

Abstract
Bartter syndrome (BS) type 1 is an autosomal recessive kidney disorder caused by loss-of-function mutations in the solute carrier family 12 member 1 (SLC12A1) gene. To date, 72 BS type 1 patients harboring SLC12A1 mutations have been documented. Of these 144 alleles studied, 68 different disease-causing mutations have been detected in 129 alleles, and no mutation was detected in the remaining 15 alleles. The mutation types included missense/nonsense mutations, splicing mutations and small insertions and deletions ranging from 1 to 4 nucleotides. A large deletion encompassing a whole exon in the SLC12A1 gene has not yet been reported. The current study initially identified an undocumented homozygous frameshift mutation (c.1833delT) by Sanger sequencing analysis of a single infant with BS type 1. However, in a subsequent analysis, the mutation was detected only in the father's DNA. Upon further investigation using a next-generation sequencing approach, a deletion in exons 14 and 15 in both the patient and patient's mother was detected. The deletion was subsequently confirmed by use of a long-range polymerase chain reaction and was determined to be 3.16 kb in size based on sequencing of the junction fragment. The results of the present study demonstrated that pathogenic variants of SLC12A1 are heterogeneous. Large deletions appear to serve an etiological role in BS type 1, and may be more prevalent than previously thought.
Keywords: Bartter syndrome type 1, deletion, heterozygous deletion, genetic heterogeneity, solute carrier family 12 member 1
Introduction
Bartter syndrome (BS) is a group of autosomal kidney disorders characterized by hypokalemic metabolic alkalosis, hypercalciuria and salt wasting (1). BS has five distinct genetic subtypes. BS types 1, 2, 3 and 4A are all autosomal recessive and are caused by loss-of-function mutations in the solute carrier family 12, member 1 (SLC12A1), potassium inwardly-rectifying channel subfamily J, member 1 (KCNJ1), chloride channel Kb (CLCNKB), and Barttin CLCNK type accessory β-subunit (BSND) genes, respectively (1). BS type 4B is caused by simultaneous heterozygous mutations in the CLCNKA and CLCNKB genes (2). BS type 5 demonstrates autosomal dominant inheritance, and is caused by a gain-of-function mutation in the CASR gene (3). Clinically, BS is classified into three types based primarily on phenotype and age of onset, including antenatal BS (BS type 1 and type 2), classic BS (BS type 3), and antenatal BS with sensorineural deafness (BS type 4A, type 4B, and type 5) (2,4). Of these subtypes, BS type 1 (Online Mendelian Inheritance in Man; no. 600839; www.omim.org/entry/600839) is clinically categorized as antenatal BS and is usually associated with life-threatening phenotypes (4,5). The SLC12A1 gene is currently the only known gene that leads to BS type 1 (6,7). This gene is located at 15q21.1 and contains 27 exons. Of these 27 exons, 26 of them are protein-encoding exons. The SLC12A1 gene has kidney-specific transcription (NM_000338.2; National Center for Biotechnology Information; www.ncbi.nlm.nih.gov) and encodes the Na-K-2Cl co-transporter. The carrier-frequency was estimated to be 1 in 360 (8). To date, 68 different BS type 1-causing mutations have been documented in the Human Gene Mutation Database (HGMD; www.hgmd.cf.ac.uk) and in the literature (7–21). The mutation types included missense/nonsense mutations, splicing mutations, small insertions and small deletions. Deletions encompassing an entire exon have not yet been reported.
In the current study of a single patient suspected to have BS type I, a novel homozygous frameshift mutation (c.1833delT) in SLC12A1 was initially detected by Sanger sequencing. However, in the parental study, the c.1833delT frameshift mutation was detected only the father but not in the mother. Upon further investigation, and after paternal segmental isodisomy 15 was ruled out, it was discovered that the proband harbored a 3.16 kb deletion spanning the region where the c.1833delT mutation was detected. Clearly, this deletion was missed by Sanger sequencing. To the best of our knowledge, the present study is the first to report an extensive deletion in the SLC12A1 gene associated with BS type 1.
Materials and methods
DNA isolation
Genomic DNA was extracted from the peripheral white blood cells of the proband and their parents using an automatic nucleic acid isolation system (QuickGene-610L; Fujifilm Holdings Corporation, Tokyo, Japan) according to the manufacturer's protocols. The DNA concentration and quality were measured using the Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE, USA) according to manufacturer's instructions.
Sanger sequencing
Polymerase chain reaction (PCR) was performed using specific primers designed and generated by Primer3 v0.4.0 software, (http://bioinfo.ut.ee/primer3/) in our laboratory (University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA; Table I) targeting the coding region and the intron-exon junctions of the SLC12A1 gene. Sanger sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and an ABI 3500xL genetic analyzer (Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. Sequences were analyzed using Mutation Surveyor software v4.0.9 (SoftGenetics LLC., State College, PA, USA).
Table I.
Primers used for Sanger sequencing of the SLC12A1 gene.
| Exon | Forward primer | Reverse primer | Product size (bp) |
|---|---|---|---|
| 2 | 5′-AGCTCCCTAATGGAAGCACA-3′ | 5′-GTAAGTAAGATGGATAGTGTTT-3′ | 531 |
| 3 and 4 | 5′-TCAATTGTTTTGATTTGCTTTGA-3′ | 5′-TGCTTATTGAATTTATGATTTACATGC-3′ | 507 |
| 5 | 5′-GAGGCATGGACCTGAAAACT-3′ | 5′-GCATCCAGCTCCTCCAAATA-3′ | 337 |
| 6 | 5′-TCAGATAGTCACAATCGTTTGGTT-3′ | 5′-TCCCTTAGTGCCCTGAGAAG-3′ | 289 |
| 7 | 5′-CCTATATGGCCCCAGGTGTA-3′ | 5′-TGCCTGCTCATTTCACCATA-3′ | 351 |
| 8 | 5′-TCTGGGTAGCAGAGACTTAACTGA-3′ | 5′-TGATGGGGATGGTGATGAG-3′ | 311 |
| 9 | 5′-GGACTAGGGAAGCCAATGGT-3′ | 5′-TTTGAATCTGTAGGGTAATATGGTCA-3′ | 318 |
| 10 | 5′-CATCAACTTGCTGTTTGCTTG-3′ | 5′-TGCTGCATTGAAAGCTCACT-3′ | 313 |
| 11 | 5′-CAGAGGCTAAGAAATGGACCTT-3′ | 5′-GCCAATAATCAATCAGTTGCAG-3′ | 363 |
| 12 | 5′-CTGGGCGATAGAGCGAGACT-3′ | 5′-ACTTGAGCCAGATGCAAACA-3′ | 401 |
| 13 | 5′-TCCCCAAATCTTCTTGTTTGA-3′ | 5′-CAAACTAAAAGGAAAGCCCTATGA-3′ | 286 |
| 14 | 5′-TGACAGATGCTCGCTATGTTTT-3′ | 5′-GGATTACGCTTCCCACTAGG-3′ | 352 |
| 15 | 5′-AGATTCTGGAACTTGGCCTAAA-3′ | 5′-ATCCACCTTACATATGCCTCA-3′ | 401 |
| 16 | 5′-ATTTGGGCACTCATCTTTGC-3′ | 5′-TTTGTATGACTGCTTATTTTTAGAATG-3′ | 354 |
| 17 | 5′-GAGAGGTTGCCCCATTTTTC-3′ | 5′-GCATGTACCTTTTCTCCTCACC-3′ | 306 |
| 18 | 5′-GGTCATCTCCAAAAGGCTGA-3′ | 5′-TGCTTGGCTGCCTAACTACA-3′ | 413 |
| 19 | 5′-TCAGGATCTTCAGAACATTACTTCA-3′ | 5′-AAGTCAGAGTACCAGGGGAATG-3′ | 353 |
| 20 | 5′-AAATGCATCAGCTCTTGGCTA-3′ | 5′-GCCCTGCTGTATCCCATAAC-3′ | 310 |
| 21 | 5′-GGTGATTTTGTCTTCTTTCATCA-3′ | 5′-TGGAGAGAAACCTTTCAGTTCCT-3′ | 301 |
| 22 | 5′-TTGTTTCTGCCCTCAAAAGC-3′ | 5′-TCCCATATACCTTCTCATGCAGT-3′ | 240 |
| 23 | 5′-ACTTAATTAAGAGCTATCAA-3′ | 5′-ATTAAAGCAACAAACCTCTGAAATG-3′ | 270 |
| 24 | 5′-CCAACCAAAAAGCCTCTGTC-3′ | 5′-TCAAGAAGTCGCTGCAGTAAA-3′ | 336 |
| 25 | 5′-GCATGATATTCAGCTCTGATTCC-3′ | 5′-TGAAAAATGCAGAAAGCTTGG-3′ | 385 |
| 26 | 5′-AAACACCATAAGTTTCTAAGCCTGA-3′ | 5′-CCTGAAGAGTCCCAAGCTTTT-3′ | 308 |
| 27 | 5′-GCTCAGAAATACTAGTGCCGTTA-3′ | 5′-TCCTCTCCAGAGGTTTGCAT-3′ | 324 |
Primers highlighted in bold were used for the long-range PCR assay. All the primers were designed in the introns and covered intron/exon junctions. The primers were designed to have polymerase chain reaction products covering each exon and a minimum 20 bp flanking region in the introns. SLC12A1, solute carrier family 12 member 1.
Uniparental disomy study (UPD)
Genomic DNA (100 ng) templates from the proband and the parents were subject to investigation by PCR amplification. This was performed using the following four pairs of short tandem repeat (STR) primer sets (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) on different loci of chromosome 15q: D15S1024, 15q21.1; D15S131, 15q23; D15S984, 15q24.3 and D15S115, 15q25.2. The amplified PCR products were subjected to fragment analysis using the 3500xL Genetic Analyzer (Thermo Fisher Scientific, Inc.). Data analysis was performed using GeneMapper Software 4.1 (Thermo Fisher Scientific, Inc.).
Ion Personal Genome Machine (PGM) next generation target sequencing
All products used for Ion PGM sequencing were supplied by Thermo Fisher Scientific, Inc., unless stated otherwise. In order to determine the mutant allele distribution and exclude the potential heterozygous deletion spanning the c.1833delT mutation as determined by Sanger sequencing, DNA samples of the proband, the mother and a normal control cell line (Coriell Institute for Medical Research, Camden, NJ, USA) were subjected to Ion PGM target sequencing to detect the possible deletion based on the method in the previous study by Faiz et al (22). Genomic DNA (10 ng) from each sample was used to prepare a barcoded library of specific amplicons using the Ion AmpliSeq Library kit 2.0 and the Ion AmpliSeq™ custom panels for the SLC12A1 gene, designed using the Ion AmpliSeq™Designer web-hosted software v3.0 (www.ampliseq.com). The libraries generated from the each DNA sample were clonally amplified with the Ion PGM™ Template OT2 200 kit and the Ion OneTouch™ 2 System for templating and enrichment prior to chip loading. Sequencing was performed using the Ion 314™ Chip with the Ion PGM™ sequencing 200 kit on the Ion PGM™ system. Conversion of the raw signal to base calls was performed on the Torrent Server using Torrent Suite Software v3.4 (Thermo Fisher Scientific, Inc.). Base-called files were aligned to the University of California Santa Cruz Genome Browser (http://genome.ucsc.edu/) hg19 reference genome to calculate run quality metrics. Sequence reads were generated and mapped to the hg19 sequence using the Torrent Suite Software v3.4. The Torrent Variant Caller utilized the mapped reads to identify single nucleotide polymorphisms, and insertion and deletion variants. The standard output files of the Ion PGM™ system, including BAM and VCF, were also analyzed with Integrative Genomics Viewer software v2.3 (www.broadinstitute.org/igv/).
Array comparative genomic hybridization (CGH)
Array CGH, using the Agilent 2×400 K chip (Agilent Technologies, Inc., Santa Clara, CA, USA), was performed to confirm the deletion in the proband identified by sequencing analysis. Human reference genomic DNA was purchased from Agilent (Agilent Technologies, Inc.). The DNA (1 µg) of the proband and the reference DNA were labeled with cyanine 3 and cyanine 5, respectively, by random priming (Agilent Technologies, Inc.). Proband DNA was combined with a normal control DNA sample and hybridized to an Agilent 2×400 K oligo microarray chip (cat. no. G4448A; Agilent Technologies, Inc.) by incubating in the Agilent Microarray Hybridization Oven (Agilent Technologies, Inc.) for 40 h at 67°C. The slide was then washed using wash buffer (Agilent Technologies, Inc.) and scanned using the NimbleGen MS 200 Microarray Scanner (Roche NimbleGen, Madison, WI, USA). The Agilent CytoGenomics 2.7 software program (Agilent Technologies, Inc.) was used for data analysis.
Long-range PCR
To confirm the deletion detected by Ion PGM study, DNA (200 ng) of the proband and the parents was subjected to long-rang PCR study. Long-range PCR using forward primers targeting intron 12 and reverse primers targeting intron 16 (Table I) was performed using the Expand Long Template PCR system (Roche Diagnostics, Indianapolis, IN, USA). The normal PCR products were expected to be 8784 bp (GRCh38.p7). The thermal cycling program was provided by the kit manual with an annealing temperature of 59°C. The PCR products were loaded on a 1% low electroendosmosis agarose gel (Lonza Rockland, Rockland, ME, USA) for electrophoresis. A 1 kb DNA (Thermo Fisher Scientific, Inc.) was used as marker.
Detection of breakpoints
Using the long-range PCR products (0.5 µl) from the proband as a template, multiple primers (generated by Primer3, v 0.4.0; purchased from Integrated DNA Technologies, Inc., Coralville, IA, USA) targeting the deletion of SLC12A1 gene were designed to determine the breakpoints of the deletion. The product from the forward primer (5′-AAGGATGACGTCTCTATATT-3′) and the reverse primer (5′-ACAGAGTTTCGCTCTTGTTG-3′) produced a clean band in the 1% low electroendosmosis agarose gel, visualized under UV light, which was subjected to Sanger sequencing (as described above) for the detection of breakpoints.
Literature review
To investigate the SLC12A1 mutation patterns, Human Gene Mutation Database (www.hgmd.cf.ac.uk) and previous English-language studies identified in PubMed (https://www.ncbi.nlm.nih.gov/pubmed) were reviewed using the search terms ‘Bartter syndrome type 1 OR SLC12A1 OR NKCC2’.
Results
Following sequencing of the entire coding region and the intron-exon boundaries of the SLC12A1 gene in the proband, a novel homozygous frameshift mutation in exon 15 (c.1833delT) was identified (Fig. 1). This deletion causes a change from phenylalanine to leucine at amino acid position 611 of the SLC12A1 gene, as well as the generation of a premature stop codon in the 32nd coding frame. The presence of this stop codon leads to protein truncation and is considered to be deleterious. In the family history, the proband's parents stated that they were non-consanguineous. As this mutation is novel and the proband's parents were unrelated, a parental mutation assay was suggested. Unexpectedly, this c.1866delT mutation was only detected in the father's DNA, but not in the mother's. The genotype of the proband and the parents indicated that the father did not have the large deletion encompassing exon 15. Therefore, heterozygous deletion encompassing exon 15 was suspected in the proband and the mother. The purpose of Ion PGM sequencing was to detect this deletion. Thus, the father was not subject to Ion PGM analysis. To rule out possible paternal segmental isodisomy 15, which may lead to the identified homozygous mutation, a UPD study using four STR markers localized to four different loci in the chromosome 15q21.1-q25.2 region was performed. The results excluded the possibility of paternal isodisomy (data not shown). Based on the genotype of the proband and the parents, heterozygous deletions encompassing exon 15 were suspected in the proband and the mother. To exclude a possible heterozygous deletion that encompassed the c.1833delT mutation, the same quantity of DNA samples from the proband, the mother, and a normal control were subjected to Ion PGM target sequencing. The Ion PGM data clearly demonstrate the presence of the c.1833delT single nucleotide deletion in the proband but not in the proband's mother. The read count of exons 14 and 15 from proband and the mother was markedly lower when compared with normal control, which indicated the presence of a potentially large deletion in this region (Fig. 2). To verify the Ion PGM findings, array CGH was performed using an Agilent 2×400 K chip. The results demonstrated that one probe located between exons 14 and 15 was deleted, whereas the other probes were in the normal base line (Fig. 3). This confirmed the Ion PGM analysis results. Long-range PCR using forward primers in intron 12 and reverse primers in intron 16 was applied to confirm the deletion in exons 14 and 15, as well as determine the size of the deleted region. The results clearly indicated that the father had one normal band with an expected size of 8.8 kb, whereas the mother and the proband had two bands that were 8.8 kb and 5.8 kb in size. This was due to the presence of a 3.0 kb deletion in one allele (Fig. 4). To determine the breakpoints of the deletion, the junction fragment was sequenced. The large deletion was observed to be c.1685–726_1941+239del, and was 3,160 bp in size (Fig. 5).
Figure 1.
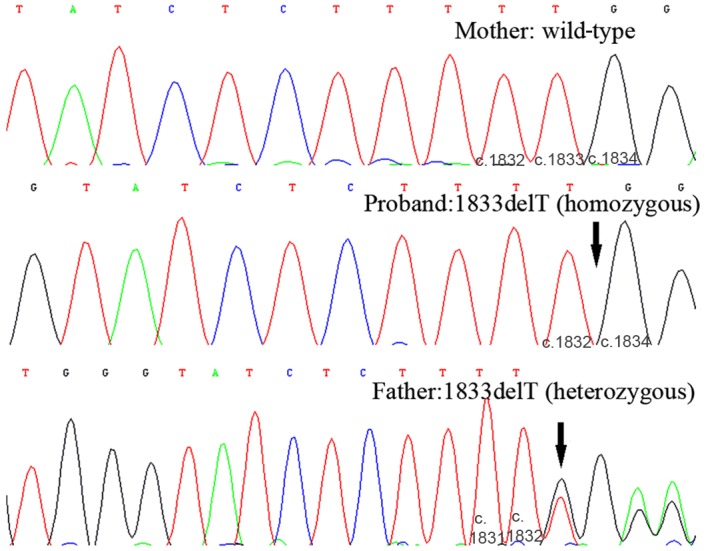
Sanger sequencing results of the SLC12A1 gene. The proband's mother demonstrated a normal wild-type sequence in both alleles (as indicated by five T-nucleotides in tandem). The proband demonstrated a homozygous mutation at c.1833delT (as indicated by four T-nucleotides in tandem). The father was heterozygous for the c.1833delT mutation. Black arrows indicate the position of the c.1833delT mutation. The numbers at the base of each plot indicated base number. SLC12A1, solute carrier family 12 member 1.
Figure 2.

Ion PGM target sequencing results. BAM files generated from Ion Torrent PGM sequencing data demonstrated that exon 14 and 15 read counts from the proband and the mother were markedly lower when compared with that of the normal control, whereas the read count of the other exons did not exhibit any obvious differences. This indicates the presence of a heterozygous deletion in the proband and the mother located in exons 14 and 15. The scale (0–400) in the top left corner of each plot indicated that the maximal reads shown in the fig. was 400. The numbered black bars at the base of the plot correspond to exons 10–16 of the SLC12A1 gene. PGM, personal genome machine; SLC12A1, solute carrier family 12 member 1.
Figure 3.
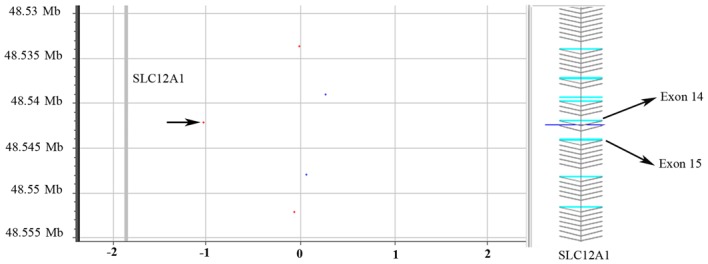
Confirmatory array CGH analysis. To verify the c.1833delT deletion in the SLC12A1 gene, the DNA sample from the proband was subject to array CGH analysis. In the confirmatory array CGH, one probe located between exons 14 and 15 of SLC12A1 (indicated by the black arrow in the plot) demonstrated the presence of a deletion in this region, whereas the other probes were observed in the normal base line. The red dots indicate the proband sample DNA; the blue dots indicate the normal control DNA. CGH, comparative genomic hybridization; SLC12A1, solute carrier family 12 member 1.
Figure 4.
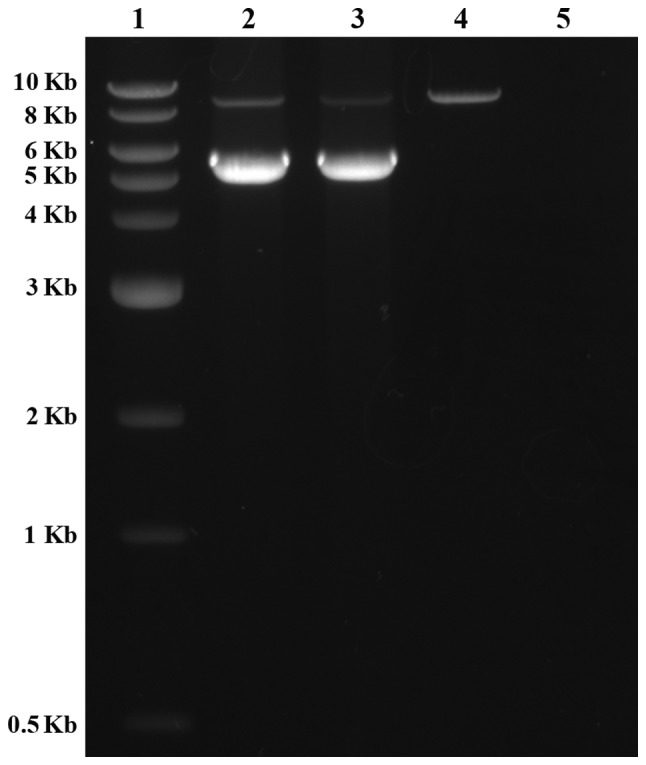
Confirmatory long-range PCR. The long-range PCR results demonstrated that one band was present in the father of the proband, whereas two bands were observed in the mother and the proband, indicating the presence of a ~3.0 kb deletion in one allele of the SLC12A1 gene. Lane 1, 1 kb DNA ladder; lane 2, the proband's mother; lane 3, the proband; lane 4, the proband's father; lane 5, control. PCR, polymerase chain reaction; SLC12A1, solute carrier family 12 member 1.
Figure 5.

Breakpoints of the large deletion in the SLC12A1 gene of the proband. To determine the breakpoints of the deletion observed in the proband, the junction fragment (sequence 48,248,661–48,252,200) was amplified and sequenced by Sanger sequencing. The large deletion was demonstrated to be c.1685-726_1941+239del, which was 3,160 bp in size. The black line in the fig. indicates the breakpoints. SLC12A1, solute carrier family 12 member 1. The upper plot is the standard normal control reference generated by the software. The lower plot was the patient's data.
Genotype of all documented BS type 1 patients
BS type 1 is a rare autosomal recessive disease caused by mutations in the SLC12A1 gene. Through examining previous reports, 72 BS type 1 patients with SLC12A1 mutations were documented (Table II). Of these, approximately 45% of patients harbored homozygous mutations, 34% were compound heterozygous mutations, and following sequencing all exons and intron-exon boundaries of the gene, and only one mutation was detected in the remaining 21% of patients. Of these 72 patients, the mutations included missense/nonsense mutations, splicing mutations, small insertions and small deletions. To date, no large deletion has been reported. To investigate SLC12A1 mutation patterns, previous studies were reviewed alongside the data of the present study which focused on the following three aspects: i) The mutation distribution patterns; ii) the possible reasons for the high proportion of homozygous mutations; iii) the possible reasons why a heterozygous mutation was identified in the present study.
Table II.
Summary of mutations in the SLC12A1 gene (NM_000338.2).
| Population origin | Nucleotide change | Amino acid change | Mutation type | Exon | Reference |
|---|---|---|---|---|---|
| USA | c.1833delT/c.1685-726_1941+239del | F611Lfs*32/Exons 14 and 15 deletion | F/D | 15/14,15 | Present case |
| Saudi Arabia | c.814G>T/c.814G>T | p.V272F/p.V272F | M homo | 6/6 | (7) |
| Saudi Arabia | c.814G>T/c.2467delC | p.V272F/p.Q823fs | M/F | 6/20 | (7) |
| Saudi Arabia | c.1519T>C/c.1519T>C | p.S507P/p.S507P | M homo | 12/12 | (7) |
| Saudi Arabia | c.1942G>T/c.1942G>T | p.D648N/p.D648N | M homo | 15/15 | (7) |
| Italy | c.904delC/c.904delC | p.R302fs/p.R302fs | F homo | 7/7 | (7) |
| Saudi Arabia | c.583-584dupA/c.583-584dupA | p.M195fs/p.M195fs | F homo | 4/4 | (7) |
| – | c.2239delT/c.2239delT | p.F747fs/p.F747fs | F homo | 18/18 | (8) |
| – | c.769G>A/c.769G>A | p.G257S/p.G257S | M homo | 6/6 | (8) |
| – | c.769G>A/c.769G>A | p.G257S/p.G257S | M homo | 6/6 | (8) |
| – | c.1663G>A/c.1663G>A | p.A555T/p.A555T | M homo | 13/13 | (8) |
| – | c.595C>T/c.1966C>T | p.R199C/p.Q656X | M/N | 4/16 | (8) |
| – | c.1576-1578delAAC/c.1576-1578delAAC | p.N526del/p.N526del | D homo | 13/13 | (8) |
| – | c.1432G>A/c.1432G>A | p.G478R/p.G478R | M homo | 11/11 | (8) |
| – | c.24-27delTGTA | p.V9fs | F/− | 2/ | (8) |
| – | c.1954G>A | p.G652S | M/− | 16/ | (8) |
| – | c.2495A>C/c.2495A>C | p.E832A/p.E832A | M homo | 21/21 | (8) |
| – | c.606G>C | p.W202C | M/− | 4/ | (8) |
| – | c.1834G>A/c.1834G>A | p.G612R/p.G612R | M homo | 15/15 | (8) |
| – | c.535T>A | p.W179R | M/− | 3/ | (8) |
| – | c.1966C>T/Intron 25 | p.Q656X/Splice mutant | N/S | 16/26 | (8) |
| 5′splice gt>at | |||||
| Costa Rican | c.1875G>A/c.1875G>A | p.W625X/p.W625X | N homo | 15/15 | (9) |
| Costa Rican | c.1875G>A/c.1875G>A | p.W625X/p.W625X | N homo | 15/15 | (9) |
| Costa Rican | c.1875G>A/c.1875G>A | p.W625X/p.W625X | N homo | 15/15 | (9) |
| Costa Rican | c.1875G>A | p.W625X | N/− | 15/ | (9) |
| Costa Rican | c.1875G>A | p.W625X | N/− | 15/ | (9) |
| German | c.924G>A/c.924G>A | p.R302Q/p.R302Q | M homo | 7/7 | (10) |
| Yugoslavia | c.1195delT/c.1195delT | F stop at 1318/F stop at 1318 | F homo | 9/9 | (10) |
| Morocco | c.1541G>A/c.1541G>A | p.A508T/p.A508T | M homo | 12/12 | (10) |
| German | c.1548C>A/c.1548C>A | p.A510D/p.A510D | M homo | 12/12 | (10) |
| Turkey | c.1595-1597del3/c.1595-1597del3 | p.526delN/p.526delN | D homo | 13/13 | (10) |
| Belgium | c.747G>A& | p.G243E | M/− | 6/ | (10) |
| German | c.1326G>A/c.614C>G | p.C436Y/p.R199G | M/M | 11/4 | (10) |
| Italy | c.1451G>A | p.G478R | M/− | 11/ | (10) |
| German | c.1541G>A | p.A508T | M/− | 12/ | (10) |
| Estonia | c.3013T>G | p.Y998X | N/− | 25/ | (10) |
| Caucasian | c.629–6A>G | S/− | 4/ | (11) | |
| Caucasian | c.629-6A>G/c.976–14C>G | S/S | 4/8 | (11) | |
| Caucasian | c.1103A>G/c.1103A>G | p.E368G/p.E368G | M homo | 9/9 | (11) |
| Morocco | c.1522G>A/c.1522G>A | p.A508T/p.A508T | M homo | 12/12 | (11) |
| Caucasian | c.577G>A/c.799G>T | p.G193R/p.A267S | M/M | 3/6 | (11) |
| Morocco | c.2281C>T/c.2281C>T | p.R761X/p.R761X | N homo | 18/18 | (11) |
| Caucasian | c.337–339dup/c.1041–1045del | p.E113dup/p.P348QfsX3 | I/F | 3/8 | (11) |
| Caucasian | c.1883C>A | p.A628D | M/− | 15/ | (11) |
| North African | c.1327G>A/c.1327G>A | p.G443R/p.G443R | M homo | 11/11 | (11) |
| Caucasian | c.1010C>T/c.3164+1G>A | p.A337V/Loss splice donor site | M/S | 8/26 | (11) |
| Mali | c.955G>A/c.955G>A | p.G319R/p.G319R | M homo | 7/7 | (11) |
| Algeria | c.2117delA/c.2117delA | p.K706RfsX23/p.K706RfsX23 | F homo | 17/17 | (11) |
| Italy | c.1381T>C/c.1381T>C | p.C461R/p.C461R | M homo | 11/11 | (12) |
| Italy | c.1062delG/c.1062delG | p.K354NfsX73/p.K354NfsX73 | F homo | 8/8 | (12) |
| Italy | c.1381T>C/c.1630C>T | p.C461R/P544S | M/M | 11/13 | (12) |
| Italy | c.904delC/c.904delC | p.R302GfsX2/p.R302GfsX2 | F homo | 7/7 | (12) |
| Italy | c.1663G>A/c.1663G>A | p.A555T/p.A555T | M homo | 13/13 | (12) |
| Italy | c.347G>A/c.1954G>A | p.R116H/p.G652S | M/M | 2/16 | (12) |
| Italy | c.551T>A/c.611T>C | p.L184Q/V204A | M/M | 3/4 | (12) |
| Italy | c.1190G>C/c.3164+1G>A | p.G397A/loss | M/S | 9/26 | (12) |
| splice donor site | |||||
| Italy | c.1493C>T/c.1522G>A | p.A498V/A508T | M/M | 12/12 | (12) |
| Italy | c.904delC/c.1493C>T | p.R302GfsX2/A498V | F/M | 7/12 | (12) |
| Italy | – | p.D12fs/p.R302fs | F/F | 2/7 | (13) |
| Italy | – | p.L522fs/− | F/− | 13/ | (13) |
| Italy | – | p.R302fs/p.R302fs | F homo | 7/7 | (13) |
| Italy | c.347G>A/c.1954G>A | p.R116H/p.G652S | M/M | 2/16 | (13) |
| German | c.530T>A/c.2751dupT | p.F177Y/p.D918fs | M/F | 4/22 | (14) |
| – | c.1411C>T | p.R471X | N/− | 11/ | (15) |
| – | c.2095G>A | p.D699N | M/− | 17/ | (15) |
| Japan | c.1664C>T/c.2426G>T | p.A555V/p.G809V | M/M | 13/20 | (16) |
| Korea | c.1277G>A/c.1679T>C | p.C436Y/p.L560P | M/M | 11/13 | (17) |
| Japan | c.724+4A>G/c.2095delG | Exon 6 skipping/p.D699fs | S/F | 6/17 | (18) |
| Japan | c.904C>T/c.2807G>A | p.R302W/p.W936X | M/N | 7/23 | (19) |
| Japan | c.577G>A/c.724+1G>A | p.G193R/exon 5 skipping | M/S | 4/6 | (19) |
| Japan | c.732-734delCTA/c.735-737delCTA | p.Y245del/p.Y245del | D homo | 6/6 | (20) |
| Japan | c.348insT/c.788G>A | p.N117X/p.G257S | N/M | 2/6 | (21) |
| Japan | c.2393-2394delGA/c.2971-2974delCAAA | p.D792fsX4/p.N984fsX26 | F/F | 19/24 | (21) |
SLC12A1, solute carrier family 12 member 1; M, missense mutation; N, nonsense mutation; F, frameshift mutation; S, splicing mutation; D, deletion mutation; I, insertion; homo, homozygous status; -, not available.
The mutation distribution pattern of the SLC12A1 gene
All previous relevant studies demonstrated that a total of 72 BS type 1 patients (patients from the same family and with the same genotype were excluded) and 144 SLC12A1 alleles have been characterized to date (Table II). In addition to the 66 mutations documented in the HGMD professional database, the literature review and the current study contributed four additional mutations to the mutation database of the SLC12A1 gene. These are D12fs, intron 25 5′- splice gt>at, c.1833delT, and c.1685–726_1941+239del. Among all 27 exons, the mutations in the SLC12A1 gene were distributed across the majority of the exons except for 5, 10, 14 and 27. Among the exons with mutations, exons 4, 6, 7, 11, 12, 13 and 15 harbored the most common mutated regions with >10 mutations in each allele. Among all 70 mutations, the R302fs mutation was the most common mutation and accounted for 5.5% of all the alleles. All of the R302fs mutations were detected in Italian population studies. At the same amino acid site, one homozygous mutation of R302Q was detected in a German population and a heterozygous mutation of R302W was detected in a Japanese population suggesting a mutation hot spot in this region. Furthermore, the mutation of W625X was detected in eight alleles accounting for 5.5% of all alleles, and were all detected in an isolated Costa Rican population, suggesting a founder effect in this population (9). The A508T mutation had been discovered in six alleles, which accounted for 4.1% of all alleles. Less common mutations, including G257S and A555T, were reported in five alleles. Mutation of N526del was reported in four alleles. Four mutations, including G625S, C461R, V272F, and G478R were detected in 3 alleles, 22 mutations were observed in two alleles, and the remaining mutations were in one allele. A total of 15 alleles lacked mutations, accounting for approximately 20.5% of all patients and 10.2% of all alleles, respectively. The mutation types included missense, nonsense, splicing mutations, small deletions and small insertions leading to frameshift mutations, and a large deletions. To the best of our knowledge, this study is the first report of a large deletion in the SLC12A1 gene.
The possible reasons for the high proportion of homozygous mutations in the SLC12A1 gene
In previous reports, homozygous mutations accounted for 45.2% of all the cases (Table II). A number of possible reasons for the high percentage of identified homozygous mutations exist. First, parental consanguinity may be a potential factor responsible for the homozygous status. Out of the 72 patients listed in Table II, 33 cases were characterized as having apparent homozygosity. Of these, approximately 14 cases had consanguineous parents (7,10,11), whereas 12 patients did not, and the status of the remaining seven cases was unknown. Among the patients from non-consanguineous families, four performed parental analysis and the presence of a heterozygous deletion was excluded (12). The remaining patients were not used for further study. In modern society, consanguineous relationships are likely to be avoided, particularly for individuals from non-isolated populations. Therefore, it is uncommon to observe a high proportion of homozygous mutations in a rare disease. The authors therefore suggest that patients from non-consanguineous families that harbor homozygous mutations should undergo a deletion test, if possible. This will provide more information regarding the potential reasons for the high percentage of homozygous mutations observed in BS type 1.
Second, the founder effect may serve a role in the observed mutation distribution. The study by Kurtz et al (9), involving BS type 1 patients from an isolated population in Costa Rica, investigated four familial and three sporadic cases. Of these cases, three of them harbored homozygous mutations, two of them demonstrated heterozygous mutations, and the remaining two cases were negative for mutations. The homozygous and heterozygous mutations identified were p.W625X, which suggests a founder effect in this Costa Rican population. In this case, due to the isolated populations, the incidence was higher when compared with other populations, and the patients shared the same few mutations (9). Therefore, although parents were not related to each other, the offspring were more likely to harbor homozygous mutations.
Regarding the patients in the cohort that were observed to harbor one heterozygous mutation or no mutation, the authors of these studies demonstrated that the methods used were not sensitive enough and may responsible for the undetected mutations (9). In addition, aside from those consanguineous parents and parents from isolated populations, non-related parents carrying the same mutation demonstrated a 25% chance of producing offspring with a homozygous mutation. This phenomenon is frequently observed in common autosomal recessive diseases that have few high-frequency mutations, such as cystic fibrosis (CF), in which the ΔF508 homozygous mutation is the most frequently observed. This mutation is present in approximately two-thirds of CF carriers (23). However, for rare autosomal recessive diseases, homozygous mutations are not commonly observed in patients from non-consanguineous parents. In the present case of a single patient with BS type I, Sanger sequencing analysis detected a frameshift mutation with apparent homozygosity. However, subsequent analyses demonstrated the presence of a compound large heterozygous deletion and frameshift mutation. Therefore, heterozygous deletions encompassing the mutant site should be excluded as homozygous when the parents are not consanguineous nor from an isolated population. This will enable clarification of the mutation pattern and should be applied in clinical practice in the future.
The possible reasons for identifying only heterozygous mutations in the SLC12A1 gene
In previous BS type 1 studies (Table II), including our case, 15 out of 73 (20.5%) patients harbored only one mutation as detected by sequencing all the coding exons and intron-exon boundaries. This is uncommon in autosomal recessive diseases. There are a number of potential reasons for this high percentage of patients with heterozygous mutations. Firstly, the methods used for mutation detection may not as sensitive enough. Three previous studies used single-strand conformation polymorphism (SSCP) as a mutation detection method (7,9,10). Of the patients reported in these three studies, 7 out of 22 (31.8%) patients harbored one mutation and all lacked mutations in the KCNJ1 gene (which leads to BS type 2), and was more challenging to distinguish BS type 1 from BS type 2 by the phenotype, whilst one patient had a second mutation discovered by Sanger sequencing in a subsequent study (12). The sensitivity of the SSCP method was estimated to be 75–98% (24). Therefore, some mutations may not have been detected due to limitations of the detection method. However, in the remaining cases analyzed by direct Sanger sequencing, which demonstrated a sensitivity of >99.9%, one mutation was detected in 9 out of 52 (17.3%) patients. Of these 9 patients, the second mutation could not have been a point mutation, small deletion, or small insertion, as these types of mutation are unlikely to be missed by Sanger sequencing. In addition, the inaccurate clinical diagnosis may have resulted in a negative genetic test result. Although it was difficult to distinguish the subtype of BS in some cases, the genetic analysis always included SLC12A1, KCNJ1, CLCNKB and/or BSND if necessary. Therefore, negative results were less likely due to an incorrect clinical diagnosis. Additional genes may also contribute to the phenotype. In a previous study, one patient demonstrating a BS type 4 phenotype, harbored no detectable mutation in the BSND gene that is responsible for BS type 4 (2). Upon further investigation, the patient was observed to harbor mutations in CLCNKA and CLCNKB genes, and was characterized as BS type 4B to distinguish from BS type 4A (2). Based on current evidence for BS type 1, the presence of mutations in genes other than SLC12A1 is less likely, however this may need to be considered for those patients with one or no detectable mutations in the SLC12A1 gene. Finally, mutations cannot be detected by the methods used for analysis due to the limitations of the methods. In the present case, the patient harbored a heterozygous deletion spanning exons 14 and 15. The deletion was 3.16 kb in size, which cannot be detected by Sanger sequencing. Therefore, for patients with heterozygous mutations, the presence of large deletions should be investigated.
Discussion
In the present study, one patient was suspected of having BS type 1, and their peripheral blood sample was sent to our genetics laboratory (University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA) for SLC12A1 gene sequencing analysis. The results of Sanger sequencing of the SLC12A1 gene indicated the presence of a novel homozygous frameshift mutation (c.1833delT) in exon 15 of the proband. Due to the rarity of this mutation and the lack of a consanguineous relationship between the parents, a parental study was subsequently performed to determine the origin of inheritance of this mutation and to exclude the possible presence of a heterozygous deletion. Notably, a heterozygous mutation (c.1833delT) was detected in the father but not in the mother. Three possible reasons were considered to be responsible for this phenomenon. The first possibility was the presence of a de novo mutation that has been reported in BS type 1 previously (21). However, this possibility was unlikely as the two mutations identified in the proband were homozygous. The second possibility was segmental paternal isodisomy of chromosome 15q encompassing the mutant gene. A similar situation has been reported in studies of Bloom syndrome, which is caused by a homozygous mutation due to maternal isodisomy (25). In the current study, as the proband was not suspected to exhibit Prader-Willi/Angelman syndromes, four STR markers located between 15q21.1 and 15q25.2 were selected to perform a UPD study. Genotyping of the microsatellites excluded the possibility that the apparent homozygous mutation was caused by paternal isodisomy. The final possibility was the presence of a previously unreported large deletion encompassing the mutant region on the maternal allele. In this case, the deleted region should at least include exon 15. Using cDNA to identify the deletion was not practical, as SLC12A1 mRNA expression is kidney-specific. To investigate the deletion in the SLC12A1 gene, parallel sequencing using Ion PGM was performed on the proband and the mother. It was revealed that the proband harbored compound heterozygous mutations, a large deletion on the maternal allele and a frameshift mutation of F611Lfs*32 on the paternal allele. The deletion was confirmed by long-range PCR and determined as c.1685-726_1941+239del by sequencing of the junction fragment.
Sanger sequencing remains as the most popular method for the detection of mutations in the SLC12A1 gene. The technique has provided the most accurate results for mutations including substitutions, small deletions and insertions, however, it is unable to detect large deletions. Therefore, in the present case, when the large deletion encompasses the mutant site, the Sanger sequencing result demonstrated a false homozygous mutation. Following the genotyping study, a deletion was suspected, therefore Ion PGM target sequencing was used to exclude this possible deletion. The results of Ion PGM sequencing indicated the presence of a large deletion encompassing exons 14 and 15 in the proband and the mother. To confirm this finding, array CGH and long-range PCR were performed, which further confirmed the presence of the deletion in the proband and his mother. By sequencing the junction fragment, the large deletion was characterized as c.1685-726_1941+239del. Although this is a single case that presented a false homozygous mutation result as determined by Sanger sequencing, in previously reported cases of BS type 1 (Table II), approximately 45% of patients were observed to harbor homozygous mutations and 21% of patients harbored only heterozygous mutations. The reasons underlying this phenomenon remain unclear. Large deletions may serve an important role in BS type 1, however, due to insufficient data, no conclusion could be reached. Further deletion analysis of patients with BS type 1 may enable clarification of this phenomenon.
In conclusion, the present study was the first to identify a large deletion encompassing exons 14 and 15 in a patient with BS type 1, which was not detected by Sanger sequencing. In addition to the previously reported types of mutations including missense, nonsense, splicing, frameshift, small deletions and small insertions, the results of the current study suggest that large deletions encompassing the whole exon(s) may serve an etiologic role in BS type 1. These types of mutations may therefore be more common than originally thought, due to the fact that 21% of reported cases with only one mutation have been observed.
Acknowledgements
The authors were grateful to the family for participating in this study and to the University of Oklahoma Health Sciences Center for supporting this study.
References
- 1.Kliegman R, Nelson WE. Nelson textbook of pediatrics. Elsevier/Saunders; Philadelphia, PA: 2011. [DOI] [Google Scholar]
- 2.Nozu K, Inagaki T, Fu XJ, Nozu Y, Kaito H, Kanda K, Sekine T, Igarashi T, Nakanishi K, Yoshikawa N, et al. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J Med Genet. 2008;45:182–186. doi: 10.1136/jmg.2007.052944. [DOI] [PubMed] [Google Scholar]
- 3.Vezzoli G, Arcidiacono T, Paloschi V, Terranegra A, Biasion R, Weber G, Mora S, Syren ML, Coviello D, Cusi D, et al. Autosomal dominant hypocalcemia with mild type 5 Bartter syndrome. J Nephrol. 2006;19:525–528. [PubMed] [Google Scholar]
- 4.Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol. 2008;4:560–567. doi: 10.1038/ncpneph0912. [DOI] [PubMed] [Google Scholar]
- 5.Bhat YR, Vinayaka G, Sreelakshmi K. Antenatal bartter syndrome: A review. Int J Pediatr. 2012;2012:857136. doi: 10.1155/2012/857136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madrigal G, Saborio P, Mora F, Rincon G, Guay-Woodford LM. Bartter syndrome in Costa Rica: A description of 20 cases. Pediatr Nephrol. 1997;11:296–301. doi: 10.1007/s004670050280. [DOI] [PubMed] [Google Scholar]
- 7.Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet. 1996;13:183–188. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 8.Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurtz CL, Karolyi L, Seyberth HW, Koch MC, Vargas R, Feldmann D, Vollmer M, Knoers NV, Madrigal G, Guay-Woodford LM. A common NKCC2 mutation in Costa Rican Bartter's syndrome patients: Evidence for a founder effect. J Am Soc Nephrol. 1997;8:1706–1711. doi: 10.1681/ASN.V8111706. [DOI] [PubMed] [Google Scholar]
- 10.Vargas-Poussou R, Feldmann D, Vollmer M, Konrad M, Kelly L, van den Heuvel LP, Tebourbi L, Brandis M, Karolyi L, Hebert SC, et al. Novel molecular variants of the Na-K-2Cl cotransporter gene are responsible for antenatal Bartter syndrome. Am J Hum Genet. 1998;62:1332–1340. doi: 10.1086/301872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brochard K, Boyer O, Blanchard A, Loirat C, Niaudet P, Macher MA, Deschenes G, Bensman A, Decramer S, Cochat P, et al. Phenotype-genotype correlation in antenatal and neonatal variants of Bartter syndrome. Nephrol Dial Transplant. 2009;24:1455–1464. doi: 10.1093/ndt/gfn689. [DOI] [PubMed] [Google Scholar]
- 12.Puricelli E, Bettinelli A, Borsa N, Sironi F, Mattiello C, Tammaro F, Tedeschi S, Bianchetti MG. Italian Collaborative Group for Bartter Syndrome: Long-term follow-up of patients with Bartter syndrome type I and II. Nephrol Dial Transplant. 2010;25:2976–2981. doi: 10.1093/ndt/gfq119. [DOI] [PubMed] [Google Scholar]
- 13.Colussi G, Bettinelli A, Tedeschi S, De Ferrari ME, Syrén ML, Borsa N, Mattiello C, Casari G, Bianchetti MG. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol. 2007;2:454–460. doi: 10.2215/CJN.02950906. [DOI] [PubMed] [Google Scholar]
- 14.Pressler CA, Heinzinger J, Jeck N, Waldegger P, Pechmann U, Reinalter S, Konrad M, Beetz R, Seyberth HW, Waldegger S. Late-onset manifestation of antenatal Bartter syndrome as a result of residual function of the mutated renal Na+-K+-2Cl- co-transporter. J Am Soc Nephrol. 2006;17:2136–2142. doi: 10.1681/ASN.2005101071. [DOI] [PubMed] [Google Scholar]
- 15.Urbanova M, Reiterová J, Stĕkrová J, Lnĕnická P, Ryšavá R. DNA analysis of renal electrolyte transporter genes among patients suffering from Bartter and Gitelman syndromes: Summary of mutation screening. Folia Biol (Praha) 2011;57:65–73. [PubMed] [Google Scholar]
- 16.Yamazaki H, Nozu K, Narita I, Nagata M, Nozu Y, Fu XJ, Matsuo M, Iijima K, Gejyo F. Atypical phenotype of type I Bartter syndrome accompanied by focal segmental glomerulosclerosis. Pediatr Nephrol. 2009;24:415–418. doi: 10.1007/s00467-008-0999-3. [DOI] [PubMed] [Google Scholar]
- 17.Lee EH, Heo JS, Lee HK, Han KH, Kang HG, Ha IS, Choi Y, Cheong HI. A case of Bartter syndrome type I with atypical presentations. Korean J Pediatr. 2010;53:809–813. doi: 10.3345/kjp.2010.53.8.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nozu K, Iijima K, Kawai K, Nozu Y, Nishida A, Takeshima Y, Fu XJ, Hashimura Y, Kaito H, Nakanishi K, et al. In vivo and in vitro splicing assay of SLC12A1 in an antenatal salt-losing tubulopathy patient with an intronic mutation. Hum Genet. 2009;126:533–538. doi: 10.1007/s00439-009-0697-7. [DOI] [PubMed] [Google Scholar]
- 19.Nozu K, Iijima K, Kanda K, Nakanishi K, Yoshikawa N, Satomura K, Kaito H, Hashimura Y, Ninchoji T, Komatsu H, et al. The pharmacological characteristics of molecular-based inherited salt-losing tubulopathies. J Clin Endocrinol Metab. 2010;95:E511–E518. doi: 10.1210/jc.2010-0392. [DOI] [PubMed] [Google Scholar]
- 20.Fukuyama S, Okudaira S, Yamazato S, Yamazato M, Ohta T. Analysis of renal tubular electrolyte transporter genes in seven patients with hypokalemic metabolic alkalosis. Kidney Int. 2003;64:808–816. doi: 10.1046/j.1523-1755.2003.00163.x. [DOI] [PubMed] [Google Scholar]
- 21.Adachi M, Asakura Y, Sato Y, Tajima T, Nakajima T, Yamamoto T, Fujieda K. Novel SLC12A1 (NKCC2) mutations in two families with Bartter syndrome type 1. Endocr J. 2007;54:1003–1007. doi: 10.1507/endocrj.K06-204. [DOI] [PubMed] [Google Scholar]
- 22.Faiz F, Allcock RJ, Hooper AJ, van Bockxmeer FM. Detection of variations and identifying genomic breakpoints for large deletions in the LDLR by Ion Torrent semiconductor sequencing. Atherosclerosis. 2013;230:249–255. doi: 10.1016/j.atherosclerosis.2013.07.050. [DOI] [PubMed] [Google Scholar]
- 23.O'Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 24.Scoggan KA, Bulman DE. Single-strand conformational polymorphism analysis (SSCP) and sequencing for ion channel gene mutations. Methods Mol Biol. 2003;217:143–151. doi: 10.1385/1-59259-330-5:143. [DOI] [PubMed] [Google Scholar]
- 25.Woodage T, Prasad M, Dixon JW, Selby RE, Romain DR, Columbano-Green LM, Graham D, Rogan PK, Seip JR, Smith A, et al. Bloom syndrome and maternal uniparental disomy for chromosome 15. Am J Hum Genet. 1994;55:74–80. [PMC free article] [PubMed] [Google Scholar]