

Abstract

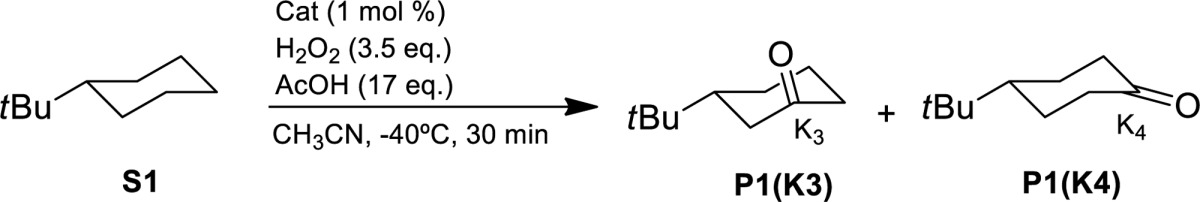
Monosubstituted cycloalkanes undergo regio- and enantioselective aliphatic C–H oxidation with H2O2 catalyzed by biologically inspired manganese catalysts. The reaction furnishes the corresponding ketones resulting from oxidation at C3 and C4 methylenic sites (K3 and K4, respectively) leading to a chiral desymmetrization that proceeds with remarkable enantioselectivity (64% ee) but modest regioselectivity at C3 (K3/K4 ≈ 2) for tert-butylcyclohexane, and with up to 96% ee and exquisite regioselectity toward C3 (up to K3/K4 > 99) when N-cyclohexylalkanamides are employed as substrates. Efficient H2O2 activation, high yield, and highly enantioselective C–H oxidation rely on the synergistic cooperation of a sterically bulky manganese catalyst and an oxidatively robust alkanoic acid. This represents the first example of nonenzymatic highly enantioselective oxidation of nonactivated methylenic sites. Furthermore, the principles of catalyst design disclosed in this work constitute a unique platform for further development of stereoselective C–H oxidation reactions.
Short abstract
Monosubstituted cycloalkanes undergo regio- and enantioselective aliphatic C−H oxidation with H2O2 catalyzed by biologically inspired manganese catalysts. The reaction furnishes the corresponding ketones resulting from oxidation at C3 and C4 methylenic sites (K3 and K4, respectively) leading to a chiral desymmetrization that proceeds with remarkable enantioselectivity (64% ee) but modest regioselectivity at C3 (K3/K4 ≈ 2) for tert-butylcyclohexane, and with up to 96% ee and exquisite regioselectity toward C3 (up to K3/K4 > 99) when N-cyclohexylalkanamides are employed as substrates.
Introduction
The oxidation of nonactivated aliphatic C–H bonds is a very powerful reaction because it can transform the inert C–H bond, ubiquitous in organic molecules, into a suitable site for further chemical elaboration.1,2 However, it also represents one of the most challenging reactions in modern synthetic organic chemistry because the multitude of aliphatic C–H bonds in a molecule makes site selective oxidation particularly difficult. This is further accentuated because of the high reactivity of the oxidizing species capable of breaking these bonds, often incompatible with chemo- and regioselective transformations. Additional challenges are encountered in enantioselective C–H oxidations.3 In the first place, reagents that are both chiral and capable of oxygenating aliphatic C–H bonds via mechanisms potentially susceptible to induce enantioselectivity are scarce. In addition, the more facile overoxidation of secondary alcohols with respect to the C–H precursor usually eliminates the chirality. Instead, enantioselective C–H oxidation is common in enzymes, where the combination of subtle interactions in the active site governs substrate orientation, and formation of the oxidizing species is finely triggered. Not surprisingly, examples of enantioselective sp3 C–H oxidation with nonenzymatic systems are rare and limited to relatively weak C–H bonds (benzylic, allylic, and adjacent to heteroatom) most commonly with low substrate conversion (Scheme 1).4−18 Examples of enantioselective oxidation of nonactivated aliphatic C–H bonds remain exclusive to enzymes.19−22
Scheme 1. Selected Precedents of Asymmetric C–H Oxidation: (a) Ref (16) (b) Ref (7) (c) Ref (5), and (d) Ref (8).

Oxidation of nonactivated aliphatic C–H bonds with biologically inspired transition metal catalysts that form high valent metal-oxo species may constitute a promising option to pursue asymmetric C–H oxidation. This class of complexes has been recently applied successfully in asymmetric olefin epoxidation23−25 and cis-dihydroxylation,26,27 but to date asymmetric C–H bond oxidation has not been described. Most interestingly, these species can hydroxylate C–H bonds with stereospecificity,28−32 and the steric and chiral properties of the first coordination sphere of the metal site have been shown to impact the C–H site selectivity of chiral molecules.33,34
Herein we describe the development of chiral manganese complexes with sterically demanding tetradentate aminopyridine ligands that catalyze the regio- and enantioselective oxidation of methylenic groups in monosubstituted cyclohexanes using H2O2 as oxidant. Oxidation occurs at positions C3 and C4 of the cyclohexane ring, producing a chiral desymmetrization in the former product, with outstanding levels of regioselectivity (>99:1 for C3 over C4) and enantioselectivity (up to 96% ee) in the reactions of N-cyclohexylalkanamides. To the best of our knowledge the current report constitutes the first example of enantioselective oxidation of a nonactivated aliphatic C–H bond by a nonenzymatic system.
Results and Discussion
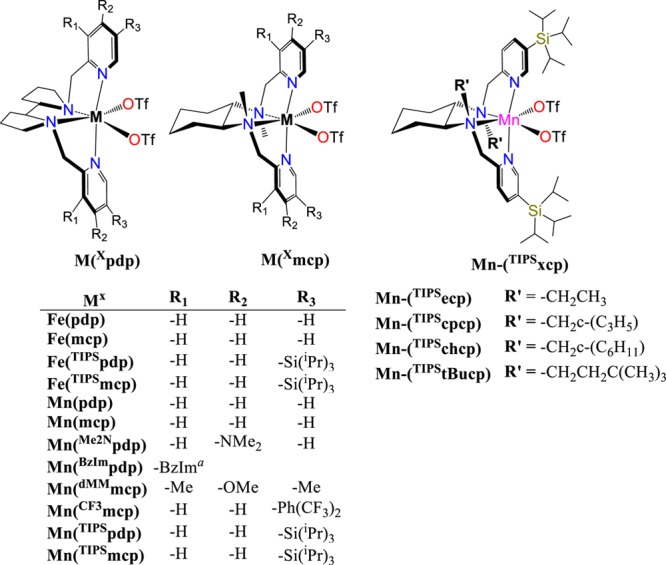
Chirality at the metal iron complexes bearing tetradentate ligands has been recently shown to exhibit site selectivity in the oxidation of methylenic units of chiral substrates, depending on the chirality of the complex.33 Therefore, we sought to explore their ability to engage in enantioselective C–H oxidation. Since secondary alcohols are rapidly oxidized by these catalysts to the corresponding achiral ketones,34 we explored the C–H oxidation of substrates that will result in an asymmetric desymmetrization.35 Parent manganese complexes have recently shown excellent catalytic activity in related aliphatic C–H oxidation reactions,36,37 and consequently, they were also included in the study. In particular chiral iron and manganese tetradentate complexes of general formula [M(CF3SO3)2(L)] (L = mcp, and pdp, mcp = N,N′-dimethyl N,N′-bis(2-pyridylmethyl)-1,2-trans-diamino cyclohexane, pdp = N,N′-bis(2-pyridylmethyl)-2,2′-bipyrrolidine, Scheme 2, complexes Fe(mcp), Fe(pdp), Mn(mcp), and Mn(pdp), respectively)32,33,38,39 were tested as catalysts in the oxidation of tert-butylcyclohexane (S1). Results are shown in Table 1 (entries 1–4). Standard conditions involved syringe pump delivery of H2O2 (3.5 equiv) to an acetonitrile solution of the catalyst (1 mol %) and AcOH (17 equiv) in CH3CN at −40 °C during 30 min. For all the catalysts oxidation of S1 occurs preferentially at C3 over C4 methylene sites to produce the corresponding ketone products (P1(K3) and P1(K4), respectively) in moderate to good product yields (31–69%). No products arising from C1 and C2 oxidation, nor from oxidation of the primary t-Bu C–H bonds were observed. Products are formed via initial hydrogen atom transfer (HAT) mediated C–H hydroxylation, followed by fast oxidation of the resulting secondary alcohol.40−42 Regioselectivity, quantified on the basis of the normalized product ratio (K3/K4), is comprised between 2.0 and 2.3, in line with the results of previous studies on the oxidation of S1 with HAT reagents such as iron and manganese complexes (K3/K4 = 1.9–2.5),41,43 methyl(trifluoromethyl)dioxirane (K3/K4 = 1.8),44 and iodanyl radicals (K3/K4 = 1.4–2.3).45,46 Poor enantioselectivities were obtained with these catalysts (<10% ee, entries 1–4), but when bulky groups like tris-(isopropyl)silyl (TIPS) were introduced at position 3 of the pyridine rings in the iron catalysts (entries 5–6), both yield and enantioselectivity increased significantly (70–71% yield, 33 and 15% ee for Fe(TIPSmcp) and Fe(TIPSpdp), respectively), while retaining regioselectivity (K3/K4 = 1.6–3.1).
Scheme 2. Diagram of the Iron and Manganese Complexes Studied.
Benzimidazole instead of pyridine.
Table 1. Oxidation of tert-Butylcyclohexane (S1) with Different Catalysts.
| entry | cat | conv (%)a | yield (%)a K3 (K4) | K3/K4b | Ee (K3) (%) |
|---|---|---|---|---|---|
| 1c | Fe(mcp) | 62 | 37 (9) | 2.1 | 2 |
| 2c | Fe(pdp) | 48 | 25 (6) | 2.0 | 6 |
| 3 | Mn(mcp) | 46 | 32 (7) | 2.3 | 9 |
| 4 | Mn(pdp) | 86 | 56 (13) | 2.2 | 3 |
| 5c | Fe(TIPSmcp) | 73 | 61 (10) | 3.1 | 33 |
| 6c | Fe(TIPSpdp) | 88 | 53 (17) | 1.6 | 15 |
| 7 | Mn(Me2Npdp) | 37 | 13 (3) | 2.2 | 8 |
| 8 | Mn(dMMpdp) | 93 | 50 (11) | 2.3 | 8 |
| 9 | Mn(BzImpdp) | 79 | 50 (11) | 2.3 | 11 |
| 10 | Mn(CF3mcp) | 55 | 22 (5) | 2.2 | 2 |
| 11 | Mn(TIPSmcp) | 77 | 53 (19) | 1.4 | 44 |
| 12 | Mn(TIPSpdp) | 51 | 22 (7) | 1.6 | 34 |
| 13d | Mn(TIPSecp) | 87 | 51 (15) | 1.7 | 43 |
| 14 | Mn(TIPScpcp) | 68 | 32 (12) | 1.3 | 32 |
| 15 | Mn(TIPSchcp) | 45 | 10 (6) | 1.0 | 6 |
| 16 | Mn(TIPStBucp) | 48 | 8 (5) | 1.0 | Rac |
Conversions and yields determined from crude reaction mixtures by GC. Ee’s determined by GC with chiral stationary phase.
Normalized ratio.
Reaction conditions: Fe catalyst (3 mol %), H2O2 (2.5 equiv), AcOH (1.5 equiv) in CH3CN at 0 °C during 30 min.
(S,S)-Mn-(TIPSecp) (2 mol %).
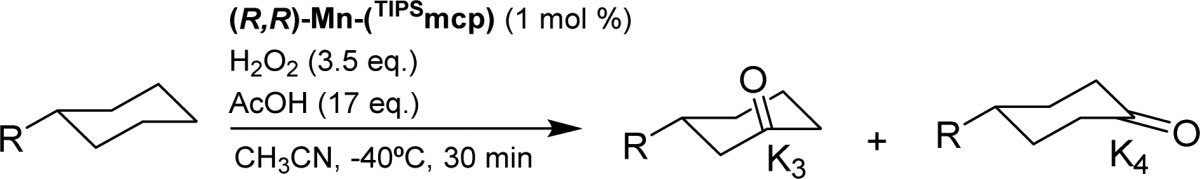
Most interestingly, use of the parent manganese complexes Mn(TIPSmcp) and Mn(TIPSpdp) (entries 11–12, full details on the synthesis and characterization of the complexes are provided in the Supporting Information) led to modest to good product yields but improved levels of enantioselectivity in ketone P1(K3) (up to 44% ee). On the other hand, the use of electron-rich manganese catalysts (Mn(Me2Npdp) and Mn(dMMpdp)), which have been recently shown to lead to high ee’s in epoxidation reactions,47,48 provided only low ee’s (entries 7–8), suggesting that the steric demand imposed by the TIPS group is the basis of the improved enantioselectivity. Further catalyst screening included related manganese complexes where the pyridine rings were replaced by benzylimidazole (BzIm) rings49 and the use of a 2,6-bis-trifluoromethylphenyl substituted mcp ligand50 (entries 9 and 10, respectively). In all the latter cases (entries 7–10), lower enantioselectivities were obtained as compared to the TIPS based catalysts. Then, different manganese catalysts based on the mcp backbone, but differing in the nature of the N-alkyl group (R′ in Scheme 2, entries 13–16 in Table 1) were prepared and tested in the model reaction. This study led to the identification of the ethyl substituted catalyst (S,S)-Mn(TIPSecp), ecp = N,N′-diethyl N,N′-bis(2-pyridylmethyl)-1,2-trans-diamino cyclohexane, as also particularly efficient in terms of product yield and enantioselectivity (entry 13). For the particular case of S1, (S,S)-Mn(TIPSecp) and (S,S)-Mn(TIPSmcp) provided analogous ee’s, but significant improvements in enantioselectivity were observed for other substrates (see below). As expected, the use of the enantiomerically related catalyst (R,R)-Mn(TIPSmcp) provides the oxidation product with comparable ee but with opposite absolute configuration.
The X-ray determined molecular structure of (S,S)-Mn(TIPSecp) is shown in Scheme 3. The complex is structurally very similar to the parent Mn(pdp)(51) and the related Fe(TIPSmcp) complexes.33 Bond distances and angles are collected in the Supporting Information. The manganese center adopts a C2-symmetric cis-α topology, with the two pyridines trans to each other. The complex exhibits chirality at the metal (Λ and Δ), in turn determined by the chirality of the cyclohexanediamine backbone (S,S and R,R, respectively). Most interestingly, TIPS groups determine a well-defined chiral clef occupied by the triflate ions, that bind cis to each other at the manganese center, trans to the two aliphatic amine sites. These sites are labile and are the place where H2O2 presumably binds and is activated, forming a high valent manganese oxo species responsible for the C–H oxidation reaction.30 Of notice is also the proximity of the N-ethyl groups to the triflate ions, suggesting that these groups can also contribute significantly in defining the structure of the C–H oxidation site.
Scheme 3. Schematic Diagram (Left) and ORTEP Diagram (Right) of the Single-Crystal X-ray Determined Structure of (S,S)-Mn(TIPSecp).
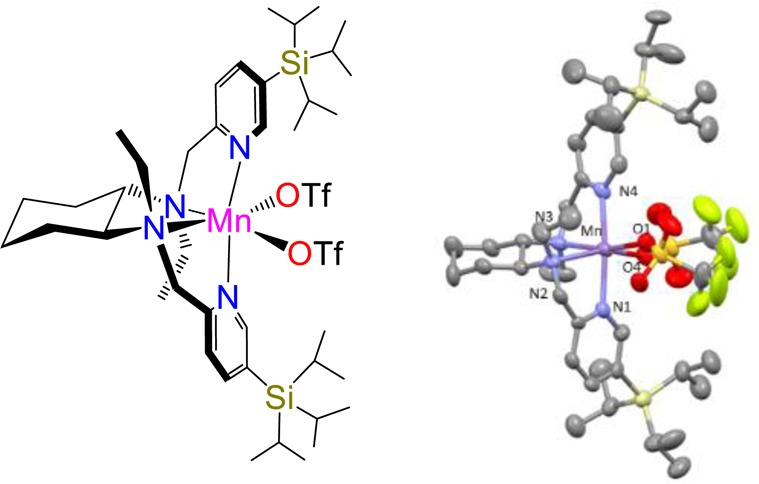
List of selected distances (Å); Mn–O(4) 2.151(6), Mn–O(1) 2.151(5), Mn–N(4) 2.186(6), Mn–N(1) 2.213(7), Mn–N(2) 2.327(7), Mn–N(3) 2.365(7).
On the basis of these results, catalyst Mn(TIPSmcp) was chosen for the oxidation of a series of monosubstituted cyclohexanes in order to estimate the effect of the substituent on yield, regioselectivity, and enantioselectivity of the reaction (Table 2). Consideration of the elements that determine regioselectivity in the oxidation of tert-butylcyclohexane (S1) serves to provide an understanding of the changes observed upon replacement of the t-Bu group for other groups. With S1, regioselectivity can be understood by considering the relative reactivity of the C–H bonds of this substrate in HAT reactions. Primary and tertiary C–H bonds of S1 are not reactive because of the high bond dissociation energy in the former, and of steric and torsional effects in the latter where in particular substrate conformation places this bond in an axial position. Tertiary axial C–H bond deactivation has been recently explained in terms of an increase in torsional strain in the HAT transition state, where planarization of the incipient carbon radical forces the bulky t-Bu group toward an unfavorable eclipsed interaction with the equatorial C–H groups on the adjacent positions.52,53 Along the same line, oxidation at C2 of S1 is disfavored because planarization of the incipient carbon radical formed following HAT forces the remaining C–H bond toward an unfavorable eclipsed interaction with the adjacent t-Bu group.52
Table 2. Oxidation of Different Cyclohexane Derivatives.
| entry | R | conv (%)a | yield (%)a K3(K4) | K3/K4b | Ee (K3) (%) |
|---|---|---|---|---|---|
| 1 | -t-Bu (S1) | 77 | 53 (19) | 1.4 | 44 |
| 2 | -OPiv (S2) | 80 | 41 (10) | 2.0 | 54 |
| 3 | -Si(Me)3 (S3) | 95 | 42 (13) | 1.6 | 23 |
| 4c | -CO2CH3 (S4) | 88 | 47 (20) | 1.2 | 11 |
| 5 | -CO2H (S5) | 45 | 20 (11) | 0.9 | 9e |
| 6c | -COCH3 (S6) | 95 | 47 (19) | 1.2 | 8 |
| 7 | -NHCOCH3 (S7) | 93 | 74 (3) | 12 | (+)63f |
| 8c | -NHCOCH3 (S7) | 94 | 75 (3) | 12 | (−)78 |
| 9 | -NHCOtBu (S8) | >99 | 90 (1) | 45 | (+)76 |
| 10d | -NHCOtBu (S8) | >99 | 90 (1) | 45 | (−)76 |
| 11c | -NHCOtBu (S8) | >99 | 90 (1) | 45 | (−)85 |
Conversions and yields determined from crude reaction mixtures by GC or 1H NMR.
Normalized ratio.
(S,S)-Mn-(TIPSecp) (2 mol %).
(S,S)-Mn-(TIPSmcp).
Ee’s determined after esterification of isolated products.
Absolute configuration was determined on the basis of the crystal structure of the product obtained from S10 (see Table 4 and Supporting Information). Ee’s determined by GC with chiral stationary phase.
The results of previous studies on the oxidation of monosubstituted cyclohexanes with methyl(trifluoromethyl)dioxirane have suggested that the K3/K4 regioselectivity observed in these reactions reflects the electronic effect of the substituent, with C3 oxidation over C4 being favored by electron releasing groups (K3/K4 = 1.8 for S1) and C4 over C3 by electron withdrawing groups (K3/K4 = 0.3 for trifluoromethylcyclohexane).44 Along this line, the regioselectivity observed in the oxidation of S1 catalyzed by Mn(TIPSmcp) (Table 2, entry 1: K3/K4 = 1.4) can be explained accordingly on the basis of the electron donating ability of the t-Bu group that activates C3 over C4 toward the electrophilic manganese-oxo species.
When the t-Bu group was replaced by a pivalate (cyclohexylpivalate (S2), Table 2, entry 2), comparable product yields and regioselectivity toward C3 (K3/K4 = 2.0) are observed, and the enantioselectivity increases up to 54% ee. Use of a trimethylsilyl (TMS) substituent as in S3 provides moderate yields and reduced enantioselectivity (ee = 23%, entry 3). Regioselectivity is not changed significantly with regard to S1 (K3/K4 = 1.6 and 1.4, for S3 and S1, respectively), in accordance with the similar electron donating abilities of the two substituents. On the other hand, with cyclohexanes bearing electron withdrawing substituents such as methyl cyclohexanecarboxylate (S4), cyclohexane carboxylic acid (S5), and cyclohexyl methyl ketone (S6) (entries 4–6, respectively), a slight decrease in the normalized K3/K4 ratio is observed (K3/K4 between 0.9 and 1.2), still reflecting however the formation of ketone K3 as the major product. This observation appears to be, at least to a certain extent, in contrast with the explanation given above, indicating that in addition to electronic effects other factors also contribute in defining regioselectivity. Very remarkably, as compared to S1 and S2, a sharp decrease in enantioselectivity is observed with the latter three substrates (S4-S6, entries 4–6, ee’s between 8 and 11%), indicating that the presence of electron withdrawing substituents can strongly influence the interaction of the substrate with the electrophilic C–H oxidation site.
Most interestingly, in the presence of a strongly Lewis basic amide substituent, as in N-cyclohexylacetamide (S7), the reaction proceeds with excellent yield, strongly improved regioselectivity toward C3 (K3/K4 = 12), and good enantioselectivity (63% ee, entry 7). Furthermore, an exquisite regioselectivity (K3/K4 = 45) and very good enantioselectivity (76% ee) are observed on moving to an amide substrate characterized by a bulky acyl moiety such as N-cyclohexylpivalamide (S8, entry 9).
These results point toward a strong interaction of these substrates with the manganese-oxo species, where substrate Lewis basicity and the presence of a bulky aliphatic group both appear to play a key role in providing an optimal substrate orientation for regioselective and enantioselective C–H oxidation. Most interestingly, with S7 and S8 enantioselectivity could be further improved to 78 and 85% ee, respectively, while retaining the excellent yield and regioselectivity, by using (S,S)-Mn(TIPSecp) as catalyst, where the N-methyl groups of the cyclohexanediamine backbone have been replaced by ethyl groups (entries 8 and 11).
The highly regioselective oxidation of S8 requires further consideration. It is well established that in the reaction of cyclohexyl amine with electrophilic HAT reagents such as oxygen centered radicals, reaction occurs selectively from the strongly activated α-C-H bond.54 Transformation of the NH2 group into a bulky amide moiety reduces the extent of activation at C1, increasing in the same time the importance of the conformation that places the tertiary C–H bond in an axial position, thus deactivating both C1 and C2 toward oxidation (see above), providing a highly efficient method for site-selective functionalization at a nonactivated methylene position.
Carboxylic acids have been recently shown to strongly impact the stereoselectivity of Mn and Fe catalyzed epoxidation reactions with related catalysts.47,51,55 These acids bind to the metal center, cis to the site where H2O2 is activated, and contribute in defining the active site.30,48 Thus, their assistance was explored in the current C–H oxidation reactions. Using (R,R)-Mn(TIPSmcp) as catalyst and amide S8 as substrate, the effect of different alkanoic acids is shown in Table 3. Almost exclusive formation of ketone K3 was observed, with the yield of ketone K4 being in all cases <1%. With the exclusion of 2-chloroethanoic acid (entry 6), cyclohexanecarboxylic acid (S5, entry 10) and carboxylic acids containing relatively long and branched alkyl chains such as 4-methylpentanoic and 2-ethylhexanoic acid (entries 11 and 12), comparable ee’s comprised between 73 and 82% were obtained for all the acids investigated. The significantly lower enantioselectivity observed with 2-chloroethanoic acid (55% ee, entry 6) reasonably reflects the electron withdrawing character of the chlorine substituent that makes this acid less prone to bind to the manganese center. The lack of or marginal conversion obtained with the latter three acids can be explained in terms of a favorable competition with the amide substrate for the oxidizing species, as specifically shown above for the case of cyclohexanecarboxylic acid (S5) (Table 2, entry 5) that is efficiently oxidized under these experimental conditions,56 clearly indicating that in order to obtain highly enantioselective C–H oxidations easily oxidizable carboxylic acids must be avoided. A chiral carboxylic acid was also studied with the aim to explore matching–mismatching effects between the chiralities of catalyst and carboxylic acid. The combination of (R,R)-Mn(TIPSmcp) and (S,S)-Mn(TIPSmcp) with (S)-(+)-2-methylbutanoic acid provided virtually identical enantioselectivities but reduced product yields (entries 15–16).
Table 3. C–H Oxidation Reactions Using Different Carboxylic Acids and N-Cyclohexylpivalamide (S8).
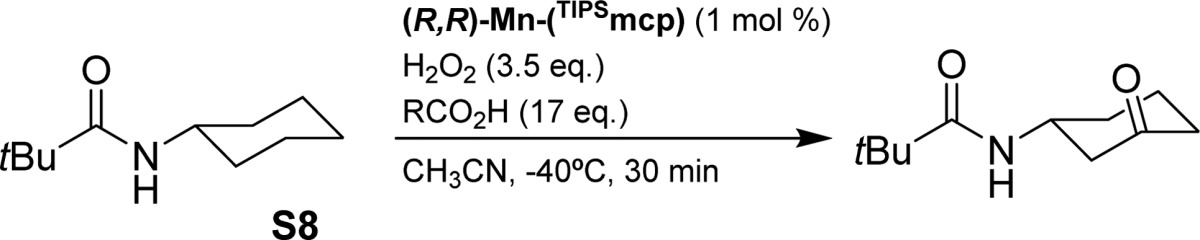
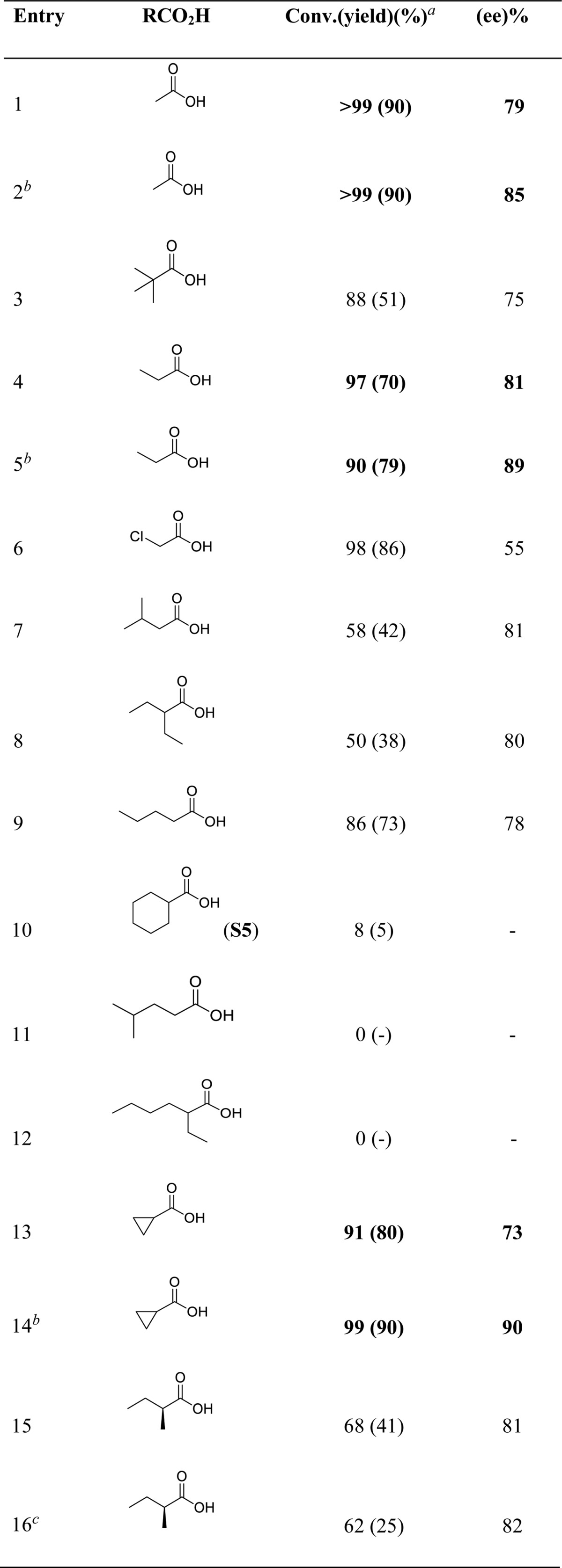
Conversions and yields (in parentheses) determined by GC.
(S,S)-Mn(TIPSecp) (2 mol %).
(S,S)-Mn(TIPSmcp). Ee’s determined by chiral GC.
Interestingly, as shown for the oxidatively robust acetic, propanoic, and cycloproanecarboxylic acids (entries 2, 5, and 14, respectively), by using (S,S)-Mn(TIPSecp) as catalyst a substantial increase in enantioselectivity was obtained (85–90% ee) while retaining the excellent yield and regioselectivity, with the rigid cyclopropanecarboxylic acid that provides the highest ee of the series (entry 14, 90% yield, 90% ee). Structural rigidity and stability toward oxidation make this acid particularly suitable to combine with the highly structured and sterically demanding, while oxidatively resistant, site of the Mn(TIPSecp) catalyst in order to create an outstandingly robust site capable of promoting a highly enantioselective oxidation of the strong aliphatic C–H bond of a nonactivated methylene group.
The role of the structure and electronic properties of the amide substrate on the regio- and enantioselectivity of the reaction was then explored under optimized conditions, using (S,S)-Mn(TIPSecp) as the catalyst and cyclopropanecarboxylic or acetic acid (17 equiv) as the acids of choice (Table 4). Secondary amides that present aliphatic acyl groups show in the most of cases high isolated yields, from good to excellent regioselectivities for oxidation at C3 and excellent ee’s (up to 96% ee, entry 5). It is possible to notice that the systematic increase in steric hindrance of the acyl group, when going from acetyl to 2-methylpropanoyl and 2,2-dimethylpropanoyl (entries 1–3), results in an increase in the enantioselectivity, which remains very high (>90% ee) as long as this group contains sterically demanding alkyl moieties (entries 3–9). A truly remarkable behavior is displayed by N-cyclohexyl-2,2-dimethylbutanamide (S10, entry 4), which combines high isolated yield (85% for product K3), excellent regioselectivity (K3/K4 = 43), and enantioselectivity (94% ee). Low yield was obtained only in the case of S12 (entry 6), suggesting that for this substrate the exceedingly high steric bulk of the acyl moiety prevents efficient oxidation. The use of an amide substrate that bears a chiral center (S14, entry 8) does not lead to any improvement in enantioselectivity; however the ee’s obtained with both (S,S)-Mn(TIPSecp) and (R,R)-Mn(TIPSecp) are excellent (91% and 86% ee, respectively). A remarkable demonstration of the important role played by electronic and steric effects in determining an optimal substrate orientation for highly regio and enantioselective C–H oxidation is provided by N-cyclohexylcyclohexanecarboxamide (S15, entry 9). Oxidation of this substrate occurs selectively at the N-cyclohexyl ring with excellent isolated yield (75%), exquisite regioselectivity (K3/K4 = 38), and very high enantioselectivity (91% ee).
Table 4. Impact of the Structure of the Acyl Moiety in Regio- and Enantioselective C–H Oxidation of N-Cyclohexyl Amides with (S,S)-Mn(TIPSecp) as Catalyst.
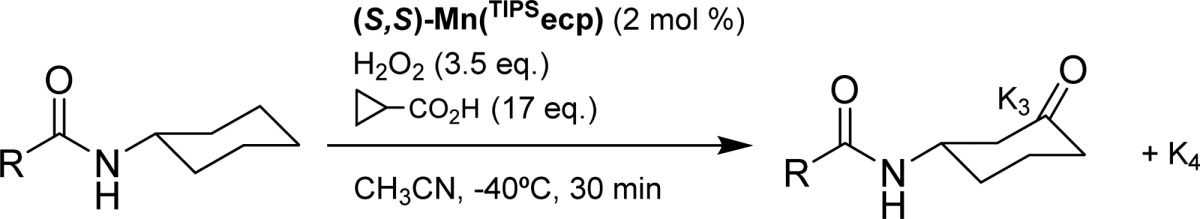
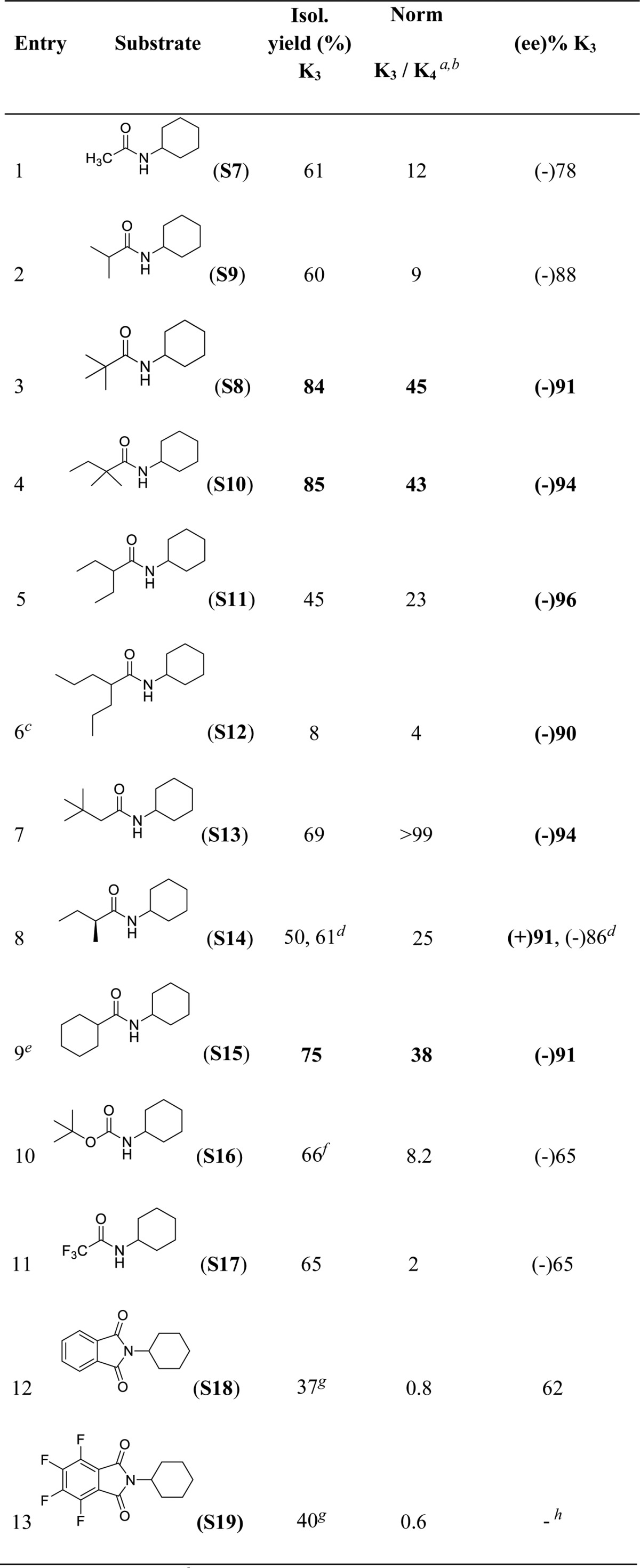
Normalized ratio.
K4 yields determined by GC.
Recovered starting material = 78%.
(R,R)-Mn(TIPSecp).
Acetic acid (17 equiv) instead of cyclopropanecarboxylic acid.
Yield determined by GC.
Products isolated as mixture of (K3 + K4).
Ee not determined. Ee’s determined by chiral GC and HPLC.
Increasing the electron withdrawing ability of the R group (S16-S19, entries 10–13) progressively decreases the K3/K4 ratio due to deactivation of proximal positions (C3) as compared to remote ones (C4), with the lowest regioselectivities for oxidation at C3 (K3/K4 = 0.6) being observed with N-cyclohexylphthalimide (S18) and N-cyclohexyltetrafluorophthalimide (S19) (K3/K4 = 0.8 and 0.6, respectively). These findings are in full agreement with the results of recent studies on the HAT-based aliphatic C–H halogenation of pentyl derivatives, where, among different electron withdrawing substituents, the phthalimido one provided the highest selectivity for the most remote methylene group.57,58 Decreased levels of enantioselectivity are also observed with these substrates (ee’s between 62 and 68%), in line with the discussion outlined above on the role played by substrate Lewis basicity on the C–H oxidation enantioselectivity.
Finally, under the optimized conditions, N-cyclohexyl amides bearing methyl substituents on the cyclohexane ring were evaluated (Scheme 4). N-(4,4-Dimethylcyclohexyl)pivalamide S20 was oxidized with excellent regioselectivity and 76% ee, while blocking positions 3 and 5 with methyl groups (N-(3,3,5,5-tetramethylcyclohexyl)pivalamide, S22) results in an inert substrate under the experimental conditions. Oxidation of a racemic mixture of N-(3,3-dimethylcyclohexyl)pivalamide S21 results in a chiral resolution, providing the ketone K3 in 39% isolated yield and 55% ee, while the starting amide is enantiomerically enriched to a moderate extent (38% ee). Under optimized conditions tert-butylcyclohexane (S1) and cyclohexyl pivalate (S2) were also oxidized in good yields and with remarkable ee’s (64 and 61% ee, respectively). The results obtained in the oxidation of S1 are particularly interesting because this substrate is characterized by a simple cycloalkane skeleton devoid of any functionality, and the relatively high enantioselectivity must be mostly based on sterics.
Scheme 4.

(a) (R,R)-Mn-(TIPSmcp) and acetic acid. (b) (S,S)-Mn(TIPSecp) and acetic acid. (c) (R,R)-Mn-(TIPSmcp) and cyclopropanecarboxylic acid. (d) Sum of K3 + K4. Isolated yields.
The significance of the levels of regio- and enantioselectivity exhibited in the current reactions must be considered in context. Cyclohexane scaffolds have been used as model substrates in studies aimed at clarifying the factors that govern selectivity in C–H oxidation. Trends in regioselectivity related to those observed for the current Mn-catalyzed oxidations, entailing preferential oxidation at C3 and C4 have been observed in the oxidation of S1 with hypervalent iodine reagents,45,46 dioxiranes,44 alkoxyl radicals,52 and iron and manganese catalysts.41,43 However, the outstanding directing role of the amide moiety is unprecedented and provides an excellent tool to consider in governing site-selectivity in C–H oxidation.
The levels of enantioselectivity observed in the current reactions deserve special discussion. To the best of our knowledge, the reactions described herein represent the first example of an enantioselective C–H oxidation of a nonactivated C–H bond carried out by a nonenzymatic system. Indeed, the current reactions are focused on methylenic sites, that, as compared to tertiary sites, are characterized by significantly stronger C–H bonds, and for which viable oxidants are limited. Previous chiral oxidations of a cyclohexane moiety have been described only after systematic mutations of a P450 enzyme,21 where enantioselective hydroxylation at C2 was obtained.
The origin of the enantioselective discrimination in the current reactions also deserves a comment. Enantioselectivity can originate from the transfer of a hydroxyl ligand belonging to a chiral Mn–OH complex to a long-lived and planar carbon centered radical (Scheme 5A) or from the initial C–H cleaving step by a chiral metal-oxo species (Scheme 5B). The high reactivity of alkyl radicals seems a priori incompatible with their use as radical relays, in a similar way as done in recent benzylic C–H functionalization reactions.17 More definitively, however, this latter mechanism is incompatible with the desymmetrization reactions described herein. In the current reactions enantioselectivity must be necessarily introduced in the C–H lysis step, which has been previously established to entail HAT from the aliphatic C–H bond to a high valent manganese-oxo species MnV(O) (carboxylate) moiety, followed by a fast hydroxyl rebound toward the incipient carbon centered radical.30 Therefore, the current reactions capture the mechanistic essence of stereoselective aliphatic C–H oxidation reactions taking place at P450 cpdI.59 Careful design of the chiral MnV(O) (carboxylate) site is necessary for reaching good product yields and high levels of stereoselection in the oxidation of these strong C–H bonds. This is achieved by embedding the manganese center in a chiral robust cavity defined by bulky TIPS groups of the ligand and the assistance of an oxidatively robust carboxylic acid. In this scenario, it must be recognized that the extraordinary role of the basic amide moiety in dictating regio- and enantioselectivity is not completely understood. The most obvious binding of the Lewis basic carbonyl moiety to the MnV(O) (carboxylate) site will require the unlikely formation of a seven coordinate manganese species. Other possibilities to consider may be the presence of a partially protonated oxo or carboxylate ligand, introducing an acidic site susceptible to engage in H-bonding with the incoming amide substrate. Precedents for a basic high valent iron-oxo species exist for chloroperoxidase.60 Elucidation of these possibilities will require a more precise understanding of the nature of the active species, maybe only possible via computational methods. On the other hand, it must be noticed that the current reactions entail breakage of strong C–H bonds, but they occur at low temperature in a remarkably efficient manner. This implies that the active species must be extraordinarily reactive, and activation barriers for the reactions must be small. Introduction of stereoselectivity in these conditions is particularly challenging, and therefore it is not surprising that ee’s appear to be quite sensitive to small changes in the nature of the catalyst, carboxylic acid, and substrate. Particular optimizations may be necessary. Indeed, we notice that structural specificity in stereoselective aliphatic C–H oxidation is a common feature of enzymatic systems and may be a fundamental drawback of this type of reaction.
Scheme 5. Description of the Possible Mechanisms at the Origin of the Enantioselectivity.

Conclusions
The present work describes unique examples of regio- and enantioselective oxidation of nonactivated aliphatic C–H bonds with small molecule manganese catalysts using hydrogen peroxide as the oxidant. A chiral sterically demanding and oxidatively robust active center can be built via careful tuning of the manganese ligands. These results represent the first examples of highly enantioselective nonenzymatic oxidation of nonactivated methylenic sites. The principles of catalyst design disclosed in this work constitute a very solid platform for further development of stereoselective C–H oxidation reactions in terms of substrate scope, but we also envision their straightforward application to other asymmetric oxygen atom transfer reactions (to olefins, amines, or sulfides, for example). Not less important, the current oxidation reactions are based on the use of an abundant first row transition metal and hydrogen peroxide as the oxidant. Therefore, they are also particularly interesting from a sustainable perspective.
Acknowledgments
We acknowledge financial support from MINECO of Spain (CTQ2015-70795-P) and the Catalan DIUE of the Generalitat de Catalunya (2009SGR637). M.C. thanks an ICREA-Academia award. We thank STR from UdG and Gabriel Peris from UJI by Xray diffraction analyses. We thank Prof. Antoni Riera from UB for kindly providing access to a polarimeter.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00368.
The authors declare no competing financial interest.
This article was published on February 8, 2017. Corrections to stereochemical designations in the article and the Supporting Information were made on February 23, 2017.
Supplementary Material
References
- Newhouse T.; Baran P. S. If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Zheng C.; You S.-L. Recent development of direct asymmetric functionalization of inert C-H bonds. RSC Adv. 2014, 4, 6173–6214. 10.1039/c3ra46996d. [DOI] [Google Scholar]
- Miyafuji A.; Katsuki T. Asymmetric desymmetrization of meso-tetrahydrofuran derivatives by highly enantiotopic selective C-H oxidation. Tetrahedron 1998, 54, 10339–10348. 10.1016/S0040-4020(98)00489-X. [DOI] [Google Scholar]
- Frost J. R.; Huber S. M.; Breitenlechner S.; Bannwarth C.; Bach T. Enantiotopos-Selective C-H Oxygenation Catalyzed by a Supramolecular Ruthenium Complex. Angew. Chem., Int. Ed. 2015, 54, 691–695. 10.1002/anie.201409224. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Viski P. Asymmetric hydroxylation, epoxidation, and sulfoxidation catalyzed by vaulted binaphthyl metalloporphyrins. J. Org. Chem. 1990, 55, 3628–3634. 10.1021/jo00298a046. [DOI] [Google Scholar]
- Groves J. T.; Viski P. Asymmetric hydroxylation by a chiral iron porphyrin. J. Am. Chem. Soc. 1989, 111, 8537–8538. 10.1021/ja00204a047. [DOI] [Google Scholar]
- Srour H.; Maux P. L.; Simonneaux G. Enantioselective Manganese-Porphyrin-Catalyzed Epoxidation and C–H Hydroxylation with Hydrogen Peroxide in Water/Methanol Solutions. Inorg. Chem. 2012, 51, 5850–5856. 10.1021/ic300457z. [DOI] [PubMed] [Google Scholar]
- Maux P. L.; Srour H. F.; Simonneaux G. Enantioselective water-soluble iron–porphyrin-catalyzed epoxidation with aqueous hydrogen peroxide and hydroxylation with iodobenzene diacetate. Tetrahedron 2012, 68, 5824–5828. 10.1016/j.tet.2012.05.014. [DOI] [Google Scholar]
- Zhang R.; Yu W. Y.; Lai T. S.; Che C.-M. Enantioselective hydroxylation of benzylic C-H bonds by D-4-symmetric chiral oxoruthenium porphyrins. Chem. Commun. 1999, 1791–1792. 10.1039/a904100a. [DOI] [Google Scholar]
- Zhang R.; Yu W.-Y.; Che C.-M. Catalytic enantioselective oxidation of aromatic hydrocarbons with D4-symmetric chiral ruthenium porphyrin catalysts. Tetrahedron: Asymmetry 2005, 16, 3520–3526. 10.1016/j.tetasy.2005.08.059. [DOI] [Google Scholar]
- Hamada T.; Irie R.; Mihara J.; Hamachi K.; Katsuki T. Highly enantioselective benzylic hydroxylation with concave type of (salen)manganese(III) complex. Tetrahedron 1998, 54, 10017–10028. 10.1016/S0040-4020(98)00603-6. [DOI] [Google Scholar]
- Hamachi K.; Irie R.; Katsuki T. Asymmetric benzylic Oxidation Using a Mn-Salen Complex as Catalyst. Tetrahedron Lett. 1996, 37, 4979–4982. 10.1016/0040-4039(96)00984-7. [DOI] [Google Scholar]
- Komiya N.; Noji S.; Murahashi S.-I. Manganese catalyzed asymmetric oxidation of alkanes to optically active ketones bearing asymmetric center at the α-position. Tetrahedron Lett. 1998, 39, 7921–7924. 10.1016/S0040-4039(98)01757-2. [DOI] [Google Scholar]
- Murahashi S.-I.; Noji S.; Hirabayashi T.; Komiya N. Manganese-catalyzed enantioselective oxidation of C–H bonds of alkanes and silyl ethers to optically active ketones. Tetrahedron: Asymmetry 2005, 16, 3527–3535. 10.1016/j.tetasy.2005.08.056. [DOI] [Google Scholar]
- Murahashi S.-I.; Noji S.; Komiya N. Catalytic Enantioselective Oxidation of Alkanes and Alkenes Using (Salen)Manganese Complexes Bearing a Chiral Binaphthyl Strapping Unit. Adv. Synth. Catal. 2004, 346, 195–198. 10.1002/adsc.200303190. [DOI] [Google Scholar]
- Zhang W.; Wang F.; McCann S. D.; Wang D.; Chen P.; Stahl S. S.; Liu G. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018. 10.1126/science.aaf7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Wang D.; Wan X.; Wu L.; Chen P.; Liu G. Enantioselective Copper-Catalyzed Intermolecular Cyanotrifluoromethylation of Alkenes via Radical Process. J. Am. Chem. Soc. 2016, 138, 15547–15550. 10.1021/jacs.6b10468. [DOI] [PubMed] [Google Scholar]
- Kille S.; Zilly F. E.; Acevedo J. P.; Reetz M. T. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 2011, 3, 738–743. 10.1038/nchem.1113. [DOI] [PubMed] [Google Scholar]
- Narayan A. R. H.; Jimenez-Oses G.; Liu P.; Negretti S.; Zhao W.; Gilbert M. M.; Ramabhadran R. O.; Yang Y.-F.; Furan L. R.; Li Z.; Podust L. M.; Montgomery J.; Houk K. N.; Sherman D. H. Enzymatic hydroxylation of an unactivated methylene C-H bond guided by molecular dynamics simulations. Nat. Chem. 2015, 7, 653–660. 10.1038/nchem.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roiban G.-D.; Agudo R.; Reetz M. T. Cytochrome P450 Catalyzed Oxidative Hydroxylation of Achiral Organic Compounds with Simultaneous Creation of Two Chirality Centers in a Single C-H Activation Step. Angew. Chem., Int. Ed. 2014, 53, 8659–8663. 10.1002/anie.201310892. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Shafer B. M.; Matthew D.; Demars I.; Stern H. A.; Fasan R. Controlled Oxidation of Remote sp3 C-H Bonds in Artemisinin via P450 Catalysts with Fine-Tuned Regio- and Stereoselectivity. J. Am. Chem. Soc. 2012, 134, 18695–18704. 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussó O.; Ribas X.; Costas M. Biologically inspired non-heme iron-catalysts for asymmetric epoxidation; design principles and perspectives. Chem. Commun. 2015, 51, 14285–14298. 10.1039/C5CC05576H. [DOI] [PubMed] [Google Scholar]
- Wang C.; Yamamoto H. Asymmetric Epoxidation Using Hydrogen Peroxide as Oxidant. Chem. - Asian J. 2015, 10, 2056–2068. 10.1002/asia.201500293. [DOI] [PubMed] [Google Scholar]
- Bryliakov K. P.; Talsi E. P. Active sites and mechanisms of bioinspired oxidation with H2O2, catalyzed by non-heme Fe and related Mn complexes. Coord. Chem. Rev. 2014, 276, 73–96. 10.1016/j.ccr.2014.06.009. [DOI] [Google Scholar]
- Zang C.; Liu Y.; Xu Z.-J.; Tse C.-W.; Guan X.; Wei J.; Huang J.-S.; Che C.-M. Highly Enantioselective Iron-Catalyzed cis-Dihydroxylation of Alkenes with Hydrogen Peroxide Oxidant via an FeIII-OOH Reactive Intermediate. Angew. Chem., Int. Ed. 2016, 55, 10253–10257. 10.1002/anie.201603410. [DOI] [PubMed] [Google Scholar]
- Chow T. W.-S.; Liu Y.; Che C.-M. Practical manganese-catalysed highly enantioselective cis-dihydroxylation of electron-deficient alkenes and detection of a cis-dioxomanganese(v) intermediate by high resolution ESI-MS analysis. Chem. Commun. 2011, 47, 11204–11206. 10.1039/c1cc11999k. [DOI] [PubMed] [Google Scholar]
- Costas M.; Olivo G.; Cussó O. Biologically Inspired C-H and C=C Oxidations with H2O2 Catalyzed by Iron Coordination Complexes. Chem. - Asian J. 2016, 11, 3148–3158. 10.1002/asia.201601170. [DOI] [PubMed] [Google Scholar]
- Oloo W. N.; Que L. Bioinspired Nonheme Iron Catalysts for C–H and C=C Bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res. 2015, 48, 2612–2621. 10.1021/acs.accounts.5b00053. [DOI] [PubMed] [Google Scholar]
- Ottenbacher R. V.; Talsi E. P.; Bryliakov K. P. Mechanism of Selective C-H Hydroxylation Mediated by Manganese Aminopyridine Enzyme Models. ACS Catal. 2015, 5, 39–44. 10.1021/cs5013206. [DOI] [Google Scholar]
- White M. C. Adding Aliphatic C-H Bond Oxidations to Synthesis. Science 2012, 335, 807–809. 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. A Predictably Selective Aliphatic C-H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- Font D.; Canta M.; Milan M.; Cussó O.; Ribas X.; Klein Gebbink R. J. M.; Costas M. Readily Accessible Bulky Iron Catalysts exhibiting Site Selectivity in the Oxidation of Steroidal Substrates. Angew. Chem., Int. Ed. 2016, 55, 5776–5779. 10.1002/anie.201600785. [DOI] [PubMed] [Google Scholar]
- Gomez L.; Canta M.; Font D.; Prat I.; Ribas X.; Costas M. Regioselective oxidation of nonactivated alkyl C-H groups using highly structured non-heme iron catalysts. J. Org. Chem. 2013, 78, 1421–33. 10.1021/jo302196q. [DOI] [PubMed] [Google Scholar]
- Borissov A.; Davies T. Q.; Ellis S. R.; Fleming T. A.; Richardson M. S. W.; Dixon D. J. Organocatalytic enantioselective desymmetrisation. Chem. Soc. Rev. 2016, 45, 5474–5540. 10.1039/C5CS00015G. [DOI] [PubMed] [Google Scholar]
- Ottenbacher R. V.; Samsonenko D. G.; Talsi E. P.; Bryliakov K. P. Highly Efficient, Regioselective, and Stereospecific Oxidation of Aliphatic C-H Groups with H2O2, Catalyzed by Aminopyridine Manganese Complexes. Org. Lett. 2012, 14, 4310–4313. 10.1021/ol3015122. [DOI] [PubMed] [Google Scholar]
- Adams A. M.; Du Bois J.; Malik H. A. Comparative Study of the Limitations and Challenges in Atom-Transfer C–H Oxidations. Org. Lett. 2015, 17, 6066–6069. 10.1021/acs.orglett.5b03047. [DOI] [PubMed] [Google Scholar]
- Canta M.; Font D.; Gómez L.; Ribas X.; Costas M. The Iron(II) Complex [Fe(CF3SO3)2(mcp)] as a Convenient, Readily Available Catalyst for the Selective Oxidation of Methylenic Sites in Alkanes. Adv. Synth. Catal. 2014, 356, 818–830. 10.1002/adsc.201300923. [DOI] [Google Scholar]
- Murphy A.; Dubois G.; Stack T. D. P. Efficient Epoxidation of Electron-Deficient Olefins with a Cationic Manganese Complex. J. Am. Chem. Soc. 2003, 125, 5250–5251. 10.1021/ja029962r. [DOI] [PubMed] [Google Scholar]
- Chen K.; Que L. Stereospecific alkane hydroxylation by non-heme iron catalysts: Mechanistic evidence for an FeV=O active species. J. Am. Chem. Soc. 2001, 123, 6327–6337. 10.1021/ja010310x. [DOI] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- Shen D. Y.; Miao C. X.; Xu D. Q.; Xia C. G.; Sun W. Highly Efficient Oxidation of Secondary Alcohols to Ketones Catalyzed by Manganese Complexes of N-4 Ligands with H2O2. Org. Lett. 2015, 17, 54–57. 10.1021/ol5032156. [DOI] [PubMed] [Google Scholar]
- Shen D.; Miao C.; Wang S.; Xia C.; Sun W. Efficient Benzylic and Aliphatic C–H Oxidation with Selectivity for Methylenic Sites Catalyzed by a Bioinspired Manganese Complex. Org. Lett. 2014, 16, 1108–1111. 10.1021/ol4037083. [DOI] [PubMed] [Google Scholar]
- González-Núñez M. E.; Castellano G.; Andreu C.; Royo J.; Báguena M.; Mello R.; Asensio G. Influence of Remote Substituents on the Equatorial/Axial Selectivity in the Monooxygenation of Methylene C–H Bonds of Substituted Cyclohexanes. J. Am. Chem. Soc. 2001, 123, 7487–7491. 10.1021/ja003667u. [DOI] [PubMed] [Google Scholar]
- Moteki S. A.; Usui A.; Zhang T.; Solorio Alvarado C. R.; Maruoka K. Site-Selective Oxidation of Unactivated C-H Bonds with Hypervalent Iodine(III) Reagents. Angew. Chem., Int. Ed. 2013, 52, 8657–8660. 10.1002/anie.201304359. [DOI] [PubMed] [Google Scholar]
- Moteki S. A.; Selvakumar S.; Zhang T.; Usui A.; Maruoka K. A Practical Approach for the Oxidation of Unactivated Csp3-H Bonds with o-Nitro(diacetoxyiodo)benzene as an Efficient Hypervalent Iodine(III)-Based Oxidizing Agent. Asian J. Org. Chem. 2014, 3, 932–935. 10.1002/ajoc.201402087. [DOI] [Google Scholar]
- Cussó O.; Garcia-Bosch I.; Font D.; Ribas X.; Lloret-Fillol J.; Costas M. Highly Stereoselective Epoxidation with H2O2 Catalyzed by Electron-Rich Aminopyridine Manganese Catalysts. Org. Lett. 2013, 15, 6158–6161. 10.1021/ol403018x. [DOI] [PubMed] [Google Scholar]
- Ottenbacher R. V.; Samsonenko D. G.; Talsi E. P.; Bryliakov K. P. Highly Enantioselective Bioinspired Epoxidation of Electron-Deficient Olefins with H2O2 on Aminopyridine Mn Catalysts. ACS Catal. 2014, 4, 1599–1606. 10.1021/cs500333c. [DOI] [Google Scholar]
- Wang X.; Miao C. X.; Wang S. F.; Xia C. G.; Sun W. Bioinspired Manganese and Iron Complexes with Tetradentate N Ligands for the Asymmetric Epoxidation of Olefins. ChemCatChem 2013, 5, 2489–2494. 10.1002/cctc.201300102. [DOI] [Google Scholar]
- Gormisky P. E.; White M. C. Catalyst-Controlled Aliphatic C–H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc. 2013, 135, 14052–14055. 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- Lyakin O. Y.; Ottenbacher R. V.; Bryliakov K. P.; Talsi E. P. Asymmetric Epoxidations with H2O2 on Fe and Mn Aminopyridine Catalysts: Probing the Nature of Active Species by Combined Electron Paramagnetic Resonance and Enantioselectivity Study. ACS Catal. 2012, 2, 1196–1202. 10.1021/cs300205n. [DOI] [Google Scholar]
- Salamone M.; Ortega V. B.; Bietti M. Enhanced Reactivity in Hydrogen Atom Transfer from Tertiary Sites of Cyclohexanes and Decalins via Strain Release: Equatorial C–H Activation vs Axial C–H Deactivation. J. Org. Chem. 2015, 80, 4710–4715. 10.1021/acs.joc.5b00636. [DOI] [PubMed] [Google Scholar]
- Zou L.; Paton R. S.; Eschenmoser A.; Newhouse T. R.; Baran P. S.; Houk K. N. Enhanced Reactivity in Dioxirane C–H Oxidations via Strain Release: A Computational and Experimental Study. J. Org. Chem. 2013, 78, 4037–4048. 10.1021/jo400350v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone M.; Carboni G.; Bietti M. Fine Control over Site and Substrate Selectivity in Hydrogen Atom Transfer-Based Functionalization of Aliphatic C–H Bonds. J. Org. Chem. 2016, 81, 9269–9278. 10.1021/acs.joc.6b01842. [DOI] [PubMed] [Google Scholar]
- Cussó O.; Garcia-Bosch I.; Ribas X.; Lloret-Fillol J.; Costas M. Asymmetric Epoxidation with H2O2 by Manipulating the Electronic Properties of Non-heme Iron Catalysts. J. Am. Chem. Soc. 2013, 135, 14871–14878. 10.1021/ja4078446. [DOI] [PubMed] [Google Scholar]
- Bigi M. A.; Reed S. A.; White M. C. Directed Metal (Oxo) Aliphatic C–H Hydroxylations: Overriding Substrate Bias. J. Am. Chem. Soc. 2012, 134, 9721–9726. 10.1021/ja301685r. [DOI] [PubMed] [Google Scholar]
- Quinn R. K.; Könst Z. A.; Michalak S. E.; Schmidt Y.; Szklarski A. R.; Flores A. R.; Nam S.; Horne D. A.; Vanderwal C. D.; Alexanian E. J. Site-Selective Aliphatic C–H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc. 2016, 138, 696–702. 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt V. A.; Quinn R. K.; Brusoe A. T.; Alexanian E. J. Site-Selective Aliphatic C–H Bromination Using N-Bromoamides and Visible Light. J. Am. Chem. Soc. 2014, 136, 14389–14392. 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. T.; Dawson J. H.; Gray H. B. Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry. Science 2004, 304, 1653–1656. 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.