

Abstract
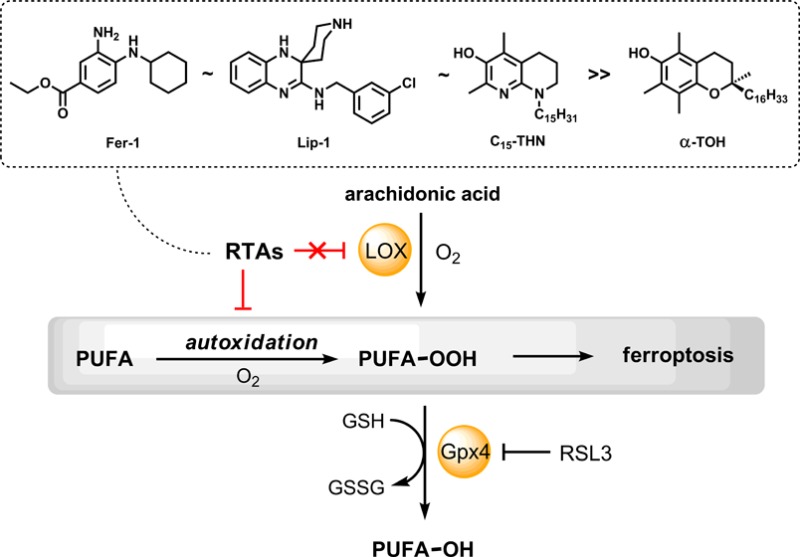
Ferroptosis is a form of regulated necrosis associated with the iron-dependent accumulation of lipid hydroperoxides that may play a key role in the pathogenesis of degenerative diseases in which lipid peroxidation has been implicated. High-throughput screening efforts have identified ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1) as potent inhibitors of ferroptosis − an activity that has been ascribed to their ability to slow the accumulation of lipid hydroperoxides. Herein we demonstrate that this activity likely derives from their reactivity as radical-trapping antioxidants (RTAs) rather than their potency as inhibitors of lipoxygenases. Although inhibited autoxidations of styrene revealed that Fer-1 and Lip-1 react roughly 10-fold more slowly with peroxyl radicals than reactions of α-tocopherol (α-TOH), they were significantly more reactive than α-TOH in phosphatidylcholine lipid bilayers − consistent with the greater potency of Fer-1 and Lip-1 relative to α-TOH as inhibitors of ferroptosis. None of Fer-1, Lip-1, and α-TOH inhibited human 15-lipoxygenase-1 (15-LOX-1) overexpressed in HEK-293 cells when assayed at concentrations where they inhibited ferroptosis. These results stand in stark contrast to those obtained with a known 15-LOX-1 inhibitor (PD146176), which was able to inhibit the enzyme at concentrations where it was effective in inhibiting ferroptosis. Given the likelihood that Fer-1 and Lip-1 subvert ferroptosis by inhibiting lipid peroxidation as RTAs, we evaluated the antiferroptotic potential of 1,8-tetrahydronaphthyridinols (hereafter THNs): rationally designed radical-trapping antioxidants of unparalleled reactivity. We show for the first time that the inherent reactivity of the THNs translates to cell culture, where lipophilic THNs were similarly effective to Fer-1 and Lip-1 at subverting ferroptosis induced by either pharmacological or genetic inhibition of the hydroperoxide-detoxifying enzyme Gpx4 in mouse fibroblasts, and glutamate-induced death of mouse hippocampal cells. These results demonstrate that potent RTAs subvert ferroptosis and suggest that lipid peroxidation (autoxidation) may play a central role in the process.
Short abstract
The potent ferroptosis inhibitors Fer-1 and Lip-1 are radical-trapping antioxidants (RTAs) superior to vitamin E, suggesting a critical role for lipid autoxidation in ferroptosis. Accordingly, potent designer RTAs are excellent ferroptosis inhibitors.
Introduction
The accumulation of lipid hydroperoxides (LOOH) has long been implicated in cell death and dysfunction, leading to aging,1,2 the onset and progression of degenerative disease,3,4 and cancer.5,6 However, only recently has the accumulation of LOOH been directly related to a specific cell death pathway, coined ferroptosis.7,8 Ferroptosis has been characterized as a form of regulated necrosis that is biochemically and morphologically distinct from apoptosis and autophagy, the more well-established cell death mechanisms.9−11 The induction of ferroptosis offers a new strategy for killing cancer cells, and disruption of the regulatory framework that keeps ferroptosis in check may contribute to the pathogenesis of degenerative diseases in which LOOH accumulation has been implicated.9,12,13
The accumulation of cellular LOOH occurs by two primary mechanisms: an iron-catalyzed spontaneous peroxyl radical-mediated process called autoxidation14,15 and enzyme-mediated processes catalyzed by (non-heme) iron-dependent lipoxygenases (LOXs).16,17 Accordingly, compounds that inhibit either or both of these processes have the potential to inhibit ferroptosis and may provide important leads for preventive and/or therapeutic agents to combat degenerative disease.
The Stockwell and Conrad groups recently independently
reported
the first potent inhibitors of ferroptosis: ferrostatin-1 (Fer-1)7 and liproxstatin-1 (Lip-1).18 Fer-1 and Lip-1 were discovered by high-throughput screening
of small molecule libraries using cell assays where ferroptosis was
induced by either deletion of the gene encoding the LOOH-detoxifying
enzyme glutathione peroxidase-4 (Gpx4)18 or pharmacological inhibition of system xc–, an antiporter that mediates the exchange of intracellular glutamate
for extracellular cystine used for glutathione (GSH) synthesis.7 Both compounds were found to suppress the accumulation
of LOOH,18,19 but the mechanism(s) by which they do so
is (are) unknown.20
Since lipid autoxidation (peroxidation) is one of the two processes that contribute directly to cellular LOOH production, compounds that trap the peroxyl radicals which propagate the radical chain reaction, i.e., radical-trapping antioxidants (RTAs),21 should be highly effective inhibitors of ferroptosis. Interestingly, both the Conrad and Stockwell groups found that α-tocopherol (α-TOH), the most biologically active form of vitamin E and Nature’s premier lipid-soluble RTA,22 is a relatively poor inhibitor of ferroptosis compared to either Fer-1 or Lip-1.18,19 These results suggest that either Fer-1 and Lip-1 are extremely potent RTAs or the inhibition of autoxidation may not be at the root of their activity. Indeed, Fer-1 and Lip-1 may be effective inhibitors of lipoxygenases, since α-TOH has been shown to be only a modest inhibitor at best.23,24 Herein we provide an assessment of both the RTA activity of Fer-1 and Lip-1 and their potency as inhibitors of human 15-lipoxygenase-1 (15-LOX-1, also sometimes referred to by its gene annotation ALOX15), the isoform recently implicated in ferroptosis.25−27
Over the years, we have made use of our comprehensive understanding of the structure–reactivity relationships in radical-trapping antioxidants to optimize the reactivity of phenolic RTAs.28−30 The tetrahydronaphthyridinols (THNs, Chart 1) are ca. 30-fold more reactive than α-TOH in organic solution,29−31 and in lipid bilayer models of cellular membranes (liposomes).32 Accordingly, if the prevention of' LOOH accumulation by autoxidation is important in subverting ferroptosis, it follows that the THNs should be highly effective inhibitors. However, to date these derivatives have not been studied in cell culture.33 Herein, we have assayed a small library of substituted THNs of varying lipid-solubility and report on their efficacy to inhibit ferroptosis in several established cell models, including (1) pharmacological inhibition of Gpx4 with (1S,3R)-RSL3,7,34 (2) deletion of the gene encoding Gpx4,18 and (3) GSH depletion via inhibition of the cystine/glutamate antiporter.27
Chart 1. Tetrahydronaphthyridinols (THNs).

Results
Fer-1 and Lip-1 Are Inherently Good, but Not Great, Radical-Trapping Antioxidants
To provide some insight into the mechanism of Fer-1 and Lip-1 as inhibitors of lipid peroxidation, their inherent RTA activities were determined by the venerable inhibited autoxidation of styrene approach pioneered by Ingold.35 Our modern twist on these experiments involves addition of an autoxidizable cosubstrate (PBD-BODIPY)36 such that the reaction can be monitored by spectrophotometry in lieu of conventional (but tedious) oxygen consumption measurements. Thus, reaction progress is monitored simply by loss of the absorbance at 591 nm due to the addition of peroxyl radicals to the 1-phenylbutadiene moiety of PBD-BODIPY (Figure 1A), which occurs with kPBD-BODIPY = 2720 M–1 s–1 in chlorobenzene at 37 °C.36 Styrene is present in order to maintain a radical chain reaction, ensuring a steady-state concentration of peroxyl radicals and enabling derivation of the rate constant for the reaction of the added RTA with peroxyl radicals (kinh) from the initial rate of the inhibited reaction (Figure 1B).36The stoichiometry (n) of the RTA–peroxyl reaction is determined from the duration of the inhibited period (tinh, also in Figure 1B).36
Figure 1.

PBD-BODIPY serves as the signal carrier in styrene autoxidations (A), enabling determination of rate constants (kinh) and stoichiometries (n) for reactions of inhibitors with chain-carrying peroxyl radicals (B). Coautoxidations of styrene (4.3 M) and PBD-BODIPY (10 μM) initiated by AIBN (6 mM) in chlorobenzene at 37 °C (black trace) and inhibited by 2 μM (red trace), 3 μM (green trace), and 4 μM (blue trace) of Fer-1 (C), Lip-1 (D), C15-THN (E), α-TOH (F), and PMHC (G). Average inhibition rate constants and stoichiometry summarized in panel H. Reaction progress was monitored by absorbance at 591 nm (ε = 139,000 M–1 cm–1). aFrom ref (29); the inhibition rate constant is greater than what can be determined from the data in panel E.
Representative results are shown for Fer-1 and Lip-1 in Figure 1C alongside corresponding data for α-TOH, its truncated analogue, 2,2,5,7,8-pentamethyl-6-hydroxychroman (PMHC), and a representative THN (C15-THN) for comparison. The traces clearly indicate that Fer-1 and Lip-1 are less reactive to peroxyl radicals than α-TOH (and PMHC). From the initial rates, rate constants for reactions of Fer-1 and Lip-1 with peroxyl radicals were determined to be (3.5 ± 0.1) and (2.4 ± 0.2) × 105 M–1 s–1, respectively − an order of magnitude lower than the rate constant determined for α-TOH of kinh = (3.6 ± 0.1) × 106 M–1 s–1. It is interesting to note that the rate constants determined for Fer-1 and Lip-1 are similar to those determined for conventional diarylamine antioxidants used as additives to petroleum-derived products,37 while the representative THN is so reactive that a rate constant for its reaction cannot be determined using this approach (using different methodology, but under similar conditions, a rate constant of (8.8 ± 3.2) × 107 M–1 s–1 was obtained).28,38 The inhibited periods indicates that while α-TOH, PMHC, and the THN each trap two peroxyl radicals, Fer-1 and Lip-1 trap roughly only one (actually 0.9 ± 0.1 and 1.3 ± 0.1, respectively). Interestingly, when the oxidizable substrate was changed from styrene to cumene, Fer-1 and Lip-1 trapped two peroxyl radicals, as do α-TOH, PMC, and the THN (see Supporting Information for the data).
Kinetic isotope effects of kinh,H/kinh,D = 2.2 ± 0.2 and 1.8 ± 0.1 were determined from autoxidations of Fer-1 and Lip-1, respectively, carried out in the presence of 1% of either MeOH or MeOD (see Supporting Information for the data). These values provide evidence for transfer of the aminic H-atom of Fer-1 and Lip-1 to chain-carrying peroxyl radicals − analogous to the mechanism of diarylamine radical-trapping antioxidants, as well as phenolic H-atom transfer from α-TOH, PMHC, and the THNs.29,39
Given the implication of H-atom transfer in the reactions of Fer-1 and Lip-1 with peroxyl radicals, we calculated the N–H bond dissociation enthalpies (BDEs) of Fer-1 and Lip-1 using the high accuracy CBS-QB3 methodology of Petersson and co-workers.40 The results are shown alongside the calculated structures of the compounds in Figure 2.41 The weakest N–H bonds in Fer-1 and Lip-1 are predicted to be 83.3 and 82.4 kcal/mol, respectively. These BDEs are significantly higher than the O−H BDEs in α-TOH and the THNs (77.7 and 75.0 kcal/mol calculated using the same methodology),42 consistent with the lower reactivity of Fer-1 and Lip-1 to peroxyl radicals. In fact, Fer-1 and Lip-1 have N–H BDEs similar to those of conventional diarylamine antioxidants (e.g., 4,4′-dialkyldiphenylamine N–H BDE = 82.2 kcal/mol),43 in line with their similar inherent reactivity.
Figure 2.
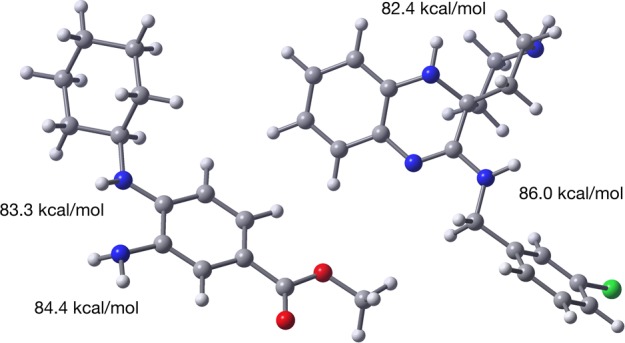
Computed (CBS-QB3) minimum energy structures of Fer-1 (left) and Lip-1 (right) and the corresponding N–H bond dissociation enthalpies of the labile aminic H atoms.
Fer-1 and Lip-1 Are Excellent Radical-Trapping Antioxidants in Phospholipid Bilayers
The reactivity of Fer-1 and Lip-1 to peroxyl radicals was also assayed in the lipid bilayers of unilamellar liposomes prepared from egg phosphatidylcholine (egg-PC). Reaction progress was followed by the competitive oxidation of STY-BODIPY (Figure 3), the more slowly oxidized analogue of PBD-BODIPY wherein the 1-phenylbutadienyl moiety is replaced with a styryl moiety.36 STY-BODIPY has been determined to react with peroxyl radicals with kp = 894 M–1 s–1 in this exact system.44 Under these conditions, Fer-1 and Lip-1 were both more reactive than α-TOH (kinh = (4.7 ± 0.4) × 103 M–1 s–1), with rate constants of kinh = (4.6 ± 0.8) × 104 and (1.2 ± 0.1) × 104 M–1 s–1, respectively. The inhibition rate constants derived for Fer-1 and Lip-1 are similar to that of PMHC (kinh= (3.7 ± 0.2) × 104 M–1 s–1), which is believed to have greater reactivity than α-TOH in lipid bilayers due to superior dynamics.45 Again, the THN was the most reactive of the compounds, with kinh = (9.3 ± 0.4) × 104 M–1 s–1. Perhaps most interestingly, while inhibited periods corresponding to the trapping of two peroxyl radicals are clear in the autoxidations inhibited by PMHC and the THN, Fer-1 and Lip-1 inhibited the autoxidations for longer. Moreover, the autoxidations inhibited by Lip-1 (and to a lesser extent Fer-1) never resume achieve the uninhibited rate, as do those inhibited by PMHC and the THN. This implies that the reactions of Fer-1 and Lip-1 with peroxyl radicals lead to compounds that remain reactive to peroxyl radicals and further retard the autoxidation past the nominal inhibition period corresponding to n ∼ 2.
Figure 3.

STY-BODIPY serves as the signal carrier in liposome oxidations (A), enabling determination of rate constants (kinh) and stoichiometries (n) for reactions of inhibitors with chain-carrying peroxyl radicals (B). Average inhibition rate constants and stoichiometry summarized in panel C. Coautoxidations of egg phosphatidylcholine lipids (1 mM) and STY-BODIPY (8 μM) suspended in phosphate-buffered saline (10 mM) at pH 7.4 initiated by MeOAMVN (0.2 mM) at 37 °C (black trace) and inhibited by 2 μM (red trace), 3 μM (green trace), and 4 μM (blue trace) of Lip-1 (D), Fer-1 (E), α-TOH (F), C15-THN (G), and PMHC (H). Reaction progress was monitored by absorbance at 565 nm (ε = 123,676 M–1 cm–1). aCould not be determined and is assumed to be 2.0.
Although it seems that improved dynamics are likely to contribute to the greater reactivity of Fer-1 and Lip-1 as RTAs in lipid bilayers compared to α-TOH, there must be more to it since they are similarly reactive to PMHC despite their far lower inherent reactivity to peroxyl radicals (as determined in chlorobenzene, vide supra). In fact, PMHC is a much poorer RTA in lipid bilayers than in hydrocarbons due to H-bonding interactions between the phenolic O–H and water and/or the phospholipid headgroups at the interface of the lipid and aqueous phases.46 When participating in a H-bond, the phenolic H-atom is no longer accessible to peroxyl radicals (Scheme 1).47 For example, upon changing solvent from chlorobenzene to DMSO, kinh for PMHC dropped 30-fold from (3.1 ± 0.2) × 106 to (1.1 ± 0.2) × 105 M–1 s–1. Fer-1 and Lip-1, being arylamines, are expected to be poorer H-bond donors than phenols, such as α-TOH or PMHC.
Scheme 1. Solvent Effects on the RTA Activity of Phenols, Such as PMHC (Shown) and a-TOH Are Larger than on Arylamines, Such as Fer-1 and Lip-1, Due to Stronger H-Bonding Interactions.

The α2H value (which quantifies H-bond donating ability)48 of a hindered monoarylamine closely related to Fer-1 and Lip-1 was determined to be 0.17,49 significantly lower than that of PMHC/α-TOH (0.37).50 Consistent with this small α2 value, the reactivity of Fer-1 and Lip-1 toward peroxyl radicals differed only by factors of 1.7 and 0.6 when measured in DMSO (an excellent H-bond accepting solvent) compared to chlorobenzene (see Supporting Information for details).
Fer-1 and Lip-1 Are Poor Inhibitors of 15-LOX-1 at Best, As Is α-TOH
The potencies of Fer-1 and Lip-1 as inhibitors of 15-LOX-1 were assessed in lysates of HEK-293 cells transfected to overexpress this isoform. Enzyme activity was determined following incubation of the cell lysate with the test compound by quantifying 15-hydroperoxyeicosatetraenoic acid (15-HPETE), the product of 15-LOX-1-catalyzed oxygenation of arachidonic acid, its natural substrate. In fact, the product was determined as the alcohol (15-HETE) by ESI-UPLC/MS/MS following reduction with TCEP and extraction from the lysate by ESI-UPLC/MS/MS. α-TOH, PMHC, and C15-THN were assayed alongside Fer-1 and Lip-1. The data are shown in Figure 4.
Figure 4.

Expression, functional characterization, and inhibition of 15-LOX-1 overexpressed in HEK-293 cells. Western blots showing the overexpression of 15-LOX-1 in transfected cells compared to wild-type (A). Structures of 5-, 12-, and 15-HETEs monitored by UPLC/MS/MS and their characteristic MS/MS transitions (B). Representative chromatograms of H(P)ETEs formed by wild-type and 15-LOX-1 overexpressing cells. (C) Effect of Fer-1, Lip-1, C15-THN, α-TOH, PMHC, and PD146176 on 15-H(P)ETE formation (D).
Lysates of wild-type HEK293 cells did not produce significant amounts of H(P)ETE when incubated with free arachidonic acid due to the low expression of 15-LOX-1 in these cells (Figure 4A–C). In contrast, enhanced formation of 15-H(P)ETE was clearly observed in the transfected cells. Fer-1 and Lip-1, which were each assayed up to 10 μM—almost 1000-fold higher than their EC50s for subverting RSL3-induced ferroptosis in these cells (15 and 27 nM, respectively)—did not exhibit significant inhibitory activity (Figure 4D). The small effect on 15-H(P)ETE levels relative to the (untreated) control is most likely due to inhibition of lipid autoxidation (which can be initiated by LOXs),51 since no dose–response is observed. α-TOH was also a poor inhibitor, in agreement with previous reports.18,34 In contrast, the 15-LOX-1 inhibitor PD146176 prevented 15-H(P)ETE production in a dose-dependent manner with similar potency as reported.52
Lipophilic THNs Are Potent Ferroptosis Inhibitors
Given the comparatively excellent RTA activity of the C15-THN relative to Fer-1 and Lip-1, the antiferroptotic activity of a library of THNs of differing lipophilicity32 was investigated. Ferroptosis was induced in Pfa-1 mouse fibroblasts by Gpx4 inhibition with (1S,3R)-RSL3,7 which was coadministered with the test compounds. The data are presented in Table 1. Although relatively hydrophilic THNs were ineffective inhibitors in these cells, lipophilic THNs were similarly potent to Fer-1 (EC50 = 45 ± 5 nM) and Lip-1 (EC50 = 38 ± 3 nM), with EC50 values of 13 ± 5 and 50 ± 2 nM for the C12- and C15-THNs, respectively. Clearly, there is an ideal side chain length for the THN, since the potency decreases as the side chain deviates from 12 carbon atoms (C15 has three tertiary sites with methyl branching, see Chart 1).
Table 1. Antiferroptotic Activity of THNs in Mouse Embryonic Fibroblastsa.
| EC50 (μM) | |
|---|---|
| C4-THN | >10 |
| C5-THN | >10 |
| C8-THN | 0.47 ± 0.16 |
| C10-THN | 0.37 ± 0.10 |
| C12-THN | 0.013 ± 0.005 |
| C15-THN | 0.050 ± 0.002 |
| C16-THN | 0.48 ± 0.09 |
| α-TOH | 1.8 ± 0.3 |
| PMHC | 0.059 ± 0.003 |
| Fer-1 | 0.045 ± 0.005 |
| Lip-1 | 0.038 ± 0.003 |
Ferroptosis was induced with (1S,3R)-RSL3 (100 nM), and cell survival was determined 6 h postinduction by AquaBluer18 assay. Data for α-TOH, Fer-1, and Lip-1 are also shown.
The THNs were also investigated in a genetic model of Gpx4 deficiency. This experiment enables resolution of any drug–drug interactions that may confound the results obtained by the pharmacological inhibition of Gpx4 (vide supra). Genetic disruption of Gpx4 was carried out in mouse embryonic fibroblasts carrying two floxed gpx4 alleles transfected to express the fusion protein MerCreMer, which is Cre recombinase (Cre) flanked by two mutated estrogen receptors (Mer) that maintain it in the cytosol.18 Upon treatment with 4-hydroxytamoxifen, Cre is released from its cytosolic complex, translocates to the nucleus, and mediates gpx4 gene deletion. The results of these experiments, shown in Figure 5A, were consistent with the trends observed upon pharmacological inhibition of Gpx4 using RSL3. That is, the lipophilic THNs were similarly potent to Fer-1 and Lip-1—all much more potent than α-TOH—whereas the hydrophilic THNs were ineffective. Interestingly, C12-THN again appeared to be the most potent THN.
Figure 5.

(A) Antiferroptotic activity of the THNs in mouse embryonic fibroblasts. Ferroptosis was initiated by tamoxifen-induced Cre-mediated deletion of the gpx4 gene, and cell survival was determined 6 h postinduction by AquaBluer assay. Corresponding data obtained for Fer-1, Lip-1, and α-TOH are also shown. (B) The THNs prevent glutamate-induced cell death in mouse HT22 hippocampal cells. Cells were treated with 5 mM glutamate and 100 nM test compound for 10 h, after which cell viability was assessed by AquaBluer assay. Corresponding data obtained for Fer-1, Lip-1, α-TOH, and β-mercaptoethanol (β-Me) are also shown. Cell viability was associated with inhibition of lipid peroxidation, as revealed by suppression of BODIPY-C11581/591 oxidation.
The cytoprotective abilities of the THNs were also compared to Fer-1 and Lip-1 in HT22 mouse hippocampal cells, wherein cell death was induced by high extracellular concentrations of glutamate. These conditions inhibit the cystine/glutamate antiporter (system xc–), depleting cells of cystine and resulting in the rapid depletion of GSH.53 Cells treated with β-mercaptoethanol (β-Me) were employed as a positive control. β-Me undergoes disulfide exchange with cystine, and the resultant mixed disulfide can be taken up by the cells independently of system xc–, thereby supplying cysteine for GSH synthesis.54 Cell viability was assayed using AquaBluer, and the data are presented in Figure 5B. Consistent with the foregoing results, the lipophilic THNs were again similarly effective to Lip-1 and Fer-1. Parallel studies indicated that cell survival coincided with the inhibition of lipid peroxidation (Figure 5B) and not with sustaining and/or restoring GSH levels (see Supporting Information).
Discussion
The mechanism by which Fer-1 and Lip-1 prevent cell death brought about by Gpx4 inhibition, gpx4 deletion, or GSH depletion has been ascribed to their ability to inhibit lipid peroxidation directly by trapping chain-carrying radicals.19 Both compounds are arylamines, and decades of research have shown that arylamines are good radical-trapping antioxidants.21,56 However, arylamines are generally only moderately reactive at ambient temperatures; they require temperatures above 100 °C to achieve the catalytic reactivity that makes them essential additives to engine lubricants, rubber, and many other petroleum-derived products. In fact, industry standard alkylated diphenylamines react ca. 20-fold more slowly with peroxyl radicals than does α-TOH at ambient temperatures (kinh = 1.8 × 105 and 3.2 × 106 M–1 s–1 for the former and latter, respectively, measured in chlorobenzene at 37 °C),30 which is inconsistent with the fact that Fer-1 and Lip-1 are much more effective than α-TOH at preventing ferroptosis. At first glance, this suggests that Fer-1 and Lip-1 possess a different mechanism of action and/or that lipid peroxidation does not contribute significantly to ferroptosis.
The kinetic studies detailed above reveal that Fer-1 and Lip-1 have inherent reactivities to peroxyl radicals that are almost indistinguishable from those of alkylated diphenylamines, with rate constants of 3.1 × 105 M–1 s–1 measured in chlorobenzene at 37 °C. Although these rate constants are still an order of magnitude lower than that measured for α-TOH under the same conditions, this difference is erased in lipid bilayers. In fact, Fer-1 and Lip-1 are both significantly more reactive than α-TOH in unilamellar phosphatidylcholine liposomes. The origin of the difference in reactivity on going from a nonpolar organic solvent (chlorobenzene) to lipid bilayers is revealed by additional experiments carried out using PMHC, an analogue of α-TOH in which the −C16H33 side chain is truncated to a methyl group. The reactivity of PMHC in lipid bilayers is roughly 10-fold greater than that of α-TOH, consistent with previous observations32,45 and the notion that the lengthy side chain leads to poorer dynamics in the bilayer. This is compounded by the fact that PMHC (and α-TOH) is significantly less reactive to peroxyl radicals in H-bond accepting media—such as the lipid/water interface of the lipid bilayers—since the phenolic H-atom can be engaged in a H-bond, precluding its transfer to peroxyl radicals. Fer-1 and Lip-1, being amines, are weaker H-bond donors, making them relatively insensitive to interactions with H-bond accepting entities and consequently more reactive to radicals.
Although weaker H-bonding interactions and better dynamics account for the greater reactivity of Fer-1 and Lip-1 to peroxyl radicals in lipid bilayers compared to α-TOH, they also suggest that the potency of α-TOH should be greater if its lipophilic side chain was truncated. Indeed, PMHC was found to have activity similar to that of Fer-1 and Lip-1 when ferroptosis was induced by Gpx4 inhibition with RSL3 (EC50 = 59 ± 3 nM compared to 45 ± 5 and 38 ± 3 nM for Fer-1 and Lip-1, respectively). While this is a useful mechanistic experiment, PMHC is ineffective in vivo because the side chain of tocopherols is essential for their bioavailability.57,58
Interestingly, Lip-1 (and, to a lesser extent, Fer-1) is more effective in subverting ferroptosis compared to PMHC despite its lower reactivity toward peroxyl radicals in lipid bilayers (smaller kinh). We wondered if this difference could be ascribed to the fact that Lip-1 (and, to a lesser extent, Fer-1) exhibits a greater capacity to trap peroxyl radicals (larger n) in lipid bilayers. Indeed, in contrast to PMHC (and α-TOH), the reaction stoichiometry measured for the reaction of Lip-1 (and Fer-1) with peroxyl radicals was highly variant with medium. That is, while PMHC and α-TOH always displayed n = 2, Lip-1 (and Fer-1) was characterized by n ∼ 1 in styrene autoxidations, n ∼ 2 in cumene autoxidations, and n ≫ 2 in phosphatidylcholine lipid bilayers. This implies a different fate of the aminyl radical that is formed following initial H-atom transfer under the different autoxidation conditions. The different behavior of Lip-1 (and Fer-1) in styrene and cumene autoxidations is reminiscent of the behavior of catechol and hydroquinone RTAs.59 In styrene autoxidations, where there is a low steady-state concentration of radicals due to rapid termination of the secondary peroxyl radicals (kt = 2.1 × 107 M–1 s–1),67 the aminyl radicals that are formed following H-atom transfer to styrylperoxyl radicals have time to react with O2, eroding n. In contrast, cumene autoxidations feature much higher steady-state concentration of radicals due to comparatively slow termination (kt = 2.3 × 104 M–1 s–1),68 preventing erosion of n.
The observation that n ≫ 2 for Lip-1 (and Fer-1) in phospholipid bilayers implies that their oxidation products are capable of trapping additional radicals. Two possibilities stemming from the key aminyl radical intermediate are illustrated in Scheme 2. In path a, H-atom transfer from the aminyl radical to a second peroxyl radical yields the highly electrophilic o-quinone diimide, which reacts with water to restore the reactive N–H bonds of Lip-1 following tautomerization. The requirement for water to both hydrate the o-quinone diimide and drive tautomerization would explain why this is not observed in the hydrocarbon autoxidations that were carried out only in the phospholipid liposome autoxidations. In path b, the intermediate aminyl radical reacts with a second peroxyl radical to produce a nitroxide. Although this is not a radical-trapping step (an alkoxyl radical is produced), the nitroxide that is formed is capable of trapping radicals in a catalytic fashion: first by reaction with a peroxyl radical and then by reaction with an alkyl radical to restore the nitroxide from the oxoammonium ion.60 The electron transfer processes key to this cycle would be facilitated in the polar interfacial region of the phospholipid liposomes compared to neat hydrocarbons, again consistent with our observations in the different media.
Scheme 2. Increased Radical-Trapping Capacity of Lip-1 (shown) and Fer-1 (not shown) Relative to Phenolic Antioxidants Such as α-TOH and PMHC Arises Due to their Ability to Form Products that Remain Highly Reactive to Peroxyl Radicals.

In order to provide support for either of these mechanisms,
we
carried out additional experiments with the dihydroquinoline 1, which features a similarly hindered aryl N–H to
Lip-1.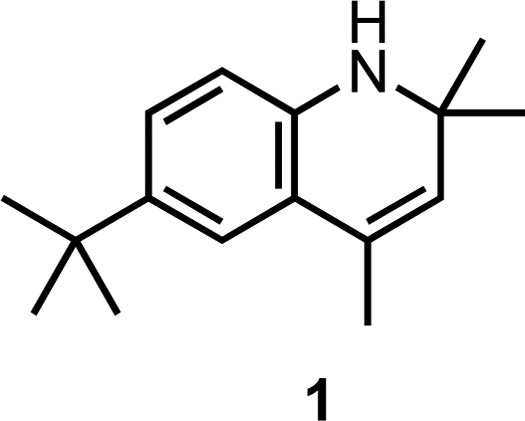
Although 1 is predicted to have an essentially identical N–H BDE to Lip-1 (calculated using CBS-QB3 to be 82.8 kcal/mol, compared to 82.7 kcal/mol for Lip-1), and can therefore be expected to transfer its aminic H-atom to a peroxyl radical in a similar fashion, it cannot undergo a second H-atom transfer reaction to form an o-quinone diimide intermediate. Nevertheless, autoxidations of phosphatidylcholine liposomes inhibited by 1 were strikingly similar to those inhibited by Lip-1, with only a slightly larger kinh of (2.2 ± 0.2) × 104 M–1 s–1, and most importantly, featuring n ≫ 2 (Figure 6A). These results suggests that path a in Scheme 2 is unlikely to be responsible for the increased radical-trapping capacity of Lip-1 compared to conventional RTAs. Moreover, 1 was effective in subverting ferroptosis in mouse fibroblasts treated with RSL3, with a potency (EC50 = 77 nM) similar to that determined for Lip-1 (EC50 = 38 nM).
Figure 6.

Coautoxidations of egg phosphatidylcholine lipids (1 mM) and STY-BODIPY (8 μM) suspended in phosphate-buffered saline (10 mM) at pH 7.4 initiated by MeOAMVN (0.2 mM) at 37 °C (black trace) and inhibited by 2 μM (red trace), 3 μM (green trace), and 4 μM (blue trace) of 1 (dotted line) and Lip-1 (solid line) (A), EPR spectrum of the nitroxide derived from 1 (B), and corresponding coautoxidations inhibited by 1 (dotted line) and the nitroxide derived therefrom (dashed line) (C). Reaction progress in the coautoxidations was monitored by absorbance at 565 nm (ε = 123,676 M–1 cm–1).
Strong evidence in support of the mechanism in path b of Scheme 2 would best include the independent preparation of the Lip-1-derived nitroxide and demonstration that it can also inhibit lipid peroxidation. Unfortunately, our attempts to independently prepare the authentic Lip-1-derived nitroxide were unsuccessful, leading to intractable mixtures of products that included at least two nitroxides, as determined by electron paramagnetic resonance (EPR) spectroscopy (see Supporting Information for example spectra). However, we were able to prepare the nitroxide derived from the dihydroquinoline 1 (see EPR spectrum in Figure 6B), and found that it was a very good RTA in this system (Figure 6C). The duration of the nominal inhibited period of the nitroxide is less than that of the amine since the amine must trap a radical prior to being converted to the nitroxide. However, the rate of oxidation following the inhibited period is indistinguishable between the amine and the nitroxide—both being less than the uninhibited rate—consistent with the formation of a common intermediate that can still retard the oxidation as suggested in Scheme 1. The nitroxide was also capable of subverting ferroptosis; we found EC50 = 99 ± 5 nM under the same conditions that yielded 77 nM for 1 and 38 nM for Lip-1. Interestingly, Wipf and co-workers recently reported that mitochondrially targeted nitroxide derivatives are good inhibitors of ferroptotic cell death,61 their best examples being slightly less effective than Fer-1, and therefore similar to the nitroxide derived from dihydroquinoline 1.
The central role of lipid peroxidation in ferroptosis—and radical-trapping antioxidants in subverting it—is further supported by the lack of significant inhibitory activity of Fer-1 and Lip-1 on lipoxygenase catalysis. Each of Fer-1, Lip-1, and α-TOH was unable to inhibit arachidonic acid oxidation to 15-H(P)ETE in lysates of HEK-293 cells transfected to overexpress 15-LOX-1 when treated up to 10 μM, almost 3 orders of magnitude greater than the concentration at which Fer-1 and Lip-1 inhibit ferroptosis in the same cells (EC50 = 15 and 27 nM, respectively). The suppression of 15-H(P)ETE formation that is observed at low concentrations of these compounds must be due to the inhibition of arachidonic acid autoxidation that occurs under the assay conditions, indicated by both the lack of a dose–response for Fer-1, Lip-1, and α-TOH that is clearly observed when 15-H(P)ETE formation is suppressed (fully) by the known 15-LOX-1 inhibitor PD146176 and also by suppression of other regioisomeric H(P)ETEs that arise from autoxidation (e.g., 5- and 12-H(P)ETE, see Supporting Information). Despite being a better 15-LOX-1 inhibitor than Fer-1, Lip-1, and α-TOH, PD146176 is a relatively poor inhibitor of ferroptosis, with no inhibition of RSL-3-induced cell death up to 10 μM in the 15-LOX-1 overexpressing cells, and an EC50 of 8.5 μM in the wild-type cells. It should be pointed out that Angeli et al. showed that 1 μM PD146176 was effective at subverting ferroptosis brought about by gpx4 deletion in mouse embryonic fibroblasts18 and Yang et al. showed that 5 μM PD146176 subverted erastin-induced ferroptosis in HT-1080 human fibrosarcoma cells.25 Consistent with our own observations, Angeli and Yang each found that Fer-1 and/or Lip-1 (and α-TOH) was effective at significantly lower concentrations under the same assay conditions.62
Although 15-LOX-1 is but one of six isoforms of lipoxygenase that occur in humans, it is the isoform that has been implicated in several recent studies linking lipoxygenase catalysis and ferroptosis.25,26,53,63 One such study suggested that α-TOH inhibits ferroptosis by LO inhibition.63 The basis for this assertion was that tocotrienols were demonstrably more effective inhibitors of ferroptosis than tocopherols, and tocotrienols were predicted to bind more strongly to 15-LOX-1 than tocopherols owing to the three unsaturations in their lipophilic side chains. An alternative explanation must exist in order to reconcile our observation that removal of α-TOH’s side chain improves its potency as an inhibitor of ferroptosis. Indeed, it is known that tocotrienols are incorporated into cells more quickly than tocopherols;64 thus, their increased potency in cell assays may simply reflect a greater concentration in the cell at the time of the assay. We wonder if alternative explanations may account for other observations that have prompted suggestions that LOXs drive ferroptosis.
Since the results of our studies on the mechanism by which Fer-1 and Lip-1 subvert ferroptosis pointed strongly at their ability to prevent lipid peroxidation by trapping chain-carrying peroxyl radicals, we surmised that the THNs should be excellent ferroptosis inhibitors. This proved to be the case. Lipophilic THNs were similarly potent to Fer-1 and Lip-1 in three different established cell models of ferroptosis (RSL3-inhibition of Gpx4, Cre-mediated deletion of gpx4, and glutamate inhibition of cystine uptake). Consistent with our recent studies on the radical-trapping antioxidant activity of the same set of THNs in lipid bilayers,32 the more hydrophilic derivatives were significantly less potent, suggesting poor cell incorporation and/or their facile autoxidation in the cytosol or cell culture medium. Similarly to Fer-1, Lip-1, and α-TOH, the THNs were not effective inhibitors of 15-LOX-1. These results reinforce the notion that, in the cell types that we have studied here, nonenzymatic lipid peroxidation drives ferroptosis. However, it must be acknowledged that lipid peroxidation and lipoxygenase catalysis may contribute differently to ferroptosis in other cell types. Further investigation is clearly necessary, as is the need to understand why phosphoethanolamine-esterified polyunsaturated fatty acids appear to play a particularly important role in ferroptosis. Regardless, we anticipate that the foregoing results, and the insights they afford on the radical-trapping mechanisms of Lip-1 and Fer-1, will enable the development of improved inhibitors of ferroptosis that may be useful as therapeutics in diseases where ferroptosis is likely to contribute.
Experimental Methods
Materials
The THNs,32 Fer-1,19 PBD-BODIPY,36 and STY-BODIPY36 were synthesized according to literature procedures, and Lip-1 was synthesized as described in the Supporting Information. Egg phosphatidylcholine, AIBN, MeOAMVN, BODIPY-C11581/591, RPMI-1640 media with/without phenol red, (D)MEM with/without phenol red, Dulbecco’s phosphate-buffered saline (DPBS), fetal bovine serum (FBS), penicillin–streptomycin, AquaBluer, and reagents for synthesis were purchased from commercial sources and used as received. Autoxidations employed HPLC grade solvents.
Inhibited Autoxidation of Styrene
These experiments were carried out in a manner similar to that described in our previous work.36 In brief, styrene (Sigma-Aldrich) was washed thrice with 1 M aqueous NaOH, dried over MgSO4, filtered, distilled under vacuum, and purified by percolating through silica, then basic alumina. To a cuvette of 1.25 mL of styrene was added 1.18 mL of chlorobenzene, and the solution equilibrated for 5 min at 37 °C. The cuvette was blanked, 12.5 μL of 2 mM PBD-BODIPY in 1,2,4-trichlorobenzene was added followed by 50 μL of 0.3 M AIBN in chlorobenzene, and the solution was thoroughly mixed. After 20 min, an aliquot of Lip-1, Fer-1, C15-THN, PMHC, or α-TOH stock solution (1 mM) in chlorobenzene was added and the loss of absorbance at 591 nm followed. The inhibition rate constant (kinh) and stoichiometry (n) were determined for each experiment according to Figure 1B (see the Supporting Information for complete details). Autoxidations were carried out with three technical replicates at each concentration, and kinetics are reported as the mean ± standard deviation.
Inhibited Autoxidation of PC Liposomes
To a cuvette of 2.34 mL of 10 mM PBS at pH 7.4 were added liposomes (125 μL of 20 mM stock in PBS at pH 7.4),32 and the solution was equilibrated for 5 min at 37 °C. The cuvette was blanked, 10 μL of 2 mM STY-BODIPY in DMSO was added followed by 10 μL of 0.05 M MeOAMVN in acetonitrile, and the solution was thoroughly mixed. After 5 min, an aliquot of Lip-1, Fer-1, C15-THN, PMHC, or α-TOH stock solution (1 mM) in DMSO was added and the loss of absorbance at 565 nm followed. The inhibition rate constant (kinh) and stoichiometry (n) were determined for each experiment according to Figure 3B (see the Supporting (see the Supporting Information for complete details). Autoxidations were carried out with three technical replicates at each concentration, and kinetics are reported as the mean ± standard deviation. Indistinguishable results were obtained in select control experiments where the antioxidant was added prior to liposome extrusion.
Cell Culture
All cell lines were cultured at 37 °C in a 5% CO2 atmosphere unless otherwise indicated. HEK-293 cells were cultured in MEM with 10% FBS, 1% 100× nonessential amino acid solution, 1 mM sodium pyruvate, and 1% penicillin–streptomycin. Pfa-1 mouse embryonic fibroblasts27 were cultured in DMEM containing 10% FBS, 10 mM glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin. HT22 cells were cultured in DMEM containing 10% FBS, 10 mM glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin in a 10% CO2 atmosphere. Cells were passaged by dissociation with 0.05% trypsin and 0.2% EDTA every other day.
Overexpression of 15-LOX-1 in HEK 293 Cells
Cells were plated to 30% confluency and were transfected with a recombinant pcDNA3/15-LOX-1 construct65 using LipoJet reagent according to the manufacturer’s recommendations. The cells were passaged to 35 mm plates after 1 day, and colonies were screened using selection medium containing 1 mg/mL Geneticin. After establishing a stable cell line, the 15-LOX-1 cells were cultured in MEM with 10% FBS, 1% 100× nonessential amino acid solution, 10 mM glutamine, and 1 mg/mL Geneticin.
Western Blot Analysis
Protein extracts were prepared by lysing either HEK 293 cells or 15-LOX-1 cells in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) protein loading buffer (62.5 mM Tris [pH 6.8], 25% glycerol, 2% SDS, 0.1% bromophenol blue, 5% 2-mercaptoethanol). After separation by 10% PAGE, the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Immuno-Blot PVDF; Bio-Rad Laboratories) and blocked in 5% dry milk, Tris-buffered saline. The membrane was probed with an antibody specific for 15-LOX-1 (Novusbio). Binding of the primary antibody was detected using a goat anti-mouse secondary antibody conjugated to horseradish peroxidase (Novusbio) and visualized by ECL chemiluminescence reaction (Pierce ECL, Thermo Scientific).
Determination of 15-LOX-1 Products by UPLC/MS/MS.62
15-LOX-1 overexpressing cells were harvested and lysed in Tris-HCl (pH 7.4) in the presence of 1% protease inhibitor cocktail (Sigma-Aldrich). The residue was vortexed at 10,000 rpm for 10 min, and the supernatants were incubated with 70 μM arachidonic acid (AA, Nu-Chek Prep) at 37 °C for 10 min. The reactions were terminated with 1 volume of methanol followed by TCEP (1.5 mg/mL, Sigma-Aldrich). The samples were stored at room temperature for 90 min. For inhibition studies, incubations with inhibitors were carried out for 10 min on ice prior to the addition of AA. Briefly, 10 μM prostaglandin B2 (Cayman Chemical) was added as internal standard along with 15 μL of 1 M HCl for acidification followed by extraction using C-18 solid phase extraction columns (Thermo Scientific) as per the manufacturer’s protocol. The eluent in methanol was evaporated to dryness and resuspended in 100 μL of methanol. Aliquots (5 μL) were injected on a Waters Acquity C18 column (2.1 mm × 50 mm, 1.7 μm particle size) in a Waters Acquity Ultra-Performance Liquid Chromatography (UPLC) system. HETEs were eluted isocratically with a mixture of acetonitrile, methanol, water, and acetic acid (42:25:33:0.007) and detected using MS/MS analysis. Eicosanoids were detected in the negative ion mode by multiple reaction monitoring of m/z 333 → 235 for PGB2, m/z 319 → 115 for 5-HETE, m/z 319 → 179 for 12-HETE, and m/z 319 → 219 for 15-HETE. Each experiment was performed with three technical replicates and reproduced independently three times with error bars representing standard deviation from the mean.
Inhibition of Ferroptosis Induced by Gpx4 Inhibition with (1S,3R)-RSL3
Pfa-1 cells (3,000 in 100 μL) were seeded in 96-well plates and cultured for 6 h, after which a mixture of linoleic acid (10 μM) and BSA (0.1%) was added and the cells were incubated overnight in order to sensitize to ferroptosis. The next day the medium was removed, the cells were washed twice with PBS, and the cells were suspended in new medium for 30 min before addition of (1S,3R)-RSL3 (100 nM) in a final volume of 100 μL. Cell viability was assessed 6 h later using the AquaBluer (MultiTarget Pharmaceuticals, LLC) assay according to the manufacturer’s instructions. Cell viability was calculated by normalizing the data to untreated controls.
Inhibition of Ferroptosis Induced by gpx4 Deletion
Tamoxifen-inducible Gpx4—/— Pfa-1 cells27 (3,000 in 100 μL) were seeded in 96-well plates and cultured for 48 h in the presence of 1 μM tamoxifen, after which the medium was replaced with medium containing the test compounds (30 or 100 nM). Cell viability was determined 6 h after the medium change using the AquaBluer assay according to the manufacturer’s instructions. Cell viability was calculated by normalizing the data to untreated controls.
Inhibition of Glutamate Toxicity and Cell Death in Mouse Hippocampal Cells
HT22 cells (30,000 in 100 μL) were seeded in 96-well plates and cultured for 24 h, after which the test compounds were added (100 nM), followed by glutamate (to 5 mM). Cell viability was assessed 10 h later using AquaBluer. Lipid peroxidation levels were determined in a similar way. Cells (100,000 in 2 mL) were seeded in 6-well plates, and glutamate and compounds were added 24 h later for 10 h. BODIPY-C11581/591 (1 μM) was then added for 30 min, and the cells were trypsinized, washed, and resuspended in PBS. Flow cytometry was performed using a 488 nm line argon laser for excitation, and emission was recorded on channels FL1 at 530 nm and FL2 at 585 nm. Data were collected from at least 30,000 cells. Total glutathione in the cells was measured by the enzymatic method described previously,66 which is based on the catalytic action of the glutathione reductase system.
Acknowledgments
We are grateful to Prof. Colin Funk of Queen’s University for providing the construct for 15-LOX-1 overexpression. This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada, the Canada Foundation for Innovation to D.A.P., and Human Frontier Science Program (HFSP) RGP0013 to M.C. D.A.P. and R.S. acknowledge support from the Canada Research Chairs program and the NSERC Post-Graduate Scholarship program, respectively.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00028.
Synthetic procedures, inhibited autoxidations of cumene in chlorobenzene and THF in DMSO, EPR spectra, computational details, and NMR spectra (PDF)
The authors declare no competing financial interest.
Due to a production error, this article published March 7, 2017 with an incorrect version of the Supporting Information. The correct version published March 8, 2017.
Supplementary Material
References
- Finkel T.; Holbrook N. J. Oxidants, Oxidative Stress and the Biology of Ageing. Nature 2000, 408, 239–247. 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Vitale G.; Salvioli S.; Franceschi C. Oxidative Stress and the Ageing Endocrine System. Nat. Rev. Endocrinol. 2013, 9, 228–240. 10.1038/nrendo.2013.29. [DOI] [PubMed] [Google Scholar]
- Di Paolo G.; Kim T.-W. Linking Lipids to Alzheimer’s Disease: Cholesterol and Beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnham K. J.; Masters C. L.; Bush A. I. Neurodegenerative Diseases and Oxidative Stress. Nat. Rev. Drug Discovery 2004, 3, 205–214. 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- Benz C. C.; Yau C. Ageing, Oxidative Stress and Cancer: Paradigms in Parallax. Nat. Rev. Cancer 2008, 8, 875–879. 10.1038/nrc2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S. P.; Hofseth L. J.; Harris C. C. Radical Causes of Cancer. Nat. Rev. Cancer 2003, 3, 276–285. 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- Dixon S. J.; Lemberg K. M.; Lamprecht M. R.; Skouta R.; Zaitsev E. M.; Gleason C. E.; Patel D. N.; Bauer A. J.; Cantley A. M.; Yang W. S.; Morrison B.; Stockwell B. R. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. S.; Stockwell B. R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M.; Angeli J. P. F.; Vandenabeele P.; Stockwell B. R. Regulated Necrosis: Disease Relevance and Therapeutic Opportunities. Nat. Rev. Drug Discovery 2016, 15, 348–366. 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trump B. F.; Berezesky I. K.; Chang S. H.; Phelps P. C. The Pathways of Cell Death: Oncosis, Apoptosis, and Necrosis. Toxicol. Pathol. 1997, 25, 82–88. 10.1177/019262339702500116. [DOI] [PubMed] [Google Scholar]
- Edinger A. L.; Thompson C. B. Death by Design: Apoptosis, Necrosis and Autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Hou W.; Song X.; Yu Y.; Huang J.; Sun X.; Kang R.; Tang D. Ferroptosis: process and function. Cell Death Differ. 2016, 23, 369–379. 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler M. M.; Stockwell B. R. Lipid Peroxidation in Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Xu L.; Porter N. A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- Porter N. A. Mechanisms for the Autoxidation of Polyunsaturated Lipids. Acc. Chem. Res. 1986, 19, 262–268. 10.1021/ar00129a001. [DOI] [Google Scholar]; Pratt D. A.; Tallman K. A.; Porter N. A. Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Acc. Chem. Res. 2011, 44, 458–467. 10.1021/ar200024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeggström J. Z.; Funk C. D. Lipoxygenase and Leukotriene Pathways: Biochemistry, Biology, and Roles in Disease. Chem. Rev. 2011, 111, 5866–5898. 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- Girotti A. W. Lipid Hydroperoxide Generation, Turnover, and Effector Action in Biological Systems. J. Lipid Res. 1998, 39, 1529–1542. [PubMed] [Google Scholar]
- Friedmann Angeli J. P.; Schneider M.; Proneth B.; Tyurina Y. Y.; Tyurin V. A.; Hammond V. J.; Herbach N.; Aichler M.; Walch A.; Eggenhofer E.; Basavarajappa D.; Radmark O.; Kobayashi S.; Seibt T.; Beck H.; Neff F.; Esposito I.; Wanke R.; Forster H.; Yefremova O.; Heinrichmeyer M.; Bornkamm G. W.; Geissler E. K.; Thomas S. B.; Stockwell B. R.; O’Donnell V. B.; Kagan V. E.; Schick J. A.; Conrad M. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouta R.; Dixon S. J.; Wang J.; Dunn D. E.; Orman M.; Shimada K.; Rosenberg P. A.; Lo D. C.; Weinberg J. M.; Linkermann A.; Stockwell B. R. Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J. Am. Chem. Soc. 2014, 136, 4551–4556. 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevertheless, the development of these compounds continues. See, e.g.:Hofmans S.; Berghe T. V.; Devisscher L.; Hassannia B.; Lyssens S.; Joossens J.; Van Der Veken P.; Vandenabeele P.; Augustyns K. Novel Ferroptosis Inhibitors with Improved Potency and ADME Properties. J. Med. Chem. 2016, 59, 2041–2053. 10.1021/acs.jmedchem.5b01641. [DOI] [PubMed] [Google Scholar]
- Ingold K. U.; Pratt D. A. Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: a Kinetics and Mechanisms Perspective. Chem. Rev. 2014, 114, 9022–9046. 10.1021/cr500226n. [DOI] [PubMed] [Google Scholar]
- Burton G. W.; Ingold K. U. Vitamin E: Application of the Principles of Physical Organic Chemistry to the Exploration of Its Structure and Function. Acc. Chem. Res. 1986, 19, 194–201. 10.1021/ar00127a001. [DOI] [Google Scholar]
- Reddanna P.; Rao M. K.; Reddy C. C. Inhibition of 5-Lipoxygenase by Vitamin-E. FEBS Lett. 1985, 193, 39–43. 10.1016/0014-5793(85)80075-2. [DOI] [PubMed] [Google Scholar]
- Grossman S.; Waksman E. G. New Aspects of the Inhibition of Soybean Lipoxygenase by Alpha-Tocopherol. Evidence for the Existence of a Specific Complex. Int. J. Biochem. 1984, 16, 281–289. 10.1016/0020-711X(84)90101-0. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; Kim K. J.; Gaschler M. M.; Patel M.; Shchepinov M. S.; Stockwell B. R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4966–E4975. 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y.; Wang S.-J.; Li D.; Chu B.; Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E6806–E6812. 10.1073/pnas.1607152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler A.; Schneider M.; Förster H.; Roth S.; Wirth E.; Culmsee C.; Plesnila N.; Kremmer E.; Rådmark O.; Wurst W.; Bornkamm G.; Schweizer U.; Conrad M. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Pratt D. A.; DiLabio G. A.; Brigati G.; Pedulli G. F.; Valgimigli L. 5-Pyrimidinols: Novel Chain-Breaking Antioxidants More Effective Than Phenols. J. Am. Chem. Soc. 2001, 123, 4625–4626. 10.1021/ja005679l. [DOI] [PubMed] [Google Scholar]
- Wijtmans M.; Pratt D. A.; Valgimigli L.; DiLabio G. A.; Pedulli G. F.; Porter N. A. 6-Amino-3-Pyridinols: Towards Diffusion-Controlled Chain-Breaking Antioxidants. Angew. Chem., Int. Ed. 2003, 42, 4370–4373. 10.1002/anie.200351881. [DOI] [PubMed] [Google Scholar]
- Valgimigli L.; Pratt D. A. Maximizing the Reactivity of Phenolic and Aminic Radical-Trapping Antioxidants: Just Add Nitrogen!. Acc. Chem. Res. 2015, 48, 966–975. 10.1021/acs.accounts.5b00035. [DOI] [PubMed] [Google Scholar]
- Nam T.-G.; Rector C. L.; Kim H.-Y.; Sonnen A. F.-P.; Meyer R.; Nau W. M.; Atkinson J.; Rintoul J.; Pratt D. A.; Porter N. A. Tetrahydro-1,8-Naphthyridinol Analogues of Alpha-Tocopherol as Antioxidants in Lipid Membranes and Low-Density Lipoproteins. J. Am. Chem. Soc. 2007, 129, 10211–10219. 10.1021/ja072371m. [DOI] [PubMed] [Google Scholar]
- Li B.; Harjani J. R.; Cormier N. S.; Madarati H.; Atkinson J.; Cosa G.; Pratt D. A. Besting Vitamin E: Sidechain Substitution Is Key to the Reactivity of Naphthyridinol Antioxidants in Lipid Bilayers. J. Am. Chem. Soc. 2013, 135, 1394–1405. 10.1021/ja309153x. [DOI] [PubMed] [Google Scholar]
- We should note that the Hecht group has extensively studied the somewhat related, but less reactive,28,29 pyridinol and pyrimidinol derivatives as inhibitors of oxidative stress and associated mitochondrial dysfunction in cell culture. See, e.g.:; Mastroeni D.; Khdour O. M.; Arce P. M.; Hecht S. M.; Coleman P. D. Novel Antioxidants Protect Mitochondria From the Effects of Oligomeric Amyloid Beta and Contribute to the Maintenance of Epigenome Function. ACS Chem. Neurosci. 2015, 6, 588–598. 10.1021/cn500323q. [DOI] [PMC free article] [PubMed] [Google Scholar]; Alam M. P.; Khdour O. M.; Arce P. M.; Chen Y.; Roy B.; Johnson W. G.; Dey S.; Hecht S. M. Cytoprotective Pyridinol Antioxidants as Potential Therapeutic Agents for Neurodegenerative and Mitochondrial Diseases. Bioorg. Med. Chem. 2014, 22, 4935–4947. 10.1016/j.bmc.2014.06.040. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; SriRamaratnam R.; Welsch M. E.; Shimada K.; Skouta R.; Viswanathan V. S.; Cheah J. H.; Clemons P. A.; Shamji A. F.; Clish C. B.; Brown L. M.; Girotti A. W.; Cornish V. W.; Schreiber S. L.; Stockwell B. R. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J. A.; Ingold K. U. The Inhibited Autoxidation Of Styrene: Part I. The Deuterium Isotope Effect For Inhibition By 2, 6-Di-Tert-Butyl-4-Methylphenol. Can. J. Chem. 1962, 40, 1851–1864. 10.1139/v62-281. [DOI] [Google Scholar]
- Haidasz E. A.; Van Kessel A. T. M.; Pratt D. A. A Continuous Visible Light Spectrophotometric Approach To Accurately Determine the Reactivity of Radical-Trapping Antioxidants. J. Org. Chem. 2016, 81, 737–744. 10.1021/acs.joc.5b02183. [DOI] [PubMed] [Google Scholar]
- Hanthorn J. J.; Amorati R.; Valgimigli L.; Pratt D. A. The Reactivity of Air-Stable Pyridine- and Pyrimidine-Containing Diarylamine Antioxidants. J. Org. Chem. 2012, 77, 6895–6907. 10.1021/jo301012x. [DOI] [PubMed] [Google Scholar]
- The increase in fluorescence during the inhibited period results from the formation of antioxidant-derived autoxidation products that absorb at this wavelength.
- Burton G. W.; Ingold K. U. Autoxidation of Biological Molecules. 1. Antioxidant Activity of Vitamin E and Related Chain-Breaking Phenolic Antioxidants in Vitro. J. Am. Chem. Soc. 1981, 103, 6472–6477. 10.1021/ja00411a035. [DOI] [Google Scholar]
- Montgomery J. A. Jr.; Frisch M. J.; Ochterski J. W.; Petersson G. A. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 1999, 110, 2822–2827. 10.1063/1.477924. [DOI] [Google Scholar]
- It should be noted that Fer-1 has an ethyl ester as opposed to the methyl ester used in the computations.
- Valgimigli L.; Bartolomei D.; Amorati R.; Haidasz E.; Hanthorn J. J.; Nara S. J.; Brinkhorst J.; Pratt D. A. 3-Pyridinols and 5-Pyrimidinols: Tailor-Made for Use in Synergistic Radical-Trapping Co-Antioxidant Systems. Beilstein J. Org. Chem. 2013, 9, 2781–2792. 10.3762/bjoc.9.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt D. A.; DiLabio G. A.; Valgimigli L.; Pedulli G. F.; Ingold K. U. Substituent Effects on the Bond Dissociation Enthalpies of Aromatic Amines. J. Am. Chem. Soc. 2002, 124, 11085–11092. 10.1021/ja026289x. [DOI] [PubMed] [Google Scholar]
- kp for STY-BODIPY in egg PC liposomes was determined by fitting data traces of α-TOH inhibited autoxidations to the corresponding kinetic scheme using the reported kinh for α-TOH (5.8 × 103 M–1 s–1)44a and Ri calculated from the inhibition time of PMHC inhibited autoxidations (Ri = (2.6 ± 0.1) × 10–9 M–1 s–1).; a Barclay L. R. C.; Baskin K. A.; Dakin K. A.; Locke S. J.; Vinqvist M. R. The Antioxidant Activities of Phenolic Antioxidants in Free Radical Peroxidation of Phospholipid Membranes. Can. J. Chem. 1990, 68, 2258–2269. 10.1139/v90-348. [DOI] [Google Scholar]
- Niki E.; Noguchi N. Dynamics of Antioxidant Action of Vitamin E. Acc. Chem. Res. 2004, 37, 45–51. 10.1021/ar030069m. [DOI] [PubMed] [Google Scholar]
- See, e.g.:Barclay L. R. C.; Edwards C. E.; Vinqvist M. R. Media Effects on Antioxidant Activities of Phenols and Catechols. J. Am. Chem. Soc. 1999, 121, 6226–6231. 10.1021/ja990878u. [DOI] [Google Scholar]
- Litwinienko G.; Ingold K. U. Solvent Effects on the Rates and Mechanisms of Reaction of Phenols with Free Radicals. Acc. Chem. Res. 2007, 40, 222–230. 10.1021/ar0682029. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Grellier P. L.; Prior D. V.; Duce P. P.; Morris J. J.; Taylor P. J. Hydrogen bonding. Part 7. A scale of solute hydrogen-bond acidity based on log K values for complexation in tetrachloromethane. J. Chem. Soc., Perkin Trans. 2 1989, 699–711. 10.1039/p29890000699. [DOI] [Google Scholar]
- The α2H value of N-t-butyl-4-t-butylaniline was determined by 1H NMR as described by Abraham et al.49a This model amine was chosen to simplify interpretation of the spectra due to the greater number of interactions possible with the multiple N-H bonds present in both Lip-1 and Fer-1.; a Abraham M. H.; Abraham R. J.; Byrne J.; Griffiths L. NMR Method for the Determination of Solute Hydrogen Bond Acidity. J. Org. Chem. 2006, 71, 3389–3394. 10.1021/jo052631n. [DOI] [PubMed] [Google Scholar]
- Valgimigli L.; Amorati R.; Petrucci S.; Pedulli G. F.; Hu D.; Hanthorn J. J.; Pratt D. A. Unexpected Acid Catalysis in Reactions of Peroxyl Radicals with Phenols. Angew. Chem., Int. Ed. 2009, 48, 8348–8351. 10.1002/anie.200903360. [DOI] [PubMed] [Google Scholar]
- Noguchi N.; Yamashita H.; Hamahara J.; Nakamura A.; Kuhn H.; Niki E. The Specificity of Lipoxygenase-Catalyzed Lipid Peroxidation and the Effects of Radical-Scavenging Antioxidants. Biol. Chem. 2002, 383, 619–626. 10.1515/BC.2002.064. [DOI] [PubMed] [Google Scholar]
- Bocan T. M. A.; Rosebury W. S.; Mueller S. B.; Kuchera S.; Welch K.; Daugherty A.; Cornicelli J. A. A Specific 15-Lipoxygenase Inhibitor Limits the Progression and Monocyte–Macrophage Enrichment of Hypercholesterolemia-Induced Atherosclerosis in the Rabbit. Atherosclerosis 1998, 136, 203–216. 10.1016/S0021-9150(97)00204-9. [DOI] [PubMed] [Google Scholar]
- Lewerenz J.; Hewett S. J.; Huang Y.; Lambros M.; Gout P. W.; Kalivas P. W.; Massie A.; Smolders I.; Methner A.; Pergande M.; Smith S. B.; Ganapathy V.; Maher P. The Cystine/Glutamate Antiporter System xc– in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid. Redox Signaling 2013, 18, 522–555. 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P. K.; Seiler A.; Perisic T.; Kolle P.; Banjac Canak A.; Forster H.; Weiss N.; Kremmer E.; Lieberman M. W.; Bannai S.; Kuhlencordt P.; Sato H.; Bornkamm G. W.; Conrad M. System xc– and Thioredoxin Reductase 1 Cooperatively Rescue Glutathione Deficiency. J. Biol. Chem. 2010, 285, 22244–22253. 10.1074/jbc.M110.121327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Wang W.; Li Y.; Xiao Y.; Cheng J.; Jia J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharm. Bull. 2015, 38, 1234–1239. 10.1248/bpb.b15-00048. [DOI] [PubMed] [Google Scholar]
- Ingold K. U. Inhibition of the Autoxidation of Organic Substances in the Liquid Phase. Chem. Rev. 1961, 61, 563–589. 10.1021/cr60214a002. [DOI] [Google Scholar]
- Burton G. W.; Traber M. G.; Acuff R. V.; Walters D. N.; Kayden H.; Hughes L.; Ingold K. U. Human plasma and tissue α-tocopherol concentrations in response to supplementation with deuterated natural and synthetic vitamin E. Am. J. Clin. Nutr. 1998, 67, 669–684. [DOI] [PubMed] [Google Scholar]
- Panagabko C.; Morley S.; Hernandez M.; Cassolato P.; Gordon H.; Parsons R.; Manor D.; Atkinson J. Ligand Specificity in the CRAL-TRIO Protein Family. Biochemistry 2003, 42, 6467–6474. 10.1021/bi034086v. [DOI] [PubMed] [Google Scholar]
- Valgimigli L.; Amorati R.; Fumo M. G.; DiLabio G. A.; Pedulli G. F.; Ingold K. U.; Pratt D. A. The Unusual Reaction of Semiquinone Radicals with Molecular Oxygen. J. Org. Chem. 2008, 73, 1830–1841. 10.1021/jo7024543. [DOI] [PubMed] [Google Scholar]
- Burton G. W.; Doba T.; Gabe E.; Hughes L.; Lee F. L.; Prasad L.; Ingold K. U. Autoxidation of biological molecules. 4. Maximizing the antioxidant activity of phenols. J. Am. Chem. Soc. 1985, 107, 7053–7065. 10.1021/ja00310a049. [DOI] [Google Scholar]
- Enes R. F.; Tomé A. C.; Cavaleiro J. A. S.; Amorati R.; Fumo M. G.; Pedulli G. F.; Valgimigli L. Synthesis and Antioxidant Activity of [60]Fullerene–BHT Conjugates. Chem. - Eur. J. 2006, 12, 4646–4653. 10.1002/chem.200501495. [DOI] [PubMed] [Google Scholar]
- Haidasz E. A.; Meng D.; Amorati R.; Baschieri A.; Ingold K. U.; Valgimigli L.; Pratt D. A. Acid Is Key to the Radical-Trapping Antioxidant Activity of Nitroxides. J. Am. Chem. Soc. 2016, 138, 5290–5298. 10.1021/jacs.6b00677. [DOI] [PubMed] [Google Scholar]
- Krainz T.; Gaschler M. M.; Lim C.; Sacher J. R.; Stockwell B. R.; Wipf P. A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Cent. Sci. 2016, 2, 653–659. 10.1021/acscentsci.6b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Since PD146176 is an arylamine, it may inhibit ferroptosis as an RTA and not as a LOX inhibitor, and the difference in potency between it and Fer-1/Lip-1 may simply reflect its lower RTA activity.
- Kagan V. E.; Mao G.; Qu F.; Angeli J. P. F.; Doll S.; St Croix C.; Dar H. H.; Liu B.; Tyurin V. A.; Ritov V. B.; Kapralov A. A.; Amoscato A. A.; Jiang J.; Anthonymuthu T.; Mohammadyani D.; Yang Q.; Proneth B.; Klein-Seetharaman J.; Watkins S.; Bahar I.; Greenberger J.; Mallampalli R.; Stockwell B. R.; Tyurina Y. Y.; Conrad M.; Bayır H. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y.; Niki E.; Noguchi N. Comparative study on the action of tocopherols and tocotrienols as antioxidant: chemical and physical effects. Chem. Phys. Lipids 2003, 123, 63–75. 10.1016/S0009-3084(02)00164-0. [DOI] [PubMed] [Google Scholar]
- Nair D. G.; Funk C. D. A cell-based assay for screening lipoxygenase inhibitors. Prostaglandins Other Lipid Mediators 2009, 90, 98–104. 10.1016/j.prostaglandins.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Sato H.; Shiiya A.; Kimata M.; Maebara K.; Tamba M.; Sakakura Y.; Makino N.; Sugiyama F.; Yagami K.; Moriguchi T.; Takahashi S.; Bannai S. Redox Imbalance in Cystine/Glutamate Transporter-deficient Mice. J. Biol. Chem. 2005, 280, 37423–37429. 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.