

Abstract
Time-lapse, deep-tissue imaging made possible by advances in intravital microscopy has demonstrated the importance of tumour cell migration through confining tracks in vivo. These tracks may either be endogenous features of tissues or be created by tumour or tumour-associated cells. Importantly, migration mechanisms through confining microenvironments are not predicted by 2D migration assays. Engineered in vitro models have been used to delineate the mechanisms of cell motility through confining spaces encountered in vivo. Understanding cancer cell locomotion through physiologically relevant confining tracks could be useful in developing therapeutic strategies to combat metastasis.
Cancer metastasis and failures to clinically treat metastases1 are responsible for the majority of patient deaths from solid tumours2. The metastatic cascade is complex and encompasses migration of cancerous cells away from the primary tumour, their intravasation into the bloodstream or lymphatic system, their transit through the circulation, their extravasation to secondary tissues and the formation of distant metastatic tumour colonies3–5. Cell migration is a pivotal step in the metastatic process3,5. However, the heterogeneous microenvironments through which cancer cells migrate in vivo6–11 and the diversity of migration mechanisms available to cancer cells10,12,13 confound efforts to abate metastasis-initiating migration in a clinical setting.
Cancer cells migrate in vivo by gradually degrading their surrounding extracellular matrix (ECM) to create their own migration tracks14,15, by following ‘leader’ cancer cells or cancer-associated stromal cells that open up paths for migration16,17 or by moving through pre-existing channel-like tracks created by anatomical structures7,11,12. The extracellular microenvironment contains confining pores (ranging from less than 1 μm to 20 μm in diameter) or fibre-like and channel-like tracks varying from less than 3 μm to 30 μm in width and from 100 μm to 600 μm in length6. When the cross-sectional area of interfibrillar pores is less than ~7 μm2, matrix degradation is required for cancer cell migration to occur18, as discussed in detail below. Evidence suggests that confinement is a physical cue that modulates intracellular signalling19, thereby altering tumour cell migration mechanisms. This Opinion article discusses confined cancer cell motility, focusing on its in vivo relevance, migration mechanisms and confinement-induced cell responses. Accounting for the entire repertoire of mechanisms that are available to cancer cells for migration in physiologically relevant microenvironments will, in our opinion, aid the development of therapeutic interventions that aim to halt metastatic spread.
Cell confinement in vivo
An important in vivo migration mode is locomotion through confining spaces. Such spaces occur as pores in the ECM of the tumour stroma6 or as tunnel-like tracks7,11. Mast cells, macrophages and cancer-associated fibroblasts in the tumour micro-environment remodel the ECM and provide both proteinases and collagen crosslinking to create pro-migratory niches and 3D longitudinal tracks16,20. Matrix remodelling occurs not only at the primary tumour but also during the development of the pre-metastatic niche21. Tracks offer ‘paths of least resistance’ for tumour cell migration7,22. An increasing amount of evidence generated using intravital microscopy reveals that migration tracks are not created solely by matrix remodelling but also occur naturally in healthy tissues6,11. Examples include tracks along ECM fibres in the interstitial space7,9,11, between muscle and nerve fibres11, along or within blood vessels23,24 and in the vasculature of target organs25,26. The different forms of migration tracks are illustrated schematically in FIG. 1.
Figure 1. Microenvironments for confined migration in vivo.
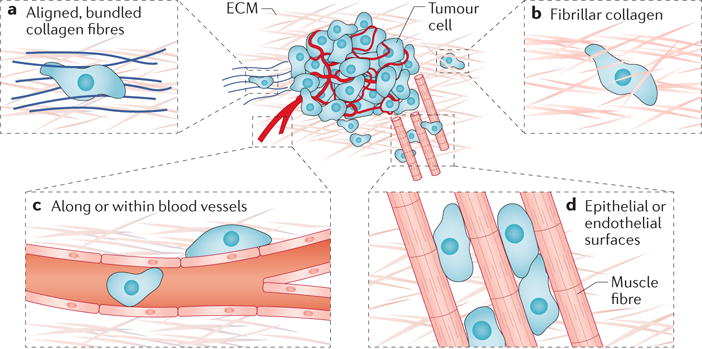
The extratumoural microenvironment offers numerous paths for confined cell migration. a | Alignment and bundling of collagen fibres at the tumour periphery provide cues for directed migration. b | Cells may also migrate through unbundled extracellular matrices (ECMs), such as fibrillar collagen, which present pore-like migration spaces. c | Microtracks also occur both intravascularly and perivascularly. d | Cells can also migrate between epithelial or endothelial surfaces, such as those found between muscle and nerve fibres
The in vivo importance of these tracks in cancer metastasis is substantiated by numerous observations. For instance, in an orthotopic rat MTLn3 xenograft model of breast cancer, tumour cells associated with a high occurrence of lung metastases in mice preferentially migrated along collagen fibres in the primary tumour27. Similar in vivo observations have been made using both mouse and human tumour cells; migration along collagen fibres has been observed in polyomavirus middle T antigen (PyMT)-derived primary mammary tumours in mice28 and in a xenograft model of primary cancer (in which human TN1 cancer cells were used to generate tumours in non-obese diabetic–severe combined immunodeficiency (NOD–SCID) mice)29. Perivascular spaces and white matter tracks in the brain also offer ‘highways’ for glioma cell migration30, and melanoma cells that have extravasated into the brain use the outer surface of blood vessels as guidance structures for continued migration and proliferation31. Intravascular migration of human HT1080 fibrosarcoma cells through the tube-like structures of capillaries has also been observed in a mouse skin-flap model after cell delivery by intracardiac injection32. It is noteworthy that a large subset of these pre-existing tracks are of the same diameter before and after tumour cell invasion, indicating non-destructive tumour cell movement11. These observations, along with the plasticity of cancer cell migration mechanisms, could help to explain the poor performance of inhibitors of matrix metalloproteinases (MMPs) in vivo33.
Cell migration along topographical features in the tissue microenvironment is clinically relevant. For example, local cell invasion from primary breast tumours is associated with bundled collagen fibres that align radially at the tumour–stroma interface22, a structural signature that is predictive of poor survival independently of other prognostic factors34. In addition, the propensity of tumour cells to migrate along topographically defined tracks has been explored in a therapeutic context in a study in which glioblastoma cells were induced to migrate along an aligned nanofibre membrane spanning the skull and out of the cranial cavity to reduce tumour volume35.
The migration microenvironments that cancer cells encounter in vivo are not fully recapitulated by biomimetic 3D ECM gels6 (BOX 1). As such, complementary assays presenting fibre-like and channel-like tracks of prescribed dimensions and stiffness have been developed to study confined migration (as reviewed in REF. 36). Engineered microenvironments enable high-throughput mechanistic studies in well-defined models of migration spaces in which the individual factors influencing migrating cells (for example, the cross-sectional area available for migration, substrate stiffness, ligand density and the presence of external gradients) are decoupled37–39. These in vitro assays providea simplified view of the in vivo setting and impose well-controlled constraints on cells, thereby enabling fine control of the microenvironment so that cell shape40, protein localization and actin polymerization41–44, as well as response to chemical stimuli45, during migration can be studied.
Box 1. In vitro migration assays.
In vitro methods enable the study of confined cell migration in environments of known physical and chemical composition. The design and fabrication of confining spaces that mimic the in vivo physical microenvironment has enabled high-throughput migration assays and the elucidation of confined migration mechanisms. These assays are described briefly below and have recently been reviewed elsewhere in detail36.
Biomimetic hydrogels: 3D gels formed of extracellular matrix proteins or chemically produced polymers. For migration assays, cells are typically encapsulated in a hydrogel material that is then polymerized. The hydrogel composition and the polymerization conditions used determine the pore sizes encountered by encapsulated cells and whether the gel can be degraded by enzymes (such as matrix metalloproteinases) that the cells secrete.
Microchannel devices: migration devices that have shaft-like spaces with rectangular cross-sections and typical widths and heights between 3 μm and 50 μm. Depending on the microchannel dimensions, cells may be laterally confined by all four microchannel walls but free to move forwards and backwards, or they may sense the microchannel wall on only one side. In most cases, microchannels are formed by polymerizing the final migration substrate (for example, polydimethylsiloxane (PDMS)39,127–130, collagen69 or polyacrylamide38,131) on a microfabricated template.
Grooved substrates: migration substrates with parallel, rectangular or trapezoidal troughs that run unidirectionally for lengths much greater than the cell diameter121. Groove depth can range from hundreds of nanometres to tens of micrometres, and the space between grooves can be adjusted so that cells span multiple grooves or sense only one. In grooved substrates, topographical cues are presented primarily at the basal surface of the cell.
Microcontact-printed and micropatterned lines: migration substrates in which thin stripes or patterns of a polymer are deposited on a 2D surface that is otherwise non-adhesive to cells. Cells are confined by adhesions to these narrow stripes and undergo ‘1D migration’ (REFS 29,75,132). In these assays, cells are not compressed, and confinement is imposed by limiting the areas in which adhesions can be formed.
Vertical confinement devices: substrates in which cells plated on a 2D substrate are sandwiched beneath a PDMS roof so that they are free to migrate laterally but are confined at their basal and apical surfaces. The height between the substrate and the PDMS roof in these devices is typically set at 3–7 μm (REF. 43).
Micropost arrays: substrates that present defined but discontinuous barriers to cell migration in the form of vertical posts. Posts are arranged such that migrating cells typically encounter several microposts simultaneously104.
Physical limits on cell motility
Nuclear size and stiffness control confined migration
Physical cues, such as confinement, strongly influence tumour cell trafficking19,46,47. As confinement increases, it becomes more challenging for a cell to deform sufficiently so that it can squeeze into narrow spaces. Accumulating evidence suggests that the nucleus, which is the stiffest cellular component, has a rate-limiting role in confined migration (FIG. 2) and prevents cell movement when the cross-sectional area of pores in the ECM is below a critical threshold18,48–50. In collagen gels, tumour cell arrest occurs at pore sizes smaller than ~7 μm2, which corresponds to ~10% of the nuclear cross-sectional area18. We refer to this threshold as the ‘nuclear limit’ on migration. Below this threshold, nuclear translocation does not occur, and thus cells cannot migrate in the absence of matrix remodelling18. However, modulation of nuclear stiffness controls the efficiency of migration through narrow pores. For instance, knockdown of lamin A, one of the key components of the nuclear lamina, decreases nuclear stiffness and enhances the transmigration of A549 lung carcinoma and U251 glioblastoma cells through narrow(~7 μm2) pores in a transwell assay48. These in vitro findings are corroborated by in vivo experiments showing that, compared with cells in the centre of a tumour xenograft, A549 cells at the periphery of the xenograft exhibit reduced lamin A levels and increased nuclear elongation48, which are indicative of a more pliable nucleus that facilitates confined migration. Moreover, ectopic expression of lamin A in A549 cells48 or leukocyte-like HL-60 cells51 decreases migration through narrow pores. Similarly, expression of progerin (a mutant form of lamin A) increases nuclear stiffness and suppresses confined cell migration52. When pores larger than ~7 μm2 are present, nuclear deformation is not required, and cell migration is not dependent on lamin A levels48.
Figure 2. Determinants of cell migration in confinement.
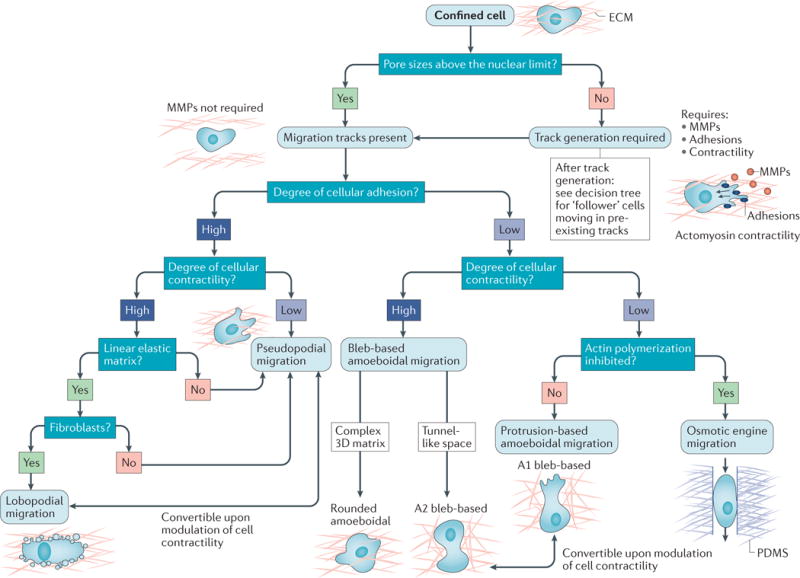
A number of intrinsic and extrinsic cues influence the migration mode used by confined cells. The expected modes of migration for a set of environmental and intrinsic factors are depicted as a decision tree. Tumour cells migrating through porous matrices with small pore sizes (less than ~7 μm2 in area, which we term the nuclear limit) migrate proteolytically through the secretion of matrix metalloproteinases (MMPs), which create microtracks for migration. Follower cells moving through these tracks and cells moving through microenvironments with pre-existing migration tracks use diverse migration mechanisms that depend on the levels of adhesion and cell contractility, and are thus dependent on both the cell and the microenvironment. When cell adhesions to the substrate are present, tumour cells migrate using a pseudopodial-based mechanism that is dependent on protrusions. Under conditions of high contractility and in linearly elastic matrices, fibroblasts can also move using a lobopodial migration mode. When cellular adhesion to the substrate is low or absent, tumour cells primarily migrate using a bleb-based mode of amoeboidal migration (rounded amoeboidal migration or A2 bleb-based migration) that is dependent on high cortical contractility. When contractility is inhibited, tumour cells may use a protrusion-based amoeboidal migration mode (A1 bleb-based migration) that is dependent on actin polymerization at the leading edge. In the absence of actin polymerization, cell movement is achievable through front-to-rear flow of water through the cell (which is termed osmotic engine migration). ECM, extracellular matrix; PDMS, polydimethylsiloxane.
Confinement imposes a mechanical stress on the nucleus48 and causes nuclear deformation, which results in localized nuclear envelope rupture and DNA damage in tumour cells53 and immune cells54. Loss of nuclear envelope integrity markedly increases in confining (20 μm2 or smaller) channels and is exacerbated by the depletion of lamins53. Nuclear envelope rupture coincides with the formation of chromatin-filled nuclear-membrane blebs that contain little or no lamins53. Although both nuclear envelope rupture and DNA damage are evident during confined migration, apoptotic events are rather infrequent owing to efficient repair of the nuclear envelope by the endosomal sorting complexes required for transport (ESCRT) III complex53,54. Additionally, Waterman and colleagues55 revealed the crucial role of formin-family actin filament nucleator FMN2 in generating a perinuclear actin and focal adhesion system that protects against nuclear damage and DNA double-strand breaks during confined migration, thereby exacerbating the extravasation and metastatic spread of melanoma cells to the lung. However, inhibition of both nuclear envelope and DNA repair mechanisms increases cell death in confinement53,54. Collectively, these findings suggest that confined migration compromises the integrity of the nucleus and causes DNA damage, which may lead to aneuploidy and genomic instability, thereby further promoting cancer progression. Although further work is needed to fully elucidate the mechanisms of confinement-induced DNA damage, it has been suggested that such damage may be attributed to the diffusion of reactive oxygen species into the nucleus during nuclear envelope rupture54.
Matrix remodelling in environments with small pores
In microenvironments with small pores that otherwise prevent cell movement (for example, pores smaller than the nuclear limit), MMPs typically work together with cell-generated contractile forces to cleave ECM fibres and bundle cleaved fibres around the cell periphery, creating migration tracks56,57. A leader cell, either a cancer-associated fibroblast or a cancer cell itself, displaying mesenchymal characteristics (that is, an elongated morphology, pseudopodial protrusions and adhesion to the substrate) generates these tracks15,16,58 through the coordinated action of MMPs and the actomyosin machinery, thereby ‘paving the way’ for the subsequent migration of follower cancer cells. The migration of follower cells through these newly formed tunnels is MMP independent15,16. The proteolytic, matrix remodelling mode of migration has been reviewed extensively elsewhere57,59.
Cancer cell invasion may also proceed in an MMP-independent manner. In this mode of invasion, MTLn3E breast cancer cells (a highly invasive subline of MTLn3 cells overexpressing the epidermal growth factor receptor (EGFR)) deform collagen via a RHO-associated protein kinase (ROCK)-dependent, proteinase-independent pathway in vitro60. These observations have been recapitulated in an animal model; cells in orthotopic MTLn3E xenograft tumours deform collagen fibres at the tumour margins, whereas the non-metastatic parental MTC cells donot. Suppression of cell contractility via ROCK inhibition in MTLn3E cells alters myosin light chain (MLC) localization and reduces cell invasion60. Interestingly, MDA-MB-231 breast cancer cells seeded in collagen gels align collagen fibres in a RHO-dependent manner61, and the proportion of motile cells is markedly reduced when pharmacological inhibitors of RHO, ROCK, MLC kinase (MLCK) or myosin II are applied62.
The ability of cancer cells to invade via an MMP-independent amoeboidal mode versus an MMP-dependent mesenchymal mode may not solely be attributed to cell-intrinsic properties but also to the 3D architecture of the local microenvironment. It has been postulated that MMP-independent cell invasion may occur in vitro in collagen gels that are devoid of the covalent crosslinks that are typically encountered in vivo63. In support of this, fibroblasts remodel collagen fibres in low-stiffness trypsinized cell-derived matrices (CDMs), whereas crosslinking of these matrices, which increases their stiffness, prevents cells from remodelling the fibres64. Interestingly, the mouse mammary gland, which is a common site of cancer cell injection in in vivo studies, contains significantly less fibrous tissue than the corresponding human tissue65. Although rat breast cancer cells deform collagen fibres in mouse mammary fat pads60, careful consideration of the physical properties of the local microenvironment is crucial when extrapolating results from in vitro studies and mouse studies to the human setting. Furthermore, although many migration mechanisms and their associated signalling pathways seem to be conserved between murine and human cancer cells (for example, migration along collagen fibres in vivo27–29), they are not necessarily identical (for example, see REF. 66), and findings from non-human cell lines should be confirmed using human cells.
Cell motility through pre-existing tracks
Cells migrating in microenvironments that contain pores, tunnels or openings with cross-sectional areas larger than the nuclear limit do not require matrix degradation to enable their movement. This is substantiated by experiments showing that cell treatment with the broad-spectrum MMP inhibitor GM6001 abrogates cell invasion from tumour spheroids in collagen gels with median pore cross-sectional areas of 8 μm2 (that is, in the range of the nuclear limit), whereas invasion proceeds when the median pore area is increased to 24 μm2 (REF. 67). Similarly, the migration speeds of single, motile membrane type 1 (MT1)-MMP-transduced HT1080 cells in the presence of GM6001 are suppressedin collagen gels with small pores but recover as pore sizes approach 25–30 μm2 (REF. 18). When MMPs are inhibited or knocked down, both human MDA-MB-231 and mouse MMT breast cancer cells are still able to invade pre-formed, patterned tracks in non-pepsinized rat tail collagen, but invasion in non-patterned 3D gels is inhibited68,69. Here, we focus on mechanisms of confined cell migration through pre-existing tracks.
Confined migration mechanisms
When migration tracks are present, the mode of cell motility is mainly determined by the interplay between cell adhesion and contractility, which, in our opinion, determines the types of protrusion (for example, actin polymerization-based versus bleb-based protrusions) that tumour cells use to migrate. Modulation of these parameters, either directly or through a pathway that feeds back on these functions70, influences the mode of migration used by a given cell. The range of interconvertible migration modes available to cells in confinement are summarized in TABLE 1 and FIG. 2 and discussed in detail below.
Table 1.
Summary of confined cell migration modes
| Migration mode | Role of cell–substrate adhesions | Role of cell contractility | Necessary environmental conditions | Characteristic cell polarization | Notes | Refs |
|---|---|---|---|---|---|---|
| Pseudopodial | Adhesions required | Migration occurs under conditions of both high or low levels of RHOA, depending on matrix pore size | Sites for cell adhesion | Polarized PIP3, RAC1 and CDC42 at the leading edge | Speed often unchanged upon inhibition of contractility | 64,72 |
| Lobopodial | Adhesions required | High RHOA levels | Linearly elastic 3D matrix | Nonpolarized cortactin, PIP3, RAC1 and CDC42 | Efficient migration following CDC42 or RAC1 knockdown | 64,80 |
| Bleb-based amoeboidal (A2 blebbing) | Low adhesion | High RHOA levels | Pore size larger than the nuclear limit on migration | Myosin II accumulation at the cell rear | Speed typically reduced upon ROCK inhibition | 43 |
| Protrusion-based amoeboidal (A1 blebbing) | Low adhesion | Low RHOA levels | Pore size larger than the nuclear limit on migration | Retrograde actin flow localized to small protrusion at the leading edge | Activation of cell contractility converts A1 blebbing to A2 blebbing | 43 |
| Osmotic engine | Low adhesion | RHOA dispensable | Full confinement | Polarized aquaporins and ion transporters at the leading edge | Occurs even if actin polymerization is disrupted | 91 |
PIP3, phosphatidylinositol-3,4,5-trisphosphate; ROCK, RHO-associated protein kinase.
Pseudopodial migration of cancer cells
Matrix adhesion is classically required for pseudopodial migration71, which we use as a general term for lamellipodial, filopodialor other migration mechanisms that rely on protrusions driven by actin polymerization72. In a 3D microenvironment, cells can migrate using thin fan-like protrusions at the distal ends of F-actin-enriched pseudopodia, adhesions to the matrix and polarization of cortactin, Wiskott–Aldrich syndrome protein (WASP), RAC1, CDC42 and phosphatidyl-inositol-3,4,5-trisphosphate (PIP)71,73 or using actin-rich, wedge-like protrusions that fill the available pore opening42. When the cellular adhesion machinery is intact and cells can remodel the matrix by exerting traction forces on the surrounding matrix, tumour cells migrate using pseudopods, often alongside MMP activity57. This mode of migration can occur in microchannels that are coated with or moulded in ECM proteins62,74, on microcontact-printed lines75 (BOX 1) and in collagen gels64.
In environments with relatively small pore sizes, cell contractility can contribute to pseudopodial migration by facilitating nuclear deformation. Inhibition of cell contractility via blebbistatin reduces fibroblast speed in 3D collagen gels, presumably because of difficulties in nuclear translocation through the matrix64. Similarly, inhibition of ROCK-mediated contractility impairs the migration of MT1-MMP-transduced HT1080 cells through gels with relatively narrow (~20 μm2) pores but not through gels with wide (55 μm2) pores18.
Pseudopodial migration can also occur in confinement when actomyosin contractilityis inhibited, particularly in well-defined microchannels76 and ECM gels that contain migration tracks or pores with cross-sectional areas larger than the nuclear limit. In ECM gels, these tracks can either occur in the architecture of the gel62 or be generated by leader cells16,77, as discussed above. The ability of cells to migrate through pre-existing tracks via an actomyosin contractility-independent manner may be due to an overall decrease in the traction forces inside these tunnels78. This reduction in traction forces correlates with the attenuation of focal adhesion size in confinement76. Similarly, the speed of MDA-MB-231 cell migration through confining microtracks in collagen gels does not correlate with levels of phosphorylated MLC but force exertion in 2D environments does, suggesting that force exertion is not directly related to speed during confined pseudopodial migration through such tracks62. Although the contractile activity of myosin motors is not necessary for confined pseudopodial migration, it does increase migration velocity79. It is noteworthy that cell contractility does, however, have a key role in amoeboidal cell migration in confinement, as discussed below.
Lobopodial migration of fibroblasts in linearly elastic matrices
The rheological properties of a matrix have a critical role in determining the mode of migration. In highly crosslinked matrices that exhibit a linearly elastic behaviour (that is, rigidity does not increase with applied force), fibroblasts can use a pressure-based lobopodial migration mode that is dependent on RHOA–ROCK–myosin II-mediated contractility64,80 and adhesions containing paxillin and vinculin64. In contrast to lamellipodial-mediated migration, lobopodially migrating fibroblasts form a blunt, cylindrical leading protrusion and intracellular pressure-based blebs that are not enriched with cortactin, and these cells migrate without polarization of the RHO GTPases RAC1 and CDC42 (REF. 64). Instead, vimentin-based connections between the nucleoskeleton and the cell membrane create compartments in the cell that become differentially pressurized, with the nucleus acting as a piston, to promote the formation of pressure-based blebs at the leading edge80. In 3D CDMs, knockdown of RHOA or inhibition of ROCK switches the migration mode of fibroblasts from lobopodial to lamellipodial without affecting migration speed64. However, evidence suggests that cancer cells may not use lobopodial migration. For instance, HT1080 cells in linearly elastic CDMs do not move using this migration mode but instead adopt amoeboidal or mesenchymal– lamellipodial morphologies64. It is currently unknown whether cancer cells are able to use a lobopodial migration mode to move through tissues or whether a certain combination of microenvironmental and cell signalling conditions might enable them to use this mechanism.
Bleb-based amoeboidal migration under conditions of high contractility
When cell adhesions to the matrix are suppressed or eliminated, tumour cells can migrate using either high-contractility amoeboidal migration driven by membrane blebbing43,81 or protrusion-based migration that occurs under conditions of low cell contractility but high confinement43 (see the next section). Amoeboidal migration is characterized by a rounded cell morphology, membrane blebs, diffuse (as opposed to focal) distribution of cell adhesion proteins and a dependence on actomyosin contractility81–83. Blebs are spherical protrusions that lack filamentous actin when they are first formed and can drive cell locomotion83. The relationship between the blebs formed during lobopodial migration and those generated during amoeboidal migration is unknown73. However, these two bleb-based migration modes are clearly distinct; whereas amoeboidal blebbing migration can occur in the absence of cell adhesions, lobopodial migration is characterized in part by the presence of these adhesions64.
Confined migration of cancer cells displaying amoeboidal blebbing is favoured by increased colocalization of contractile machinery (studied using phosphorylation of MLC as a marker) with actin–plasma membrane linkages that are mediated by ezrin–radixin–moesin (ERM) proteins84. Interestingly, on 2D planar surfaces, colocalization of actomyosin and these linkages between the actin cortex and cell membrane promotes bleb formation at the expense of lamellipodium formation and suppresses cell speed84. Forces exerted during ‘adhesion-free’ blebbing migration of rat Walker 256 carcinosarcoma cells are several orders of magnitude lower than those exerted during traction-driven migration, and they are directed outwards from the cell body, generating friction to enable actin-driven cell migration85. Amoeboidal bleb-based migration of MT1-MMP-transduced HT1080 cells82 and A375 melanoma cells86 has been observed in vivo in mouse xenograft models.
Both computational modelling and in vitro experiments reveal that bleb-based migration is most efficient in confinement and in environments of low adhesiveness86. Although cells can migrate by blebbing in the absence of adhesions, cell adhesions do not necessarily have to be blocked for blebbing to occur; instead, overexpressing cell contractility proteins or blocking protease activity seems to switch cells towards blebbing migration81,82. Inhibition of the actin-related protein 2/3 (ARP2/3) actin nucleator complex also promotes a switch to bleb-based protrusions from protrusions formed by actin polymerization, without modulating cell adhesions, in both HL-60 cells migrating in 5 μm × 5 μm microchannels42 and in an adherent subline of Walker 256 cells migrating on a 2D surface87. Moreover, an absence of proteolysis is not sufficient for amoeboidal migration. In melanoma cells, a rounded cell morphology is associated with increased secretion of MMP9, which is itself dependent on active RHO–ROCK and Janus kinase (JAK)–signal transducer and activator of transcription 3 (STAT3) signalling88.
HeLa cervical cancer cells under conditions of vertical confinement (BOX 1) and low adhesion rapidly migrate using a stable bleb migration mechanism (termed A2 blebbing) in which a large rear uropod-shaped body drags behind the cell, myosin II and actin localize at the cell rear immediately anterior to the uropod, and rapid retrograde flow of actin and myosin II occurs in the central part of the cell; the proportion of cells using this mode of migration is increased under conditions of high contractility that are induced by cell treatment with calyculin A43, a broad-spectrum serine/threonine phosphatase inhibitor that causes an increase in the phosphorylation of MLC, as well as that of other proteins such as vimentin89. Interestingly, the uropod resembles that found in neutrophils and functions as a frictional ‘brake’ (REF. 43). The A2 blebbing mode of migration also occurs in vivo during the embryonic development of zebrafish and is dependent on high contractility90. Future work should aim to verify the prevalenceof A2 blebbing in in vivo models of cancer metastasis.
Protrusion-based amoeboidal migration under low contractility
Under conditions of both low cell adhesion to the substrate and low contractility, cell movement is dependent on the activity of protrusions at the leading edge. When cells are vertically confined to a height of 3 μm and plated on substrates with low levels of integrin ligands, rapid retrograde flow of actin in a small protruding region at the leading edge of the cell can drive migration at much faster speeds than achieved by cells migrating in 2D43. HeLa cells in low-adhesion environments and confined to a height of 3 μm use this mode of migration, termed A1 blebbing, when ROCK signalling is inhibited43. These cells switch to A2 blebbing migration when contractility is enhanced, indicating that these mechanisms are plastic and depend on the contractile state of the cell43. It will be interesting to determine whether A1 blebbing, which is promoted in environments of low cell contractility, occurs in vivo, given that a decrease in amoeboid cell migration is typically noted upon inhibition of contractility.
The osmotic engine model
Protrusions generated by local water permeation across the plasma membrane can lead to a mode of cell migration under confining conditions that we term the ‘osmotic engine model’. When confined in narrow microchannels with a cross-sectional area of 30 μm2, several tumour cell types can migrate even when actin polymerization is inhibited76,91. The osmotic engine model also predicts that migration can occur independently of actomyosin contractility and posits that the polarized uptake and expulsion of water at the leading and trailing edges of confined cells, respectively, drive cell locomotion91. Indeed, RNA interference (RNAi)-mediated reduction or pharmacological inhibition of aquaporins or ion transporters (for example, sodium–hydrogen exchanger 1 (NHE1)) to suppress or modulate water uptake and expulsion represses the confined migration of tumour cells91. Although this migration mode is not dependent on contractility, it is linked to other volume-change-based migration modes. For example, water transport has been linked to bleb formation, as knockdown of aquaporins prevents cell volume changes and bleb generation92. As such, blebs can form as a result of cellular volume changes driven by water influx, instead of simple propulsion of the cell membrane92.
Actin polymerization seems to be necessary for establishing the polarization of aquaporins and ion transporters during cell entry into confining microchannels. However, once this polarization is set up, actin polymerization is dispensable for confined cell migration91. Interestingly, actin is also required for the repolarization of aquaporins and ion transporters in cells migrating inside narrow channels in response to an external cue91. The overexpression of aquaporins and ion transporters in numerous metastatic tumour cell lines and in resected human tumour tissue specimens93–95 may cause the osmotic engine model to be more evident in these tumour cells than in normal cells. It is noteworthy that results supporting the osmotic engine model were obtained using a microfabricated device in which the leading and trailing edges of the cell were exposed to media while the cell periphery was surrounded by liquid-impermeable stiff polydimethylsiloxane (PDMS) (BOX 1). It is currently unknown whether this mode of migration occurs in other in vitro microenvironments, such as channels with stiffnesses that are similar to those found in human tissue, or in porous 3D matrices, or in vivo contexts in which the cell membrane encounters solutes and water around the entire periphery as a result of diffusion through the matrix pores. Moreover, how this migration mode is related or convertible to other migration modes has yet to be determined. We speculate that ROCK1, which phosphorylates MLCs to induce actomyosin contractility and is an upstream activator of NHE1 (REF. 96), may function as a linker in the switch between actin polymerization-driven migration and osmotic engine migration97.
Cell navigation through complex topographies
In vivo, cells migrate through complex topographies that impose directional choices on cells. However, little is known about the inputs of a directional decision-making process in regions that present cells with different paths for cell migration. Chemical-gradient sensing has a role in neutrophil-like cell navigation of microfabricated mazes98. In the absence of CDC42 activity, dendritic cells are unable to migrate in vivo, but are still able to polarize and form protrusions in response to chemotactic cues99. However, the presence of multiple competing protrusions upon Cdc42 knockout, as opposed to a single leading-edge protrusion in the presence of CDC42, abrogates their actual movement by trapping them in complex 3D topographies99. Rapidly moving neutrophil-like cells confined in narrow (18 μm2) microchannels push water as they move forwards100. As such, these cells, when they reach an intersection, ‘decide’ to follow the path of least hydraulic resistance, meaning that cells choose the shortest path or the path with wider microchannels. Similarly, MDA-MB-231 cells confined in narrow (30 μm2) channels preferentially enter the wider branches at asymmetrical bifurcations40. In wider (200 μm2) 3D tracks, cell elongation and alignment along either the left or right side wall of the feeder channel manifests in persistent cell migration through the phenomenon termed contact guidance40. As such, when they reach an asymmetrical bifurcation, cells enter both narrow (30 μm2) and wide (200 μm2) branches with an equal probability40. Increases in the persistenceof contact-guided cells are due, at least in part, to a decrease in the occurrence of nascent protrusions perpendicular to the cell poles in matrices with aligned fibres or topographical features101; this is possibly due to myosin II-mediated contractility, which minimizes cell-surface curvature102.
Contact guidance along local topography is generally decreased when cell contractility is inhibited. However, contact guidance can be promoted upon inhibition of CDC42 (REF. 40). MDA-MB-231 cells can also efficiently migrate through microchannel mazes in the absence of an external chemotactic stimulus using self-generated EGF gradients, although this process is probably dependent on the presence of limited avenues for EGF diffusion in the microchannel network, which establishes local gradients at the leading edge of migrating cells103. This navigation mechanism bears some resemblance to the autocrine colony-stimulating factor 1 (CSF1) signalling loop that promotes the invasion of cells from MDA-MB-231 xenograft tumours in mice66. In summary, we postulate that physical characteristics such as track size, tissue stiffness and adhesion molecule expression could similarly drive cell trafficking to, at least partially, guide metastatic tropism in vivo.
Physical confinement effects
Multicellular to single cell transitions
Cells can transition between collective migration and single cell migration when moving through confining spaces12. For example, topographical barriers, which restrict the extent of cell–cell contact, cause dispersal of cells from a collective sheet104. Increasing micropost spacing in a micropost array (BOX 1) suppresses individual cell scattering and promotes collective migration104, thereby demonstrating the influence of the local microenvironment on the plasticity of cancer cell migration modes. In collagen gels, MV3 melanoma cells and HT1080 cells transition from single cell to collective migration as matrix pore or track size decrease, owing to the piling up of follower cells; when large pore or track sizes are present, cells move individually67. This switch is not affected by changes in ligand density or matrix stiffness, but is instead driven solely by the physical size of the pores in the gel67, indicating how the type of physical confinement experienced by groups of cells can change how these cells migrate.
Confinement to microcontact-printed lines of various widths also affects the mode of collective migration. On 20 μm-wide fibronectin strips, Madin–Darby canine kidney (MDCK) epithelial cells display a contraction-relaxation or caterpillar-like mode of migration with well-coordinated push-and-pull force patterns within the cell chain105. By contrast, on wider fibronectin strips (that are more than 100 μm wide), these cells move as a single sheet that is maintained under tension by cells leading the collectively migrating group, and the magnitude of traction forces decays away from the cell front105. These migration patterns, on both narrow and wide strips, require intact cell–cell contact and actomyosin contractility105. Changes in the distance over which individual cell movements are correlated with the movements of neighbouring or near-neighbouring cells in the collective group are also a function of the extent of physical confinement, cell–cell contact and cell–substrate adhesion105,106, further suggesting that the mode of collective cell migration isa function of the extent of cell confinement. Whether such changes occur in collectively migrating groups of cancer cells in vivo remains to be seen.
Cell signaling
Confinement is a physical stimulus that is capable of initiating and regulating an intracellular cascade of signalling events, thereby modulating cell migration mechanisms. Cells tune the signalling input to achieve an optimal balance between RAC1 and RHOA– myosin II signalling, such that there is a strong RAC1 output for efficient cell locomotion on unconfined 2D surfaces and a strong RHOA–myosin II output for confined migration70. The underlying mechanisms by which cells sense and adapt to different physical microenvironments remain to be determined. Different mechanisms have been proposed and involve three major classes of mechanosensors: stretch-activated ion channels107, elements of the cytoskeleton and nuclear matrix108 and components of adhesion complexes and ECM109. It was recently reported19 that confinement of A375 cells induces an increase in intracellular calcium levels via the stretch-activated cation channel PIEZO1 (REF. 19). This confinement-induced increase in intracellular calcium levels negatively regulates the activity of protein kinase A (PKA) via a phospho diesterase 1 (PDE1)-dependent pathway19. Interestingly, confinement-induced changes in PKA activity are abolished only upon dual, but not individual, inhibition of PIEZO1 and myosin II, suggesting that the PIEZO1–PDE1–PKA and myosin II signalling modules represent two independent confinement-sensing mechanisms19. Thus, signals activated by PIEZO1 and myosin II in response to confinement are integrated in a signalling circuit that optimizes cell locomotion. Of note, components of adhesion complexes, such as α4 and α5 integrins, are not required for confinement sensing19. Myosin II has also been implicated in fibroblast mechanosensing of surface topography110 and stiffness111, and it may also be important in regulating tumour cell responses to topographical cues19,40.
Recent work suggests that the nucleus can itself function as a mechanosensor through a purely mechanical process that is mediated by tension in the nuclear envelope. In both HeLa cells in culture and live embryonic zebrafish, swelling or mechanical compression of the nucleus promotes calcium-dependent accumulation of cytosolic phospholipase A2 (cPLA2) on the inner nuclear membrane, which consequently signals cell damage through the release of pro-inflammatory eicosanoids112. Although the relevance of this pathway to cancer progression has yet to be established, it provides an interesting example of how physical cues can directly influence cell signalling, and a similar pathway could potentially be used by cancer cells during migration through confining matrices when nuclear deformation occurs50.
Cell division and gene expression
Confinement can itself drive cell division defects and changes in gene expression, which could exacerbate tumorigenesis by increasing genetic instability, as has been observed in the stiffer, compressive environment of a primary tumour113,114. Division of taxol (microtubule-stabilizing drug)-resistant MDA-MB-231 cells confined within straight microchannels tapering from 15 μm to 3 μm in width results in asymmetrical daughter cell sizes115. In addition, vertical confinement induced by a low PDMS roof (3–7 μm in height) increases the number of aberrant divisions (that is, divisions resulting in three or more daughter cells), the differences in volume between daughter cells and the time required for division, as well as the rate of cell death116,117. These defects are probably due to problems in microtubule spindle assembly. When mitotic HeLa cells are unable to become round owing to vertical confinement, chromosomes are spread more widely throughout the cell, with some out of reach of astral microtubules, which results in delayed spindle formation118. In some cases, this leads to multipolar spindle formation, improper chromosomal segregation and/or cell death118. Of note, the combination of confinement in epithelial tissue and inhibition of actin polymerization in vivo causes defects in cell rounding during mitosis and leads to asymmetrical cell divisions and defects in skin stratification119, further suggesting the importance of cell rounding in cell division. Although the skin effectively functions as a model of epithelial cell confinement in vivo, it remains to be seen whether similar effects occur in the crowded environment of a developing tumour.
Vertical confinement ruptures the nuclear lamina and induces the differential expression of genes involved in inflammation, stress responses and membrane synthesis, specifically those involved in the DNA damage response and the nuclear factor-κB (NF-κB) pathway120. Similar changes ingene expression have been observed upon confinement within microgrooves121 and on microcontact-printed islands122 (BOX 1). Cell responses to confinement are likely to be a function of both short-term and long-term signals. Isolated nuclei can respond to force, not through chromatin or nuclear actin responses, but by using a pathway that requires an intact nuclear lamina and the inner nuclear membrane protein emerin123. Thus, the nucleus functions as an important mechanosensor in confinement by inducing signalling changes upon nuclear lamina rupture or shape change.
Outlook and perspectives
Cell-intrinsic properties coupled with the heterogeneity of the tissue micro-environment in vivo enable tumour cells to use a wide repertoire of migration modes during metastasis. This great plasticity of migration mechanisms available to tumour cells confounds efforts to abate metastasis in the clinical setting. Indeed, the ability of tumour cells to use multiple modes of migration makes it difficult to predict whether modulation of a given signalling pathway associated with migration in one context will actually inhibit cell movement in the diverse microenvironments found in vivo. This is particularly true for pathways that modulate cell adhesion and contractility, which are differentially required across migration modes. For example, under conditions of environmental confinement and low adhesiveness, high and low levels of contractility promote migration in a number of cancer cell lines, whereas intermediate contractility leads to nonpolarized bleb formation and limited cell movement43,86. Interestingly, in normal rat liver epithelial cells plated on 2D substrates, an intermediate level of inhibition of myosin-mediated contractility frees actin from the actomyosin cortex and results in cell polarization and migration, whereas complete myosin inactivation or conditions of high contractility inhibit cell polarization such that cells remain stationary124. Decreasing the adhesiveness of the surface alone without changing cell contractility is insufficient to induce the migration of these cells124. To reconcile these disparate results, future work should entail careful characterization of both the extracellular microenvironment (for example, stiffness, adhesion ligand concentration and pore size) and cell-intrinsic properties (specifically, the degree of cell contractility, which should be matched to quantitative traction force measurements) to more definitively elucidate the role of myosin II-mediated contraction in cancer cell migration.
Although genetically engineered mouse models recapitulate essential features of metastatic disease, they have profound limitations. Most notably, they lack the high-throughput capacity that will allow the acquisition of accurate quantitative results from different microenvironments of prescribed physical properties (for example, stiffness or confinement) and biochemical properties (for example, low or high adhesion). Further advances in deep-tissue imaging will be required to enable precise quantification of confining dimensions in different tissues and accurate cell visualization in order to delineate the diverse cellular phenotypes that are present during migration in vivo. Although patient-derived breast cancer cells have been shown to migrate both individually and as multicellular ‘streams’ in mouse models17, we are not aware of any direct observations of cell motility in patient-derived xenograft (PDX) tumours. As PDX tumours should retain the original tumour architecture, it will be important to delineate the primary modes of migration in PDX models in the future. To better refine the exact conditions in which a given migration mode occurs, various complementary, reductionist in vitro models have been developed that recapitulate key aspects of the local microenvironment. These engineered model systems include biomimetic 3D ECM gels, as well as fibre-like and channel-like tracks of prescribed dimensions and stiffnesses (BOX 1). However, migration mechanisms are at times difficult to compare across platforms owing to non-uniformities in matrix pore size, matrix adhesion ligands, substrate stiffness and cell contractility levels47. Careful integration of cell biology and bioengineering approaches will enable precise control of each of these factors in order to better delineate modes of cell migration and elucidate strategies to inhibit cell trafficking in the context of cancer metastasis.
We anticipate that knowledge of confined migration will lead to an improved understanding of cancer metastasis by providing better models to use for studying the role of cell heterogeneity in cancer, which will enable high-throughput and physiologically relevant drug and cell line screens. Ultimately, confining environments consisting of both fibrillar matrices and track-like migration spaces should be integrated on organ-on-a-chip devices to recapitulate the in vivo architecture of tissue structures in in vitro models of tumour growth and metastasis. Accommodating for the diverse mechanisms available to confined tumour cells, the interaction of tumour cells with associated stromal cells29, environmental conditions such as hypoxia125 and the interplay between chemotaxis and contact inhibition41 at the bench stage of drug discovery will increase the clinical success rate of new anticancer drugs.
We envision that targeting the migratory spread of cancer cells will be useful in an adjuvant setting for treating patients with aggressive or locally invasive disease but with no evidence of distant metastasis and in preventing the recurrence of cancer after treatment and/or resection of a primary tumour. Such clinical interventions should be tested alongside treatment of a primary tumour to evaluate their effectiveness in suppressing or halting the presentation of metastatic disease. Interestingly, intravital imaging of mouse colorectal cancer cells colonizing the liver has revealed that cells are highly motile following extravasation and that the metastatic burden can be reduced by inhibiting the motility of these cells before they form a micrometastasis126. Thus, targeting cancer cell motility may be an important therapeutic option, even if some cells have already disseminated by the time of treatment. Identification of the full repertoire of migration modes will aid the development of a multimodal therapeutic approach to inhibit cancer cell migration via the numerous interchangeable modes and thus combat metastasis.
Acknowledgments
The authors gratefully acknowledge support from the US National Institutes of Health (grants R01CA183804, R01CA186286, R01GMS114675 and U54CA210173).
Glossary
- 3D longitudinal tracks
Tunnel-like spaces in which cells are confined either at their basal and apical surfaces or around their periphery, but encounter open space at the cell front and rear
- Collective migration
Cell migration in which groups of cells migrate while in physical contact and in the same net direction. This is in contrast to single cell migration in which cells move individually and are not in physical contact with other motile cells
- Contact guidance
The tendency of cells (or groups of cells) to align and polarize along topographical features, such as microchannel walls or aligned collagen fibres
- Contact inhibition
The tendency of cells to suppress forward movement upon leading-edge contact with another cell
- Matrix metalloproteinases
(MMPs). Soluble or membrane bound enzymes that sever extracellular matrix proteins to mediate matrix remodelling, cell migration or cell signalling
- Organ-on-a-chip
Engineered devices that attempt to recapitulate the major functions and anatomical organization of an organ on a miniaturized scale
- Polydimethylsiloxane
(PDMS). A silicone rubber polymer that is frequently used to fabricate microfluidic devices. PDMS is optically transparent, allows diffusion of oxygen and can be coated with various extracellular matrix proteins to promote cell adhesion
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201–218. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Cancer Society. Cancer facts and figures 2016. American Cancer Society. 2016 http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2016/
- 3.Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolf K, et al. Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol. 2009;20:931–941. doi: 10.1016/j.semcdb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexander S, Koehl GE, Hirschberg M, Geissler EK, Friedl P. Dynamic imaging of cancer growth and invasion: a modified skin-fold chamber model. Histochem Cell Biol. 2008;130:1147–1154. doi: 10.1007/s00418-008-0529-1. [DOI] [PubMed] [Google Scholar]
- 8.Alexander S, Weigelin B, Winkler F, Friedl P. Preclinical intravital microscopy of the tumour-stroma interface: invasion, metastasis, and therapy response. Curr Opin Cell Biol. 2013;25:659–671. doi: 10.1016/j.ceb.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Gritsenko PG, Ilina O, Friedl P. Interstitial guidance of cancer invasion. J Pathol. 2012;226:185–199. doi: 10.1002/path.3031. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt S, Friedl P. Interstitial cell migration: integrin-dependent and alternative adhesion mechanisms. Cell Tissue Res. 2010;339:83–92. doi: 10.1007/s00441-009-0892-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weigelin B, Bakker G-J, Friedl P. Intravital third harmonic generation microscopy of collective melanoma cell invasion: principles of interface guidance and microvesicle dynamics. IntraVital. 2012;1:32–43. doi: 10.4161/intv.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 13.Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011;21:736–744. doi: 10.1016/j.tcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Bremer C, Tung CH, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nat Med. 2001;7:743–748. doi: 10.1038/89126. [DOI] [PubMed] [Google Scholar]
- 15.Fisher KE, et al. MT1-MMP- and Cdc42-dependent signaling co-regulate cell invasion and tunnel formation in 3D collagen matrices. J Cell Sci. 2009;122:4558–4569. doi: 10.1242/jcs.050724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaggioli C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 17.Patsialou A, et al. Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. IntraVital. 2013;2:e25294. doi: 10.4161/intv.25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf K, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. 2013;201:1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hung WC, et al. Confinement-sensing and signal optimization via Piezo1/PKA and myosin II pathways. Cell Rep. 2016;15:1430–1441. doi: 10.1016/j.celrep.2016.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goetz JG, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–163. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costa-Silva B, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–826. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Provenzano PP, et al. Collagen reorganization at the tumor–stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lugassy C, et al. Angiotropism, pericytic mimicry and extravascular migratory metastasis in melanoma: an alternative to intravascular cancer dissemination. Cancer Microenviron. 2014;7:139–152. doi: 10.1007/s12307-014-0156-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bentolila LA, et al. Imaging of angiotropism/vascular co-option in a murine model of brain melanoma: implications for melanoma progression along extravascular pathways. Sci Rep. 2016;6:23834. doi: 10.1038/srep23834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lugassy C, Barnhill RL. Angiotropic melanoma and extravascular migratory metastasis: a review. Adv Anat Pathol. 2007;14:195–201. doi: 10.1097/PAP.0b013e31805048d9. [DOI] [PubMed] [Google Scholar]
- 26.Naumov GN, et al. Cellular expression of green fluorescent protein, coupled with high-resolution in vivo videomicroscopy, to monitor steps in tumor metastasis. J Cell Sci. 1999;112:1835–1842. doi: 10.1242/jcs.112.12.1835. [DOI] [PubMed] [Google Scholar]
- 27.Sahai E, et al. Simultaneous imaging of GFP, CFP and collagen in tumors in vivo using multiphoton microscopy. BMC Biotechnol. 2005;5:14. doi: 10.1186/1472-6750-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang W, et al. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007;67:3505–3511. doi: 10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- 29.Sharma VP, et al. Reconstitution of in vivo macrophage–tumor cell pairing and streaming motility on one-dimensional micro-patterned substrates. IntraVital. 2012;1:77–85. doi: 10.4161/intv.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuddapah VA, Robel S, Watkins S, Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci. 2014;15:455–465. doi: 10.1038/nrn3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kienast Y, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16:116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 32.Yamauchi K, et al. Real-time in vivo dual-color imaging of intracapillary cancer cell and nucleus deformation and migration. Cancer Res. 2005;65:4246–4252. doi: 10.1158/0008-5472.CAN-05-0069. [DOI] [PubMed] [Google Scholar]
- 33.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 34.Conklin MW, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain A, et al. Guiding intracortical brain tumour cells to an extracortical cytotoxic hydrogel using aligned polymeric nanofibres. Nat Mater. 2014;13:308–316. doi: 10.1038/nmat3878. [DOI] [PubMed] [Google Scholar]
- 36.Paul CD, Hung WC, Wirtz D, Konstantopouos K. Engineered models of confined cell migration. Annu Rev Biomed Eng. 2016;18:159–180. doi: 10.1146/annurev-bioeng-071114-040654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irimia D, Charras G, Agrawal N, Mitchison T, Toner M. Polar stimulation and constrained cell migration in microfluidic channels. Lab Chip. 2007;7:1783–1790. doi: 10.1039/b710524j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pathak A, Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc Natl Acad Sci USA. 2012;109:10334–10339. doi: 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tong Z, et al. Chemotaxis of cell populations through confined spaces at single-cell resolution. PLoS ONE. 2012;7:e29211. doi: 10.1371/journal.pone.0029211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paul CD, et al. Interplay of the physical microenvironment, contact guidance, and intracellular signaling in cell decision making. FASEB J. 2016;30:2161–2170. doi: 10.1096/fj.201500199R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin B, Yin T, Wu YI, Inoue T, Levchenko A. Interplay between chemotaxis and contact inhibition of locomotion determines exploratory cell migration. Nat Commun. 2015;6:6619. doi: 10.1038/ncomms7619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson K, et al. Mechanisms of leading edge protrusion in interstitial migration. Nat Commun. 2013;4:2896. doi: 10.1038/ncomms3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu YJ, et al. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell. 2015;160:659–672. doi: 10.1016/j.cell.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Maiuri P, et al. Actin flows mediate a universal coupling between cell speed and cell persistence. Cell. 2015;161:374–386. doi: 10.1016/j.cell.2015.01.056. [DOI] [PubMed] [Google Scholar]
- 45.Lin B, et al. Synthetic spatially graded Rac activation drives cell polarization and movement. Proc Natl Acad Sci USA. 2012;109:E3668–E3677. doi: 10.1073/pnas.1210295109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stroka KM, Konstantopoulos K. Physical biology in cancer. 4. Physical cues guide tumor cell adhesion and migration. Am J Physiol Cell Physiol. 2014;306:C98–C109. doi: 10.1152/ajpcell.00289.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Charras G, Sahai E. Physical influences of the extracellular environment on cell migration. Nat Rev Mol Cell Biol. 2014;15:813–824. doi: 10.1038/nrm3897. [DOI] [PubMed] [Google Scholar]
- 48.Harada T, et al. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J Cell Biol. 2014;204:669–682. doi: 10.1083/jcb.201308029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beadle C, et al. The role of myosin II in glioma invasion of the brain. Mol Biol Cell. 2008;19:3357–3368. doi: 10.1091/mbc.E08-03-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davidson PM, Denais C, Bakshi MC, Lammerding J. Nuclear deformability constitutes a rate-limiting step during cell migration in 3D environments. Cell Mol Bioeng. 2014;7:293–306. doi: 10.1007/s12195-014-0342-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rowat AC, et al. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J Biol Chem. 2013;288:8610–8618. doi: 10.1074/jbc.M112.441535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Booth-Gauthier EA, et al. Hutchinson–Gilford progeria syndrome alters nuclear shape and reduces cell motility in three dimensional model substrates. Integr Biol. 2013;5:569–577. doi: 10.1039/c3ib20231c. [DOI] [PubMed] [Google Scholar]
- 53.Denais CM, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352:353–358. doi: 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raab M, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352:359–362. doi: 10.1126/science.aad7611. [DOI] [PubMed] [Google Scholar]
- 55.Skau CT, et al. FMN2 makes perinuclear actin to protect nuclei during confined migration and promote metastasis. Cell. 2016;167:1–15. doi: 10.1016/j.cell.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedl P, Wolf K. Proteolytic interstitial cell migration: a five-step process. Cancer Metastasis Rev. 2009;28:129–135. doi: 10.1007/s10555-008-9174-3. [DOI] [PubMed] [Google Scholar]
- 58.Wolf K, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9:893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- 59.Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008;68:7247–7249. doi: 10.1158/0008-5472.CAN-08-0784. [DOI] [PubMed] [Google Scholar]
- 60.Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr Biol. 2006;16:1515–1523. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- 61.Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008;95:5374–5384. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carey SP, et al. Comparative mechanisms of cancer cell migration through 3D matrix and physiological microtracks. Am J Physiol Cell Physiol. 2015;308:C436–C447. doi: 10.1152/ajpcell.00225.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus-independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol. 2009;185:11–19. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petrie RJ, Gavara N, Chadwick RS, Yamada KM. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol. 2012;197:439–455. doi: 10.1083/jcb.201201124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parmar H, Cunha GR. Epithelial–stromal interactions in the mouse and human mammary gland in vivo. Endocr Relat Cancer. 2004;11:437–458. doi: 10.1677/erc.1.00659. [DOI] [PubMed] [Google Scholar]
- 66.Patsialou A, et al. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009;69:9498–9506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haeger A, Krause M, Wolf K, Friedl P. Cell jamming: collective invasion of mesenchymal tumor cells imposed by tissue confinement. Biochim Biophys Acta. 2014;1840:2386–2395. doi: 10.1016/j.bbagen.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 68.Ilina O, Bakker GJ, Vasaturo A, Hofmann RM, Friedl P. Two-photon laser-generated microtracks in 3D collagen lattices: principles of MMP-dependent and -independent collective cancer cell invasion. Phys Biol. 2011;8:015010. doi: 10.1088/1478-3975/8/1/015010. [DOI] [PubMed] [Google Scholar]
- 69.Kraning-Rush CM, Carey SP, Lampi MC, Reinhart-King CA. Microfabricated collagen tracks facilitate single cell metastatic invasion in 3D. Integr Biol. 2013;5:606–616. doi: 10.1039/c3ib20196a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hung WC, et al. Distinct signaling mechanisms regulate migration in unconfined versus confined spaces. J Cell Biol. 2013;202:807–824. doi: 10.1083/jcb.201302132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Petrie RJ, Yamada KM. At the leading edge of three-dimensional cell migration. J Cell Sci. 2012;125:5917–5926. doi: 10.1242/jcs.093732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Friedl P, Sahai E, Weiss S, Yamada KM. New dimensions in cell migration. Nat Rev Mol Cell Biol. 2012;13:743–747. doi: 10.1038/nrm3459. [DOI] [PubMed] [Google Scholar]
- 73.Petrie RJ, Yamada KM. Fibroblasts lead the way: a unified view of 3D cell motility. Trends Cell Biol. 2015;25:666–674. doi: 10.1016/j.tcb.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang P, et al. Fluid shear promotes chondrosarcoma cell invasion by activating matrix metalloproteinase 12 via IGF-2 and VEGF signaling pathways. Oncogene. 2015;34:4558–4569. doi: 10.1038/onc.2014.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doyle AD, Wang FW, Matsumoto K, Yamada KM. One-dimensional topography underlies three-dimensional fibrillar cell migration. J Cell Biol. 2009;184:481–490. doi: 10.1083/jcb.200810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balzer EM, et al. Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 2012;26:4045–4056. doi: 10.1096/fj.12-211441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fischer RS, Gardel M, Ma X, Adelstein RS, Waterman CM. Local cortical tension by myosin II guides 3D endothelial cell branching. Curr Biol. 2009;19:260–265. doi: 10.1016/j.cub.2008.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raman PS, Paul CD, Stroka KM, Konstantopoulos K. Probing cell traction forces in confined microenvironments. Lab Chip. 2013;13:4599–4607. doi: 10.1039/c3lc50802a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hawkins RJ, et al. Pushing off the walls: a mechanism of cell motility in confinement. Phys Rev Lett. 2009;102:058103. doi: 10.1103/PhysRevLett.102.058103. [DOI] [PubMed] [Google Scholar]
- 80.Petrie RJ, Koo H, Yamada KM. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science. 2014;345:1062–1065. doi: 10.1126/science.1256965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 82.Wolf K, et al. Compensation mechanism in tumor cell migration: mesenchymal–amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Paluch EK, Raz E. The role and regulation of blebs in cell migration. Curr Opin Cell Biol. 2013;25:582–590. doi: 10.1016/j.ceb.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Madsen CD, et al. STRIPAK components determine mode of cancer cell migration and metastasis. Nat Cell Biol. 2015;17:68–80. doi: 10.1038/ncb3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bergert M, et al. Force transmission during adhesion-independent migration. Nat Cell Biol. 2015;17:524–529. doi: 10.1038/ncb3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tozluoglu M, et al. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol. 2013;15:751–762. doi: 10.1038/ncb2775. [DOI] [PubMed] [Google Scholar]
- 87.Bergert M, Chandradoss SD, Desai RA, Paluch E. Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc Natl Acad Sci USA. 2012;109:14434–14439. doi: 10.1073/pnas.1207968109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Orgaz JL, et al. Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun. 2014;5:4255. doi: 10.1038/ncomms5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chartier L, et al. Calyculin-A increases the level of protein phosphorylation and changes the shape of 3T3 fibroblasts. Cell Motil Cytoskeleton. 1991;18:26–40. doi: 10.1002/cm.970180104. [DOI] [PubMed] [Google Scholar]
- 90.Ruprecht V, et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell. 2015;160:673–685. doi: 10.1016/j.cell.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stroka KM, et al. Water permeation drives tumor cell migration in confined microenvironments. Cell. 2014;157:611–623. doi: 10.1016/j.cell.2014.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Taloni A, et al. Volume changes during active shape fluctuations in cells. Phys Rev Lett. 2015;114:208101. doi: 10.1103/PhysRevLett.114.208101. [DOI] [PubMed] [Google Scholar]
- 93.Chae YK, et al. Expression of aquaporin 5 (AQP5) promotes tumor invasion in human non-small-cell lung cancer. PLoS ONE. 2008;3:e2162. doi: 10.1371/journal.pone.0002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jung HJ, Park JY, Jeon HS, Kwon TH. Aquaporin-5: a marker protein for proliferation and migration of human breast cancer cells. PLoS ONE. 2011;6:e28492. doi: 10.1371/journal.pone.0028492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Machida Y, et al. Relationship of aquaporin 1, 3, and 5 expression in lung cancer cells to cellular differentiation, invasive growth, and metastasis potential. Hum Pathol. 2011;42:669–678. doi: 10.1016/j.humpath.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 96.Tominaga T, Ishizaki T, Narumiya S, Barber D. L p160ROCK mediates RhoA activation of Na–H exchange EMBO J. 1998;17:4712–4722. doi: 10.1093/emboj/17.16.4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stroka KM, Gu Z, Sun SX, Konstantopoulos K. Bioengineering paradigms for cell migration in confined microenvironments. Curr Opin Cell Biol. 2014;30:41–50. doi: 10.1016/j.ceb.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Skoge M, et al. A worldwide competition to compare the speed and chemotactic accuracy of neutrophil-like cells. PLoS ONE. 2016;11:e0154491. doi: 10.1371/journal.pone.0154491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lammermann T, et al. Cdc42-dependent leading edge coordination is essential for interstitial dendritic cell migration. Blood. 2009;113:5703–5710. doi: 10.1182/blood-2008-11-191882. [DOI] [PubMed] [Google Scholar]
- 100.Prentice-Mott HV, et al. Biased migration of confined neutrophil-like cells in asymmetric hydraulic environments. Proc Natl Acad Sci USA. 2013;110:21006–21011. doi: 10.1073/pnas.1317441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Riching KM, et al. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys J. 2014;107:2546–2558. doi: 10.1016/j.bpj.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Elliott H, et al. Myosin II controls cellular branching morphogenesis and migration in three dimensions by minimizing cell-surface curvature. Nat Cell Biol. 2015;17:137–147. doi: 10.1038/ncb3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Scherber C, et al. Epithelial cell guidance by self-generated EGF gradients. Integr Biol. 2012;4:259–269. doi: 10.1039/c2ib00106c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wong IY, et al. Collective and individual migration following the epithelial–mesenchymal transition. Nat Mater. 2014;13:1063–1071. doi: 10.1038/nmat4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vedula SR, et al. Emerging modes of collective cell migration induced by geometrical constraints. Proc Natl Acad Sci USA. 2012;109:12974–12979. doi: 10.1073/pnas.1119313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garcia S, et al. Physics of active jamming during collective cellular motion in a monolayer. Proc Natl Acad Sci USA. 2015;112:15314–15319. doi: 10.1073/pnas.1510973112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Coste B, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dupont S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 109.Roca-Cusachs P, Iskratsch T, Sheetz MP. Finding the weakest link: exploring integrin-mediated mechanical molecular pathways. J Cell Sci. 2012;125:3025–3038. doi: 10.1242/jcs.095794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Frey MT, Tsai IY, Russell TP, Hanks SK, Wang YL. Cellular responses to substrate topography: role of myosin II and focal adhesion kinase. Biophys J. 2006;90:3774–3782. doi: 10.1529/biophysj.105.074526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wong S, Guo WH, Wang YL. Fibroblasts probe substrate rigidity with filopodia extensions before occupying an area. Proc Natl Acad Sci USA. 2014;111:17176–17181. doi: 10.1073/pnas.1412285111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Enyedi B, Jelcic M, Niethammer P. The cell nucleus serves as a mechanotransducer of tissue damage-induced inflammation. Cell. 2016;165:1160–1170. doi: 10.1016/j.cell.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Potapova TA, Zhu J, Li R. Aneuploidy and chromosomal instability: a vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 2013;32:377–389. doi: 10.1007/s10555-013-9436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Padera TP, et al. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- 115.Mak M, Reinhart-King CA, Erickson D. Elucidating mechanical transition effects of invading cancer cells with a subnucleus-scaled microfluidic serial dimensional modulation device. Lab Chip. 2013;13:340–348. doi: 10.1039/c2lc41117b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tse HT, Weaver WM, Di Carlo D. Increased asymmetric and multi-daughter cell division in mechanically confined microenvironments. PLoS ONE. 2012;7:e38986. doi: 10.1371/journal.pone.0038986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kittur H, Weaver W, Di Carlo D. Well-plate mechanical confinement platform for studies of mechanical mutagenesis. Biomed Microdevices. 2014;16:439–447. doi: 10.1007/s10544-014-9846-4. [DOI] [PubMed] [Google Scholar]
- 118.Lancaster OM, et al. Mitotic rounding alters cell geometry to ensure efficient bipolar spindle formation. Dev Cell. 2013;25:270–283. doi: 10.1016/j.devcel.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 119.Luxenburg C, Pasolli HA, Williams SE, Fuchs E. Developmental roles for Srf, cortical cytoskeleton and cell shape in epidermal spindle orientation. Nat Cell Biol. 2011;13:203–214. doi: 10.1038/ncb2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Le Berre M, Aubertin J, Piel M. Fine control of nuclear confinement identifies a threshold deformation leading to lamina rupture and induction of specific genes. Integr Biol. 2012;4:1406–1414. doi: 10.1039/c2ib20056b. [DOI] [PubMed] [Google Scholar]
- 121.Dalby MJ, Riehle MO, Yarwood SJ, Wilkinson CD, Curtis AS. Nucleus alignment and cell signaling in fibroblasts: response to a micro-grooved topography. Exp Cell Res. 2003;284:274–282. doi: 10.1016/s0014-4827(02)00053-8. [DOI] [PubMed] [Google Scholar]
- 122.Thomas CH, Collier JH, Sfeir CS, Healy KE. Engineering gene expression and protein synthesis by modulation of nuclear shape. Proc Natl Acad Sci USA. 2002;99:1972–1977. doi: 10.1073/pnas.032668799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guilluy C, et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway within the nucleus. Nat Cell Biol. 2014;16:376–381. doi: 10.1038/ncb2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lomakin AJ, et al. Competition for actin between two distinct F-actin networks defines a bistable switch for cell polarization. Nat Cell Biol. 2015;17:1435–1445. doi: 10.1038/ncb3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang Y, Wen J, Zhou L, Qin L. Utilizing a high-throughput microfluidic platform to study hypoxia-driven mesenchymal-mode cell migration. Integr Biol. 2015;7:672–680. doi: 10.1039/c5ib00059a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ritsma L, et al. Intravital microscopy through an abdominal imaging window reveals a pre-micrometastasis stage during liver metastasis. Sci Transl Med. 2012;4:158ra145. doi: 10.1126/scitranslmed.3004394. [DOI] [PubMed] [Google Scholar]
- 127.Rolli CG, Seufferlein T, Kemkemer R, Spatz JP. Impact of tumor cell cytoskeleton organization on invasiveness and migration: a microchannel-based approach. PLoS ONE. 2010;5:e8726. doi: 10.1371/journal.pone.0008726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Breckenridge MT, Egelhoff TT, Baskaran H. A microfluidic imaging chamber for the direct observation of chemotactic transmigration. Biomed Microdevices. 2010;12:543–553. doi: 10.1007/s10544-010-9411-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Irimia D. Cell migration in confined environments. Methods Cell Biol. 2014;121:141–153. doi: 10.1016/B978-0-12-800281-0.00010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vargas P, Terriac E, Lennon-Dumenil AM, Piel M. Study of cell migration in microfabricated channels. J Vis Exp. 2014;84:e51099. doi: 10.3791/51099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pathak A, Kumar S. Transforming potential and matrix stiffness co-regulate confinement sensitivity of tumor cell migration. Integr Biol. 2013;5:1067–1075. doi: 10.1039/c3ib40017d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]