

Abstract
Background
Ring chromosome 6 is a rare sporadic chromosomal abnormality, associated with extreme variability in clinical phenotypes. Most ring chromosomes are known to have deletions on one or both chromosomal arms. Here, we report an atypical and unique ring chromosome 6 involving both a distal deletion and a distal duplication on the different arms of the same chromosome.
Case presentation
In a patient with intellectual disability, short stature, microcephaly, facial dysmorphology, congenital heart defects and renovascular disease, a ring chromosome 6 was characterised using array-CGH and dual-colour FISH. The de-novo ring chromosome 6 involved a 1.8 Mb terminal deletion in the distal short arm and a 2.5 Mb duplication in the distal long arm of the same chromosome 6. This results in monosomy for the region 6pter to 6p25.3 and trisomy for the region 6q27 to 6qter. Analysis of genes in these chromosomal regions suggests that haploinsufficiency for FOXC1 and GMDS genes accounts for the cardiac and neurodevelopmental phenotypes in the proband. The ring chromosome 6 reported here is atypical as it involves a unique duplication of the distal long arm. Furthermore, the presence of renovascular disease is also a unique feature identified in this patient.
Conclusion
To the best of our knowledge, a comparable ring chromosome 6 involving both a distal deletion and duplication on different arms has not been previously reported. The renovascular disease identified in this patient may be a direct consequence of the described chromosome rearrangement or a late clinical presentation in r(6) cases. This clinical finding may further support the implicated role of FOXC1 gene in renal pathology.
Keywords: Ring chromosome 6, Molecular cytogenetics, Dysmorphology, Renovascular disease, FOXC1 gene
Background
Constitutional ring chromosomes are rare sporadic chromosomal abnormalities and can involve any of the 22 pairs of autosomes as well as the sex chromosomes. They arise from breaks in the two arms of a chromosome, followed by fusion of the two broken ends or of one broken end with the opposite telomere region [1]. Alternatively, two dysfunctional telomeres from the same chromosome fuse, resulting in the formation of a complete ring with no deletions. The clinical phenotype associated with ring chromosomes is highly variable, depending on the extent of the deletion and the chromosome involved.
Ring chromosome 6 is a rare sporadic chromosomal abnormality, first described by Moore et al in 1973 in a female infant who exhibited varying dysmorphic features [2]. The phenotypic features are very variable and range from an almost normal phenotype to severe malformations and intellectual disability. Peeden et al reviewed the variability of phenotypic features in 14 cases of ring chromosome 6 [3]. The most frequent clinical features include failure to thrive, congenital heart defects, intellectual disability, microcephaly and facial dysmorphology [4]. Also reported are various abnormalities in the ocular, auditory and central nervous systems.
We report a unique de novo ring chromosome 6 characterised by array-CGH and FISH involving a deletion in the distal short arm and a duplication in the distal long arm of the same chromosome.
Case presentation
The proband was referred to the genetics clinic at the age of 49 years, during a hospital admission for a chest infection. He is the 4th in a sibship of 5 offspring of non-consanguineous Caucasian parents. He was born by normal vaginal delivery following an uneventful term pregnancy. At birth he was found to have a small atrial septal defect. He was a slow feeder and early motor, cognitive and developmental milestones were delayed. At the age of 10 years he was diagnosed with bilateral hearing loss which worsened progressively. A CT scan of the brain performed at the age of 21 years, revealed the presence of a Dandy Walker variant, normal internal auditory maeti and a normal sella turcica. His medical history also includes chronic venous insufficiency and hypertension. The parents gave no relevant family history.
Physical examination at the genetics clinic revealed an adult male with moderate intellectual disability, short stature (height of 141 cms) and microcephaly (head circumference of 51.6 cms). Dysmorphic features included a right-sided hair whorl, a small nose, a high arched palate, a nuchal hump, mild scoliosis, brachydactyly and overlapping toes on the left foot. The skin was very dry and there was considerable venous insufficiency of the lower limbs.
An echocardiogram revealed borderline concentric left ventricular hypertrophy, mild right ventricular hypertrophy, a patent foramen ovale and a very small atrial septal defect, for which he subsequently underwent cardiac surgery. Blood investigations revealed renal pathology but renal imaging was unremarkable. Subsequently a renal biopsy revealed long-standing renovascular disease but no vasculitis.
Methods
Array comparative genomic hybridisation (array-CGH) was performed at the Wessex Regional Genetics Laboratory using an Oxford Gene Technology (OGT, Oxford, UK) CytosureTM Constitutional v3 custom 8x60K oligo array, manufactured by Agilent Technologies [Agilent Technologies, Santa Clara, CA].
Dual-colour fluorescence in situ hybridisation (FISH) was performed with 2 bacterial artificial chromosomes (BACs) chosen from the Sanger 30K cloneset using the Ensembl genome browser (http://www.ensembl.org/Homo_sapiens/) together with the chromosome 6 specific centromere probe, D6Z1. The probes used were the 6p25.3 specific BAC, RP11-13J16 (GRCh37 bp 1296996 – 1357295) and the 6q27 specific BAC, RP11-417E7 (GRCh37 bp 169417007 – 16959015).
Results
Array CGH identified an approximately 1.8 Mb terminal deletion involving the distal short arm of a chromosome 6, with the breakpoint within a 68kb interval in 6p25.3 (arr(GRCh37) 6p25.3(164360_1773695)x1) (Fig. 1a). The array also detected an approximately 2.5 Mb duplication involving the distal long arm of a chromosome 6, with the breakpoint within a 125kb interval in 6q27 (arr(GRCh37) 6q27(168682830_170923504)x3) (Fig. 1b). The proband is, therefore, monosomic for the region 6pter to 6p25.3 and trisomic for the region 6q27 to 6qter.
Fig. 1.
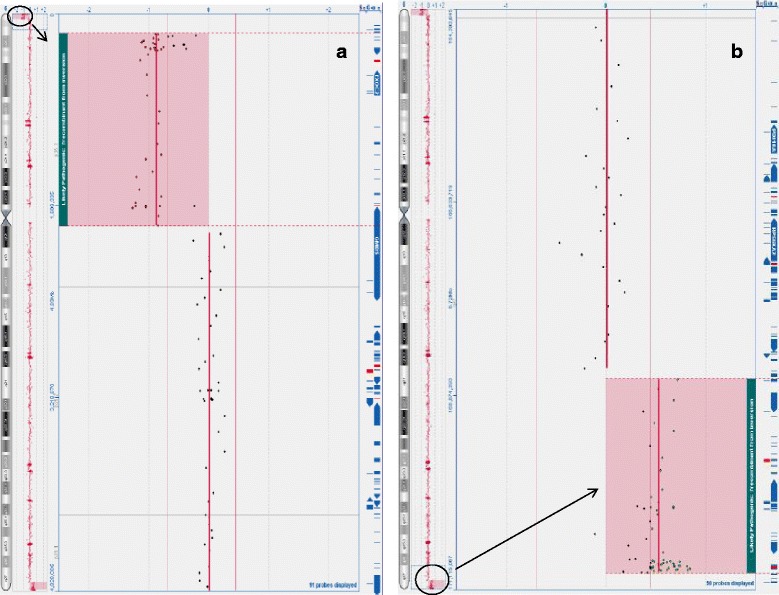
Array-CGH plots, analysed using Oxford Gene Technology (OGT) CytosureTM Constitutional v3 oligo array, showing. a the 1.8 Mb terminal 6p25.3 deletion with a log2 ratio of -1.0 at genomic coordinates 164360_1773695 (NCBI build 37, February 2009, hg19) and (b) the 2.5 Mb 6q27 duplication with a log2 ratio of +0.58 at genomic coordinates 168682830_170923504 (deleted/duplicated regions circled on chromosome 6 ideogram and magnified and highlighted in main image as indicated by an arrow)
To characterise the imbalances further and to determine whether they occurred in cis or trans, FISH studies were undertaken on peripheral blood metaphase chromosomes from the proband and both his parents using the 6p25.3 specific BAC RP11-13J16 and the 6q27 specific BAC RP11-417E7 as well as a 6 centromere probe (D6Z1).
FISH confirmed both the 6p deletion and 6q duplication identified by array-CGH and showed that the imbalances are present in the form of a ring chromosome 6, r(6), which replaces a normal chromosome 6. The r(6) was shown to be monocentric, deleted for the short arm of 6p (6p25.3) and duplicated for the distal long arm of 6q (6q27) (Fig. 2). This r(6) represents an atypical and unique ring chromosome in that the duplicated material is located in a different arm of the chromosome to that of the deleted material. FISH studies on both parents showed that they had two normal chromosomes 6 and therefore the r(6) had arisen de novo in the proband.
Fig. 2.
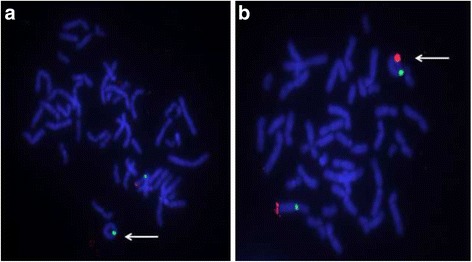
FISH with the chromosome 6 specific centromere probe D6Z1 (green signal), the 6p25.3 BAC RP11-13J16 (a; red signal), and the 6q27 BAC RP11-417E7 (b; red signal) showing a monocentric r(6) chromosome (arrow) with (a) 6p deletion and (b) 6q duplication
Discussion
Ring chromosome 6 is an exceptionally rare cytogenetic rearrangement that usually arises de novo and is associated with extreme inter-individual variability in clinical phenotypes. A number of reports have described the clinical features in r(6) patients. However, to our knowledge this is the first case of r(6) involving both a distal 6p deletion and a distal 6q duplication.
At least two case reports have described a r(6) involving a comparable deletion at 6p25.3 [4, 5]. Both patients had psychomotor delay, cerebral ventriculomegaly, a prominent forehead and malformed ears. Furthermore, Zhang et al report a deletion of identical size to the present case (1.78Mb at 6p25.3) and additional clinical features that include, microcephaly, hydrocephalus, epilepsy and hearing loss [5].
Submicroscopic deletions involving the 6p25 subtelomeric region is a distinct clinical syndrome. The clinical phenotypes described include developmental delay, intellectual disability, language impairment, hearing loss, and various ophthalmic, cardiac and craniofacial anomalies [6]. As the clinical phenotypes observed in ring chromosomes overlap with those of deletion of one or both ends of the respective chromosome syndromes, we explored karyotype-phenotype associations by reviewing literature that describes the function of genes located in the 6p25 and 6q27 regions.
A number of genes located in the 6p25-pter deletion interval have OMIM entries, namely DUSP22, IRF4, EXOC2, HUS1B, FOXQ1, FOXF2, FOXCUT, FOXC1 and GMDS, as well as a number of uncharacterised genes. Of these, IRF4 and FOXC1 are human disease genes. Interferon regulatory factor 4 (IRF4) is a transcription factor essential for the development of T helper 2 (Th2), Th17 and Th9 cells, and the rs872071 variant in its 3-prime untranslated region is a susceptibility locus for chronic lymphocytic leukemia [7]. IRF4 is also required for hair, skin and eye pigmentation [8].
The forkhead gene cluster at the 6p25.3-pter region includes FOXQ1, FOXF2, FOXCUT and FOXC1 genes. These transcription factors are essential for development and morphogenesis. FOXC1 and FOXC2 genes are expressed during cardiac development, and mutations in these genes have been associated with a wide range of cardiac abnormalities [9]. Zhang et al suggest that the congenital cardiac anomalies observed in r(6) is due to haploinsufficiency of FOXC1 [5].
Developmental delay and varying degrees of neurological defects are consistent features in 6p25 deletion syndromes. FOXC1, FOXF2 and GMDS are involved in central nervous system development and function. Animal studies have shown that mice homozygous for a null allele of Mf1 (the murine homolog of FOXC1) show congenital hydrocephalus and eye defects [10]. Haploinsufficiency for FOXC1 has been associated with hydrocephalus in humans [6]. Abnormalities in the posterior cranial fossa, including Dandy-Walker malformation, mega-cisterna magna and cerebellar vermis hypoplasia have also been associated with homozygous FOXC1 mutations [11]. Any deviation from normal FOXC1 gene dosage results in central nervous system (CNS) vascular anomalies. Similarly, FOXQ1 gene mediates neurite growth and neuronal differentiation [12]. FOXF2 is expressed in murine pericytes, and FOXF2 knockout mice develop intracranial hemorrhage, perivascular oedema and a leaky blood-brain barrier, further highlighting the role of these transcription factors in CNS development and function [13].
The GMDS gene encodes GDP-mannose 4,6-dehydratase, an enzyme that catalyzes the first step in protein fucosylation. This is an essential post translational modification in members of the Notch family of transmembrane receptors [12]. Notch signaling is essential for neuronal and glial cell differentiation, maturation, learning and memory. Song et al have described a zebrafish model that harbours a missense mutation in GMDS resulting in defective fucosylation of Notch receptors that leads to defects in neuronal development and synapse branching [13]. These studies strongly suggest that the varying degree of intellectual disability observed in r(6) cases is due to haploinsufficiency of the FOXC1 and GMDS genes. Further evidence supporting the role of these two genes in neuronal function is provided by Hockner et al [14]. Here, the authors describe a case of de-novo r(6) in a 25-year-old female with short stature and only minor dysmorphic features, but with normal psychomotor development. Cytogenetic analysis had identified a 6p breakpoint telomeric to the DUSP22 gene, with no disruption of either FOXC1 or GMDS coding sequences at 6p. The short stature in this case report was attributed to mitotic instability of the ring chromosome.
The 6q duplication interval contains at least ten OMIM genes, of which four have OMIM Morbid entries (SMOC2, THBS2, ERMARD and TBP). Burnside et al report extensive CNS developmental abnormalities in a two-month old infant with a duplication of THBS2. This gene encodes thrombospondin 2, an astrocyte-secreted protein essential for synaptogenesis and neurite growth [15].
The r(6) outlined in this report is atypical in that no functional genetic material from the long arm has been lost, but material from the distal long arm (6q27) has been duplicated. Guilherme et al studied breakpoints and mechanisms of ring formation in fourteen cases of ring chromosomes, and reported two cases of ring chromosome 13 where terminal deletions of 13q were associated with duplications near the breakpoints on the same chromosome arm [1]. Similarly, Knijnenburg et al describe a case of ring chromosome 14 shown to have a terminal 14q32.33 deletion and an inverted duplication of 14q32.12 to 14q32.32 [16]. Comparably, Rossi et al investigated 33 probands with ring chromosomes using both array-CGH and FISH, and identified seven cases where duplications were also detected in ring chromosomes 13, 15, 18, 21 and 22 [17]. It is unusual for the duplicated material to be located on a different arm of the chromosome to that of the deletion. No similar findings involving ring chromosome 6 have been reported in the literature. However, additional variants and imbalances of ring chromosome 6 have been described. In a male infant with a number of dysmorphic features and a large patent ductus arteriosus, Lee et al report a mosaic karyotype with a dicentric ring chromosome (46, XY, r(6)(p25q27)/46, XY, dic r(6;6)(p25q27;p25q27) [18]. Birnbacher et al report a r(6) with an 6q26-qter deletion and no 6p subtelomeric deletion, and Nishigaki et al a similar r(6) with a 1.5Mb deletion at 6q27 [19, 20].
The clinical features described in this proband, in particular the cardiac and neurological anomalies are largely in common with other case reports of r(6). Haploinsufficiency for the forkhead gene cluster at 6p appears to be the major driver leading to development of these features. Of particular interest is the renovascular disease finding reported in this proband. While FOXC1 disruption has been implicated in congenital anomalies of the kidney and urinary tract in both animal and human models, there is no reported link between this gene and the development of renovascular disease [21]. Renovascular disease was not described in any of the published r(6) patients. This may suggest that either this is a direct consequence of the unique r(6) presented here or that this is a late onset clinical feature. It may therefore be advisable to screen r(6) patients for renal pathology.
Conclusion
This report provides a detailed characterisation of a novel r(6), involving a distal deletion at 6p25.3 and a distal duplication at 6q27. The learning disability, hearing loss, cardiac and CNS defects observed in the proband, can be attributed to hemizygous expression of FOXC1, FOXF2 and GMDS genes on 6p25, and possibly also to the partial duplication of the distal 6q region. Screening of r(6) patients for renal pathology is advisable not only for the medical management of these individuals, but also to further explore the possible role of FOXC1 gene in renovascular disease and secondary hypertension. Despite the genomic associations outlined in the literature, the correlation with phenotypes and clinical severity in r(6) cases remains highly variable and complex.
Acknowledgements
We thank the proband and his family for their collaboration in this study.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Authors’ contributions
All authors contributed equally to this work. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the proband’s parents for the publication of this case report.
Ethics approval and consent to participate
The patient’s parents has gave written informed consent for participation in this study.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- Array-CGH
Array comparative genomic hybridisation
- BAC
Bacterial artificial chromosome
- CNS
Central nervous system
- CT scan
Computed tomography
- FISH
Fluorescence in situ hybridisation
- OMIM
Online Mendelian Inheritance in Man
- R(6)
Ring chromosome 6
Contributor Information
Nikolai Paul Pace, Email: nikolai.p.pace@um.edu.mt.
Frideriki Maggouta, Email: frideriki.maggouta@salisbury.nhs.uk.
Melissa Twigden, Email: melissa.twigden@salisbury.nhs.uk.
Isabella Borg, Phone: 00356 2545 6325, Email: isabella.borg@um.edu.mt.
References
- 1.Guilherme RS, Ayres Meloni VF, Kim CA, Pellegrino R, Takeno SS, Spinner NB, et al. Mechanisms of ring chromosome formation, ring instability and clinical consequences. BMC Med Genet. 2011;12:171. doi: 10.1186/1471-2350-12-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore CM, Heller RH, Thomas GH. Developmental Abnormalities Associated with a Ring Chromosome 6. J Med Genet. 1973;10:299–303. doi: 10.1136/jmg.10.3.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peeden JN, Scarbrough P, Taysi K, Wilroy RS, Finley S, Luthardt F, et al. Ring chromosome 6: Variability in phenotypic expression. Am J Med Genet. 1983;16:563–573. doi: 10.1002/ajmg.1320160413. [DOI] [PubMed] [Google Scholar]
- 4.Ciocca L, Surace C, Digilio MC, Roberti MC, Sirleto P, Lombardo A, et al. Array-CGH characterization and genotype-phenotype analysis in a patient with a ring chromosome 6. BMC Med Genomics. 2013;6:3. doi: 10.1186/1755-8794-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang R, Chen X, Li P, Lu X, Liu Y, Li Y, et al. Molecular characterization of a novel ring 6 chromosome using next generation sequencing. Mol Cytogenet. 2016;9. doi:10.1186/s13039-016-0245-9 [DOI] [PMC free article] [PubMed]
- 6.Gould DB, Jaafar MS, Addison MK, Munier F, Ritch R, MacDonald IM, et al. Phenotypic and molecular assessment of seven patients with 6p25 deletion syndrome: relevance to ocular dysgenesis and hearing impairment. BMC Med Genet. 2004;5:17. doi: 10.1186/1471-2350-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Bernardo MC, Crowther-Swanepoel D, Broderick P, Webb E, Sellick G, Wild R, et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2008;40:1204–1210. doi: 10.1038/ng.219. [DOI] [PubMed] [Google Scholar]
- 8.Gathany AH, Hartge P, Davis S, Cerhan JR, Severson RK, Cozen W, et al. Relationship between interferon regulatory factor 4 genetic polymorphisms, measures of sun sensitivity and risk for non-Hodgkin lymphoma. Cancer Causes Control CCC. 2009;20:1291–1302. doi: 10.1007/s10552-009-9348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seo S, Kume T. Forkhead transcription factors, Foxc1 and Foxc2, are required for the morphogenesis of the cardiac outflow tract. Dev Biol. 2006;296:421–436. doi: 10.1016/j.ydbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998;93:985–996. doi: 10.1016/S0092-8674(00)81204-0. [DOI] [PubMed] [Google Scholar]
- 11.Delahaye A, Khung-Savatovsky S, Aboura A, Guimiot F, Drunat S, Alessandri J-L, et al. Pre- and postnatal phenotype of 6p25 deletions involving the FOXC1 gene. Am J Med Genet A. 2012;158A:2430–2438. doi: 10.1002/ajmg.a.35548. [DOI] [PubMed] [Google Scholar]
- 12.Smith PL, Myers JT, Rogers CE, Zhou L, Petryniak B, Becker DJ, et al. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 2002;158:801–815. doi: 10.1083/jcb.200203125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song Y, Willer JR, Scherer PC, Panzer JA, Kugath A, Skordalakes E, et al. Neural and Synaptic Defects in slytherin, a Zebrafish Model for Human Congenital Disorders of Glycosylation. PLoS ONE. 2010;5. doi:10.1371/journal.pone.0013743 [DOI] [PMC free article] [PubMed]
- 14.Höckner M, Utermann B, Erdel M, Fauth C, Utermann G, Kotzot D. Molecular characterization of a de novo ring chromosome 6 in a growth retarded but otherwise healthy woman. Am J Med Genet A. 2008;146A:925–929. doi: 10.1002/ajmg.a.32251. [DOI] [PubMed] [Google Scholar]
- 15.Burnside MN, Pyatt RE, Hughes A, Baker PB, Pierson CR. Complex brain malformations associated with chromosome 6q27 gain that includes THBS2, which encodes thrombospondin 2, an astrocyte-derived protein of the extracellular matrix. Pediatr Dev Pathol Off J Soc Pediatr Pathol Paediatr Pathol Soc. 2015;18:59–65. doi: 10.2350/14-06-1516-CR.1. [DOI] [PubMed] [Google Scholar]
- 16.Knijnenburg J, van Haeringen A, Hansson KBM, Lankester A, Smit MJM, Belfroid RDM, et al. Ring chromosome formation as a novel escape mechanism in patients with inverted duplication and terminal deletion. Eur J Hum Genet. 2007;15:548–555. doi: 10.1038/sj.ejhg.5201807. [DOI] [PubMed] [Google Scholar]
- 17.Rossi E, Riegel M, Messa J, Gimelli S, Maraschio P, Ciccone R, et al. Duplications in addition to terminal deletions are present in a proportion of ring chromosomes: clues to the mechanisms of formation. J Med Genet. 2008;45:147–154. doi: 10.1136/jmg.2007.054007. [DOI] [PubMed] [Google Scholar]
- 18.Lee SJ, Han DK, Cho HJ, Cho YK, Ma JS. Mosaic Ring Chromosome 6 in an Infant with Significant Patent Ductus Arteriosus and Multiple Congenital Anomalies. J Korean Med Sci. 2012;27:948–952. doi: 10.3346/jkms.2012.27.8.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birnbacher R, Chudoba I, Pirc-Danoewinata H, König M, Kohlhauser C, Schnedl W, et al. Microdissection and reverse painting reveals a microdeletion 6(q26qter) in a de novo r(6) chromosome. Ann Genet. 2001;44:13–18. doi: 10.1016/S0003-3995(00)01033-9. [DOI] [PubMed] [Google Scholar]
- 20.Nishigaki S, Hamazaki T, Saito M, Yamamoto T, Seto T, Shintaku H. Periventricular heterotopia and white matter abnormalities in a girl with mosaic ring chromosome 6. Mol Cytogenet. 2015;8:54. doi: 10.1186/s13039-015-0162-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Motojima M, Kume T, Matsusaka T, Ichikawa I. Foxc1 and Foxc2 cooperate in maintaining glomerular podocytes. FASEB J. 2015;29(1 Supplement):880.8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.