

Abstract
Skeletal bone formation and maintenance requires coordinate functions of several cell types, including bone forming osteoblasts and bone resorbing osteoclasts. Gsα, the stimulatory subunit of heterotrimeric G proteins, activates downstream signaling through cAMP and plays important roles in skeletal development by regulating osteoblast differentiation. Here, we demonstrate that Gsα signaling also regulates osteoclast differentiation during bone modeling and remodeling. Gnas, the gene encoding Gsα, is imprinted. Mice with paternal allele deletion of Gnas (Gnas+/p−) have defects in cortical bone quality and strength during early development (bone modeling) that persist during adult bone remodeling. Reduced bone quality in Gnas+/p− mice was associated with increased endosteal osteoclast numbers, with no significant effects on osteoblast number and function. Osteoclast differentiation and resorption activity was enhanced in Gnas+/p− cells. During differentiation, Gnas+/p− cells showed diminished pCREB, β-catenin and cyclin D1, and enhanced Nfatc1 levels, conditions favoring osteoclastogenesis. Forskolin treatment increased pCREB and rescued osteoclast differentiation in Gnas+/p− by reducing Nfatc1 levels. Cortical bone of Gnas+/p− mice showed elevated expression of Wnt inhibitors sclerostin and Sfrp4 consistent with reduced Wnt/β-catenin signaling. Our data identify a new role for Gsα signaling in maintaining bone quality by regulating osteoclast differentiation and function through cAMP/PKA and Wnt/β-catenin pathways.
Embryonic and early postnatal skeletal growth predominantly occurs through bone modeling, with bone resorption and formation occurring independently of each other to control bone size and shape. After skeletal development is complete, bone remodeling occurs throughout life with resorption and formation balanced at the bone surfaces in order to repair and maintain skeletal homeostasis1,2,3. However, during inflammation, diseases such as osteoporosis, and cancer metastasis, the balance between bone formation by osteoblasts and resorption by osteoclasts is shifted to increased resorption leading to reduced or poor quality bone4,5. Therefore, it is imperative to understand how osteoclast differentiation and function are regulated in order to improve therapeutic approaches.
Bone modeling and remodeling are precisely coordinated through interactions and regulation by multiple genes and signaling pathways including GNAS, which encodes the α-subunit of stimulatory G-protein (Gsα) of adenylyl cyclase and activates cAMP signaling. Gsα deletion in osteogenic lineage cells expressing osterix and col1a has demonstrated that Gsα plays important roles during skeletal development by regulating mesenchymal cell commitment, osteoblast differentiation, and mineralization without affecting osteoclast function6,7,8,9. Recent studies have also suggested that cAMP levels are regulated by several mechanisms including Wnt signaling, adenylyl cyclase, and Ca2+/Calmodulin-dependent kinases to delicately control osteoclastogenesis10,11. In addition, in fibrous dysplasia, a clinical condition in which activating Gsα mutations cause elevated cAMP levels, increased osteoclasts occur primarily in response to osteoblast secretion of interleukin-612,13. There has been no evidence of a direct role for Gsα in osteoclast differentiation and function, and the impact of Gsα on skeletal bone quality during modeling and remodeling remains insufficiently understood.
The GNAS gene is a complex locus that expresses multiple transcripts14,15,16. The GNAS locus is imprinted, expressing some transcripts specifically from the paternally-inherited allele and others from the maternally-inherited allele. Gsα mRNA is bi-allelically expressed in most cells17. These different allele-specific expression patterns are reflected in human diseases and mouse models that are phenotypically distinct depending on the allele carrying a GNAS inactivating mutation18,19. Mice with paternally-inherited heterozygous deletion of either Gsα exon 1 or exon 2 have shorter body lengths and lower body weights, while mice with maternal inheritance of the deletion of either of the exons are short but obese20. These data suggested that heterozygous GNAS inactivation impacts skeletal bone by affecting growth plate development and bone formation. But how Gsα deletion affects skeletal bone quality during modeling and remodeling, and whether these effects differ with paternal and maternal inheritance of the mutation, has not been examined.
In this study, we investigated the effects of heterozygous GNAS inactivation in mice on skeletal bone during modeling and remodeling. We examined trabecular and cortical bone from mice with paternal or maternal inheritance of Gsα deletion, and the roles of Gsα signaling on osteoblast and osteoclast formation and function. Our data reveal that heterozygous Gnas inactivation negatively affects cortical bone quality, with mutation of the paternal allele causing more severe effects than maternal mutations. We further determined that paternally inherited Gsα deletion alters cortical bone maintenance not through effects on osteoblasts and bone formation, but through enhanced osteoclast differentiation and increased bone resorption, and that these effects are mediated by Gsα signaling via cAMP/PKA and Wnt/β-catenin pathways.
Results
Paternal inheritance of heterozygous Gnas inactivation adversely affects cortical bone quality during bone modeling and remodeling
To determine the role of Gsα signaling on skeletal bone quality, we used an established mouse model of heterozygous Gnas inactivation20. Since Gnas is an imprinted locus, we examined mice with inactivation of the paternally-inherited Gnas allele (Gnas+/p−) and the maternally-inherited Gnas allele (Gnasm−/+). qRT-PCR of mRNA from cortical bone confirmed significant reduction in Gsα expression in both Gnas+/p− and Gnasm−/+ mice (Fig. 1a).
Figure 1. Paternal but not maternal heterozygous Gnas deletion causes reduction in cortical bone quality in 9 month old mice.

(a) mRNA expression of Gsα in cortical bone was reduced in heterozygous Gnas mutants compared to WT by qRT-PCR. There was no statistical difference between Gnas+/p− and Gnasm−/+. (b) Representative 3D μCT images of (top) trabecular and (bottom) cortical bone. (c) No differences in trabecular bone volume fraction were observed between the groups. Decreased (d) cortical bone volume fraction, (e) cortical thickness and (g) stiffness and (h) peak load in Gnas+/p− mice but not Gnasm−/+ mice as compared to WT. (f) Endosteal circumference (Endo) measured at the femoral mid-shaft is increased with no change in periosteal circumference (Peri) in Gnas+/p− mice while both are increased in Gnasm−/+ mice. Data represent mean ± SD. N = 13 WT, 7 Gnas+/p− and 5 Gnasm−/+ animals. *p < 0.05, **p < 0.01.
In most mouse strains, rapid accrual of bone occurs until 3 months of age, stabilizes by 6–8 months, and then is maintained by remodeling21. To evaluate the effects of heterozygous Gnas deletion on adult bone remodeling, we analyzed mice at 3 and 9 months of age. At both ages, Gnas+/p− mice weighed less and were shorter (body and femur length were measured; femur length is reported in this study) than WT controls while Gnasm−/+ mice had increased body weight with no difference in femur length (Supplementary Table 1), consistent with reported results20. Previously, histomorphometry of distal femurs in 3-month-old animals had identified no differences vs. controls in trabecular bone volume fraction or osteoblast function in either Gnas+/p− or Gnasm−/+ mice20. Trabecular microarchitecture was not investigated in detail and no analysis of cortical bone was performed20. To examine trabecular microarchitecture, distal metaphyseal femurs of 3- (Supplementary Table 1) and 9-month-old mice were examined by μCT (Fig. 1 and Supplementary Table 1). Overall, trabecular bone volume fraction (BV/TV) and microarchitecture (Fig. 1b,c and Supplementary Table 1) in distal femurs showed no significant differences at both ages in Gnas+/p− and Gnasm−/+ mice compared to littermate controls, consistent with previous results20, however trends of marginal reduction, notably trabecular BV/TV and trabecular thickness, were observed in Gnas+/p− mice. The trabecular bone of lumbar vertebrae (data not shown) also appeared unaffected in both paternally- and maternally-inherited Gnas mutants compared to controls.
Cortical bone μCT analyses of mid-diaphyses of adult 3- and 9-month-old Gnasm−/+ femurs revealed significantly increased total cortical bone volume as well as greater periosteal and endosteal circumference, consistent with the increased body weight of these mutants (Fig. 1f and Supplementary Table 1) relative to WT. No differences in cortical thickness or cortical bone volume fraction were observed (Fig. 1b,d,e). In contrast, Gnas+/p− mice showed significant reductions in total cortical bone volume, cortical thickness, and cortical bone volume fraction at both 3 (data not shown) and 9 months (Fig. 1b,d,e and Supplementary Table 1). A larger endosteal circumference with no change in periosteal circumference suggested an increase in endosteal resorption in Gnas+/p− mice compared to WT (Fig. 1f). Cortical porosity was marginally increased in the Gnas+/p− mice at both ages, but not statistically significant (Supplementary Table 1). Gnas+/p− femurs at both ages were weaker by three-point bending tests, with significant reductions in peak load and stiffness compared to WT; bone strength was unaffected in Gnasm−/+ mice at either age (Fig. 1g,h and Supplementary Table 1). These results indicated that paternal-allele inactivation of Gnas affects cortical bone quality during stages of adult bone remodeling.
To determine effects of Gnas inactivation on early skeletal development and bone modeling, we examined cortical bone in 2-week-old mice by μCT and mechanical testing. As in older mice, no statistically significant differences in trabecular bone were found although trends of reduced BV/TV and trabecular thickness were observed in Gnas+/p− mice. However, in contrast to older mice (Fig. 1), both Gnas+/p− and Gnasm−/+ young mice showed cortical bone defects, with significant decreases in cortical thickness and bone volume fraction (Fig. 2a–c, Supplementary Table 1). At this age, both Gnas+/p− and Gnasm−/+ mice were lower in body weight and femur length compared to WT, with paternally inherited mutants showing greater reduction than the maternal allele mutants. Using three-point bending tests, femurs from both mutants showed significant reductions in stiffness and peak load (Supplementary Table 1) consistent with the μCT data.
Figure 2. Cortical bone defects in Gnas+/p− mice are not due to defects in osteoblast numbers and function in Gnas+/p− mice.

(a) Representative 3D μCT images of cortical bone from mice at 2 weeks of age. (b) Cortical bone volume fraction and (c) cortical thickness measurements by μCT scans at the mid-diaphyseal region of femurs were significantly reduced in both Gnas+/p− and Gnasm−/+ 2-week-old mice. (d) H&E staining of cortical bone shows osteoblasts (arrows) lining the endocortical surface. (f) Double labeling of cortical bone surface with calcein and xylenol orange in 2-week-old mice. Quantification of osteoblast number (e) and mineral apposition rate (g) along the endocortical surface showed reduction in Gnasm−/+ but not Gnas+/p− when compared to WT mice. Data represent mean ± SD. N = 7–11 animals per group for μCT and 5–6 animals per group for histology. *p < 0.05, **p < 0.01.
Collectively, these results show that although deletion of either parental Gnas allele affects cortical bone during modeling, only inactivation of the paternally-inherited allele impacts cortical bone quality during remodeling.
Osteoblast numbers and function are unaffected in Gnas +/p− mice
Several studies demonstrated that ablation of Gsα signaling causes skeletal defects, including lower trabecular and cortical bone volume, due to effects on osteoblasts and bone formation6,8. These data were mainly observed in mice with Gsα deletion specifically in cells of the osteoblast lineage.
To investigate skeletal bone effects of heterozygous Gnas inactivation in osteoblasts, we used conditional Gnas heterozygous mice with paternal or maternal allele Gsα deletion and Cre expression driven by the early osteoblast marker osterix (Osx-Cre; Gnas+/p− and Osx-Cre; Gnasm−/+). Unlike previous studies in Gnas null osteoblasts, μCT analyses revealed no differences at 6 weeks of age in either trabecular or cortical bone (Supplementary Figure 1a,b) in these mutants compared to control mice (Gnas+/+, Gnasfl/+ and Osx-Cre; Gnas+/+).
To determine potential contributions of osteoblast numbers and function to our observed cortical bone defects in 2-week-old mutant mice, we quantified osteoblasts and mineral apposition rates (MAR). While both parameters in Gnas+/p− mice were similar to WT controls, Gnasm−/+ mice showed a marginal but statistically significant decrease in both osteoblast number and MAR compared to WT controls (Fig. 2d–g). This suggested that paternally and maternally inherited Gsα mutants affect cortical bone during development differently and support that cortical bone defects in Gnas+/p− mice were not due to effects in osteoblasts but possibly to effects on other cell types.
Gnas+/p− mice show increased endosteal osteoclasts during remodeling
Cortical bone remodeling is a balance between osteoblast and osteoclast activity. Since osteoblast numbers and function were unaffected in paternal Gnas mutants, we hypothesized that cortical bone defects in these mice could be due to altered osteoclasts. To determine osteoclast numbers, femurs from WT and Gnas+/p− and Gnasm−/+ mutants at 2 weeks, and 3 and 9 months of age were stained with TRAP and numbers of TRAP positive multi-nucleated osteoclasts along the cortical surface were quantified. At 2 weeks of age, when bone modeling is occurring, TRAP staining was primarily observed on the periosteal surface, and no differences among the genotypes were detected (data not shown). At 3 months of age, the numbers of TRAP positive osteoclasts were significantly elevated in Gnas+/p− mice (Fig. 3a,c) while Gnasm−/+ were similar to WT. At 9 months of age (Fig. 3b), the numbers of TRAP positive osteoclasts observed in all genotypes along the endosteal surface were higher than at 3 months. Gnasm−/+ femurs showed no differences in numbers of TRAP positive multi-nucleated osteoclasts compared to WT as was found at 3 months, while Gnas+/p− mice showed a dramatic increase in endosteal osteoclast numbers (Fig. 3c), consistent with the cortical bone defects detected in Gnas+/p− mice. No differences among the genotypes were observed for osteoclast numbers on trabecular bone surfaces (Supplementary Figure 2). These data suggest that inactivation of the paternally-inherited allele of Gnas increases osteoclast numbers and enhances endosteal resorption resulting in cortical bone loss.
Figure 3. Mice with paternal inheritance of heterozygous deletion of Gnas have elevated numbers of endosteal osteoclasts.

At (a) 3 months and (b) 9 months of age, Gnas+/p− exhibit increased endocortical osteoclasts (arrows) detected by TRAP staining compared to WT and Gnasm−/+ mice. (c) Quantification of endocortical osteoclast number at the diaphyseal region in WT, Gnas+/p− and Gnasm−/+ mice at 3 and 9 months of age. Data represent mean ± SD. N = 4–8 animals per group. *p < 0.05. EN – endosteum, BM – bone marrow.
Paternal allele Gnas inactivation enhances osteoclast differentiation and resorption in vitro
Since only Gnas+/p− but not Gnasm−/+ mice showed persistent defects in cortical bone quality concomitant with increase in endosteal osteoclasts, we investigated whether paternal allele Gnas inactivation has a direct effect on either the osteoclast progenitor population or osteoclast differentiation and function. In order to study the progenitor population, osteoclast precursors were counted from the bone marrow of WT and Gnas+/p− mice at 7–9 weeks of age by flow cytometry22. The numbers of osteoclast precursor cells, namely the CD3− CD45R− CD11b−/low CD115high population, were the same in mutant and WT bone marrow (Supplementary Figure 3) indicating that cortical bone defects in Gnas+/p− mice are not caused by increased osteoclast precursors in the bone marrow.
Upon differentiation to osteoclasts in vitro, bone marrow macrophages (BMM) from Gnas+/p− produced significantly more TRAP positive multi-nucleated (≥3 nuclei) osteoclasts at 2 and 3 days of differentiation compared to WT (Fig. 4a and b). qRT-PCR confirmed that Gsα expression was significantly decreased in the cells from Gnas+/p− during osteoclast differentiation relative to WT (Fig. 4e). cAMP levels showed trends of reduction in Gnas+/p− cells relative to WT (Supplementary Figure 4f). In addition, osteoclasts from Gnas+/p− mice on dentine slices formed larger resorption pits compared to WT osteoclasts (Fig. 4c,d). When BMMs from Gnasm−/+ were cultured, they produced more mature osteoclasts that formed larger resorption pits than WT littermate controls (Supplementary Figure 5a,b), however these differences in Gnasm−/+ cells were less than in Gnas+/p− cells, with a 40% increase in osteoclast differentiation and resorption in Gnas+/p− relative to controls vs. only 15% increase in these parameters in Gnasm−/+. These results support that reduced Gsα signaling enhances osteoclast differentiation and osteoclast resorption activity, with greater effects from deletion of the paternal Gnas allele than the maternal allele.
Figure 4. Paternal inheritance of Gnas inactivation enhances osteoclast differentiation and resorption activity of osteoclasts.

(a) Differentiation of bone marrow macrophages (BMMs) from 7–9 week old WT and Gnas+/p− mice into osteoclasts. (b) Quantitation of TRAP+ multi-nucleated cells (≥3 nuclei) at days 1–3 of differentiation shows increased Gnas+/p− osteoclasts at days 2 and 3 of differentiation. (c) Osteoclasts differentiated from BMMs were seeded on bone slices for 48 h and (d) the relative resorption area measured; resorption activity was greater with Gnas+/p− osteoclasts compared to WT. (e) mRNA expression of Gsα was reduced over time in Gnas+/p− cells during osteoclast differentiation. β2-microglobulin was used for normalization and WT values were set to 1. (f) pCREB was lower during osteoclast differentiation in Gnas+/p− cells as compared to WT cells. (g,h) Nfatc1 from whole cell lysate was significantly elevated in Gnas+/p− cells as compared to WT cells at day 3 of osteoclast differentiation. Data represent mean ± SD. Experiments were performed at least 3 times with n = 2–3 animals per group per experiment. Total of 5–7 animals per group from 3 experiments used for quantification. For osteoclast differentiation and pit formation, cells were seeded in triplicates in 96-well plate and on bone slices respectively. For pit formation, resorption area mean of WT was set to 1. *p < 0.05; **p < 0.01.
Decreased cAMP/PKA pathway, increased Nfatc1 and enhanced osteoclast differentiation in Gnas+/p− mice
Gsα signals primarily through adenylyl cyclase and the cAMP/PKA pathway23,24,25. Recent studies implicated this pathway as inhibitory to osteoclast differentiation by phosphorylating Nfatc1, an important transcription factor for osteoclast differentiation11,26. Adenylyl cyclase activity is decreased in Gnas+/p− mice20 raising the possibility that decreased cAMP leads to decreased phospho-Nfatc1 and increased osteoclasts. To investigate this mechanism, we analyzed protein levels of pCREB, a downstream activator of the cAMP/PKA pathway during osteoclast differentiation. We found that pCREB protein levels were marginally reduced at day 0 and significantly reduced at day 1 of osteoclast differentiation in cells from Gnas+/p− mice (Fig. 4f and Supplementary Figures 4a,b). We investigated whether this decreased cAMP signaling is associated with altered Nfatc1 levels and determined that both total and nuclear Nfatc1 protein was highly up-regulated at day 3 of osteoclast differentiation in Gnas+/p− osteoclasts compared to WT (Fig. 4g,h and Supplementary Figures 4d,e) suggesting that paternal Gnas allele deletion reduces pCREB during early differentiation and enhances Nfatc1 at a protein level to impact osteoclastogenesis.
Forskolin inhibits Nfatc1-induced osteoclastogenesis by elevating adenylyl cyclase and PKA activity11. We therefore hypothesized that treatment with forskolin would rescue the increased osteoclast differentiation by Gnas+/p− cells. In order to test this, we induced osteoclast differentiation of WT and Gnas+/p− osteoclast precursor cells in the presence or absence of forskolin. Treatment of Gnas+/p− cells with forskolin abolished the increase in TRAP+ multi-nucleated osteoclasts to levels comparable to WT (Fig. 5a,b). In addition, forskolin increased pCREB (Fig. 5c) and decreased Nfatc1 (Fig. 5d) to levels comparable to WT. Together, these results demonstrate that activation of PKA rescues pCREB, and regulates Nfatc1 levels and osteoclast differentiation in Gnas+/p− cells.
Figure 5. Forskolin (Fsk) rescues the osteoclast differentiation phenotype of cells from Gnas+/p− mice.

(a,b) Forskolin treatment during osteoclast differentiation rescued the increased osteoclast numbers in Gnas+/p− mice. (c,d) Forskolin treatment of Gnas+/p− cells increased pCREB (c) and reduced Nfatc1 (d) to levels comparable to WT. Experiments were performed at least 3 times with n = 1–2 animals per group per experiment. Total of 5 animals per group used for quantification. *p < 0.05.
β-catenin and its target cyclin D1 are reduced in Gnas+/p− mice
Wnt signaling inhibits osteoclast differentiation through canonical β-catenin-dependent27 and non-canonical10 pathways. β-catenin and its transcriptional target cyclin D1 have been shown to play biphasic roles during osteoclast proliferation and differentiation28. Their expression is required for osteoclast precursor proliferation, and both are activated by M-CSF but suppressed by RANKL during osteoclast differentiation. Since mice with Gsα deletion in osteoblasts6 and osteocytes29 exhibit decreased Wnt/β-catenin signaling, we examined whether reduction in Wnt/β-catenin signaling in Gnas+/p− mice could lead to increased osteoclast differentiation. To determine the levels of Wnt/β-catenin signaling, total β-catenin from cell lysates during osteoclast differentiation were detected by immunoblot. Under normal circumstances, RANKL suppresses β-catenin and cyclin D1 during osteoclast differentiation. Indeed, we found that both WT and Gnas+/p− cells showed reduction in β-catenin (data not shown) and cyclin D1 (Fig. 6b) in response to RANKL. However, comparison of WT and Gnas+/p− cells showed that β-catenin and cyclin D1 were more greatly decreased in Gnas+/p− cells than WT (Fig. 6a,b), supporting that inappropriately low levels of β-catenin and its downstream cyclin D1 enhance osteoclast differentiation in Gnas+/p− mice.
Figure 6. Wnt/β-catenin-mediated cyclin D1 signaling is downregulated in Gnas+/p− mice.
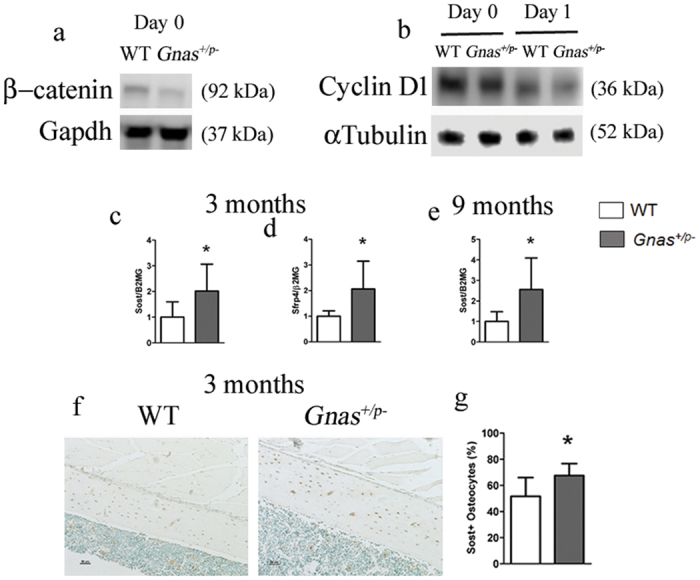
Western blots of (a) β-catenin and (b) cyclin D1 at days 0 and 1 during osteoclast differentiation showed reduced levels in Gnas+/p− cells during osteoclast differentiation. mRNA expression of the Wnt pathway inhibitors Sost (c) and Sfrp4 (d) at 3 months and Sost at 9 months (e) is increased in cortical bone from Gnas+/p− mice as compared to WT by qRT-PCR. β2-microglobulin was used for normalization and WT values were set to 1. (f) Immunohistochemistry of sclerostin in the mid-diaphyseal region of femurs from 3-month old WT and Gnas+/p−mice. (g) Quantitation of IHC detection shows increase in Sclerostin positive osteocytes in Gnas+/p− cortical bone as compared to WT. Data represent mean ± SD. Western blots were performed twice with n = 2–3 animals per group per experiment. N = 5–8 animals per group for real time PCR. N = 6 animals per group for IHC. *p < 0.05.
To determine whether Wnt/β-catenin signaling is altered in skeletal bone cells of Gnas+/p− mice, expression of Wnt targets and inhibitors from the diaphyses of femurs and tibiae from 3- and 9- month-old mice was quantified. While there was no difference in expression of the Wnt target gene Lef1 (data not shown), mRNA levels of the Wnt inhibitors Sost and Sfrp4 were up-regulated in both 3 and 9-month-old Gnas+/p− mice (Fig. 6c–e). A higher percent of osteocytes positive for Sclerostin in femurs of 3-month-old Gnas+/p− mice was detected by immunohistochemistry (Fig. 6f,g). Collectively, these data support that an overall increase in Wnt inhibitors causes a decrease in β-catenin/cyclin D1 leading to increased osteoclastogenesis in paternally-inherited Gnas inactivation mutants.
Discussion
Human diseases caused by GNAS mutations provide insight into the roles of GNAS in bone formation and have identified regulatory functions in osteoblasts. In progressive osseous heteroplasia (POH) and pseudohypoparathyroidisms 1A (PHP1A), heterozygous GNAS/Gnas inactivation and decreased Gsα signaling cause heterotopic ossification (extra-skeletal bone formation) in soft tissues such as subcutaneous fat and muscle15,24,30,31. Fibrous dysplasia (FD), caused by somatic activating mutations in GNAS, is characterized by woven bone lesions in skeletal bone due to defective osteoblast differentiation32,33,34; FD also shows increased osteoclastic bone resorption at the site of these lesions that is not a direct effect of Gsα signaling in osteoclasts but is a response by osteoclasts to increased interleukin-6 secretion by osteoblasts12.
In this study, we investigated the effects of heterozygous GNAS inactivation on skeletal bone during modeling and remodeling and unexpectedly identified a novel requirement for Gsα signaling to maintain bone quality through regulation of osteoclast differentiation via cAMP/PKA/pCREB and Wnt/β-catenin signaling pathways. Although we cannot exclude effects of the mutation on trabecular bone, we identified a stronger effect in cortical bone vs. trabecular bone and therefore focused our investigations on the mechanisms through which Gnas+/p− causes decreased cortical bone quality. Cortical bone forms 80% of our skeletal mass and 80% of fractures occur at cortical bone sites35,36, therefore, it is clinically relevant to understand the signaling mechanisms that regulate cortical bone maintenance during normal remodeling as well as aging and disease.
The GNAS gene locus encodes several transcripts, with expression patterns dependent on the parent of origin of each allele15,16. In mice and humans, inactivation or deletion of the paternally vs. maternally-inherited GNAS allele results in different phenotypes. For example, PHP1A is caused by mutation of the maternally-inherited GNAS allele and leads to subcutaneous heterotopic ossification and hormone resistance. In contrast, POH which is mainly associated with paternally-inherited allele mutation, presents with more severe progression of heterotopic ossification into deep connective tissues such as the muscle and without hormone resistance14,18,37,38. The underlying mechanisms for these phenotypic differences are incompletely understood, and may be influenced by genomic and tissue-specific imprinting at the GNAS locus and/or differences in GNAS transcript expression.
In our study, heterozygous Gnas knockout mice with paternal (Gnas+/p−) vs. maternal (Gnasm−/+) inheritance of a Gnas deletion showed similar defects in cortical bone quality and strength during early development (bone modeling). This reduction in bone quality persisted during adult bone remodeling in Gnas+/p− mice, however Gnasm−/+ mice recovered, showing bone qualities similar to Gnas+/+ control mice at adult bone remodeling stages.
Gsα signaling activates cAMP which phosphorylates PKA and CREB24,39. Previous studies of homozygous deletion of Gsα in osterix-expressing osteoblast precursors revealed adverse effects on both trabecular and cortical bone due to defective mesenchymal progenitor commitment to osteoblasts and terminal osteoblast differentiation6,9. Gsα deletion in col1α1-expressing osteoblasts showed decreased TRAP positive osteoclasts at the endocortical bone surface leading to an increase in cortical thickness; however, this analyses was limited to the bone modeling stage in newborn mice9. Furthermore, activation of cAMP and phosphorylation of PKA and CREB in osteoblasts was found to activate osteoclasts by increasing RANKL and inhibiting OPG (a decoy receptor for RANKL that impedes osteoclastogenesis)40,41. None of these studies examined the effects of Gsα signaling on osteoclast differentiation and function or its impact on postnatal bone quality during bone remodeling.
Given this previous evidence that Gsα affects osteoblasts and bone formation, we examined whether the reduced skeletal bone quality in Gnas mutant mice was due to impaired osteoblast numbers or function. We found no significant effects on osteoblast number and function in the paternal mutants while maternal mutants unexpectedly showed a marginal reduction in both parameters. Although statistically significant, these effects in maternal Gnas mutants are small and insufficient to draw conclusions about functional effects. Future studies to further investigate proliferation and osteoblast differentiation with cells from WT and Gnasm−/+ are necessary to determine the mechanisms underlying these in vivo osteoblast changes.
By contrast, endosteal osteoclast numbers were increased in vivo in Gnas+/p− but not in Gnasm−/+ mice when compared to WT. However, in vitro differentiation of either Gnas+/p− or Gnasm−/+ bone marrow macrophages to osteoclasts was enhanced. Gnas mutant osteoclasts also had increased bone resorption activity in vitro, with paternal mutants showing a more dramatic increase than the maternal mutants. These results suggest that haploinsufficiency of Gsα leads to increase in osteoclastogenesis in vitro, but additional factors in vivo [such as sclerostin which is increased in Gnas+/p− cortical bone (Fig. 6c–g) and decreased in Gnasm−/+ cortical bone (Supplementary Figure 6b)] and imprinting effects may contribute to the persistent cortical bone phenotype and increased endosteal osteoclasts that we observe only in the paternal mutants. Although our in vitro experiments suggest a cell-autonomous role for Gsα in osteoclasts, future studies ablating Gnas specifically in the osteoclast lineage (using LysozymeM-Cre42 or RANK-Cre10) would further elucidate the role of Gnas in osteoclast differentiation and function as well as its impact on skeletal development and remodeling.
Interactions between the Wnt signaling pathways and cAMP/PKA have been reported previously43,44. Wnt signaling has been established to inhibit osteoclast differentiation via canonical10,45 (β-catenin) and non-canonical (cAMP/PKA) pathways10. Wnt signaling inhibits osteoclastogenesis by stabilizing β-catenin10, a primary component of the canonical pathway, and also by increasing levels of OPG27,45,46. In addition, Wnt also enhances cAMP and PKA phosphorylation, and suppresses Nfatc1 by inhibiting its nuclear translocation and autoamplification of Nfatc110. However, these reports did not directly demonstrate a role for Gsα in osteoclast differentiation. Here, we show that reduced Gnas/Gsα increased osteoclast differentiation and resorption function via cAMP/PKA and the Wnt/β-catenin pathway. Our results not only support an important role for Gsα signaling in osteoclastogenesis but, together with previous reports10, also suggest crosstalk between Gsα/cAMP/PKA and canonical Wnt/β-catenin signaling pathways to regulate osteoclast differentiation.
We did not detect significant trabecular bone effects in either Gnas+/p− or Gnasm−/+ mice, however our data suggested that trabecular bone may also be effected, although more mildly. The trabecular bone effects in our heterozygous germline deletion of Gnas were more subtle compared to the stronger effects in cell-specific Gnas null mouse models6,8,9,29 and could be due to differences in Gsα levels in cells that interact to regulate bone. Additionally or alternatively, locally-acting factors could differentially affect cortical and trabecular bone. We found that expression of Sost, a Wnt inhibitor that was recently determined to induce osteoclast formation and activity47, was increased in the cortical bone of Gnas+/p− mice, however was extremely low in trabecular bone and could not be reliably quantified (Supplementary Figure 6a); this finding has been consistently noted in published48 and unpublished data (J. Wu et al). If Sost levels are indeed much lower in trabecular bone, this could be an explanation for the stronger cortical bone phenotype. Further analyses of these mutants will provide information to delineate the roles of Gsα in cortical and trabecular bone.
Our data suggest that osteoclast cell autonomous effects of Gnas mutation together with extrinsic factors that act on osteoclasts influence the observed in vivo bone phenotype. This study reveals important roles for Gnas/Gsα signaling in bone remodeling through regulation of osteoclastogenesis and osteoclast resorption activity, with potential implications for drug development approaches to treating diseases that affect cortical bone quality.
Materials and Methods
Animals
Mice with heterozygous deletion of maternal and paternal alleles of Gnas were described previously20. To generate mice with paternal inheritance of the deletion (Gnas+/p−), male mice carrying a heterozygous deletion in exon 1 of Gnas (maintained on a SvEv background) were crossed to female CD1 wild-type mice. Mice with maternal inheritance of the Gnas deletion (Gnasm−/+) were generated by crossing female mutant mice to male wild-type mice. In both breeding schemes, Gnas+/+ littermates were used as wild-type controls in all experiments. Only males were used for all experiments to minimize variability and because males were previously reported to have a more consistent phenotype20. All animal experiments were performed in accordance with the relevant regulations and guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC), University of Pennsylvania.
Osx1-GFP::Cre49 and Gsα(fl/fl)50 mice were described previously. Because these mice have a mixed genetic background (C57BL/6 and CD1), littermates were used as controls. Female conditional heterozygous mice [Osx1-GFP::Cre + ;Gsαfl/+] were mated to control males (mixed background) to generate heterozygous mice with disruption of the maternal allele (Osx1-GFP::Cre + ;Gsαm−/+ referred to as Osx-Cre;Gnasm−/+ in this study). Male conditional heterozygous mice were mated to wild-type females to generate heterozygous mice with disruption of the paternal allele (Osx1-GFP::Cre + ;Gsα+/p− referred to as Osx-Cre; Gnas+/p− in this study). Control mice with Osx1-GFP::Cre [Osx1-GFP::Cre + ; Gsα+/+ referred to as Osx-Cre; Gnas+/+ in this study] and without [Gsαfl/+ and Gsα+/+ referred to Gnasfl/+ and Gnas+/+ in this study] were also analyzed to ensure that the Osx1-GFP::Cre transgene did not confer a phenotype. Genotyping used genomic DNA isolated from tails and previously published protocols6,8,51. These animals were housed in the Center for Comparative Medicine at the Massachusetts General Hospital, and all experiments were approved by the hospital’s Subcommittee on Research Animal Care.
Microcomputed tomography (μCT)
Femurs from 2 week, 3 month and 9 month old mice were harvested and scanned by μCT (μCT35, SCANCO Medical AG, Brüttisellen, Switzerland). For trabecular bone analysis, scans were performed at the distal femoral metaphysis 0.4 mm proximal to the growth plate. For cortical bone, mid-diaphysis of femurs were scanned. All scans were performed at a resolution of 6 μm per slice using a X-ray energy of 55 kvp and an integration time of 300 ms. A total of 125 slices were analyzed using the instrument’s software.
Mechanical testing
Femurs from 2 week, 3 month and 9 month old mice were subjected to mechanical testing by three-point bending using a custom-made fixture52 on an Instron machine. Load was applied until failure and stiffness and peak load were calculated from the load-displacement data53. 2D cortical bone μCT images were incorporated into a custom developed Matlab program to determine the moment of inertia.
Histology
Limbs were fixed in 4% paraformaldehyde for 24–48 hours, decalcified in 10% EDTA for 7 days, processed, and embedded in paraffin. Sections of 5 μm thickness were cut for staining. TRAP staining used the leukocyte acid phosphatase kit (Sigma 387 A). Multi-nucleated osteoclasts (≥3 nuclei) were counted along the endosteal surface at the femoral diaphysis and at the distal femur proximal to the growth plate. Osteoblasts were identified based on their morphology after H&E staining and counted along the endosteum at the diaphyseal region. To determine mineral apposition rate (MAR), mice were injected with calcein (15 μg/g body weight) on postnatal day 10 and xylenol orange (100 μg/g body weight) on postnatal day 13 and sacrificed 24 hours later. MAR was calculated as the distance between the two labels along the femoral mid-diaphyseal region by the number of days between the injections.
Immunohistochemistry
Paraffin embedded sections were detected with Sost antibody (anti-goat; R&D systems, AF1589). Antigen retrieval was performed by treating the sections with Proteinase K (20 μg/ml, Roche) at 37 °C for 15–20 minutes. Sections were then treated with 3% hydrogen peroxide for 10 minutes followed by blocking with 5% BSA and 10% donkey serum in 1X PBST. Sections were incubated with the Sost antibody at 1:100 dilution overnight, then donkey anti-goat IgG-HRP linked antibody (Santa Cruz Biotechnology) at 1:1200 for 1 hour and developed using DAB chromogen (Life Technologies). Three to five images were taken along the mid-diaphyses of femur, starting from 2 mm proximal to the growth plate, and Sost positive cells were quantified.
Real-time PCR
Femur and tibiae from mice were stripped of soft tissue, ends were cut and bone marrow was flushed to obtain only the diaphyseal bone region. Cortical bone pieces were frozen in liquid nitrogen and crushed with stainless steel beads (7 mm diameter, Qiagen) using a tissue lyser (TissueLyser LT, Qiagen). RNA was then extracted with Trizol (Thermo Fisher Scientific) using manufacturer’s instructions. cDNA was prepared using High-Capacity RNA-to-cDNA kit (Thermo Fisher Scientific). Real-time qRT-PCR was performed using SYBR Green method. Primer sequences are as follows: Gsα: 5′-GCGCGAGGCCAACAAAAAGAT and 5′-TGCCAGACTCTCCAGCACCCAG; Sost: 5′-CCAGGGCTTGGAGAGTACC and 5′-GCAGCTGTACTCGGACACATC; Sfrp4: 5′-AGAAGGTCCATACAGTGGGAAG and 5′-GTTACTGCGACTGGTGCGA.
Osteoclast differentiation and function
Following our standard procedures54, bone marrow from 6–8 week old mice was flushed from femurs and tibiae using a 26-G needle into α-MEM (Gibco) with 10% fetal bovine serum and 1X antibiotics. After lysis of RBCs, cells were cultured overnight with recombinant M-CSF (Peprotech, NJ) at 5 ng/ml. Next day, non-adherent cells were harvested and reseeded with M-CSF at 30 ng/ml for 3 additional days to acquire bone marrow macrophages (BMM). After 3 days, BMM were collected and cultured with M-CSF (30 ng/ml) and RANKL (Peprotech, 150 ng/ml) at 5 × 105 cells per well in a 6-well plate for RNA or protein and 5 × 103 cells per well in a 96-well plate for TRAP staining.
For pit formation assays54, BMM were first differentiated into osteoclasts with M-CSF (30 ng/ml) and RANKL (150 ng/ml) for 3 days. Osteoclasts were then seeded at 1 × 104 cells on dentine slices for 48 hours and percent resorption area was determined. Mean of resorption area for WT was set to 1.
Flow cytometry
Bone marrow was flushed from 6–8 week old mice using 1X PBS with 2% FBS. Red blood cells were lysed using 1X RBC lysis buffer (Biolegend). Cells were washed twice in 1X PBS with 0.5% BSA, then blocked using rat IgG (TruStain fcX, Biolegend) for 10 minutes on ice. Cells (1 × 106 cells in 100 μl) were then detected with antibodies at 0.2 μg per million cells for 30 minutes on ice protected from light. The antibodies used for flow cytometry to detect osteoclast precursors were the following: anti-mouse CD3-BV421 (BD Biosciences #564008), anti-mouse CD45R-Alexa Fluor488 (BD Biosciences #557669), anti-mouse CD11b-APC (BD Biosciences #553312), anti-mouse CD115-PE (BD Biosciences #565249). Flow cytometry was conducted with a LSR II (BD Biosciences) and data were analyzed using FlowJo software.
Western blot
Whole cell extracts were obtained using RIPA buffer (Sigma) while nuclear fraction was isolated using NE-PER nuclear and cytoplasmic extraction kit (ThermoFisher Scientific) along with proteinase (Sigma) and phosphatase inhibitor cocktails (Sigma). Protein samples were separated by 4–10% SDS-PAGE and transferred to nitrocellulose membranes. Blots were blocked with 5% milk in 1 X TBST, then incubated with primary antibodies overnight at 4 °C. Primary antibodies used in this study were pCREB, β-catenin, cyclin D1, GAPDH, Histone H3 (all from Cell Signaling, MA, USA) and Nfatc1 (Santa Cruz Biotechnology, CA, USA); all were used at 1:1000 dilution. Gapdh and Histone H3 were used at 1:5000 dilution. Membranes were washed 3 times in TBST and incubated in secondary antibodies conjugated to HRP at 1:6000 dilution for 1 hour at room temperature. Blots were developed and densitometry was quantified with a LI-COR C-Digit blot scanner.
cAMP Measurement
Intracellular cAMP levels were measured in macrophages from WT and Gnas+/p− mice using a cAMP EIA system (GE Healthcare, Little Chalfont, UK). The assay was performed according to the mannufacturer’s instructions. Data were normalized to total protein and WT was set to 1.
Statistical analyses
One-way ANOVA with Tukey post-hoc tests was used for comparing WT (Gnas+/+), Gnas+/p− and Gnasm−/+ groups for μCT and mechanical testing data. WT data from paternal and maternal litters were pooled. Two-way ANOVA with Bonferroni post-hoc tests was used for analyzing osteoclast differentiation assays. Student’s t-test or one-way ANOVA was performed to compare real time qRT-PCR, pit formation data and sclerostin immunohistochemistry data after setting littermate controls to 1 when comparing two or more than two groups respectively. p < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Ramaswamy, G. et al. Gsα Controls Cortical Bone Quality by Regulating Osteoclast Differentiation via cAMP/PKA and β-Catenin Pathways. Sci. Rep. 7, 45140; doi: 10.1038/srep45140 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by Progressive Osseous Heteroplasia Association (POHA), the Center for Research in FOP and Related Disorders, the Ian Cali Endowment for FOP Research, the Cali-Weldon Professorship of FOP Research (EMS), the Penn Center for Musculoskeletal Diseases (P30-AR06919) and US National Institute of Health grant R01-AR046831 (EMS) and K08-AR054741 (JYW). We thank Dr. Sherry Liu and Wei-Ju Tseng for their expertise with μCT scans. We thank Dr. Mike Hast for help with mechanical testing and MATLAB analysis.
Footnotes
The authors declare no competing financial interests.
Author Contributions G.R. and E.M.S. designed the experiments; G.R., H.K., D.Z., V.L. and J.Y.W. performed the experiments and analyzed the data; G.R. and E.M.S. wrote the manuscript with contributions from J.Y.W., F.S.K., R.J.P. and Y.C. All the authors read and approved the final version of the manuscript.
References
- Eriksen E. F. Cellular mechanisms of bone remodeling. Reviews in Endocrine and Metabolic Disorders 11, 219–227 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman E. Bone modeling and remodeling. Critical Reviews in Eukaryotic Gene Expression 19, 219–233 (2009). [DOI] [PubMed] [Google Scholar]
- Sims N. A. & Martin T. J. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. BoneKEy reports 3, 481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Amer Y. Inflammation, cancer, and bone loss. Current Opinion in Pharmacology 9, 427–433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D., Glimcher L. H. & Aliprantis A. O. Osteoimmunology at the nexus of arthritis, osteoporosis, cancer, and infection. Journal of Clinical Investigation 121, 2534–2542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. Y. et al. Gsα enhances commitment of mesenchymal progenitors to the osteoblast lineage but restrains osteoblast differentiation in mice. Journal of Clinical Investigation 121, 3492–3504 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha P. et al. Loss of Gsα early in the osteoblast lineage favors adipogenic differentiation of mesenchymal progenitors and committed osteoblast precursors. Journal of Bone and Mineral Research 29, 2414–2426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha P. et al. Loss of Gsα in the postnatal skeleton leads to low bone mass and a blunted response to anabolic parathyroid hormone therapy. Journal of Biological Chemistry 291, 1631–1642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto A. et al. Deficiency of the G-protein α-subunit Gsα in osteoblasts leads to differential effects on trabecular and cortical bone. Journal of Biological Chemistry 280, 21369–21375 (2005). [DOI] [PubMed] [Google Scholar]
- Weivoda M. M. et al. Wnt Signaling Inhibits Osteoclast Differentiation by Activating Canonical and Noncanonical cAMP/PKA Pathways. Journal of Bone and Mineral Research 31, 65–75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S. H., Ryu J. Y., Lee Y., Lee Z. H. & Kim H. H. Adenylate cyclase and calmodulin-dependent kinase have opposite effects on osteoclastogenesis by regulating the PKA-NFATc1 pathway. Journal of Bone and Mineral Research 26, 1217–1229 (2011). [DOI] [PubMed] [Google Scholar]
- Riminucci M. et al. Osteoclastogenesis in fibrous dysplasia of bone: In situ and in vitro analysis of IL-6 expression. Bone 33, 434–442 (2003). [DOI] [PubMed] [Google Scholar]
- Chapurlat R. D. & Orcel P. Fibrous dysplasia of bone and McCune-Albright syndrome. Best Practice and Research: Clinical Rheumatology 22, 55–69 (2008). [DOI] [PubMed] [Google Scholar]
- Pignolo R. J., Ramaswamy G., Fong J. T., Shore E. M. & Kaplan F. S. Progressive osseous heteroplasia: diagnosis, treatment, and prognosis. 37–48 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turan S. & Bastepe M. The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene. Hormone Research in Paediatrics 80, 229–241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastepe M. The GNAS Locus: Quintessential Complex Gene Encoding Gsalpha, XLalphas, and other Imprinted Transcripts. Current genomics 8, 398–414 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagge A., Kelsey G. & Germain-Lee E. I. Physiological functions of the imprinted Gnas locus and its protein variants Gαs and XLαs in human and mouse. Journal of Endocrinology 196, 193–214 (2008). [DOI] [PubMed] [Google Scholar]
- Adegbite N. S., Xu M., Kaplan F. S., Shore E. M. & Pignolo R. J. Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification. American Journal of Medical Genetics, Part A 146, 1788–1796 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun M. et al. Progressive osseous heteroplasia: A model for the imprinting effects of GNAS inactivating mutations in humans. Journal of Clinical Endocrinology and Metabolism 95, 3028–3038 (2010). [DOI] [PubMed] [Google Scholar]
- Germain-Lee E. L. et al. A mouse model of albright hereditary osteodystrophy generated by targeted disruption of exon 1 of the Gnas gene. Endocrinology 146, 4697–4709 (2005). [DOI] [PubMed] [Google Scholar]
- Ke H. Z. In vivo characterization of skeletal phenotype of genetically modified mice. Journal of Bone and Mineral Metabolism 23, 84–89 (2005). [DOI] [PubMed] [Google Scholar]
- Jacquin C., Gran D. E., Lee S. K., Lorenzo J. A. & Aguila H. L. Identification of Multiple Osteoclast Precursor Populations in Murine Bone Marrow. Journal of Bone and Mineral Research 21, 67–77 (2005). [DOI] [PubMed] [Google Scholar]
- Kronenberg H. M. Gs signaling in osteoblasts and hematopoietic stem cells. Annals of the New York Academy of Sciences 1192, 327–329 (2010). [DOI] [PubMed] [Google Scholar]
- Regard J. B. et al. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nature Medicine 19, 1505–1512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Kaplan F. S. & Shore E. M. Different roles of GNAS and cAMP signaling during early and late stages of osteogenic differentiation. Horm Metab Res. 44, 724–731 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi H. The role of NFAT in osteoclast formation. Annals of the New York Academy of Sciences 1116, 227–237 (2007). [DOI] [PubMed] [Google Scholar]
- Kramer I. et al. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol 30, 3071–3085 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W. et al. Biphasic and Dosage-Dependent Regulation of Osteoclastogenesis by -Catenin. Molecular and Cellular Biology 31, 4706–4719 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulzele K. et al. Myelopoiesis is regulated by osteocytes through Gsα-dependent signaling. Blood 121, 930–939 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns D. M. et al. Somitic disruption of GNAS in chick embryos mimics progressive osseous heteroplasia. Journal of Clinical Investigation 123, 3624–3633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huso D. L. et al. Heterotopic ossifications in a mouse model of Albright hereditary osteodystrophy. PLoS ONE 6, 5–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regard J. B. et al. Wnt/β-catenin signaling is differentially regulated by Gα proteins and contributes to fibrous dysplasia. Proceedings of the National Academy of Sciences of the United States of America 108, 20101–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riminucci M., Robey P. G., Saggio I. & Bianco P. Skeletal progenitors and the GNAS gene: Fibrous dysplasia of bone read through stem cells. Journal of Molecular Endocrinology 45, 355–364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piersanti S. et al. Transfer, analysis, and reversion of the fibrous dysplasia cellular phenotype in human skeletal progenitors. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 25, 1103–1116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman E. et al. Microarchitectural deterioration of cortical and trabecular bone: differing effects of denosumab and alendronate. Journal of Bone and Mineral Research 25, 1886–1894 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H. et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nature medicine 20, 1270–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani G., Elli F. M., Spada A. & Unit D. GNAS Epigenetic Defects and Pseudohypopara- thyroidism: Time for a New Classification? 44, 716–723 (2012). [DOI] [PubMed] [Google Scholar]
- Kaplan F. S. & Shore E. M. Progressive Osseous Heteroplasia. Journal of Bone and Mineral Research 15, 2084–2094 (2000). [DOI] [PubMed] [Google Scholar]
- Rosenberg D. et al. Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Annals of the New York Academy of Sciences 968, 65–74 (2002). [DOI] [PubMed] [Google Scholar]
- Fu Q., Jilka R. L., Manolagas S. C. & O’Brien C. A. Parathyroid hormone stimulates receptor activator of NFκB ligand and inhibits osteoprotegerin expression via protein kinase a activation of cAMP-response element-binding protein. Journal of Biological Chemistry 277, 48868–48875 (2002). [DOI] [PubMed] [Google Scholar]
- Fu Q. et al. Parathyroid Hormone Controls Receptor Activator of NF- κ B Ligand Gene Expression via a Distant Transcriptional Enhancer Parathyroid Hormone Controls Receptor Activator of NF-κ B Ligand Gene Expression via a Distant Transcriptional Enhancer. 26, 6453–6468 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen B. E., Burkhardt C., Reith W., Renkawitz R. & Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Research 8, 265–277 (1999). [DOI] [PubMed] [Google Scholar]
- Chen A. E., Ginty D. D. & Fan C.-M. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature 433, 317–322 (2005). [DOI] [PubMed] [Google Scholar]
- Zhang M. et al. Protein Kinase A Activation Enhances b -Catenin Transcriptional Activity through Nuclear Localization to PML Bodies. PLoS ONE 9, e109523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass D. A. et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Developmental Cell 8, 751–764 (2005). [DOI] [PubMed] [Google Scholar]
- Movérare-Skrtic S. et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nature medicine 20, 1279–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijenayaka A. R. et al. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wein M. N. et al. HDAC5 Controls MEF2C-Driven Sclerostin Expression in Osteocytes. 30, 400–411 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda S. J. & McMahon A. P. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231–3244 (2006). [DOI] [PubMed] [Google Scholar]
- Chen M. et al. Increased glucose tolerance and reduced adiposity in the abscence of fasting hypoglycemia in mice with liver-specific Gsα deficiency. Journal of Clinical Investigation 115, 3217–3227 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. Y. et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proceedings of the National Academy of Sciences of the United States of America 105, 16976–81 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney S. I. et al. Exercise protocol induces muscle, tendon, and bone adaptations in the rat shoulder Corresponding author. 4, 413–419 (2014). [PMC free article] [PubMed] [Google Scholar]
- Schriefer J. L. et al. A comparison of mechanical properties derived from multiple skeletal sites in mice. 38, 467–475 (2005). [DOI] [PubMed] [Google Scholar]
- Kim H. et al. Tmem64 modulates calcium signaling during RANKL-mediated osteoclast differentiation. 17, 249–260 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.