the alternating catalytic sites scheme.23,24
Although high-resolution structures have offered great insights on the function of ABC transporters, they only reflect snapshots of the complex catalytic cycle, and it remains to be examined how closely they reflect physiologically relevant conformations. Double electron–electron resonance (DEER) spectroscopy experiments with spin label pairs introduced at strategic locations of the MsbA, BmrCD, and TM287/288 transporters have provided information on the conformation of the NBDs and TMDs under turnover conditions.33,34Table S1 summarizes the conformations provided by X-ray structures and those deduced from DEER experiments for different transporters. These data illustrate the wide range of conformations that are observed but not fully accounted for in currently proposed models. Hence, the hypothesis that association/dissociation of NBDs drives conformational changes in the TMD for drug transport is still open to question. Mechanistic insights at the atomistic level were provided by molecular dynamics simulations based on the high-resolution structure of Sav.35−40 Aittoniemi et al. highlighted the asymmetry of the X-ray structure, notably at the two NBDs, suggesting that only one ATP is hydrolyzed per drug transported.36 In different simulations, nucleotides found in the X-ray structures have been removed,35,38 or replaced by ADP or ATP36,37,39 to investigate their influence on the TMD. Although conformational changes of key loops were observed, no signaling pathway linking the NBD to the TMD has been described yet, and the stoichiometry of ATP hydrolyzed per drug transported still could not be determined conclusively.
To investigate the communication pathway between the NBDs and TMDs, we ran simulations of Sav in seven different nucleotide occupancy states. Interconversions between outward-closed and outward-open conformations of TMD were observed. The conformational change is related to ATP binding and hydrolysis, and communicated to the TMD through a route involving the Q-loop and X-loop. The simulations support that only one ATP is hydrolyzed per transport cycle and provide basis for the unidirectionality of transport in ABC exporters.
Results
Outward-Closed and Outward-Open Conformations of the TMD
Seven different nucleotide occupancy states were set up based on the X-ray structure of Sav: 2ATP, 2ADP, 2apo, ATPI_ADPII, ATPI_apoII, ADPI_ATPII, and apoI_ATPII, indicating that the two NBSs are occupied by ATP or ADP, or are empty (apo). These account for most of the states postulated by two mechanisms under debate, the alternating catalytic sites mechanism,23 and the processive clamp mechanism.31 Both mechanisms assume a state in which two ATP molecules are bound to the exporter. Hydrolysis at one NBS leads to asymmetric occupancy states (ATPI_ADPII, ATPI_apoII, ADPI_ATPII, and apoI_ATPII). From these states, the alternating catalytic sites mechanism predicts that the remaining ATP would not be hydrolyzed, and another ATP would eventually bind to the free site. The processive clamp mechanism rather predicts hydrolysis at the second NBS, eventually leading to the 2ADP state and possibly the 2apo state (the combinations of ADP and apo were not modeled).
The different nucleotide occupancy states lead to either of two possible conformations of the TMD: A first one, similar to the X-ray structure with an outward facing cavity, and a second one in which the cavity has collapsed (Figure 2A). To characterize these two conformations, the volume of the cavity at the level of the outer membrane leaflet (outer cavity) was calculated (Figure 2B). When the two NBSs are found in the same nucleotide occupancy state, i.e., 2ATP, 2ADP, and 2apo (symmetric cases), the volume of the outer cavity is small (less than 200 Å3), indicating that the TMD is in an outward-closed conformation as shown in Figure 2A (right panel). On the other hand, when the two NBSs are in different states, i.e., ATPI_ADPII, ATPI_apoII, ADPI_ATPII, and apoI_ATPII (asymmetric cases), the volume of the outer cavity is larger, in the range of 1000 to 2000 Å3, indicating an outward-open conformation as illustrated in Figure 2A (left panel). The volume of the cavity at the level of the intracellular leaflet (inner cavity, Figure 2C) also varies, but to a lesser extent. The inner cavity in the outward-open conformation is slightly larger (500 to 1000 Å3) than in the outward-closed conformation (<500 Å3). Interestingly, in the outward-closed conformation, particularly in the 2ATP state, the volume of the inner cavity is higher than that of the outer cavity.
Figure 2.
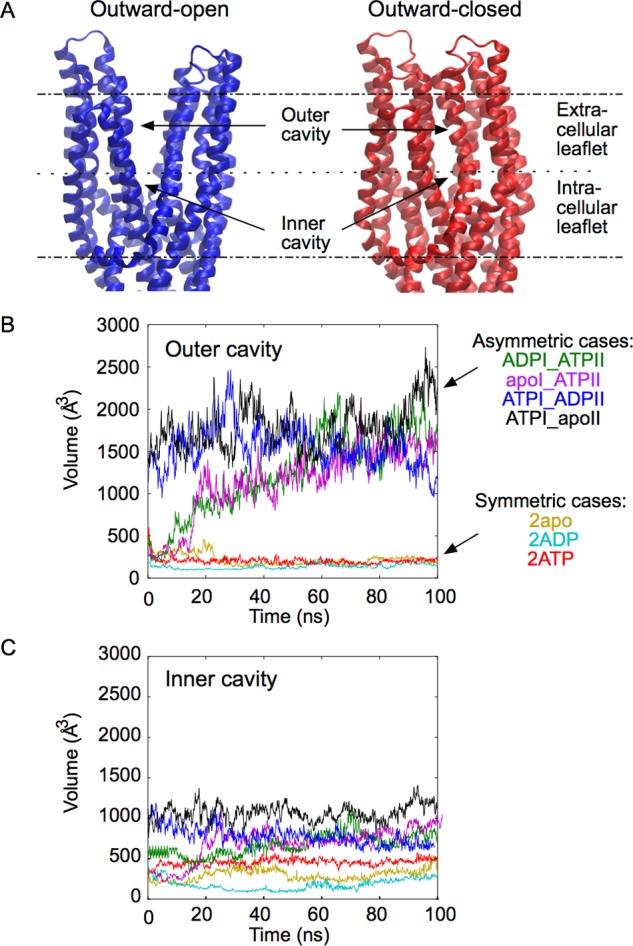
Two different conformations of the TMDs. A: Outward-open and outward-closed conformation of TMDs, taken at t = 100 ns in the simulations of the ATPI_ADPII and 2ATP states, respectively. Membrane boundaries are indicated by dashed lines. B, C: Volume of the cavity formed by the TMDs at the level of the extracellular leaflet (B) and intracellular leaflet (C) for different nucleotide occupancy states.
In the outward-open state, the TMD cavity is hydrated and the polar head of 2 or 3 lipids diffuses into the interstice between helices 1 and 6 of the two subunits. By rotation of helix 1 and helix 6 of both subunits (see Figure S1), the outward-open conformation is converted into an outward-closed conformation, in which the outer segments of these four helices come together to form a hydrophobic core at the level of the extracellular leaflet of the membrane (Figure S2A). Two pairs of residues, Thr276-Asp42 and Thr279-Lys38, form H-bonds between helix 1 and helix 6, stabilizing the closed conformation of the TMD (Figure S2B). Under these conditions water molecules are excluded from the upper leaflet hydrophobic bundle, while the inner cavity, which contains many polar side chains, remains largely hydrated (Figure S2A).
ATP Hydrolysis Induces Perturbation at the NBS
The signature motif and Walker A play a central role in ATP binding. When both NBSs are occupied by ATP (2ATP), the NBS binding distances (i.e., the distance between the Walker A of one subunit and the signature motif from the other subunit) remain quite stable at 9 Å, whereas when ADP replaces ATP at one site, the NBS binding distance increases at the corresponding site (Figure 3A). It is observed that, in the 2ATP state, two hydrogen bonds are formed, one between the beta phosphate of ATP and Lys380 in Walker A, the other between the gamma phosphate and Gly481 in the signature motif, which are also seen in the X-ray structure of MJ0796-E171Q with ATP,41 HlyB-NBD-H662A with Mg-ATP,32 and maltose transporter with AMP-PNP.42 When ATP is replaced by ADP (as after ATP hydrolysis), the second hydrogen bond is missing, and hence the signature motif diffuses away, resulting in the increase of the NBS binding distance. In all occupancy states the distances remain <14 Å (see also Figure S3C). Since the X-loop is directly connected to the signature motif (see Figure 1D), the hydrolysis also impacts on the X-loop conformation. The graphs presented in Figure 3B show that the distances between the X-loop and Walker A are correlated with the NBS binding distance. Ultimately, the hydrolysis of ATP leads to a displacement of about 2 Å at the level of the X-loop, allowing for the formation of new interactions between the X-loop and TMDs. The X-loop residue Arg474 notably destabilizes the H-bond formed by the two Gln208 residues at the bottom of helices 4 (see Figures S4 and S5).
Figure 3.
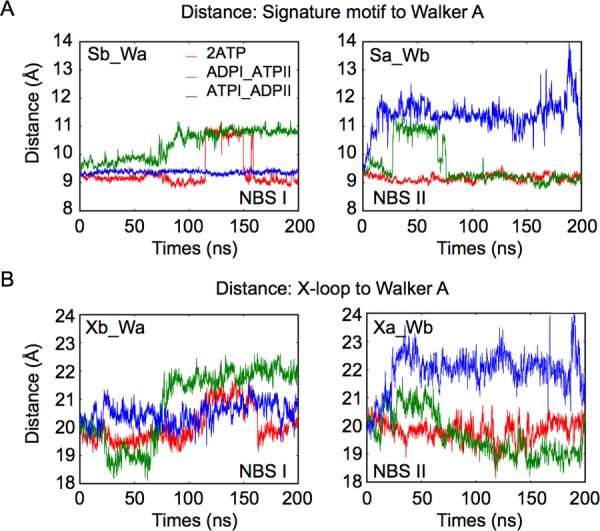
ATP hydrolysis disrupts the nucleotide binding site and allows movement of X-loop. A: Time-series of distance between signature motif and Walker A for different nucleotide occupancy states. Sa, Sb: signature motif from subunit A(a) and B(b) (see Figure 1C). Wa, Wb: Walker A from the two subunits. Sb_Wa: distance between Sb and Wa, at NBS I. Sa_Wb: distance between Sa and Wb, at NBS II. See the scheme in Figure 1E. B: Time-series of distances between X-loop and Walker A. Xa, Xb: X-loop from subunit A(a) and B(b) (see Figure 1C). Xb_Wa: distance between Xb and Wa, at NBS I. Xa_Wb: distance between Xa and Wb, at NBS II.
NBS Perturbations Impact on the Cohesion of Helices 3 and 4
Next, we investigated the relation between the disruption of the H-bond between the two Gln208 residues and the TMD conformational changes. We first calculated the distance between the backbone center-of-mass of the two Gln208 residues (Figures 4 and S6). When two ATPs are bound, the distance between the two Gln208 is approximately 13 Å. When ADP replaces one of the ATP molecules, the distance increases to about 15 Å (Figure 4B), which implies that the two helices 4 are moving away from each other, as illustrated by a superposition of structures taken from the simulations of the 2ATP and ATPI_ADPII states at t = 100 ns (Figure 4A,C–D). The plots in Figure 4E show that the distance between residues Gly118 and Gly201 also increases, indicating that the separation between helix 3 and helix 4 becomes larger. The increase of the separation distance between Gly118 and Gly201 is larger than that between the two Gln208 residues, suggesting that the rupture of the Gln208–Gln208 interaction leads to even larger changes along helix 3 and helix 4, and might contribute to the opening of the TMD toward the extracellular side.
Figure 4.
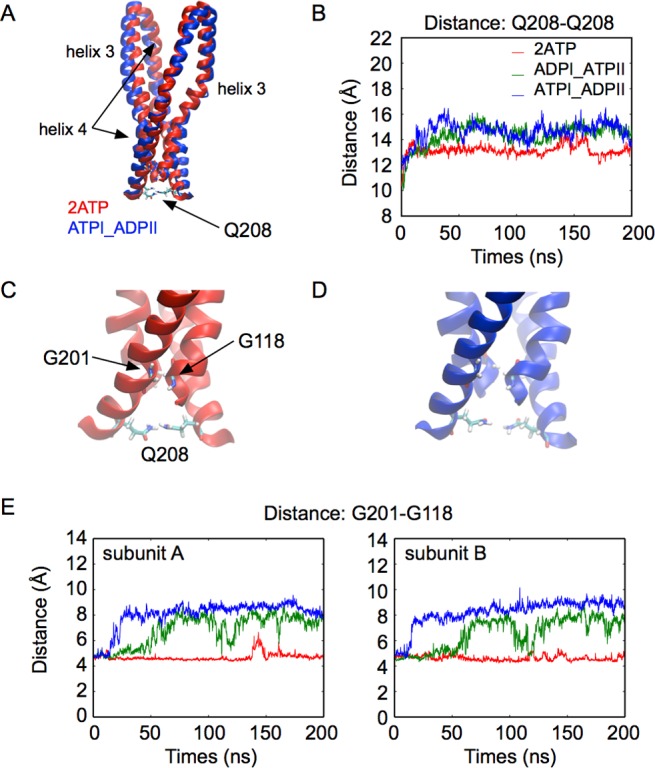
ATP hydrolysis at one binding site influences helix 3 and helix 4 in both subunits. A: Helix 3 and helix 4 in a superposition of snapshots taken at t = 100 ns in simulations of the 2ATP (red) and ATPI_ADPII (blue) states. B: Time-series of the distance between the Q208 backbone center-of-mass of the two subunits. C, D: Close-up view of the bottom of helix 3 and helix 4. (C: 2ATP. D: ATPI_ADPII.) E: Time-series of the distance between G118 and G201.
As seen in Figure 5A, helix 3 and helix 4 form a hairpin and are kinked at the level of residues Gly138 and Gly183, which are found near the intracellular membrane–water interface. These kink angles (Figure 5B,C, Figure S7), particularly that in helix 4 induced by residue Gly183, are strongly correlated to the NBD occupancy state. The plots in Figure 5C show that the kink angle in helix 4 is maximized when 2 ATPs are bound to the NBSs. After hydrolysis at either NBS, the kink angle of helix 4 is reduced in both subunits (see also Figure 5A, right panel). A straight helix 4 involves that the hairpin formed by helices 3 and 4 orients away from the central cavity (Figure 6A), while a kinked helix 4 implies an inward displacement of the hairpin (Figure 6B). Thus, the presence of these kinks at these specific positions provides structural flexibility that allows the hairpin formed by helices 3 and 4 to move toward the central cavity when two ATPs are bound, corresponding to the outward-closed conformation, and away from it when one ATP is hydrolyzed, corresponding to the outward-open conformation (Figure 6A,B). Similar kinks, resulting from the presence of glycine or proline residues, are notably observed in helix 4 of the outward-closed structures of the related ABC exporters McjD43 and PglK.44
Figure 5.
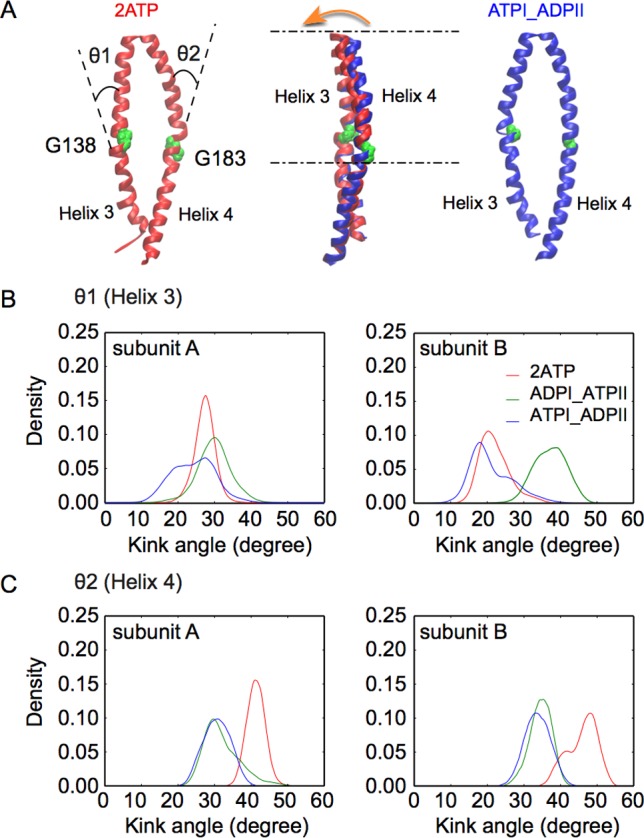
Kink angle in helix 4 characterizes the outward-closed and outward-open conformations of the TMD. A: Comparison of the helix 3/helix 4 hairpin in the outward-closed (red) and outward-open (blue) conformations. Gly138 and Gly183 allow for a kink in the middle of helix 3 and helix 4, respectively. The orange arrow points toward the TMD central cavity. B, C: Density histogram of the kink angle for different nucleotide occupancy states. (B: θ1 in helix 3. C: θ2 in helix 4.)
Figure 6.
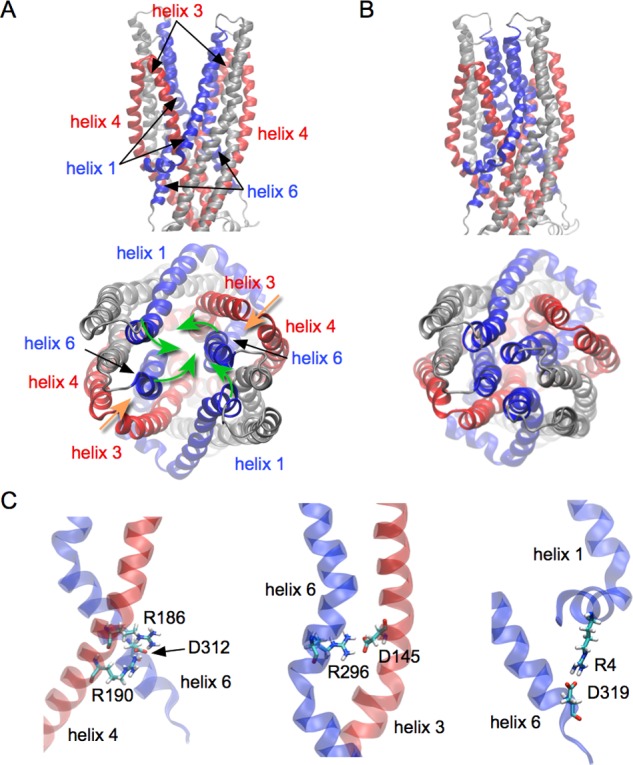
A network formed by helix 1, helix 3, helix 4, and helix 6. A: Conformations of the four helices in the outward-open conformation. (Up: side view. Down: top view.) B: Conformations of the four helices in the outward-closed conformation (Up: side view. Down: top view.) Rotation of helix 1 and helix 6 (green arrows) is central to the transition from the outward-open conformation to the outward-closed conformation. The hairpin formed by helix 3 and helix 4 moves together with helix 1 and helix 6 (orange arrows). C: Stable salt-bridges contribute to the mechanical coherence between the different helices. (Left: between helix 4 and helix 6. Middle: between helix 3 and helix 6. Right: between helix 1 and helix 6.) For clarity, helix 1 and helix 6 are highlighted in blue, while helix 3 and helix 4 are in red.
In addition to the interactions between helices 3 and 4 described above, there is a network of salt-bridges linking helices 1, 3, 4, and 6: Arg186 and Arg190 from helix 4 form salt-bridges with Asp312 from helix 6 (Figure 6C, left); helix 3 and helix 6 are linked by a salt-bridge between Asp145 and Arg296 (Figure 6C, center); helix 1 and helix 6 are connected through a salt-bridge formed by Arg4 and Asp319 (Figure 6C, right). Time-series analyses show that these salt-bridges are stable along the simulations (Figure S8), suggesting that such a network could contribute to the transmission of the conformational changes among these helices. Whether the movement of the hairpin formed by helices 3 and 4 induces or is a consequence of the movement of helices 1 and 6 remains to be understood.
Discussion
Structural data of ABC exporters suggest a wide range of conformations (Table S1) that need to be unambiguously associated with functional states. Our simulations of Sav1866 under different nucleotide occupancy states provide a novel signaling route between the NBDs and TMDs at the atomistic level. Conformational asymmetries of the NBDs appear to be central for the regulation of binding and hydrolysis of ATP.45,46 Our simulations further reveal that the release of the signature motif following ATP hydrolysis at only one of the two NBSs is a central process in the communication between the NBDs and TMDs (Figure 3). The displacement of the signature motif is correlated with the displacement of the X-loop, which perturbs the tetrahelix bundle formed by helices 3 and 4, and in turn impacts on the conformation of the TMD. Such a signaling route is supported by the fact that mutation of charged residues at the tetrahelix bundle formed by helices 3 and 4 in another ABC exporter, MsbA, reduces transport activity, and was thought to impair conformational changes of the TMDs.47
Departing from the outward-open X-ray structure of Sav with two nucleotides bound, the simulations of the 2ATP state show a tendency toward an outward-closed conformation. This conformation is coherent with the X-ray structures of McjD, another bacterial homodimeric transporter, in complex with AMP-PNP,48 and with cross-link studies of Pgp that suggest close interaction of helices 6 and 12 in the prehydrolysis state (i.e., in the presence of 2 ATP or 2 AMP-PNP molecules).49−51 The Sav outward-closed conformation, also observed in the 2ADP simulation, is reminiscent of the outward-occluded X-ray structure of PglK, an ATP dependent flippase, in complex with ADP.44 Overall, the conformations observed in our MD simulations for given nucleotide occupancy states agree well with the conformational state distributions revealed by X-ray crystallography and DEER spectroscopy of different homodimeric and heterodimeric transporters (Table S1).
In the simulations, the spontaneous closure of the TMD cavity, which remains closed in the case of symmetric nucleotide occupancy states and reopens in the case of asymmetric occupancy states (Figure 2B), suggests that the Sav conformation observed in the X-ray structure might correspond to a transition state. Key interactions between helices 3 and 4 that are shown in our simulations to stabilize the outward-closed state are present in the X-ray structures, which nevertheless show the TMD in an outward-open state. In the crystal of Sav, the TMDs from neighboring proteins form contacts that could artificially stabilize the outward-open conformation (Figure S9). Variation of crystal contacts under different crystallization conditions have been suggested to explain the wide distribution of conformations observed in ABC transporter.7−9,44 The detergent octaethylene glycol monododecyl ether (C12E8), which was used in the crystallization of Sav, was demonstrated to be an allocrite for Sav11 and Pgp,52 and could also contribute to the stabilization of the outward-open conformation. Our simulations further suggest that lipids diffusing into the TMD cavity could stabilize the outward-open conformation.
Interestingly, the H-bonds between helix 1 and helix 6, which stabilize the outward-closed conformation, are reminiscent of H-bonds seen in the inward-open crystal structures of CmABCB1 (PDB ID: 3WME)53 and TM287–288 (PDB ID: 4Q4A).54 The fact that the hydrophobic bundle formed by helices 1 and 6 is found exclusively at the level of the extracellular leaflet of the membrane represents a key feature. It implies that, upon closure of the TMD, allocrites found in the outer cavity would be squeezed out toward the extracellular leaflet of the membrane, while allocrites in the inner cavity would be less affected, creating an asymmetry between the two leaflets and potentially giving rise to a directional transport mechanism.
We thus propose a unidirectional model of active transport in ABC exporters (Figure 7), comprising the outward-closed conformation (states 1 and 2) and the outward-open conformation (states 3 and 4). Given that the concentration of ATP in cells is typically in the millimolar range, CATP = 1–10 mM,55,56 and that its dissociation constant is in the low millimolar range for Sav1866 and in the micromolar range for Pgp,11 the apo state in either binding site is considered to be transient, and hence is not explicitly included in the model. A corollary is that the outward-closed conformation in the 2ADP state is unlikely to occur in the transport cycle and is thus not included as well. (1) State 1 shows an outward-closed conformation with two ATP bound, allowing allocrites partial access to the inner cavity. (2) ATP hydrolysis at one NBS removes key electrostatic interactions and favors an outward-open conformation, reached by stochastic diffusion (and potentially accelerated by allocrite binding). (3) With the opening of the TMDs, allocrites can flop between the inner and outer cavities. (4) As the cytosolic ATP concentration is high, a new ATP binds as soon as the hydrolysis product ADP is released. The electrostatic interactions between ATP and notably the signature motif stabilize the NBD and remove the perturbations at the TMD, thus favoring the outward-closed conformation. Allocrites that have not left the outer cavity at this stage are squeezed out toward the extracellular leaflet of the membrane, generating an outward flow. This model features TMD opening/closing without full dissociation of the NBDs and supports a unidirectional transport of allocrites.
Figure 7.
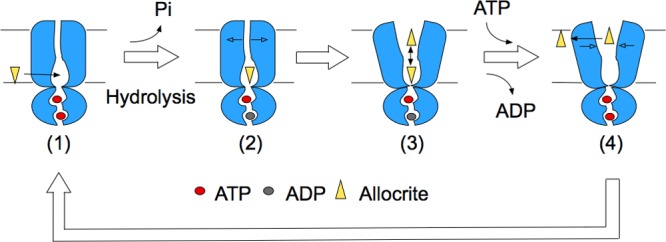
Unidirectional transport cycle in ABC exporters. (1) With two ATP bound to the NBDs, the TMDs are in an outward-closed state. (2) Hydrolysis of ATP and release of the inorganic phosphate (Pi) leads to an asymmetric occupation state of the NBDs, which initiates the opening of the TMDs. (3) The TMDs adopt an outward-open conformation, which allows water molecules to fill in and leaves room for allocrite flopping. (4) The binding of ATP to the empty NBS restores the symmetric occupancy state and favors the outward-closed conformation, which leads the allocrites on the extracellular side of the cavity between the TMDs to be squeezed out to the membrane.
The present data are consistent with the early model proposed by Senior and co-workers,23,24 suggesting transport of one allocrite per one ATP hydrolyzed, and provide the first mechanistic insight into this process. However, under conditions of fast ATP rebinding, our model suggests that hydrolysis at the NBSs is rather a stochastic than a strictly alternating process. The fact that hydrolysis takes place at only one NBS involves that the NBDs remain closely associated through the transport cycle, in opposition to the alternating access mechanism. Models in which the NBDs remain in contact were recently proposed for flipping of allocrites by the heterodimeric BmrCD33 and of lipid-linked oligosaccharides by the homodimeric PglK.44 While our model and the PglK model proposed by Perez et al. agree on the conformations adopted by the transporter throughout the transport cycle, our model brings rationale to support that hydrolysis takes place at only one NBS and not at both as the PglK model proposes.
In conclusion, our simulations revealed two stable states of the Sav1866 exporters: outward-closed when two ATP molecules are bound and outward-open when hydrolysis has taken place at one NBS. The structural differences between the two states suggest a putative communication pathway establishing a link between the nucleotide binding state at the NBD and the conformation of the TMD, allowing for a unidirectional transport mechanism. The driving force provided by ATP binding is thought to favor key interactions on the intracellular side of the TMD, but it remains unclear how such a driving force is propagated to the outermost segments of the TMD. Reciprocally, the opening of the TMD is thought to follow ATP hydrolysis at a single NBS and to arise from stochastic diffusion. The model provides a framework to address important remaining questions notably on the selectivity and transport of allocrites, as well as on the role played by allocrite binding in the regulation of ATP hydrolysis.
Methods
System Preparations
In order to explore the conformational changes of the Pgp and Sav structures, different molecular systems were assembled using the CHARMM-GUI web service.57,58 A first system was set up based on the crystal structure of apo-Pgp (PDB ID: 3G5U). Two Sav1866 structures are available (PDB ID: 2HYD, 2ONJ). We chose the one solved with two ADP molecules bound to its nucleotide-binding domain (PDB ID: 2HYD), which has a higher resolution (3.0 Å). Seven different systems of Sav were set up with different nucleotide occupancy states: 2ATP, 2ADP, 2apo, ATPI_ADPII, ATPI_apoII, ADPI_ATPII, and apoI_ATPII. The first three labels correspond to symmetric nucleotide binding states, while the latter four cases correspond to asymmetric states, i.e., the two binding sites are in different states. The binding site constituted by Walker A of subunit A is called NBS I, while the one constituted by Walker A of subunit B is called NBS II.
In all the systems with nucleotides bound, a Mg2+ ion is included next to each nucleotide. ADP coordinates are directly taken from the Sav structure with ADP.8 The positions of ATP and Mg2+ are determined by alignment of the NBD with the X-ray structure of MJ0796-E171Q (PDB: 1L2T),41 which includes ATP molecules and sodium ions at positions where magnesium ions should be found.
The TMDs of Pgp and Sav are embedded in a lipid bilayer composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid molecules. The membrane is positioned according to OPM (orientation of proteins in membrane database). The systems were solvated by about 24300 water molecules, and potassium/chloride ions were added to neutralize the systems and mimic a 150 mM KCl solution.
Molecular Simulations
The membrane systems were first equilibrated with CHARMM v3659 according to a protocol provided by CHARMM-GUI57,58 using the CHARMM36 force field.60 Productions were carried out using NAMD 2.961 with the same force field. All the systems were set up under periodic boundary conditions in an NPT ensemble. The cutoff for both the Lennard-Jones and Coulomb interactions was set to 12 Å, with a switching function beginning at 10 Å. The nonbonded pair list, which includes all the pairs within 16 Å, was updated every 10 steps. Long-ranged electrostatic interactions were calculated using the particle mesh Ewald (PME) method with an interpolation order of 6. Langevin dynamics was used on non-hydrogen atoms to maintain the temperature at 323.15 K with a damping coefficient of 1/ps, whereas pressure was maintained at 1 atm using a Nosé–Hoover Langevin piston with an oscillation period of 50 fs and an oscillation decay time of 25 fs. All bonds involving hydrogen atoms are constrained using the SHAKE algorithm. The time step for all simulations was 2 fs.
Structural Analysis
OpenStructure62 was used for most of the analyses of the conformational changes along the simulation trajectories. The NBS “binding distance” was defined as the distance between the signature motif (residues 478 to 482) from one NBD and Walker A (residues 374 to 381) from the other NBD. The X-loop consists of residues 473 and 474 as shown in Figure 1B. All the distances are calculated using the center-of-mass of the backbone of the involved residues. The helical segments used for the analyses of conformational changes of helices 1, 3, 4, and 6 are shown in Tables S2 and S3.
HOLE63 was used to calculate the radius along the cavity of the TMD after the protein structures taken from the simulations were aligned to the Sav1866 X-ray structure (PDB ID: 2HYD) using VMD.64 The volume of the TMD cavity was calculated based on the radii obtained for different positions across the membrane.
Acknowledgments
S.B. acknowledges support by the Swiss Foundation for Excellence and Talent in Biomedical Research, the Swiss National Science Foundation (SNF Professorship No. PP00P3_139205), and the FP 7 European Union Human Brain Project (Grant No. 604102). Calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing core facility at University of Basel and at the Swiss National Supercomputing Centre (CSCS) under project ID s421. Y.X. acknowledges support from the China Scholarship Council and the Novartis-University of Basel Excellence Scholarship for Life Sciences program.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00068.
Complementary structural analyses of the molecular dynamic simulations, and a summary table of experimental conformations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Holland I. B.; Blight M. A. ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol. 1999, 293, 381–399. 10.1006/jmbi.1999.2993. [DOI] [PubMed] [Google Scholar]
- Szakács G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discovery 2006, 5, 219–234. 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Wong K.; Ma J.; Rothnie A.; Biggin P. C.; Kerr I. D. Towards understanding promiscuity in multidrug efflux pumps. Trends Biochem. Sci. 2014, 39, 8–16. 10.1016/j.tibs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jaimes K. F.; Aller S. G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckford P. D.; Sharom F. J. Functional characterization of Escherichia coli MsbA: interaction with nucleotides and substrates. J. Biol. Chem. 2008, 283, 12840–12850. 10.1074/jbc.M708274200. [DOI] [PubMed] [Google Scholar]
- Ward A.; Reyes C. L.; Yu J.; Roth C. B.; Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19005–19010. 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R. J.; Locher K. P. Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- Dawson R. J.; Locher K. P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007, 581, 935–938. 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- Velamakanni S.; Yao Y.; Gutmann D. A.; van Veen H. W. Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus. Biochemistry 2008, 47, 9300–9308. 10.1021/bi8006737. [DOI] [PubMed] [Google Scholar]
- Beck A.; Aänismaa P.; Li-Blatter X.; Dawson R.; Locher K.; Seelig A. Sav1866 from Staphylococcus aureus and P-glycoprotein: similarities and differences in ATPase activity assessed with detergents as allocrites. Biochemistry 2013, 52, 3297–3309. 10.1021/bi400203d. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Pastan I.; Ambudkar S. V. P-glycoprotein and multidrug resistance. Curr. Opin. Genet. Dev. 1996, 6, 610–617. 10.1016/S0959-437X(96)80091-8. [DOI] [PubMed] [Google Scholar]
- Li-Blatter X.; Beck A.; Seelig A. P-glycoprotein-ATPase modulation: the molecular mechanisms. Biophys. J. 2012, 102, 1383–1393. 10.1016/j.bpj.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkadi B.; Price E. M.; Boucher R. C.; Germann U. A.; Scarborough G. A. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem. 1992, 267, 4854–4858. [PubMed] [Google Scholar]
- Nervi P.; Li-Blatter X.; Aänismaa P.; Seelig A. P-glycoprotein substrate transport assessed by comparing cellular and vesicular ATPase activity. Biochim. Biophys. Acta, Biomembr. 2010, 1798, 515–525. 10.1016/j.bbamem.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Ling V. Extraction of Hoechst 33342 from the cytoplasmic leaflet of the plasma membrane by P-glycoprotein. Eur. J. Biochem. 1997, 250, 122–129. 10.1111/j.1432-1033.1997.00122.x. [DOI] [PubMed] [Google Scholar]
- Omote H.; Al-Shawi M. K. A novel electron paramagnetic resonance approach to determine the mechanism of drug transport by P-glycoprotein. J. Biol. Chem. 2002, 277, 45688–45694. 10.1074/jbc.M206479200. [DOI] [PubMed] [Google Scholar]
- Seelig A. The role of size and charge for blood-brain barrier permeation of drugs and fatty acids. J. Mol. Neurosci. 2007, 33, 32–41. 10.1007/s12031-007-0055-y. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Ling V. Stoichiometry of coupling of rhodamine 123 transport to ATP hydrolysis by P-glycoprotein. Eur. J. Biochem. 1998, 254, 189–193. 10.1046/j.1432-1327.1998.2540189.x. [DOI] [PubMed] [Google Scholar]
- Ambudkar S. V.; Cardarelli C. O.; Pashinsky I.; Stein W. D. Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein. J. Biol. Chem. 1997, 272, 21160–21166. 10.1074/jbc.272.34.21160. [DOI] [PubMed] [Google Scholar]
- Urbatsch I. L.; Sankaran B.; Bhagat S.; Senior A. E. Both P-glycoprotein nucleotide-binding sites are catalytically active. J. Biol. Chem. 1995, 270, 26956–26961. 10.1074/jbc.270.45.26956. [DOI] [PubMed] [Google Scholar]
- Urbatsch I. L.; Sankaran B.; Weber J.; Senior A. E. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 1995, 270, 19383–19390. 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- Senior A. E.; al-Shawi M. K.; Urbatsch I. L. The catalytic cycle of P-glycoprotein. FEBS Lett. 1995, 377, 285–289. 10.1016/0014-5793(95)01345-8. [DOI] [PubMed] [Google Scholar]
- Tombline G.; Muharemagić A.; White L. B.; Senior A. E. Involvement of the ″occluded nucleotide conformation″ of P-glycoprotein in the catalytic pathway. Biochemistry 2005, 44, 12879–12886. 10.1021/bi0509797. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Davidson A. L. Vanadate-induced trapping of nucleotides by purified maltose transport complex requires ATP hydrolysis. J. Bacteriol. 2000, 182, 6570–6576. 10.1128/JB.182.23.6570-6576.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauna Z. E.; Smith M. M.; Müller M.; Kerr K. M.; Ambudkar S. V. The mechanism of action of multidrug-resistance-linked P-glycoprotein. J. Bioenerg. Biomembr. 2001, 33, 481–491. 10.1023/A:1012875105006. [DOI] [PubMed] [Google Scholar]
- Rosenberg M. F.; Velarde G.; Ford R. C.; Martin C.; Berridge G.; Kerr I. D.; Callaghan R.; Schmidlin A.; Wooding C.; Linton K. J.; Higgins C. F. Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 2001, 20, 5615–5625. 10.1093/emboj/20.20.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins C. F.; Linton K. J. The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 2004, 11, 918–926. 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- Dawson R. J.; Hollenstein K.; Locher K. P. Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol. Microbiol. 2007, 65, 250–257. 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- Chen J.; Lu G.; Lin J.; Davidson A. L.; Quiocho F. A. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell 2003, 12, 651–661. 10.1016/j.molcel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Janas E.; Hofacker M.; Chen M.; Gompf S.; van der Does C.; Tampé R. The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J. Biol. Chem. 2003, 278, 26862–26869. 10.1074/jbc.M301227200. [DOI] [PubMed] [Google Scholar]
- Zaitseva J.; Jenewein S.; Jumpertz T.; Holland I. B.; Schmitt L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 2005, 24, 1901–1910. 10.1038/sj.emboj.7600657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S.; Verhalen B.; Stein R. A.; Wen P. C.; Tajkhorshid E.; Mchaourab H. S. Conformational dynamics of the nucleotide binding domains and the power stroke of a heterodimeric ABC transporter. eLife 2014, 3, e02740. 10.7554/eLife.02740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timachi M. H.; Hutter C. A.; Hohl M.; Assafa T.; Böhm S.; Mittal A.; Seeger M. A.; Bordignon E. Exploring conformational equilibria of a heterodimeric ABC transporter. eLife 2017, 6, e20236. 10.7554/eLife.20236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J. P.; Van Bambeke F.; Tulkens P. M.; Prévost M. Dynamics and structural changes induced by ATP binding in SAV1866, a bacterial ABC exporter. J. Phys. Chem. B 2010, 114, 15948–15957. 10.1021/jp1038392. [DOI] [PubMed] [Google Scholar]
- Aittoniemi J.; de Wet H.; Ashcroft F. M.; Sansom M. S. Asymmetric switching in a homodimeric ABC transporter: a simulation study. PLoS Comput. Biol. 2010, 6, e1000762. 10.1371/journal.pcbi.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A. S.; Baptista A. M.; Soares C. M. Conformational changes induced by ATP-hydrolysis in an ABC transporter: a molecular dynamics study of the Sav1866 exporter. Proteins: Struct., Funct., Genet. 2011, 79, 1977–1990. 10.1002/prot.23023. [DOI] [PubMed] [Google Scholar]
- Jones P. M.; George A. M. Molecular-dynamics simulations of the ATP/apo state of a multidrug ATP-binding cassette transporter provide a structural and mechanistic basis for the asymmetric occluded state. Biophys. J. 2011, 100, 3025–3034. 10.1016/j.bpj.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyimesi G.; Ramachandran S.; Kota P.; Dokholyan N. V.; Sarkadi B.; Hegedus T. ATP hydrolysis at one of the two sites in ABC transporters initiates transport related conformational transitions. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 2954–2964. 10.1016/j.bbamem.2011.07.038. [DOI] [PubMed] [Google Scholar]
- St-Pierre J. F.; Bunker A.; Róg T.; Karttunen M.; Mousseau N. Molecular dynamics simulations of the bacterial ABC transporter SAV1866 in the closed form. J. Phys. Chem. B 2012, 116, 2934–2942. 10.1021/jp209126c. [DOI] [PubMed] [Google Scholar]
- Smith P. C.; Karpowich N.; Millen L.; Moody J. E.; Rosen J.; Thomas P. J.; Hunt J. F. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 2002, 10, 139–149. 10.1016/S1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M. L.; Chen J. Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 15152–15156. 10.1073/pnas.1108858108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury H. G.; Tong Z.; Mathavan I.; Li Y.; Iwata S.; Zirah S.; Rebuffat S.; van Veen H. W.; Beis K. Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9145–9150. 10.1073/pnas.1320506111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C.; Gerber S.; Boilevin J.; Bucher M.; Darbre T.; Aebi M.; Reymond J. L.; Locher K. P. Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature 2015, 524, 433–438. 10.1038/nature14953. [DOI] [PubMed] [Google Scholar]
- Siarheyeva A.; Liu R.; Sharom F. J. Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: implications for catalysis. J. Biol. Chem. 2010, 285, 7575–7586. 10.1074/jbc.M109.047290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aittoniemi J.; de Wet H.; Ashcroft F. M.; Sansom M. S. Asymmetric switching in a homodimeric ABC transporter: a simulation study. PLoS Comput. Biol. 2010, 6, e1000762. 10.1371/journal.pcbi.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi R.; Ali A.; Shi W.; Freeman E. V.; Fagg L. A.; van Veen H. W. Molecular disruption of the power stroke in the ATP-binding cassette transport protein MsbA. J. Biol. Chem. 2013, 288, 6801–6813. 10.1074/jbc.M112.430074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury H. G.; Tong Z.; Mathavan I.; Li Y.; Iwata S.; Zirah S.; Rebuffat S.; van Veen H. W.; Beis K. Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9145–9150. 10.1073/pnas.1320506111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Drug-stimulated ATPase activity of human P-glycoprotein requires movement between transmembrane segments 6 and 12. J. Biol. Chem. 1997, 272, 20986–20989. 10.1074/jbc.272.34.20986. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. Nucleotide binding, ATP hydrolysis, and mutation of the catalytic carboxylates of human P-glycoprotein cause distinct conformational changes in the transmembrane segments. Biochemistry 2007, 46, 9328–9336. 10.1021/bi700837y. [DOI] [PubMed] [Google Scholar]
- Stockner T.; de Vries S. J.; Bonvin A. M.; Ecker G. F.; Chiba P. Data-driven homology modelling of P-glycoprotein in the ATP-bound state indicates flexibility of the transmembrane domains. FEBS J. 2009, 276, 964–972. 10.1111/j.1742-4658.2008.06832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Blatter X.; Nervi P.; Seelig A. Detergents as intrinsic P-glycoprotein substrates and inhibitors. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 2335–2344. 10.1016/j.bbamem.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Kodan A.; Yamaguchi T.; Nakatsu T.; Sakiyama K.; Hipolito C. J.; Fujioka A.; Hirokane R.; Ikeguchi K.; Watanabe B.; Hiratake J.; Kimura Y.; Suga H.; Ueda K.; Kato H. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 4049–4054. 10.1073/pnas.1321562111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl M.; Hürlimann L. M.; Böhm S.; Schöppe J.; Grütter M. G.; Bordignon E.; Seeger M. A. Structural basis for allosteric cross-talk between the asymmetric nucleotide binding sites of a heterodimeric ABC exporter. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 11025–11030. 10.1073/pnas.1400485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beis I.; Newsholme E. A. The contents of adenine nucleotides, phosphagens and some glycolytic intermediates in resting muscles from vertebrates and invertebrates. Biochem. J. 1975, 152, 23–32. 10.1042/bj1520023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataullakhanov F. I.; Vitvitsky V. M. What determines the intracellular ATP concentration. Biosci. Rep. 2002, 22, 501–511. 10.1023/A:1022069718709. [DOI] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Lee J.; Cheng X.; Swails J. M.; Yeom M. S.; Eastman P. K.; Lemkul J. A.; Wei S.; Buckner J.; Jeong J. C.; Qi Y.; Jo S.; Pande V. S.; Case D. A.; Brooks C. L.; MacKerell A. D.; Klauda J. B.; Im W. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L.; Mackerell A. D.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E.; Mittal J.; Feig M.; Mackerell A. D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M.; Schmidt T.; Bienert S.; Mariani V.; Studer G.; Haas J.; Johner N.; Schenk A. D.; Philippsen A.; Schwede T. OpenStructure: an integrated software framework for computational structural biology. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 701–709. 10.1107/S0907444913007051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O. S.; Neduvelil J. G.; Wang X.; Wallace B. A.; Sansom M. S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graphics 1996, 14, 354–360. 10.1016/S0263-7855(97)00009-X. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.