

Abstract
Hyperlipidemia is a risk factor for Alzheimer's disease (AD) and other neurodegenerative diseases. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a lipid regulatory gene involved in cell apoptosis. However, the function and mechanism of PCSK9 in neuronal apoptosis following hyperlipidemia remains to be elucidated. The present study established a hyperlipidemic mouse model by feeding a high-fat diet (HFD) to 6-week-old apoE(−/−) mice. Plasma lipid levels, hippocampal lipid accumulation, hippocampal histology, and hippocampal neuronal apoptosis were all monitored for changes. The expression levels of PCSK9, β-secretase 1 (BACE1), B-cell lymphoma 2 (Bcl-2), Bcl-2-associated X protein (Bax), and caspase-3 in hippocampal CA3 and CA1 neurons were also measured. Results demonstrated that a HFD increased the lipid accumulation in the CA3 hippocampus and the levels of plasma lipids, including triglycerides, total cholesterol, low-density lipoprotein, and high-density lipoprotein. In addition, CA3 neurons in the HFD group indicated apparent injuries and increased neuronal apoptosis, which are associated with the expression of Bcl-2, Bax, and caspase-3. A HFD also increased the expression levels of PCSK9 and BACE1. BACE1 promotes cleavage of amyloid precursor proteins to generate β-amyloid peptide (Aβ), which induces neuronal apoptosis. Protein levels of Aβ are associated with the observation of amyloid plaques in the hippocampus of the HFD group. The results suggest that hyperlipidemia regulates neuronal apoptosis by increasing PCSK9 and BACE1 expression. Overall, the current study may elucidate the role of lipid metabolism disorder in AD pathogenesis.
Keywords: hyperlipidemia, apoptosis, Alzheimer's disease, proprotein convertase subtilisin/kexin type 9, β-secretase 1, amyloid β-protein
Introduction
Alzheimer's disease (AD) is characterized by a widespread functional disturbance of the human brain. AD pathogenesis is driven by the production and deposition of β-amyloid peptides (Aβs) (1). Aβs are peptides composed of 36–43 amino acids, which are the predominant components of the amyloid plaques observed in the brains of AD patients. Aβ is formed from the cleavage of amyloid precursor protein (APPs) by β- and γ-secretases. The formation of amyloid plaques is a major factor in AD, thus, determining the underlying mechanism by which Aβ induces neuronal cell death is crucial. A recent study demonstrated that dying cells in AD brains and cultures of neurons exposed to Aβ exhibit the characteristics of apoptosis (2). However, the specific signaling pathways by which Aβ triggers cell apoptosis have not been well defined.
Hyperlipidemia is a risk factor for atherosclerosis, as well as for neurodegenerative diseases, including AD, as evidenced by epidemiological, clinical, and animal studies. Kivipelto et al (3) and Solomon et al (4) selected 1449 residents for a 21-year follow-up study and demonstrated that total cholesterol was an independent risk factor for AD in middle-aged adults. Lipid-lowering treatment can reduce the risk of neurodegenerative diseases (5–7). Chan et al (8) observed that sphingomyelin and cholesterol esters were notably increased in AD patients and transgenic AD mouse brain tissues. Kosari et al (9) demonstrated that hippocampus-dependent memory was markedly impaired in rats fed with a HFD for 12 weeks. Furthermore, El-Sayyad et al (10) observed a higher rate of neuronal apoptosis in rats with hyperlipidemia compared with those fed a normal diet.
Despite years of intensive research, the link between hyperlipidemia and AD has yet to be established, although it has previously been suggested that genes regulating lipid metabolism may also be important in AD (11). Proprotein convertase subtilisin/kexin type 9 (PCSK9) encodes a protein formerly termed neuronal apoptosis-regulated convertase-1, a proprotein convertase belonging to the subtilase subfamily (12). PCSK9 is highly expressed in the liver, where it negatively regulates the low-density lipoprotein (LDL) receptor (LDLR) in hepatocytes and, thus, is important in controlling circulating levels of LDL-cholesterol (LDL-C) (13). PCSK9 affects neural development and participates in neuronal apoptosis (12). Our previous study reported that high PCSK9 expression levels, induced by oxidized LDL (oxLDL), were positively associated with a high apoptotic ratio in human umbilical vein endothelial cells (14).
The present study investigated whether PCSK9 was involved in the effects of hyperlipidemia on hippocampal neuronal apoptosis by establishing an apoE(−/−) hyperlipidemic mouse model. Furthermore, the current study aimed to investigate a novel association between lipid metabolism and AD.
Materials and methods
Animals and diets
A total of 18 male apoE(−/−) mice (age, 6 weeks; average weight, 21±2.7 g), were purchased from Nanjing Qingzilan Technology Co., Ltd. (Nanjing, China) and maintained in a temperature-controlled environment (22–25°C, 45% humidity) with a 12 h light-dark cycle. Mice were randomly assigned to one of two groups: HFD group, fed with food consisting of 2% (w/w) cholesterol, 10% (w/w) lard, and 0.5% (w/w) cholic acid; and a normal diet (ND) group. All mice were allowed ad libitum access to their designated diet for 12 weeks. All animal procedures were conducted in accordance with the International Guidelines for the Ethical Use of Laboratory Animals and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (15).
Plasma lipid analysis
At the end of the feeding period, animals were anesthetized with 10% chloral hydrate (350 mg/kg body weight). Blood was collected using cardiac puncture into tubes containing 0.1% EDTA following an overnight fast. Blood was then centrifuged at 5,500 × g for 12 min at 4°C to separate the plasma. The plasma was immediately used to determine the levels of LDL-C, high-density lipoprotein cholesterol (HDL-C), total cholesterol (TC), and triglyceride (TG) using an automatic biochemistry analyzer (AU480 Chemistry system; Beckman Coulter, Inc., Brea, CA, USA).
Tissue preparation for morphological analyses
Following reaching surgical tolerance, the anesthetized animals were sacrificed by cervical dislocation, and were transcardially perfused with physiological saline for 60 sec, followed by perfusion with a fixative (4% w/v formaldehyde) for 15 min at 22°C. Following perfusion, the brains were rapidly dissected without the olfactory bulb and cerebellum. The brains were post-fixed overnight (18–22 h) at 4°C in the formaldehyde solution used for perfusion supplemented with 20% (w/v) sucrose. Tissues were then immersed in 30% sucrose solution with 0.1 M sodium cacodylate buffer at pH 7.3 for an additional day at 4°C. Frozen hippocampal slices were cryosectioned into 8-µm thick sections.
Hematoxylin and eosin staining
Frozen hippocampal sections were stained with hematoxylin and eosin (H&E) and examined under a light microscope to observe cellular morphology changes in the mouse hippocampus.
Oil red O staining
Frozen brain sections were stained with Oil Red O solution [60% Oil Red O stock solution (5 mg/ml isopropanol)/40% water] for 15 min. Following thoroughly washing twice with distilled water, sections were counterstained with hematoxylin solution for 30 sec. The staining of lipid droplets in the brain was quantified using a phase contrast microscope and ImagePro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Immunohistochemistry
Brain hippocampal tissues were obtained from the HFD and ND groups in accordance with the protocols approved by the Animal Investigative Review Committee of the University of South China. The primary antibodies used were against PCSK9 (1:100; cat. no. 55206-1-AP; Proteintech Group, Inc., Chicago, IL, USA), β-secretase 1 (BACE1; 1:50; cat. no. ab108394; Abcam, Cambridge, MA, USA), caspase-3 (1:50; cat. no. 19677-1-AP; Proteintech Group, Inc.), B-cell lymphoma 2 (Bcl-2; 1:50; cat. no. BS1031; Bioworld Technology, Inc., St. Louis Park, MN, USA), and Bcl-2-associated X protein (Bax; 1:50; cat. no. 60267-1-Ig; Proteintech Group, Inc.). Incubation without the primary antibodies served as the negative control. The frozen 8-µm thick sections were blocked with endogenous peroxidase blocking agent (Fuzhou Maixin Biotech, Co., Ltd., Fuzhou, China) for 10 min at room temperature and subsequently incubated with the different primary antibodies for 30 min at room temperature. Following washing in distilled water, the sections were incubated with biotinylated secondary antibodies (cat. nos. KIT-0105R and KIT-0105M; Fuzhou Maixin Biotech, Co., Ltd.) and streptavidin-peroxidase (Proteintech Group, Inc.) for 10 min at room temperature. Subsequently, 3,3′-diaminobenzidine peroxidase substrate solution (Fuzhou Maixin Biotech, Co., Ltd.) was added until the desired color (brown) was developed. Apoptotic cells in the mouse hippocampus were detected using a Hoechst 33258 Staining kit (Beyotime Institute of Biotechnology, Haimen, China) in accordance with the manufacturer's protocols.
Modified Bielschowsky staining for substance P (SP)
Frozen slides were immersed in 2% silver nitrate solution for 30 min at 37°C in the dark. Subsequently, the silver nitrate solution was removed and the slides were washed three times in distilled water for 3 min. The stain was deoxidized with 10% reducing agent until the color of the slides turned pale yellow. The slides were rinsed again three times in distilled water, and excess water was removed. Ammoniacal silver solution was added to each slide for 30 sec. The excess solution was removed, and the slides were immersed in 10% reducing agent for 2 min. The slides were rotated a number of times until the yellow dye was stabilized. The slides were then rinsed in tap water, fixed in 5% sodium thiosulfate for 3 min, air-dried, cleared in xylene, and finally coverslipped for light microscopic examinations.
Statistical analysis
Experimental results were expressed as the mean ± standard error of the mean. Statistical analyses were conducted using unpaired Student's t-test assuming unequal variance unless otherwise indicated. Statistical analyses were conducted using SPSS 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Comparison of plasma lipid profiles between ND and HFD mice
The apoE(−/−) mouse has been established as a suitable model to investigate diet-induced hyperlipidemia. ApoE(−/−) mice (age, 6 weeks) were randomly assigned to receive either ND or HFD for 12 weeks. The plasma lipid profiles, including TC, TG, LDL-C, and HDL-C contents, were determined for the two groups of mice. The data for plasma lipid levels are presented in Table I. Plasma TG, TC, LDL-C, and HDL-C concentrations were significantly increased in HFD-fed mice compared with ND-fed mice. These results indicated that a diet-induced hyperlipidemic model was successfully established.
Table I.
Plasma concentrations of lipids in apoE(−/−) mice given normal diet and high-fat diet.
| Serum lipid | Normal diet (n=9) | High-fat diet (n=9) |
|---|---|---|
| TG (mmol/l) | 0.70±0.12 | 1.33±0.09a |
| TC (mmol/l) | 8.75±0.52 | 27.89±4.56a |
| HDL-C (mmol/l) | 1.63±0.15 | 11.48±1.97a |
| LDL-C (mmol/l) | 1.47±0.08 | 8.57±2.56a |
Data are presented as mean ± standard error of the mean, where n is number of animals.
P<0.05 vs. the normal diet group. TG, triglycerides; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein C.
Effects of HFD on lipid accumulation in the hippocampus of ApoE(−/−) mice
Lipids are essential to brain functions, including membrane morphology, signal transduction, membrane fluidity, and cell survival. Lipid abnormalities in the brain may contribute to AD risk or severity. However, the mechanism by which hyperlipidemia affects lipid accumulation in the hippocampus, which is a central target of AD plaque pathology, remains to be elucidated. In the present study, hippocampal sections were stained with Oil Red O to identify the sites of lipid accumulation in apoE(−/−) mice fed with ND and HFD. The majority of the Oil Red O-stained particles were distributed as clusters in the hippocampus tissues. Lipid accumulation increased in hippocampus CA3 neurons in HFD-fed apoE(−/−) mice compared with ND-fed apoE(−/−) mice. However, the difference in lipid contents between ND- and HFD-fed mice was not notable in hippocampus CA1 of apoE(−/−) mice (Fig. 1). The results indicated that the lipid contents of the hippocampus can be increased by HFD-induced hyperlipidemia.
Figure 1.
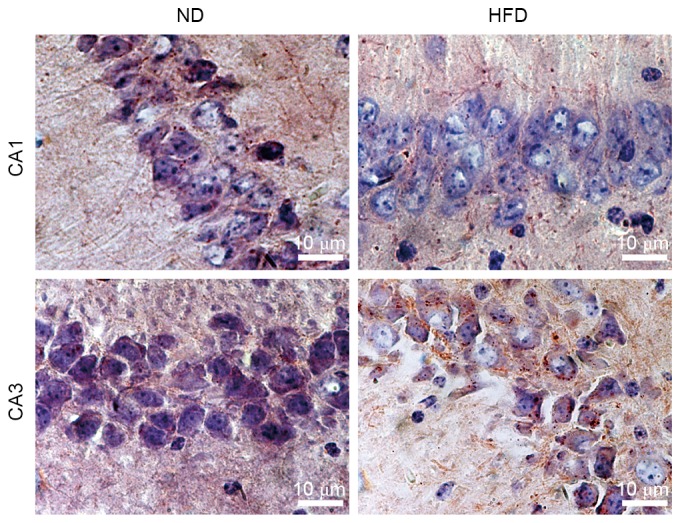
Lipid contents of the hippocampus may be increased by HFD-induced hyperlipidemia. Oil Red O-stained hippocampus CA1 and CA3 neurons of apoE(−/−) mice fed with ND or HFD for 12 weeks (magnification, ×1000). HFD hippocampus exhibited increased lipid accumulation in hippocampus CA3 neurons compared with ND-fed mice and similar lipid accumulation in CA1. ND, normal diet; HFD, high-fat diet.
Effects of HFD on histology of the hippocampus in apoE(−/−) mice
The histological changes in the hippocampus were observed in the two groups of mice following H&E staining of the brain sections. ND-fed apoE(−/−) mice exhibited normal histological appearance and neuronal distribution in CA1 and CA3 hippocampal areas. Degenerative changes were observed in CA3 hippocampal areas of HFD-fed apoE(−/−) mice, which exhibited pyknotic cells with reduced neuron count. Enlarged intercellular spaces between CA1 and CA3 pyramidal cells were frequently observed in HFD-fed apoE(−/−) mice. Furthermore, a number of cells in the CA3 area of HFD-fed apoE(−/−) mice lost typical cell structure compared with ND-fed mice (Fig. 2). These results suggested that hyperlipidemia may result in structural damage and neuronal loss in the hippocampus of HFD-fed apoE(−/−) mice.
Figure 2.
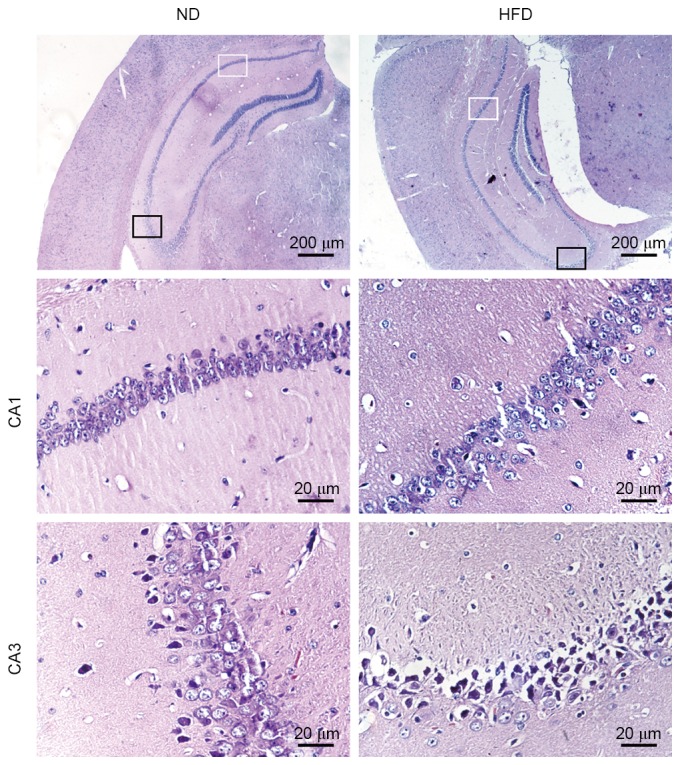
Hyperlipidemia results in structural damage and neuronal loss in the hippocampus of HFD-fed apoE(−/−) mice. Hematoxylin and eosin stained hippocampus CA1 and CA3 neurons from apoE(−/−) mice fed with ND or HFD for 12 weeks. CA1 (magnification, ×400) indicated by the white rectangular frame (magnfication, ×40) and CA3 (magnification, ×400) indicated by the black rectangular frame (magnification, ×40). ND, normal diet; HFD, high-fat diet.
Effects of HFD on neuronal apoptosis in the hippocampus of apoE(−/−) mice
Marked neuronal loss in the hippocampus is often observed in AD brains. To observe whether neuronal apoptosis may be induced by hyperlipidemia, a Hoechst 33258 staining assay was performed to detect cell apoptosis in the hippocampus. Neuronal apoptosis was observed in a number of cells in the hippocampus CA3 of ND-fed apoE(−/−) mice. By contrast, a large number of hippocampus CA3 neuronal cells underwent apoptosis in HFD-fed apoE(−/−) mice. Although individual apoptotic cells were observed in hippocampus CA1 of HFD-fed apoE(−/−) mice, no notable difference was observed between the percentage of apoptotic cells in hippocampus CA1 between ND- and HFD-fed mice (Fig. 3). These results demonstrated that apoptosis was the predominant cause of neuronal death in the hippocampus, and hyperlipidemia increases neuronal apoptosis in hippocampus CA3 of apoE(−/−) mice.
Figure 3.
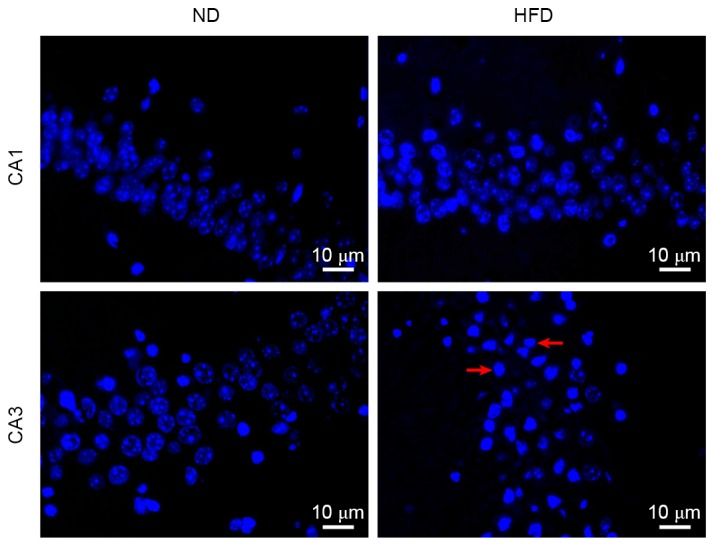
Hyperlipidemia increases neuronal apoptosis in hippocampal CA3 neurons. Hoechst 33258-stained hippocampus CA1 and CA3 from apoE(−/−) mice fed with ND or HFD for 12 weeks (magnification, ×400). Cells with nuclei exhibited marked chromatin condensation and nuclear fragmentation (indicated by the red arrows) were considered to be apoptotic. ND, normal diet; HFD, high-fat diet.
Effects of HFD on Bcl-2, Bax, and caspase-3 expression levels in the hippocampus of apoE(−/−) mice
To investigate the underlying molecular mechanisms of hyperlipidemia-induced apoptosis of hippocampal neurons, apoptosis-associated protein expression in the hippocampus of apoE(−/−) mice was observed via immunohistochemistry. The expression of caspase-3, which is the major executioner caspase during the demolition phase of apoptosis, was markedly higher in CA3 of HFD-fed mice than in ND-fed mice, and pro-apoptotic protein Bax also increased in CA1 and CA3 of HFD-fed apoE(−/−) mice. Notably, anti-apoptotic protein Bcl-2 slightly increased in CA3 of HFD-fed apoE(−/−) mice (Fig. 4). These results indicated that hyperlipidemia-induced hippocampal neuronal apoptosis of apoE(−/−) mice may be controlled via the Bcl-2/Bax-caspase-3 signaling pathway.
Figure 4.

Hyperlipidemia-induced hippocampal neuronal apoptosis of apoE(−/−) mice may be controlled via the Bcl-2/Bax-caspase-3 pathway. Immunohistochemistry of Bcl-2, Bax, and caspase-3 in hippocampus CA1 and CA3 of apoE(−/−) mice fed with ND or HFD for 12 weeks (magnification, ×400). Brown particles in the cytoplasm indicate protein expression. ND, normal diet; HFD, high-fat diet; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein.
Effects of HFD on PCSK9 and BACE1 expression in the hippocampus of ApoE(−/−) mice
PCSK9, an apoptosis-associated protein, may degrade BACE1, the predominant enzyme cleaving APP to generate Aβ. Aβ is another key apoptosis inducer in neurodegenerative diseases. To determine whether PCSK9 and BACE1 are involved in hippocampal neuronal apoptosis, changes in PCSK9 and BACE1 expression levels in the hippocampus of apoE(−/−) mice were observed by immunohistochemistry. PCSK9 expression in CA1 and CA3 of HFD-fed apoE(−/−) mice notably increased compared with ND-fed apoE(−/−) mice. Furthermore, BACE1 expression in CA3 of HFD-fed apoE(−/−) mice notably increased (Fig. 5). These results suggested that PCSK9 and BACE1 may be associated with hyperlipidemia-induced hippocampal neuronal apoptosis.
Figure 5.

PCSK9 and BACE1 may be correlated with hyperlipidemia-induced hippocampal neuronal apoptosis. Immunohistochemistry of PCSK9 and BACE1 in hippocampus CA1 and CA3 of apoE(−/−) mice fed with ND or HFD for 12 weeks (400x). ND, normal diet; HFD, high-fat diet; PCSK9, proprotein convertase subtilisin/kexin type 9; BACE1, β-secretase 1.
Effects of HFD on amyloid plaques in the hippocampus of apoE(−/−) mice
To further elucidate the role of BACE1 in hyperlipidemia-induced hippocampal neuronal apoptosis, the changes in levels of SP in the brains in ND- and HFD-fed apoE(−/−) mice were compared using modified Bielschowsky staining, which allowed visualization of diffuse plaques in the brains. Small amyloid plaques were observed in the hippocampus of HFD-fed apoE(−/−) mice but not in ND-fed apoE(−/−) mice (Fig. 6). This finding was consistent with the expression pattern of BACE1.
Figure 6.
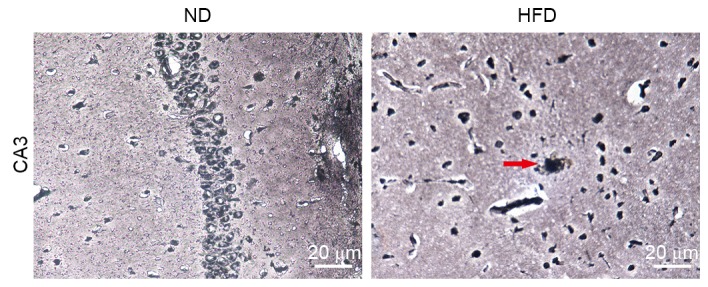
Small amyloid plaques were observed in the hippocampus of HFD-fed apoE(−/−) mice. Modified Bielschowsky stain of amyloid plaques in hippocampus CA3 of apoE(−/−) mice fed with ND or HFD for 12 weeks (magnification, ×400). Amyloid plaques are indicated by the red arrows. ND, normal diet; HFD, high-fat diet.
Discussion
Neurodegenerative diseases, which affect tens of millions of people annually worldwide, are characterized by the physical decay and eventual loss of neurons. AD is one of the most common neurodegenerative diseases, which predominantly affects the bilateral frontotemporal lobe and hippocampus. Histopathology indicates deposition of senile plaques, neurofibrillary tangles, lost neurons, and glial hyperplasia. Neuronal apoptosis is one of the major pathological characteristics of AD. The majority of cases of AD have a genetic element, although sporadic cases of this disease have been reported. To date, no specific treatment is able to reverse the progression of AD. Limiting this harmful disease is one of the most critical challenges in current medicine.
Hyperlipidemia is a risk factor of AD and other neurodegenerative diseases, as evidenced by epidemiological (3,4), clinical, and experimental studies (8–10). In the present study, 18 six-week-old apoE(−/−) mice were randomly divided into two groups: ND and HFD. After 12 weeks of feeding, hippocampal tissues were collected for frozen sectioning. Compared with the ND group, pyramidal cells in the hippocampus of apoE(−/−) mice fed with HFD were disordered. In addition, intercellular spaces increased, cells swelled, and nuclei became smaller and hyperchromatic, exhibiting karyopyknosis, particularly in CA3 areas. By contrast, CA1 indicated no notable changes. These results suggested that hyperlipidemia results in structural damage in the hippocampus of HFD-fed apoE(−/−) mice. Oil red O staining demonstrated that HFD-fed apoE(−/−) mice exhibited slightly increased lipid accumulation levels compared with ND-fed apoE(−/−) mice in hippocampus CA3. This staining also demonstrated that plasma lipids can pass through the blood-brain barrier (BBB) into the brain tissues when hyperlipidemia occurs, resulting in increased lipid deposits in the pyramidal cells of the hippocampus. Stranahan et al (16) also observed that free cholesterol levels in the hippocampus were markedly increased following induction of hyperlipidemia in Sprague-Dawley (SD) rats by feeding with high-fat and high-sugar diets for 3 months. Generally, blood lipids cannot completely pass through BBB, however, previous studies have indicated that the permeability of BBB in apoE(−/−) mice is 3.7 times of ordinary mice (17,18). This increase in permeability results in the passage of nonessential small molecules into the brain, leading to brain function disorders. This may be attributed to HFD-induced AD of apoE(−/−) mice (17). The increased raw material for lipid synthesis in the blood passes through the BBB freely, possibly increasing the lipid contents in the brain. Furthermore, lysophosphatidic acid, the predominant bioactive lipid of oxLDL, may damage BBB, leading to the occurrence of AD (19).
Although hyperlipidemia has been identified as a risk factor of AD, the mechanism of hyperlipidemia-induced AD has not been completely elucidated. It has been demonstrated that following feeding C57BL/6 and LDLR-/− mice with a HFD for 8 weeks, the expression levels of a number of cytokines, including tumor necrosis factor-α, interleukin-1, interleukin-6, nitric oxide synthase-2, and cyclooxygenase-2, increased along with BACE1 expression, suggesting that hypercholesterolemia results in nerve inflammation and influences the APP metabolic pathway, resulting in neurodegenerative diseases (20). Stranahan et al (16) observed that the levels of lipid peroxidation products 4-hydroxynonenal-lysine and 4-hydroxynonenal-histidine were locally increased in the hippocampus subsequent to feeding SD rats high-fat and high-sugar diets for 3 months to induce hyperlipidemia, indicating cell membrane-associated oxidative stress. On the basis of the above results, the present study suggests that hyperlipidemia can increase lipid deposition and promote cell apoptosis, leading to neuronal degeneration.
Hoechst 33258 staining results indicated that pyramidal cells in hippocampus CA3 of HFD-fed apoE(−/−) mice exhibited greater apoptosis than ND-fed apoE(−/−) mice, indicating that hyperlipidemia increased neuronal apoptosis in the hippocampus. Furthermore, the expression of PCSK9, BACE1, caspase-3, and Bax significantly increased, whereas Bcl-2 expression increased only marginally, indicating that hyperlipidemia resulted in neuronal apoptosis in the hippocampus of apoE(−/−) mice possibly via the Bcl-2/Bax-caspase-3 signaling pathway.
PCSK9 is a newly identified gene associated with blood cholesterol metabolism. Its predominant biological function is the degradation of LDLR in hepatic cells. PCSK9 function has been comprehensively investigated, revealing its regulatory functions, such as regulation of neuronal apoptosis in addition to lipid metabolism (21). A previous study demonstrated that when cerebellar granule neurons were damaged, PCSK9 expression increased, activating the caspase-3 and caspase-9 signaling pathways, possibly leading to apoptosis (21). The current study observed that PCSK9 expression in hippocampal CA3 of HFD-fed apoE(−/−) mice was significantly increased compared with that in the ND-fed group. Combined with previous experimental findings, the results of the present study suggest that PCSK9 is important in neuronal apoptosis of HFD-fed apoE(−/−) mice.
Numerous studies have confirmed that the large number of Aβ deposits in the brain is an important risk factor for the occurrence of AD (22,23). As previously mentioned, BACE1 is the predominant enzyme producing Aβ from APP. Aβ-induced neuronal apoptosis leads to AD. BACE1 protein expression levels and enzyme activities increased in the majority of sporadic AD patients. The present study also compared the changes of substance P levels in the brains of ND- and HFD-fed apoE(−/−) mice using modified Bielschowsky staining, and a slightly increased SP deposition was observed in the hippocampus of HFD-fed apoE(−/−) mice. This is consistent with previous studies that have observed that HFD can lead to an increase in SP deposition (24–26). Although numerous studies have verified the association between lipids and Aβ, the underlying mechanism remains to be elucidated.
PCSK9 expression was demonstrated to be negatively associated with BACE1 level in certain studies (27,28), however, other reports have indicated no association between PCSK9 and BACE1 (29). Thus, the correlation between PCSK9 and BACE1 requires further elucidation. The present study investigated the association between PCSK9 and BACE1 and observed that PCSK9 and BACE1 expression increased in HFD-fed apoE(−/−) mice. Furthermore, BACE1 mRNA and protein levels increased subsequent to feeding C57BL/6 and LDLR-/−mice a HFD for 8 weeks (20). Thus, the association between PCSK9 and BACE1 requires further investigation at a cellular level.
In conclusion, the present study demonstrated that hyperlipidemia can induce apoptosis of hippocampal neurons in apoE(−/−) mice, and that PCSK9 may be involved in this process.
Acknowledgements
The present study was supported by grants from the National Natural Science Foundation of China (grant nos. 81370376 and 81200217), the Visiting Scholar Foundation of Key Laboratory of Biorheological Science and Technology (Chongqing University), Ministry of Education (grant nos. CQKLBST- 2014–002 and CQKLBST-2015-004), the Construct Program of the Key Discipline in Hunan Province (grant no. 2011001), and the ‘225’ High-level Health Professionals Grant in Hunan Province (grant no. 201313).
References
- 1.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akhter R, Sanphui P, Das H, Saha P, Biswas SC. The regulation of p53 up-regulated modulator of apoptosis by JNK/c-Jun pathway in β-amyloid-induced neuron death. J Neurochem. 2015;134:1091–1103. doi: 10.1111/jnc.13128. [DOI] [PubMed] [Google Scholar]
- 3.Kivipelto M, Helkala EL, Laakso MP, Hänninen T, Hallikainen M, Alhainen K, Iivonen S, Mannermaa A, Tuomilehto J, Nissinen A, Soininen H. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med. 2002;137:149–155. doi: 10.7326/0003-4819-137-3-200208060-00006. [DOI] [PubMed] [Google Scholar]
- 4.Solomon A, Kåreholt I, Ngandu T, Winblad B, Nissinen A, Tuomilehto J, Soininen H, Kivipelto M. Serum cholesterol changes after midlife and late life cognition: Twenty-one-year follow-up study. Neurology. 2007;68:751–756. doi: 10.1212/01.wnl.0000256368.57375.b7. [DOI] [PubMed] [Google Scholar]
- 5.Shepardson NE, Shankar GM, Selkoe DJ. Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch Neurol. 2011;68:1239–1244. doi: 10.1001/archneurol.2011.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pregelj P. Involvement of cholesterol in the pathogenesis of Alzheimer's disease: Role of statins. Psychiatr Danub. 2008;20:162–167. [PubMed] [Google Scholar]
- 7.Longenberger J, Shah ZA. Simvastatin and other HMG-CoA reductase inhibitors on brain cholesterol levels in Alzheimer's disease. Curr Alzheimer Res. 2011;8:434–442. doi: 10.2174/156720511795745393. [DOI] [PubMed] [Google Scholar]
- 8.Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem. 2012;287:2678–2688. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kosari S, Badoer E, Nguyen JC, Killcross AS, Jenkins TA. Effect of western and high fat diets on memory and cholinergic measures in the rat. Behav Brain Res. 2012;235:98–103. doi: 10.1016/j.bbr.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 10.El-Sayyad HI, El Sherbiny MA, Sobh MA, El Naga AM Abou, Ibrahim MA, Mousa SA. Protective effects of Morus alba leaves extract on ocular functions of pups from diabetic and hypercholesterolemic mother rats. Int J Biol Sci. 2011;7:715–728. doi: 10.7150/ijbs.7.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JH, Hsieh CJ, Huang YL, Chen YC, Chen TF, Sun Y, Wen LL, Yip PK, Chu YM. Genetic polymorphisms of lipid metabolism gene SAR1 homolog B and the risk of Alzheimer's disease and vascular dementia. J Formos Med Assoc. 2016;115:38–44. doi: 10.1016/j.jfma.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100:928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, et al. NARC-1/PCSK9 and its natural mutants: Zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 14.Wu CY, Tang ZH, Jiang L, Li XF, Jiang ZS, Liu LS. PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax caspase9-caspase3 pathway. Mol Cell Biochem. 2012;359:347–358. doi: 10.1007/s11010-011-1028-6. [DOI] [PubMed] [Google Scholar]
- 15.National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals, corp-author. Guide for the Care and Use of Laboratory Animals. 8th edition. National Academies Press (US); Washington (DC): 2011. [Google Scholar]
- 16.Stranahan AM, Cutler RG, Button C, Telljohann R, Mattson MP. Diet-induced elevations in serum cholesterol are associated with alterations in hippocampal lipid metabolism and increased oxidative stress. J Neurochem. 2011;118:611–615. doi: 10.1111/j.1471-4159.2011.07351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moghadam A Hafezi, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am J Physiol Cell Physiol. 2007;292:C1256–C1262. doi: 10.1152/ajpcell.00563.2005. [DOI] [PubMed] [Google Scholar]
- 18.Methia N, André P, Hafezi-Moghadam A, Economopoulos M, Thomas KL, Wagner DD. ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med. 2001;7:810–815. [PMC free article] [PubMed] [Google Scholar]
- 19.Frisardi V, Panza F, Seripa D, Farooqui T, Farooqui AA. Glycerophospholipids and glycerophospholipid-derived lipid mediators: A complex meshwork in Alzheimer's disease pathology. Prog Lipid Res. 2011;50:313–330. doi: 10.1016/j.plipres.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Thirumangalakudi L, Prakasam A, Zhang R, Bimonte-Nelson H, Sambamurti K, Kindy MS, Bhat NR. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem. 2008;106:475–485. doi: 10.1111/j.1471-4159.2008.05415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bingham B, Shen R, Kotnis S, Lo CF, Ozenberger BA, Ghosh N, Kennedy JD, Jacobsen JS, Grenier JM, DiStefano PS, et al. Proapoptotic effects of NARC 1 (=PCSK9), the gene encoding a novel serine proteinase. Cytometry A. 2006;69:1123–1131. doi: 10.1002/cyto.a.20346. [DOI] [PubMed] [Google Scholar]
- 22.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: The solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/S0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 23.Fernández-Vizarra P, Fernández AP, Castro-Blanco S, Serrano J, Bentura ML, Martínez-Murillo R, Martínez A, Rodrigo J. Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer's disease. Histol Histopathol. 2004;19:823–844. doi: 10.14670/HH-19.823. [DOI] [PubMed] [Google Scholar]
- 24.Shie FS, Jin LW, Cook DG, Leverenz JB, LeBoeuf RC. Diet-induced hypercholesterolemia enhances brain A beta accumulation in transgenic mice. Neuroreport. 2002;13:455–459. doi: 10.1097/00001756-200203250-00019. [DOI] [PubMed] [Google Scholar]
- 25.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0366. [DOI] [PubMed] [Google Scholar]
- 26.Umeda T, Tomiyama T, Kitajima E, Idomoto T, Nomura S, Lambert MP, Klein WL, Mori H. Hypercholesterolemia accelerates intraneuronal accumulation of Aβ oligomers resulting in memory impairment in Alzheimer's disease model mice. Life Sci. 2012;91:1169–1176. doi: 10.1016/j.lfs.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 27.Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 2008;9:916–922. doi: 10.1038/embor.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko MH, Puglielli L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J Biol Chem. 2009;284:2482–2492. doi: 10.1074/jbc.M804901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu M, Wu G, Baysarowich J, Kavana M, Addona GH, Bierilo KK, Mudgett JS, Pavlovic G, Sitlani A, Renger JJ, et al. PCSK9 is not involved in the degradation of LDL receptors and BACE1 in the adult mouse brain. J Lipid Res. 2010;51:2611–2618. doi: 10.1194/jlr.M006635. [DOI] [PMC free article] [PubMed] [Google Scholar]