

Abstract
Metal aquo ions occupy central roles in all equilibria that define metal complexation in natural environments. These complexes are used to establish thermodynamic metrics (i.e., stability constants) for predicting metal binding, which are essential for defining critical parameters associated with aqueous speciation, metal chelation, in vivo transport, and so on. As such, establishing the fundamental chemistry of the actinium(III) aquo ion (Ac-aquo ion, Ac(H2O)x3+) is critical for current efforts to develop 225Ac [t1/2 = 10.0(1) d] as a targeted anticancer therapeutic agent. However, given the limited amount of actinium available for study and its high radioactivity, many aspects of actinium chemistry remain poorly defined. We overcame these challenges using the longer-lived 227Ac [t1/2 = 21.772(3) y] isotope and report the first characterization of this fundamentally important Ac-aquo coordination complex. Our X-ray absorption fine structure study revealed 10.9 ± 0.5 water molecules directly coordinated to the AcIII cation with an Ac–OH2O distance of 2.63(1) Å. This experimentally determined distance was consistent with molecular dynamics density functional theory results that showed (over the course of 8 ps) that AcIII was coordinated by 9 water molecules with Ac–OH2O distances ranging from 2.61 to 2.76 Å. The data is presented in the context of other actinide(III) and lanthanide(III) aquo ions characterized by XAFS and highlights the uniqueness of the large AcIII coordination numbers and long Ac–OH2O bond distances.
Short abstract
The actinium aquo complex has been characterized using Ac L3-edge X-ray absorption spectroscopy and molecular dynamics density functional theory.
Introduction
Because metal aquo ions, M(H2O)xn+, are both ubiquitous and chemically important, their structures and chemical properties serve as fundamental benchmarks in exploring trends across the periodic table. Characterization of aqueous speciation provides a foundation for advances throughout chemistry and biology. For example, understanding the chemistry of metal aquo ions is essential for solving technical problems relevant to biomedical applications, metal ions in the environment, extraction, food chemistry, and so on. In this sense, metal aquo ions occupy central roles in all chemical equilibria that define complexation properties of a metal by a particular ligand in aqueous media (eq 1).
| 1 |
Historically, one of the first critical steps in characterizing the chemical behavior of any element involved establishing its aqueous coordination chemistry. These results provided a foundation for determining critical metrics (i.e., stability constants) and metal solution behavior (i.e., complexation, precipitation, etc.) that enabled predictive capability for metal binding affinity. These days, the aquo ion identities and behaviors for many elements are taken for granted, as they have been well established for decades.
Motivated by the recent global efforts to exploit radioactive decay from 225Ac as a promising anticancer therapeutic agent, we set out to explore actinium’s chemical binding properties in support of chelator design.1,2 Unfortunately, an insufficient understanding of actinium coordination chemistry has considerably hindered development of an appropriate actinium chelator.1 As an example, even something as fundamental as the actinium aquo ion, Ac(H2O)x3+ (referred hereafter as Ac-aquo), remains poorly defined. Closing this gap is one of the first steps toward establishing thermodynamic data needed for predicting actinium behavior in biologically relevant media.
Gathering experimental information about actinium is difficult, primarily because all of its isotopes are highly radioactive. The most stable actinium isotopes—225Ac and 227Ac—have very short half-lives (t1/2) of 10.0(1) d and 21.772(3) y,3 respectively. Additionally, only very small quantities of these isotopes are available for research. As a result, many basic properties associated with actinium have yet to be defined. For instance, consider that the first actinium bond distance was not measured until 2016.4 Perhaps the most well-defined aspect of actinium chelation chemistry is the realization that actinium’s affinities for binding certain donor atoms are difficult to predict.5 This deficiency, as well as the implications of the large actinium ionic radius on chelation, severely hampers ligand design efforts for stabilizing actinium in targeted α-therapy applications.
Herein, we overcame the sample handling and spectroscopic obstacles associated with studying the actinium(III) (5f0 6d0) ion in aqueous media and report the first characterization of arguably the most fundamentally important actinium coordination complex, namely, the Ac-aquo ion, Ac(H2O)x3+. In this study, we made use of X-ray absorption fine structure (XAFS) spectroscopy and molecular dynamics density functional theory (MD-DFT) calculations to evaluate actinium speciation in dilute triflic acid. These data were compared to previous actinium XAFS studies and presented in the context of what is currently understood for the other actinide(III) aquo ions.4,6−16 Overall, the results highlight the uniqueness of the AcIII ion, most notably in terms of the large coordination numbers and very long Ac–OH2O bond distance.
Results and Discussion
Sample Preparation
The synthesis of the Ac-aquo ion was inspired from methodology developed previously for other actinides (Scheme 1).4,13,17−22 In general, the three step procedure involved (1) chemical and radiochemical purification to generate an 227Ac stock solution, (2) preparation of the Ac-aquo ion, and (3) recovery of the valuable AcIII reagent. The procedure started by chemically and radiochemically purifying a sample of actinium—recovered from a previous scientific campaign—using a combination of anion exchange (AG1-X8) and liquid/liquid extraction chromatography (branched DGA; Eichrom). In the course of our studies we found it important to add a cation exchange column to further purify AcIII from organic contaminants that followed through the anion exchange and liquid/liquid extraction steps. Details associated with the first anion exchange and liquid/liquid extraction columns have been described previously.4 In the cation exchange procedure, AcIII dissolved in dilute nitric acid (HNO3; 0.05 M) was loaded onto an AG50W-X8 resin. Under these conditions the resin bound AcIII and organic contaminants were removed with copious HNO3 (0.05 M) washing. Elution of the AcIII in HNO3 (6 M) provided a chemically pure stock solution that was suitable for subsequent chemical transformations. The overall purification procedure shown in Scheme 1 was attractive because it enabled recovery of AcIII from numerous inorganic and organic contaminants. Additionally, the process reduced the dose rate by removing the radioactive daughters, namely, 227Th and 223Ra [t1/2 = 18.68(9) d, 11.43(5) d, respectively].4,17−23,5,24,25 Finally, this procedure provided a means to recycle the valuable and rare 227Ac analyte for subsequent experimentation.
Scheme 1. Schematic Showing the Synthetic Methodology for Preparing the Ac-aquo Ion and Recovering Actinium from Previous Scientific Campaigns.
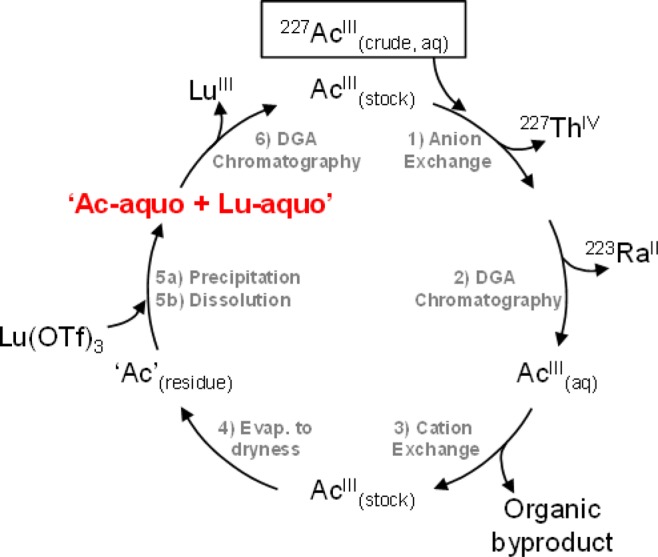
From the chemically and radiochemically purified actinium stock solution (described above) the Ac-aquo ion was prepared using a modification of the synthesis reported for the curium(III) aquo ion, [Cm(H2O)9](CF3SO3)3.13 This previously published Cm(H2O)93+ procedure involved precipitating CmIII from aqueous solutions with sodium hydroxide (NaOH). The presumed curium(III) hydroxide was subsequently washed with water and dissolved in dilute triflic acid (HO3SCF3; 1.67 M). Unfortunately, in our laboratory, this hydroxide precipitation was not directly transferrable to our small actinium sample. For example, in the CmIII study, there was sufficient mass (15 mg) to separate the curium(III) hydroxide precipitate from the supernate by centrifugation. In contrast, the AcIII transformations were carried out on a microscale (only 30 μg). Hence, it was not possible to isolate the actinium(III) hydroxide—the most soluble f-element hydroxide5—using conventional separation methods from the aqueous solvent.
To overcome this technical challenge, we introduced stable lutetium as a macroscopic carrier to facilitate precipitation and isolation of actinium(III) hydroxide. Here, the purified AcIII stock was heated under a stream of argon until the solvent evaporated. Care was taken to avoid bubble formation, splattering, and aerosolizing radioactive particles outside of the flask. After achieving a soft dryness, the residue was dissolved in water. Then, a macroscopic quantity of Lu(CF3SO3)3 was added (0.5 mg) to the solution, and actinium(III) and lutetium(III) hydroxides were coprecipitated using NaOH (2 M). Centrifugation of the mixture generated a substantial pellet, from which the supernate was easily decanted and discarded. After washing the pellet and dissolving the solid in dilute triflic acid, a solution suitable for XAFS spectroscopy that contained macroscopic quantities of the Lu-aquo ion, Lu(H2O)x3+,26−31 with trace amounts of the Ac-aquo ion was obtained.
Several factors contributed to the success of this preparative method. First and foremost, the chemical behavior of LuIII loosely mimics that of AcIII, which ensured quantitative coprecipitation of the respective hydroxides. Additionally, there had to be a method to remove the LuIII from AcIII after the experiment, so that the precious actinium sample could be recycled for future studies. Consistent with many actinium/lanthanide separation studies,17,5,24,25,32−38 we observed that liquid/liquid extraction chromatography (branched DGA; Eichrom) was effective for purifying AcIII from LuIII. As an example, “proof-of-principle” experiments using stable LuIII (0.5 mg) spiked with a radioactive 173Lu tracer [t1/2 = 1.37(1) y; 5.8 kBq; 3.6 × 1011 atoms]3 and short-lived 225Ac [t1/2 = 10.0(1) d; 8.8 kBq; 1.1 × 1010 atoms] demonstrated feasibility.3 These 173Lu and 225Ac isotopes were employed because their radioactive decay properties provided a convenient method for characterizing LuIII and AcIII separations using γ-spectroscopy. The representative chromatogram in Figure 1 shows that complete separation of 225Ac from 173Lu was achieved and that no cross contamination occurred in the representative 225Ac and 173Lu fractions. Moreover, this procedure was successfully applied in recovering 227Ac after the XAFS experiment. Although similar elution profiles were previously reported using trace quantities of actinium and lanthanides,4,17,18,36,38−44 our separation demonstrated that the DGA process accommodated a large mass range. This spanned picogram (pg) to microgram (μg) quantities with successful separations both at the tracer level (225Ac = 1.1 × 1010 atoms; 173Lu = 3.6 × 1011 atoms) and using large masses (227Ac = 7.4 × 104 kBq, 7.4 × 1016 atoms, 2.8 × 10–5 g; stable Lu = 1.7 × 1018 atoms, 5.0 × 10–4 g).
Figure 1.
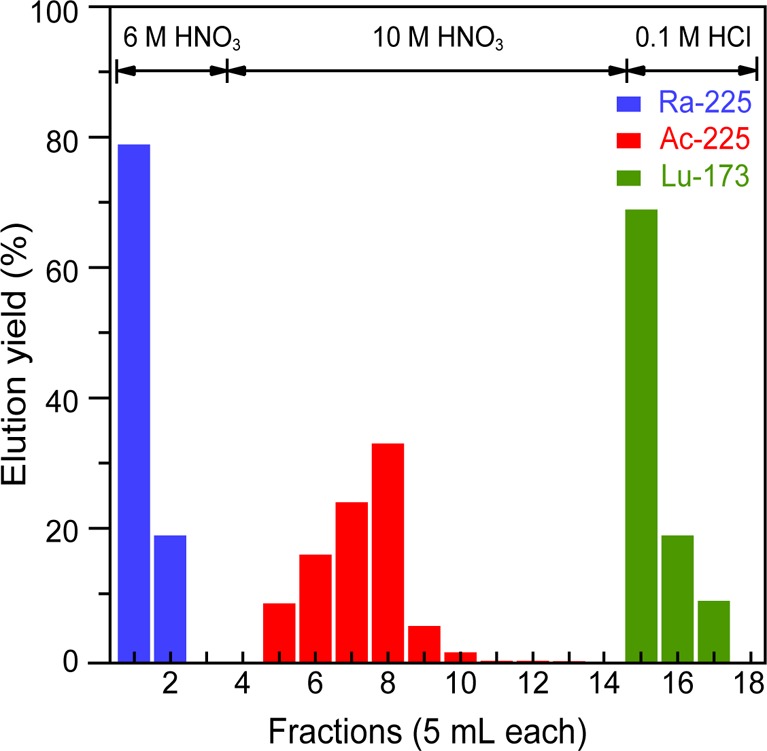
Elution profile of 225Ra, 225Ac, and 173Lu separations using DGA (Eichrom) on a Bio-Rad column (1 mL of resin in 10 mL column).
Ac L3-Edge XANES
To experimentally characterize Ac-aquo speciation, we exploited the element specific properties associated with X-ray absorption fine structure spectroscopy (XAFS). This spectroscopic approach has the crucial capability to probe low levels of actinium among large quantities of lutetium carrier. For instance, the Ac L3-absorption edge is well separated in energy from the Lu K- and L-edges, as well as the corresponding X-ray emission lines used for fluorescence detection.45Figure 2 compares the background subtracted and normalized XANES spectrum from Ac-aquo (obtained in this study) with a spectrum of AcIII dissolved in HCl (AcIII in HCl; 11 M; Ac-HCl) from our previous work.4 These two spectra are the only XANES measurements reported for actinium to date. The spectra were similar, each having a pronounced edge peak superimposed on an absorption threshold. From the perspective of the free ion, the edge feature has been crudely described as originating from electric-dipole allowed transitions from the actinide 2p-orbitals to unoccupied states that contain actinium 6d-character, i.e., 2p6 ... 5f0 6d0 → 2p5 ... 5f0 6d1.46,47 The inflection point in the Ac-aquo spectrum was found at 15874.3 eV, as determined graphically where the second derivative of the data equaled zero. This value was 0.4 eV lower in energy than the analogous inflection point 15873.9 eV reported previously for Ac-HCl.4 It was tempting to attribute this energy difference to electronic changes accompanying substitution of inner sphere Cl1– ligands in Ac-HCl for H2O ligands in Ac-aquo. However, given the uncertainty associated with actinide L-edge XANES inflection point determination, these energy differences were only marginally relevant statistically.
Figure 2.
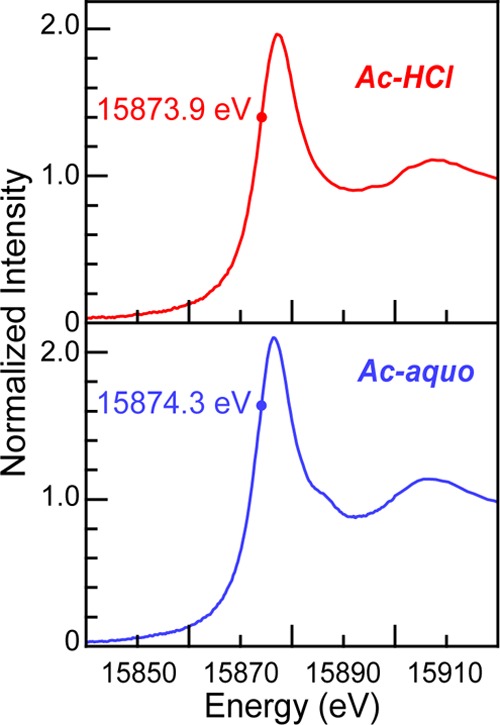
Room temperature background subtracted and normalized AcIII L3-edge XANES spectra obtained from Ac-aquo (HO3SCF3, 0.11 M; blue trace, bottom) and Ac-HCl (HCl, 11 M; red trace, top).
Ac-aquo MD-DFT
Before discussing the Ac L3-edge EXAFS data, we found it instructive to first present results from the molecular dynamics density functional theory (MD-DFT) calculations. These computational results were used to guide the spectral interpretations by providing a glimpse into actinium species potentially present during the EXAFS experiment. In these calculations, the speciation of a single AcIII ion with 64 H2O molecules was modeled within a box that had dimensions of 12.54 Å × 12.45 Å × 12.68 Å. To keep the system neutral, the charge on the box was constrained to be uniformly −3. Prior to the simulation, the temperature was elevated (498 K) to randomize the box components. Next, the system was returned to 298 K and the molecular dynamics (MD) modeled for 8 ps. Results from the calculation are depicted graphically in Figure 3, where an averaged H2O occupation was displayed as a function of the mean distance from the AcIII ion over the course of the simulation. This plot showed a tight distribution of H2O molecules at 2.689 ± 0.11 Å in the first actinium coordination sphere (Figure 4). Second and third shells of water molecules were subsequently observed near 5 and 7 Å, respectively, and linked to the first water shell through dynamic and intricate hydrogen bonding networks. However, over the course of the entire calculation only 9 H2O molecules were ever observed in this first actinium coordination sphere. Attempts to add or subtract H2O molecules failed, as a tenth H2O molecule quickly disassociated and an eight coordinate Ac(H2O)83+ ion rapidly picked up an extra H2O ligand.
Figure 3.
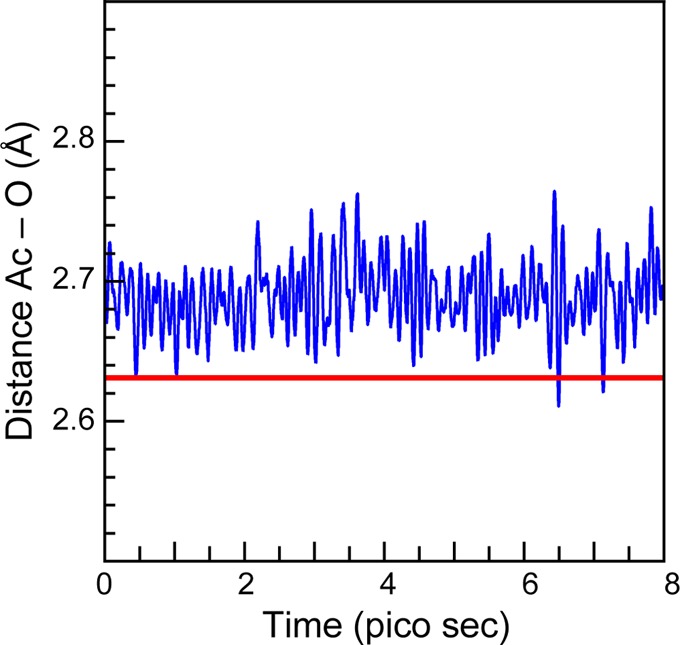
A comparison of the experimental Ac–OH2O bond distance (red trace) determined by EXAFS spectroscopy and the calculated mean distance (blue trace) between AcIII and water molecules within 3 Å from the actinium center during the 8 ps MD-DFT calculation.
Figure 4.
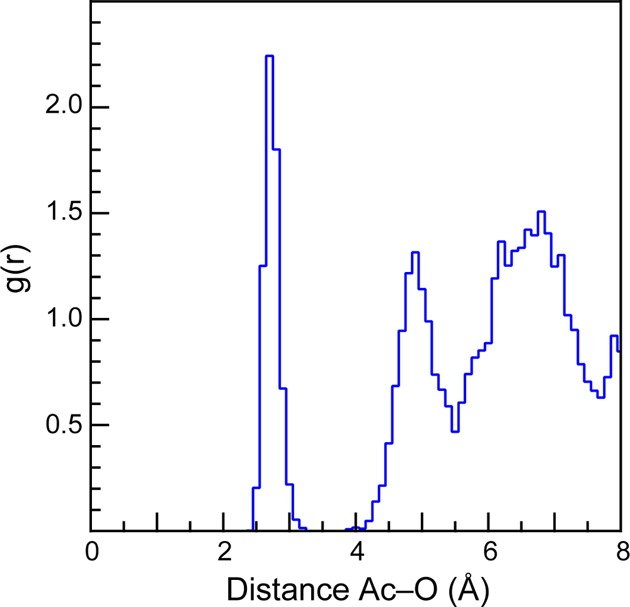
MD-DFT average radial distribution function of water molecules versus the calculated mean Ac–OH2O distance during the 8 ps MD-DFT calculation.
Ac L3-Edge EXAFS
Structural information for the Ac-aquo ion was experimentally determined from the k3χ(k) EXAFS solution phase measurements shown in Figure 5 and Figure 6. In spite of the low analyte concentration, high quality data with a reasonable signal-to-noise ratio was obtained to approximately 8.5 k. As a result, the data confidently represented the actinium first coordination shell. Meanwhile, longer scattering pathways were obscured and not considered, i.e., the second water shell and multiple scattering pathways like Ac–H2O···H2O. The data was modeled using established methods based on the EXAFS equation,48 where the coordination number (CN) and the bond length (R) variables were allowed to converge to reasonable values. Unfortunately, a reasonable model of the data could not be obtained when the Debye–Waller factor (σ2) was a free variable. An unconstrained σ2 model produced spectral fits with unreasonably large coordination numbers, or S02 values >1 when coordination numbers were fixed. To overcome this challenge, σ2 was constrained to the reasonable value of 0.009. As shown in the Supporting Information, this value was determined through extrapolation based on an anticipated linear relationship between σ2 and the atomic number for the actinide aquo complexes (U to Cf)6−16,49,50 listed in Table 1.
Figure 5.
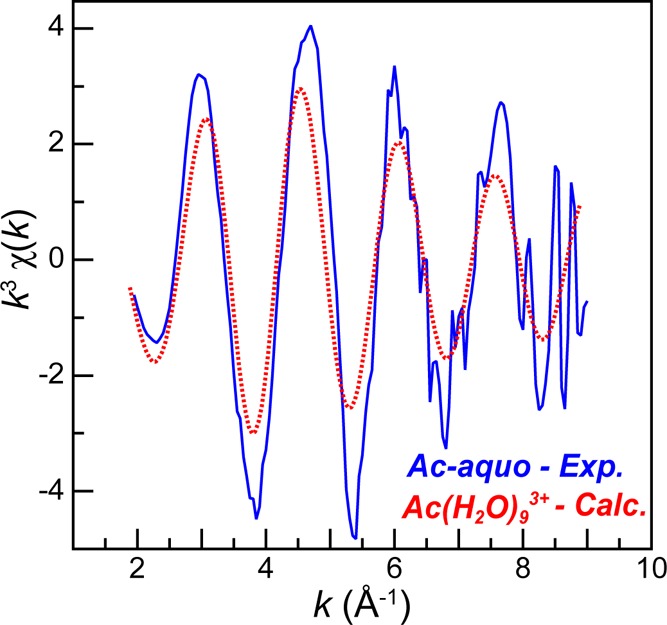
Room temperature solution-phase AcIII L3-edge EXAFS function k3χ(k) from the Ac-aquo ion (HO3SCF3, 0.11 M) (solid blue trace) and a FEFF8 model for Ac(H2O)93+ (dashed red trace), whose coordinates were obtained from a single frame of the MD-DFT calculation.
Figure 6.

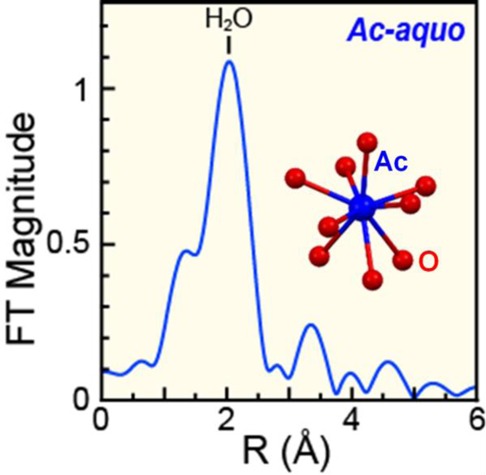
Left: The room temperature solution-phase AcIII L3-edge EXAFS function k3χ(k) from the Ac-aquo ion (HO3SCF3, 0.11 M) (solid blue trace). Fit of the data is shown as a dashed black trace. Right: Fourier transform of k3-EXAFS spectra from the Ac-aquo ion (HO3SCF3, 0.11 M; blue trace) and its real part shown as the red trace. Fits to the data have been provided as dashed black traces.
Table 1. Comparison of Lanthanide and Actinide +3 Aquo Ions Analyzed by EXAFSa.
| compound | metal soln concn | matrix | ionic radius (CN = 6)63 | CN | R [Å] | σ2 [Å2] | ΔE0 [eV] |
|---|---|---|---|---|---|---|---|
| Ac-aquo | 0.264 mM | 0.11 M HO3SCF3 | 1.12 | 10.9 ± 0.5 | 2.63(1) | 0.009 | -3.9 |
| LaIII(H2O)y31 | 0.2 M | pH 1 HO3SCF3 | 1.032 | 6.0 ± 0.5 | 2.560(9) | 0.007(1) | |
| 3.0 ± 0.5 | 2.66(2) | 0.0065(22) | |||||
| LaIII(H2O)y9 | 2–3 M | 0.25 M HCl | 1.032 | 9.2 ± 0.37 | 2.54(3) | 0.0090 | –7.3 |
| LaIII(H2O)y31 | 0.2 M | pH 1 HO3SCF3 | 1.032 | 6.0 ± 1.0 | 2.50(4) | 0.014(6) | |
| 3.0 ± 1.2 | 2.57(4) | 0.009(7) | |||||
| UIII(H2O)y6 | 1–10 mM | pH 0 HCl | 1.025 | 9.1 ± 0.6 | 2.52(1) | 0.009(1) | 12.8 |
| UIIIaq7 | 1 M HCl | 1.025 | 8.7 ± 0.9 | 2.56(1) | 0.10(1) | 2.1 | |
| NpIII(H2O)y6 | 0.5–2 mM | pH 0 HCl | 1.01 | 10.0 ± 1.2 | 2.51(1) | 0.009(1) | 7.2 |
| NpIIIaq7 | 1 M HCl | 1.01 | 9.8 ± 0.9 | 2.52(1) | 0.10(1) | 3.8 | |
| NpIIIaq8 | 4.7 mM | 1 M HClO4 | 1.01 | 9.0 ± 1.0 | 2.48(2) | ||
| PuIII(H2O)y49 | 20 mM | 0.01 M LiCl | 1 | 10.2 ± 1.1 | 2.510(6) | 0.010 | –10.4 |
| PuIII(H2O)y9 | 10 mM | 0.01 M LiCl | 1 | 9.2 ± 0.33 | 2.510(6) | 0.010 | –10.4 |
| PuIII(H2O)y6 | 0.8–2 mM | pH 0 HCl | 1 | 9.9 ± 0.3 | 2.49(1) | 0.009(1) | 7.0 |
| PuIIIaq10 | 0.01 mM | 1 M HClO4 | 1 | 8.6 ± 0.2 | 2.50(2) | 0.0083 | 7.16 |
| PuIIIaq7 | 1 M HCl | 1 | 9.9 ± 0.9 | 2.51(1) | 0.10(1) | 2.3 | |
| PuIIIaq50 | 1 | 8–9 | 2.48 | ||||
| NdIII(H2O)y9 | 2–3 M | 0.25 M HCl | 0.983 | 9.5 ± 0.37 | 2.49(3) | 0.0090 | –8.2 |
| NdIII(H2O)y52 | HClO4 | 0.983 | 9.5 | 2.51 | 0.0091 | ||
| Am-aquo4 | 4.8 mM | 0.11 M HO3SCF3 | 0.975 | 9.5 ± 0.87 | 2.48(1) | 0.0088(9) | –4.71 |
| AmIII(H2O) y(11) | 1 mM | 0.025 M HClO4 | 0.975 | 8.3 ± 0.4 | 2.473(4) | 0.0071(6) | –12.2 |
| AmIII(H2O) y(9) | 10 mM | 0.25 M HCl | 0.975 | 10.3 ± 0.33 | 2.480(6) | 0.009 | –8.7 |
| AmIII(H2O) y(12) | 7.9 mmol kg–1 | 0.03 M NaClO4 (pH 3.5) | 0.975 | 9.0 ± 0.0 | 2.47(1) | 0.0074(5) | 7.2 |
| AmIIIaq7 | 1 M HCl | 0.975 | 9.5 ± 0.9 | 2.51(1) | 0.10(1) | 1.0 | |
| CmIII(H2O)y9 | 10 mM | 0.25 M HCl | 0.97 | 10.2 ± 0.33 | 2.450(6) | 0.009 | –13.0 |
| CmIII(H2O)y13 | 0.523 M | 1 M HClO4 | 0.97 | 7.0 ± 0.4 | 2.469(7) | 0.0071(8) | –2.0 |
| BkIII(H2O)y14 | 0.47 mM | 1 M HClO4 1 M | 0.96 | 9.0 ± 0.6 | 2.43(2) | 0.009(2) | 2.7 |
| SmIII(H2O)y52 | HClO4 | 0.958 | 9.3 | 2.45 | 0.0077 | ||
| CfIIIaq7 | 1 M HCl | 0.95 | 9.5 ± 0.9 | 2.44(1) | 0.10(1) | 2.5 | |
| CfIII(H2O)y15 | 2.2 mM | 0.1 M HClO4 | 0.95 | 8.0 ± 0.0 | 2.42(1) | 0.0077(1) | 1.76 |
| CfIII(H2O)y16 | 1.67 M | 1 M HCl | 0.95 | 8.5 ± 1.5 | 2.42(2) | 0.0095(1) | 1.4 |
| GdIII(H2O)y52 | HClO4 | 0.938 | 7.6 | 2.41 | 0.0066 |
Data in bold are from this study, and data are presented in the order of decreasing ionic radius.63
To guide interpretations of the EXAFS data from Ac-aquo, we calculated (using FEFF851) an EXAFS spectrum for a stable configuration of the actinium(III) nona-aquo trication, Ac(H2O)93+, based on atomic coordinates obtained from one “frame” of the MD-DFT calculations described above (see Figure 5). The experimental and calculated spectra were nearly superimposable, except at low k, owing to differences in the Debye–Waller factors (none were applied to the DFT model). Each spectrum was best described as having a single frequency whose amplitude methodically dampened with increased k. The frequencies were in good agreement, which suggested that the experimental Ac–OH2O distances approximated those in the Ac(H2O)93+ model.
In the FEFF8 calculation (coordinates obtained from MD-DFT), the nine Ac–OH2O scattering pathways ranged from 2.544 to 2.845 Å, spanning 0.301 Å. Given that the experimental data was of high quality between k of 2.6 and 8.5, the experimental resolution was calculated to be 0.266 Å (π/2Δk). Hence, we refrained from attempting to experimentally resolve multiple Ac–OH2O bond distances within the first water shell. A high quality fit with low residual factors and a reduced chi-squared value were obtained using an “averaged” single H2O shell, as displayed in Figure 6 for the k3χ(k) data and its corresponding Fourier transform. In this model a shell of H2O molecules was observed at a 2.63(1) Å distance (Table 1, Figure 6). This value was quite similar to the mean 2.66 ± 0.09 Å Ac–O distance for the static Ac(H2O)93+ structure obtained from the single “frame” of the MD-DFT simulation, which was used as the initial atomic coordinates guess in modeling the EXAFS data. We remind the reader of the MD-DFT results shown in Figure 3, where the average Ac–OH2O distances for the nine water molecules varied between ∼2.61 and ∼2.76 Å [mean 2.689 ± 0.11 Å] over an 8 ps dynamic simulation. These calculated values were similar to the experimentally determined distance. Additionally, this 2.63(1) Å experimental distance was quite similar to the only other reported Ac–OH2O distance, specifically the 2.59(3) Å distance determined recently by EXAFS from HCl solutions containing AcIII, AcCl3.2(1)(H2O)6.6(2).4 As these values are the only bond distances reported for actinium to date, their agreement is important in establishing confidence in the Ac–OH2O bond length being approximately 2.6 Å. Most notably, this AcIII–OH2O distance was more than 0.1 Å longer than analogous distances in other actinide–aquo complexes, as determined by EXAFS analysis (see Table 1 and Figure 7).
Figure 7.
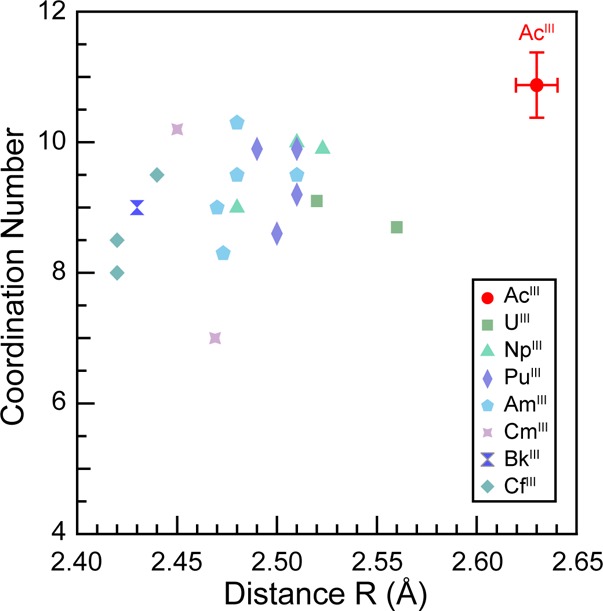
Coordination numbers and An–OH2O bond distances determined by EXAFS for the Ac-aquo ion and actinide(III) aquo ions previously reported.6−16,49
The major difference between the EXAFS calculation from Ac(H2O)93+ and the experimental data was associated with wave amplitudes, which were larger in the experiment (Figure 5). This increase suggested that the experimental coordination number was greater than the calculation (coordination numbercalc = 9). The fit of the experimental data showed 10.9 ± 0.5 oxygen atoms in the inner AcIII coordination sphere. The error with this coordination number is only associated with the data fitting model and does not encompass subjective decisions made during data processing (background subtraction, normalization, spline range, fitting range, Fourier transform range, choice of Debye–Waller factor, etc.). Having 10.9 ± 0.5 inner sphere water molecules seemed reasonable for the large AcIII ion, especially because this value overlapped with coordination numbers determined by EXAFS for other +3 actinide and lanthanide aquo ions (Table 1 and Figure 7). For the +3 actinides, these values ranged from 7.0 to 10.3 for U, Np, Pu, Am, Cm, Bk, and Cf6−16,49,50 and averaged 9.3 ± 0.9. Similarly, for the +3 lanthanides, these values ranged from 7.6 to 9.5 for La, Nd, Sm, and Gd and averaged 9.0 ± 0.7.9,31,52 We note that many of the coordination numbers determined by EXAFS for the actinide and lanthanide +3 aquo ions were larger than those observed by single crystal X-diffraction, where coordination numbers larger than 9 have yet to be reported.13,26,30,53−62
Outlook
Herein, we characterized the Ac-aquo complex by Ac L3-edge XAFS spectroscopy and MD-DFT calculations. Experimentally, we observed approximately 10.9 ± 0.5 water molecules in the inner actinium coordination sphere at an average distance of 2.63(1) Å. These results were in reasonable agreement with the MD-DFT results, which predicted exactly nine water molecules at a 2.689 ± 0.11 Å distance. The good agreement between experiment and theory is impressive when one accounts for the inherently large uncertainties typically associated with EXAFS coordination number determination alongside limitations associated with the GGA functionals and the difficulty in capturing key variables that influence speciation within the calculations. We anticipate that access to more experimental data, through studies like this, will reveal unique chemical and physical properties associated with the actinium element and enhance overall predictive capabilities in actinium chemistry. The characterization of the Ac-aquo ion presented here represents a fundamental benchmark in the development of actinium coordination chemistry. For example, with this data in hand, credible efforts to characterize actinium speciation in more relevant biological and natural media can proceed. In addition, characterization of the Ac-aquo ion will facilitate future efforts to study displacement of H2O by chelating ligands.
While the Ac-aquo ion coordination number was slightly higher than the coordination numbers of the other actinide and lanthanide aquo ions, the incredibly long Ac–OH2O distance exemplified how actinium is the largest +3 cation known (ionic radius: 1.12 Å63). While it remains unclear how actinium’s unique size influences coordination numbers and preferences for binding certain donor atoms, the ion’s size should be a major consideration when designing ligand architectures for actinium chelation. Hence, our current efforts are focused on characterizing the impact of actinium’s large ionic radius on complexation by chelators relevant to biomimetic applications, i.e., DOTA, EDTA, HEHA, PEPA, DTPA, NOTA, etc.2,64−69 These studies, in combination with the data reported here, will heavily influence our foray into developing new actinium chelators that support use of actinium in targeted α-therapy.
Experimental Section
General Consideration
CAUTION! The 225Ac, 227Ac, and 173Lu isotopes present serious health threats, due to their (and their daughters’) α-, β-, and γ-radiation. As such, this research was conducted in a radiological facility within certified fume hoods and monitored with appropriate α-, β-, and γ-particle detecting instruments. The 227Ac isotope was supplied by the United States Department of Energy Office of Science Isotope Program in the Office of Nuclear Physics. The 173Lu isotope was produced and purified at Los Alamos.70 The 225Ac isotope was purified from a clean 229Th source, which had been achieved in the LANL isotope inventory many years ago.71 Trifluoromethanesulfonic acid and sodium hydroxide were obtained commercially (Fisher Scientific). Water was purified to 18.2 MΩ/cm resistivity using Thermo-Scientific Barnstead Nanopure or Millipore Nanopure water purification systems. Resins used for separations—DOWEX AG1-X8 (BioRad; 100–200 mesh; Cl1– form), branched DGA (Eichrom; 50 μm), and DOWEX AG50W-X8 (BioRad; 100–200 mesh; H+ form)—were obtained commercially, suspended in water, and the fines were decanted prior to use. Separations were characterized using γ-spectroscopy using an EG&G Ortec model GMX-35200-S HPGe detector system in combination with a Camberra model 35-Plus multichannel analyzer associated with Gamma Vision software.
AcIII Purification
Actinium was radiochemically and chemically purified as follows. A crude solution containing AcIII in nitric acid, HNO3 (∼10 mL; 8 M), was loaded onto a Biorad column (10 mL) filled with DOWEX AG1-X8 (1 mL; BioRad; 100–200 mesh; Cl1– form) that had been conditioned prior with H2O (3 × 5 mL), HNO3 (3 × 5 mL; 8 M), H2O (3 × 5 mL), and HNO3 (3 × 5 mL; 8 M). The AcIII and 223Ra daughter (+2 oxidation state) passed directly through the column, while the 227Th daughter (+4 oxidation state) was retained. This effluent was diluted with water so that the final HNO3 concentration was 6 M. This AcIII containing solution was then loaded onto a second Biorad column (10 mL) containing a DGA (1 mL; Eichrom) resin that had been conditioned with H2O (3 × 10 mL) followed by HNO3 (1 × 10 mL; 6 M). Under these conditions the RaII passed through the column while AcIII was retained. The column was washed with HNO3 (6 × 2 mL; 6 M). Subsequently AcIII was eluted using HNO3 (2 × 5 mL; 0.05 M). To remove any organic compounds that followed AcIII through the DGA resin, the DGA column effluent was loaded directly onto a Biorad column (10 mL) containing DOWEX AG50W-X8 resin (1 mL) that had been conditioned with H2O (3 × 10 mL) and HNO3 (1 × 10 mL; 0.05 M). After washing of the column with copious amounts of HNO3 (2 × 5 mL; 0.05 M), AcIII was eluted with HNO3 (2 × 8 mL; 6 M). The 227Ac fractions were combined. Subsequently 5 mL of the solution was assayed by γ-spectroscopy. We note that when assaying 227Ac there is a large uncertainty (>3%) associated with γ-spectroscopy results, as the 227Ac γ-emission is complicated by low relative intensities (0.001–0.006% at energies higher than 100 keV). Hence, more rigorous evaluation of the actinium concentration was achieved using the Bateman equation and monitoring ingrowth of the 227Ac daughters, namely 227Th and 223Ra.72
Synthesis of Ac-aquo
The radiochemically and chemically purified AcIII stock solution described above was evaporated to a soft dryness in a conical shaped glass vial on a hot plate under a slow stream of argon. The resulting residue was dissolved in Millipore H2O (0.200 mL). Subsequently, sodium hydroxide, NaOH (0.100 mL; 2 M) was added and the solution was capped and agitated. To ensure quantitative precipitation and effective separation of actinium hydroxide precipitate from the supernate, lutetium(III) tris-triflate, Lu(O3SCF3)3 (0.5 mg in 0.010 mL), was added as a stable carrier. The solution was then centrifuged (3 min at 6000 rpm) and the supernate was removed from the fluffy solid. The solid was washed with NaOH (1 × 0.100 mL; 10 mM) and with Millipore H2O (1 × 0.050 mL). The solid precipitate was then dissolved in trifluoromethanesulfonic acid (HO3SCF3; 0.5 mL; 0.11 M) and transferred to an XAFS holder. The holder was triply contained, which protected against release of radiological material during shipment and XAFS experiments, as described below.
AcIII Separation from LuIII
A solution containing AcIII and LuIII was evaporated to a soft dryness in a conical shaped glass vial on a hot plate under a slow stream of air. The resulting residue was dissolved in HNO3 (3 × 2 mL; 8 M). The resulting solution was loaded onto a Biorad column (10 mL) filled with a DGA (1 mL; Eichrom; 50 μm) resin that had been conditioned with H2O (3 × 10 mL) followed by HNO3 (3 × 10 mL; 6 M). Under these conditions both AcIII and LuIII were retained on the resin. Actinium(III) was selectively eluted with HNO3 (5 × 10 mL; 10 M). The 227Ac fractions were combined. Then, the solution was evaporated in a conical shaped glass vial on a hot plate under a slow stream of argon to a soft dryness. The residue was subsequently dissolved in a minimal amount of HNO3 (8 M).
XAFS Measurements
The Ac L3-edge XAFS measurements were made on samples loaded into XAFS cells that were triply contained. The XAFS holder consisted of a plastic body with a 2 mm well equipped with a set of Teflon windows (1 mil) and a Kapton window (1 mil). Solutions were introduced into the holder through an injection hole sealed with a Teflon gasket that was held in place by an aluminum plate. The sample cell holder was then transferred into the secondary and the tertiary container, which are best described as a set of nested aluminum holders equipped with Kapton windows (2 mil).
The XANES and EXAFS were measured at the Stanford Synchrotron Radiation Lightsource (SSRL) under dedicated operating conditions (3.0 GeV, 5%, 500 mA) on end station 11-2. This beamline was equipped with a 26-pole and a 2.0 T wiggler. Using a liquid nitrogen cooled double-crystal Si[220] (Φ = 0°) monochromator and employing collimating and focusing mirrors, a single energy was selected from the incident white beam. Although the crystals were run fully tuned, higher harmonics from the monochromatic light were removed using a 370 mm Rh coated harmonic rejection mirror. The Rh coating was 50 nm with 20 nm seed coating, and the substrate was Zerodur. Vertical acceptance was controlled by slits positioned before the monochromator. The harmonic rejection cutoff was set by the mirror angle, thereby controlling which photons experience total external reflection. The samples were attached to the beamline 11-2 XAFS rail. The rail was equipped with three ionization chambers through which nitrogen gas was continually flowed. One chamber was positioned before the sample holder, to monitor the incident radiation (I0, 10 cm). The second chamber was positioned after the sample holder, such that sample transmission (I1, 30 cm) could be evaluated against I0, while a third chamber (I2, 30 cm) was positioned downstream from I1 so that the XANES of a calibration foil could be measured in situ during the XAFS experiments against I1. Actinium solution samples were measured in fluorescence mode using a solid-state 100-element Ge detector against the incident radiation (I0). The 100-element Ge detector was windowed on the Ac Lα emission line (12.652 keV). Low energy contributions to the fluorescence signal were removed using a bromine filter (3 path lengths). Prior to conducting the measurements, dead time correction experiments were performed at approximately 400 eV above the element edge of the filter. The dead time correction curve corresponds to the plot of the windowed counts of the emission line of interest versus the total of incoming counts in the solid-state detector. This procedure was performed on a Se filter.
XAFS Data Analysis
Data manipulation and analysis was conducted as previously described.4,48 First the data were dead time corrected and calibrated to the energy of the first inflection point of a rubidium(II) chloride, RbCl, pellet, diluted with boron nitride, BN, to a 1 absorption length thickness. The energy for the first inflection point for RbCl was determined in comparison to the Bi LII-edge of a bismuth foil (15711 eV) to be 15874.3 eV. The energy of the calibration pellet was monitored before and after each Ac L3-edge measurement. No energy drift during the experiment time was observed. The XAFS data were analyzed by fitting a line to the pre-edge region, which removed the background from experimental data in the spectra. Then a second to third order polynomial fitting was chosen for the postedge region. The difference between pre- and postedge lines was set to unity at the first inflection point, normalizing the absorption jump to 1.0. Samples were measured for several hours resulting in the collection of 31 scans. Fittings using Athena and Artemis73 were performed using atomic coordinates from the MD-DFT calculations (see below) and FEFF8 calculations.51 The spectra were fit using only single scattering paths obtained from FEFF8. The adjustments of spectra were performed in 2.6 < k < 8.5 Å–1 and 1.25 < R < 3 Å. For the fitting procedure, the coordination number (CN) and distance (R) variables were allowed to vary. In generating the model, the Debye–Waller factor (σ2) was fixed to 0.009, which was extrapolated from the anticipated linear relationship between σ2 and the atomic number anticipated for U–Cf aquo complexes (see the Supporting Information). A single value of energy shift (ΔE0) was used for all scattering paths. The amplitude reduction factor (S02) was set at 0.9 based on initial fits. Results were compared to data published previously for actinium in concentrated HCl (Ac-HCl).4
MD-DFT Calculations
The Born–Oppenheimer molecular dynamics (MD) simulations in the Helmholtz ensemble (NVT) were performed using the computer code VASP (Vienna Ab-initio Simulations Package)74 version 5.35. In this code the forces on the ions are calculated from the electronic structure of the whole system computed using density functional theory at the generalized gradient approximation (GGA) level using the functional by Purdue–Burke–Enzerhof (PBE).75 A simulation box of (12.54 × 12.45 × 12.68 Å3) was used, including the metal ion (Ac3+) surrounded by 64 water molecules while a uniform background charge of −3 was added to keep the neutrality of the simulation box. The basis set consists of an expansion into plane wave functions. Due to the large size of the simulation box the k-space representation included only the Γ point. The energy cutoff for the plane-wave expansion was set at 500 eV, and scalar relativistic effects were included using the PAW–PBE potentials.76 Initially the metal ion and the closest neighboring molecules and counterions were kept frozen, and the solvent plus remaining counterion atoms were heated up to 498 K to be thermalized for 1 ps. After that a 1 ps run was done at 298 K with all the degrees of freedom released to thermalize the complex with the solvent. Finally 8 ps of data collection was performed where we monitored the solvent and ion dynamics.
Acknowledgments
We gratefully recognize the United States Department of Energy, Office of Science, Isotope Development and Production for Research and Application subprogram within Office of Nuclear Physics for their support in supplying the 227Ac isotope. The work was funded under the LANL LDRD program (Berg, Birnbaum, Engle, Redman) and work under the Heavy Element Chemistry Program by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy and the U.S. Department of Energy (Batista, Kozimor). Portions of this work were supported by postdoctoral Fellowships from the Glenn T. Seaborg Institute (Ferrier, Stein). Los Alamos National Laboratory is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (Contract DE-AC52-06NA25396). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). We thankfully acknowledge the computational resources for this project from the Environmental Molecular Science Laboratory of PNNL, in the cascade supercomputer. Some of the calculations were carried out with computational support of LANL’s institutional computers, wolf cluster. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00356.
A figure describing our determination for the Ac Debye–Waller factor (σ2) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kim Y. S.; Brechbiel M. W. An Overview of Targeted Alpha Therapy. Tumor Biol. 2012, 33, 573–590. 10.1007/s13277-011-0286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier O.; Supiot S.; Degraef-Mougin M.; Faivre-Chauvet A.; Carlier T.; Chatal J.-F.; Davodeau F.; Cherel M. Cancer Radioimmunotherapy with Alpha-Emitting Nuclides. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 601–614. 10.1007/s00259-005-1803-2. [DOI] [PubMed] [Google Scholar]
- Brookhaven National Laboratory. National Nuclear Data Center. http://www.nndc.bnl.gov/chart/.
- Ferrier M. G.; Batista E. R.; Berg J. M.; Birnbaum E. R.; Cross J. N.; Engle J. W.; La Pierre H. S.; Kozimor S. A.; Lezama Pacheco J. S.; Stein B. W.; Stieber S. C. E.; Wilson J. J. Spectroscopic and Computational Investigation of Actinium Coordination Chemistry. Nat. Commun. 2016, 7, 12312. 10.1038/ncomms12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby H. W.; Morss L. R.. Actinium. In The Chemistry of the Actinide and Transactinide Elements; Springer: Dordrecht, 2006; pp 18–51. [Google Scholar]
- Brendebach B.; Banik N. L.; Marquardt C. M.; Rothe J.; Denecke M. A.; Geckeis H. X-Ray Absorption Spectroscopic Study of Trivalent and Tetravalent Actinides in Solution at Varying pH Values. Radiochim. Acta 2009, 97, 701–708. 10.1524/ract.2009.1674. [DOI] [Google Scholar]
- David F.; Fourest B.; Hubert S.; Le Du J. F.; Revel R.; Den Auwer C.; Madic C.; Morss L. R.; Ionova G.; Mikhalko V.; Vokhmin V.; Nikonov M.; Berthet J. C.; Ephritikhine M.. Aquo Ions of Some Trivalent Actinides: EXAFS Data and Thermodynamics Consequences. In Speciation Techniques and Facilities for Radioactive Materials at Synchrotron Light Sources; Edelstein N., Nitshe H., Reich T., Eds.; Nuclear Energy Agency: Organization for Economic Co-Operation and development: Grenoble, France, 1998; pp 95–100. [Google Scholar]
- Antonio M. R.; Soderholm L.; Williams C. W.; Blaudeau J.-P.; Bursten B. E. Neptunium Redox Speciation. Radiochim. Acta 2001, 89, 17–25. 10.1524/ract.2001.89.1.017. [DOI] [Google Scholar]
- Allen P. G.; Bucher J. J.; Shuh D. K.; Edelstein N. M.; Craig I. Coordination Chemistry of Trivalent Lanthanide and Actinide Ions in Dilute and Concentrated Chloride Solutions. Inorg. Chem. 2000, 39, 595–601. 10.1021/ic9905953. [DOI] [PubMed] [Google Scholar]
- Kirsch R.; Fellhauer D.; Altmaier M.; Neck V.; Rossberg A.; Fanghänel T.; Charlet L.; Scheinost A. C. Oxidation State and Local Structure of Plutonium Reacted with Magnetite, Mackinawite, and Chukanovite. Environ. Sci. Technol. 2011, 45, 7267–7274. 10.1021/es200645a. [DOI] [PubMed] [Google Scholar]
- Stumpf T.; Hennig C.; Bauer A.; Denecke M. A.; Fanghänel T. An EXAFS and TRLFS Study of the Sorption of Trivalent Actinides onto Smectite and Kaolinite. Radiochim. Acta 2004, 92, 133–138. 10.1524/ract.92.3.133.30487. [DOI] [Google Scholar]
- Marques Fernandes M.; Scheinost A. C.; Baeyens B. Sorption of Trivalent Lanthanides and Actinides onto Montmorillonite: Macroscopic, Thermodynamic and Structural Evidence for Ternary Hydroxo and Carbonato Surface Complexes on Multiple Sorption Sites. Water Res. 2016, 99, 74–82. 10.1016/j.watres.2016.04.046. [DOI] [PubMed] [Google Scholar]
- Skanthakumar S.; Antonio M. R.; Wilson R. E.; Soderholm L. The Curium Aqua Ion. Inorg. Chem. 2007, 46, 3485–3491. 10.1021/ic061798b. [DOI] [PubMed] [Google Scholar]
- Antonio M. R.; Williams C. W.; Soderholm L. Berkelium Redox Speciation. Radiochim. Acta 2002, 90, 851–856. 10.1524/ract.2002.90.12_2002.851. [DOI] [Google Scholar]
- Galbis E.; Hernández-Cobos J.; Den Auwer C.; Le Naour C.; Guillaumont D.; Simoni E.; Pappalardo R. R.; Sánchez Marcos E. Solving the Hydration Structure of the Heaviest Actinide Aqua Ion Known: The californium(III) Case. Angew. Chem. 2010, 122, 3899–3903. 10.1002/ange.200906129. [DOI] [PubMed] [Google Scholar]
- Revel R.; Den Auwer C.; Madic C.; David F.; Fourest B.; Hubert S.; Le Du J. F.; Morss L. R. First Investigation on the L Edges of the 249Cf Aquo Ion by X-Ray Absorption Spectroscopy. Inorg. Chem. 1999, 38, 4139–4141. 10.1021/ic990214l. [DOI] [Google Scholar]
- Radchenko V.; Engle J. W.; Wilson J. J.; Maassen J. R.; Nortier F. M.; Taylor W. A.; Birnbaum E. R.; Hudston L. A.; John K. D.; Fassbender M. E. Application of Ion Exchange and Extraction Chromatography to the Separation of Actinium from Proton-Irradiated Thorium Metal for Analytical Purposes. J. Chromatogr. A 2015, 1380, 55–63. 10.1016/j.chroma.2014.12.045. [DOI] [PubMed] [Google Scholar]
- Horwitz E. P.; McAlister D. R.; Bond A. H.; Barrans R. E. J. Novel Extraction of Chromatographic Resins Based on Tetraalkyldiglycolamides: Characterization and Potential Applications. Solvent Extr. Ion Exch. 2005, 23, 319–344. 10.1081/SEI-200049898. [DOI] [Google Scholar]
- Zielinska B.; Apostolidis C.; Bruchertseifer F.; Morgenstern A. An Improved Method for the Production of Ac-225/Bi-213 from Th-229 for Targeted Alpha Therapy. Solvent Extr. Ion Exch. 2007, 25, 339–349. 10.1080/07366290701285108. [DOI] [Google Scholar]
- Kotovskii A. A.; Nerozin N. A.; Prokof’ev I. V.; Shapovalov V. V.; Yakovshchits Y. A.; Bolonkin A. S.; Dunin A. V. Isolation of Actinium-225 for Medical Purposes. Radiochemistry 2015, 57, 285–291. 10.1134/S1066362215030091. [DOI] [Google Scholar]
- Boll R. A.; Malkemus D.; Mirzadeh S. Production of Actinium-225 for Alpha Particle Mediated Radioimmunotherapy. Appl. Radiat. Isot. 2005, 62, 667–679. 10.1016/j.apradiso.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Alhassanieh O.; Abdul-Hadi A.; Ghafar M.; Aba A. Separation of Th, U, Pa, Ra and Ac from Natural Uranium and Thorium Series. Appl. Radiat. Isot. 1999, 51, 493–498. 10.1016/S0969-8043(99)00068-8. [DOI] [Google Scholar]
- Aliev R. A.; Ermolaev S. V.; Vasiliev A. N.; Ostapenko V. S.; Lapshina E. V.; Zhuikov B. L.; Zakharov N. V.; Pozdeev V. V.; Kokhanyuk V. M.; Myasoedov B. F.; Kalmykov S. N. Isolation of Medicine-Applicable Actinium-225 from Thorium Targets Irradiated by Medium- Energy Protons. Solvent Extr. Ion Exch. 2014, 32, 468–477. 10.1080/07366299.2014.896582. [DOI] [Google Scholar]
- Hagemann F. T.The Chemistry of Actinium. In National Nuclear Energy Series, Manhattan Project Technical Section, Division 4: Plutonium Project 14A; 1954; pp 14–44. [Google Scholar]
- Gmelin L.; Alleluia I. B.; Eberle S. H.; Keller C.; Kirby H. W.; Munzel H.; Seidel A.. Gmelin Handbook of Inorganic Chemistry Ac Supplement Vol.1, System num.; Kugler H. K., Kelier C., Eds.; Springer-Verlag: Berlin, Heidelberg, New York, 1981. [Google Scholar]
- Harrowfield J. M.; Kepert D. L.; Patrick J. M.; White A. H. Structure and Stereochemistry in “F-Block” Complexes of High Coordination Number. VIII The [M(unidentate)9] System. Crystal Structures of [M(OH2)9] [CF3S03]3, M = La, Gd, Lu, Y. Aust. J. Chem. 1983, 36, 483–492. 10.1071/CH9830483. [DOI] [Google Scholar]
- Chatterjee A.; Maslen E. N.; Watson K. J. The Effect of the Lanthanoid Contraction on the nonaaqualanthanoid(III) Tris(trifluoromethanesulfonates). Acta Crystallogr., Sect. B: Struct. Sci. 1988, 44, 381–386. 10.1107/S0108768188001764. [DOI] [Google Scholar]
- Abbasi A.; Lindqvist-Reis P.; Eriksson L.; Sandström D.; Lidin S.; Persson I.; Sandström M. Highly Hydrated Cations: Deficiency, Mobility, and Coordination of Water in Crystalline Nonahydrated scandium(III), yttrium(III), and lanthanoid(III) Trifluoromethanesulfonates. Chem. - Eur. J. 2005, 11, 4065–4077. 10.1002/chem.200401339. [DOI] [PubMed] [Google Scholar]
- Albertsson J.; Elding I. The Geometry of the Nonaaqualanthanoid(3+) Complex in the Solid Bromates and Ethyl Sulphates. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1977, 33, 1460–1469. 10.1107/S0567740877006293. [DOI] [Google Scholar]
- Gerkin R. E.; Reppart W. J. The Structures of the Lanthanide Ethyl Sulfate Enneahydrates, M(C2H5SO4)3.9H2O [M = La - Lu (except Pm)], at 171 K. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1984, 40, 781–786. 10.1107/S0108270184005710. [DOI] [Google Scholar]
- D’Angelo P.; De Panfilis S.; Filipponi A.; Persson I. High-Energy X-Ray Absorption Spectroscopy: A New Tool for Structural Investigations of Lanthanoids and Third-Row Transition Elements. Chem. - Eur. J. 2008, 14, 3045–3055. 10.1002/chem.200701282. [DOI] [PubMed] [Google Scholar]
- Kosynkin V. D.; Moiseev S. D.; Vdovichev V. S. Cleaning Rare Earth Elements from Actinium. J. Alloys Compd. 1995, 225, 320–323. 10.1016/0925-8388(94)07132-2. [DOI] [Google Scholar]
- Sekine T.; Koike Y.; Hasegawa Y. Studies of Actinium(III) in Various Solutions. II. Distribution Behavior of Lanthanum(III) and Actinium(III) in Some Chelate Extraction Systems. Bull. Chem. Soc. Jpn. 1969, 42, 432–436. 10.1246/bcsj.42.432. [DOI] [Google Scholar]
- Danon J. Anion-Exchange Studies with Actinium and Lanthanides in Nitrate Solutions. J. Inorg. Nucl. Chem. 1958, 7, 422–424. 10.1016/0022-1902(58)80254-7. [DOI] [Google Scholar]
- Filosofov D. V.; Lebedev N. a.; Radchenko V.; Rakhimov A. V.; Happel S.; Roesch F. Behavior of Actinium, Alkaline, and Rare Earth Elements in Sr-Resin/Mineral Acid Systems. Solvent Extr. Ion Exch. 2015, 33, 496–509. 10.1080/07366299.2015.1046293. [DOI] [Google Scholar]
- Horwitz E. P.; McAlister D. R.; Thakkar A. H. Synergistic Enhancement of the Extraction of Trivalent Lanthanides and Actinides by Tetra-(N-Octyl)Diglycolamide from Chloride Media. Solvent Extr. Ion Exch. 2008, 26, 12–24. 10.1080/07366290701779423. [DOI] [Google Scholar]
- Horwitz E. P.; Bloomquist C. A. A. Chemical Separations for Super-Heavy Element Searches in Irradiated Uranium Targets. J. Inorg. Nucl. Chem. 1975, 37, 425–434. 10.1016/0022-1902(75)80350-2. [DOI] [Google Scholar]
- Ostapenko V.; Vasiliev A.; Lapshina E.; Ermolaev S.; Aliev R.; Totskiy Y.; Zhuikov B.; Kalmykov S. Extraction Chromatographic Behavior of Actinium and REE on DGA, Ln and TRU Resins in Nitric Acid Solutions. J. Radioanal. Nucl. Chem. 2015, 306, 707–711. 10.1007/s10967-015-4331-y. [DOI] [Google Scholar]
- Lumetta G. J.; Gelis A. V.; Carter J. C.; Niver C. M.; Smoot M. R. The Actinide-Lanthanide Separation Concept. Solvent Extr. Ion Exch. 2014, 32, 333–347. 10.1080/07366299.2014.895638. [DOI] [Google Scholar]
- Sasaki Y.; Sugo Y.; Morita K.; Nash K. L. The Effect of Alkyl Substituents on Actinide and Lanthanide Extraction by Diglycolamide Compounds. Solvent Extr. Ion Exch. 2015, 33, 625–641. 10.1080/07366299.2015.1087209. [DOI] [Google Scholar]
- Sasaki Y.; Tsubata Y.; Kitatsuji Y.; Sugo Y.; Morita Y.; Kimura T. Extraction Behavior of Metal Ions by TODGA, DOODA, MIDOA, and NTAamide Extractants from HNO3 to N-Dodecane. Solvent Extr. Ion Exch. 2013, 31, 401–415. 10.1080/07366299.2013.800431. [DOI] [Google Scholar]
- Pourmand A.; Dauphas N. Distribution Coefficients of 60 Elements on TODGA Resin: Application to Ca, Lu, Hf, U and Th Isotope Geochemistry. Talanta 2010, 81, 741–753. 10.1016/j.talanta.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Ansari S. A.; Pathak P. N.; Husain M.; Prasad A. K.; Parmar V. S.; Manchanda V. K. Extraction Chromatographic Studies of Metal Ions Using N, N, N′, N′ -Tetraoctyl Diglycolamide as the Stationary Phase. Talanta 2006, 68, 1273–1280. 10.1016/j.talanta.2005.07.042. [DOI] [PubMed] [Google Scholar]
- Husain M.; Ansari S. A.; Mohapatra P. K.; Gupta R. K.; Parmar V. S.; Manchanda V. K. Extraction Chromatography of Lanthanides Using N, N, N′, N′-tetraoctyl Diglycolamide (TODGA) as the Stationary Phase. Desalination 2008, 229, 294–301. 10.1016/j.desal.2007.10.016. [DOI] [Google Scholar]
- Thompson A.; Attwood D.; Gullikson E.; Howells M.; Kim K.-J.; Kirz J.; Kortright J.; Lindau I.; Pianetta P.; Robinson A.; Scofield J.; Underwood J.; Vaughan D.; Williams G.; Winick H.. Center for X-Ray Optics and Advanced Light Source X-Ray Data Booklet, 2nd ed.; Lawrence Berkeley National Laboratory, University of California: Berkeley, CA, 2001. [Google Scholar]
- Teo B. K.EXAFS: Basic Principles and Data Analysis; Springer-Verlag: New York, Berlin, 1986. [Google Scholar]
- Stohr J.NEXAFS Spectroscopy, 1st ed.; Springer-Verlag: Berlin, 1992. [Google Scholar]
- Calvin S.XAFS for Everyone; CRC Press Tayor and Francis Group: Boca Raton, FL, 2013. [Google Scholar]
- Allen P. G.; Bucher J. J.; Shuh D. K.; Edelstein N. M.; Reich T. Investigation of Aquo and Chloro Complexes of UO22+, NpO2+, Np4+, and Pu3+ by X-Ray Absorption Fine Structure Spectroscopy. Inorg. Chem. 1997, 36, 4676–4683. 10.1021/ic970502m. [DOI] [PubMed] [Google Scholar]
- Conradson S. D. Application of X-Ray Absorption Fine Structure Spectroscopy to Materials and Environmental Science. Appl. Spectrosc. 1998, 52, 252A–279A. 10.1366/0003702981944599. [DOI] [Google Scholar]
- Ankudinov A.; Conradson S.; Mustre de Leon J.; Rehr J. Relativistic XANES Calculations of Pu Hydrates. Phys. Rev. B: Condens. Matter Mater. Phys. 1998, 57, 7518–7525. 10.1103/PhysRevB.57.7518. [DOI] [Google Scholar]
- Yamaguchi T.; Nomura M.; Wakita H.; Ohtaki H. An Extended X-Ray Absorption Fine Structure Study of Aqueous Rare Earth Perchlorate Solutions in Liquid and Glassy States. J. Chem. Phys. 1988, 89, 5153–5159. 10.1063/1.455633. [DOI] [Google Scholar]
- Glaser J.; Johansson G. Crystal Strcutures of the Isomorphous Perchlorate Hexahydrates of Some Trivalent Metal Ions (M = La, Tb, Er, Tl). Acta Chem. Scand. 1981, 35a, 639–644. 10.3891/acta.chem.scand.35a-0639. [DOI] [Google Scholar]
- Habenschuss A.; Spedding F. H. The Coordination (Hydration) of Rare Earth Ions in Aqueous Chloride Solutions from X Ray Diffraction. II. LaCl3, PrCl3, and NdCl3. J. Chem. Phys. 1979, 70, 3758–3763. 10.1063/1.437928. [DOI] [Google Scholar]
- Habenschuss A.; Spedding F. H. The Coordination (Hydration) of Rare Earth Ions in Aqueous Chloride Solutions from X Ray Diffraction. III.SmCl3, EuCl3, and Series Behavior. J. Chem. Phys. 1980, 73, 442–450. 10.1063/1.439895. [DOI] [Google Scholar]
- Paiva Santos C. O.; Castellano E. E.; Machado L. C.; Vicentini G. Crystal Structures of Neodymium and Holmium Trifluoromethanesulfonate Enneahydrated. Inorg. Chim. Acta 1985, 110, 83–86. 10.1016/S0020-1693(00)84560-0. [DOI] [Google Scholar]
- Helmholz L. The Crystal Structure of Neodymium Bromate Enneahydrate, Nd(BrO3)3.9H2O. J. Am. Chem. Soc. 1939, 61, 1544–1550. 10.1021/ja01875a062. [DOI] [Google Scholar]
- Berthet J. C.; Lance M.; Nierlich M.; Ephritikhine M. Synthesis of the Uranium Triflates U(OTf)3 and U(OTf)4 – Crystal Structure of [U(OTf)2(OPPh3)4][OTf]. Eur. J. Inorg. Chem. 1999, 1999, 2005–2007. . [DOI] [Google Scholar]
- Natrajan L.; Mazzanti M.; Bezombes J.-P.; Pecaut J. Practical Synthetic Routes to Solvates of U(OTf)3: X-Ray Crystal Structure of [U(OTf)3(MeCN)3]n, a Unique U(III) Coordination Polymer. Inorg. Chem. 2005, 44, 6115–6121. 10.1021/ic0505652. [DOI] [PubMed] [Google Scholar]
- Matonic J. H.; Scott B. L.; Neu M. P. High-Yield Synthesis and Single-Crystal X-Ray Structure of a Plutonium(III) Aquo Complex: [Pu(H2O)9][CF3SO3]3. Inorg. Chem. 2001, 40, 2638–2639. 10.1021/ic015509p. [DOI] [PubMed] [Google Scholar]
- Lindqvist-Reis P.; Apostolidis C.; Rebizant J.; Morgenstern A.; Klenze R.; Walter O.; Fanghänel T.; Haire R. G. The Structures and Optical Spectra of Hydrated Transplutonium Ions in the Solid State and in Solution. Angew. Chem., Int. Ed. 2007, 46, 919–922. 10.1002/anie.200603947. [DOI] [PubMed] [Google Scholar]
- Apostolidis C.; Schimmelpfennig B.; Magnani N.; Lindqvist-Reis P.; Walter O.; Sykora R.; Morgenstern A.; Colineau E.; Caciuffo R.; Klenze R.; Haire R. G.; Rebizant J.; Bruchertseifer F.; Fanghänel T. [An(H2O)9](CF3SO3)3 (An = U-Cm, Cf): Exploring Their Stability, Structural Chemistry, and Magnetic Behavior by Experiment and Theory. Angew. Chem., Int. Ed. 2010, 49, 6343–6347. 10.1002/anie.201001077. [DOI] [PubMed] [Google Scholar]
- Shannon R. D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, 32, 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Davis I. A.; Glowienka K. A.; Boll R. A.; Deal K. A.; Brechbiel M. W.; Stabin M.; Bochsler P. N.; Mirzadeh S.; Kennel S. J. Comparison of 225Actinium Chelates: Tissue Distribution and Radiotoxicity. Nucl. Med. Biol. 1999, 26, 581–589. 10.1016/S0969-8051(99)00024-4. [DOI] [PubMed] [Google Scholar]
- Chappell L. L.; Deal K. A.; Dadachova E.; Brechbiel M. W. Synthesis, Conjugation, and Radiolabeling of a Novel Bifunctional Chelating Agent for 225Ac Radioimmunotherapy Applications. Bioconjugate Chem. 2000, 11, 510–519. 10.1021/bc990153f. [DOI] [PubMed] [Google Scholar]
- McDevitt M. R.; Ma D.; Lai L. T.; Simon J.; Borchardt P.; Frank R. K.; Wu K.; Pellegrini V.; Curcio M. J.; Miederer M.; Bander N. H.; Scheinberg D. A. Tumor Therapy with Targeted Atomic Nanogenerators. Science 2001, 294, 1537–1540. 10.1126/science.1064126. [DOI] [PubMed] [Google Scholar]
- McDevitt M. R.; Ma D.; Simon J.; Frank R. K.; Scheinberg D. A. Design and Synthesis of 225Ac Radioimmunopharmaceuticals. Appl. Radiat. Isot. 2002, 57, 841–847. 10.1016/S0969-8043(02)00167-7. [DOI] [PubMed] [Google Scholar]
- Deal K. A.; Davis I. A.; Mirzadeh S.; Kennel S. J.; Brechbiel M. W. Improved in Vivo Stability of Actinium-225 Macrocyclic Complexes. J. Med. Chem. 1999, 42, 2988–2992. 10.1021/jm990141f. [DOI] [PubMed] [Google Scholar]
- Wilbur S. D. Chemical and Radiochemical Considerations for Radiolabelling with Alpha-Emitting Radionuclides. Curr. Radiopharm. 2011, 4, 214–247. 10.2174/1874471011104030214. [DOI] [PubMed] [Google Scholar]
- Taylor W. A.; Rundberg R. S.; Bond E. M.; Nortier F. M.; Vieira D. J. Production of a 173Lu Target for Neutron Capture Cross Section Measurements. J. Radioanal. Nucl. Chem. 2009, 282, 391–394. 10.1007/s10967-009-0278-1. [DOI] [Google Scholar]
- Wilson J. J.; Ferrier M.; Radchenko V.; Maassen J. R.; Engle J. W.; Batista E. R.; Martin R. L.; Nortier F. M.; Fassbender M. E.; John K. D.; Birnbaum E. R. Evaluation of Nitrogen-Rich Macrocyclic Ligands for the Chelation of Therapeutic Bismuth Radioisotopes. Nucl. Med. Biol. 2015, 42, 428–438. 10.1016/j.nucmedbio.2014.12.007. [DOI] [PubMed] [Google Scholar]
- Bateman H. The Solution of a System of Differential Equations Occuring in the Theory of Radioactive Transformations. Proc. Cambridge Philos. Soc. 1910, 15, 423–428. [Google Scholar]
- Ravel B.; Newville M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47, 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.