

Abstract
Methamphetamine (MA) abuse is a major public health and safety concern throughout the world and a growing burden on healthcare costs. The purpose of the present study was to investigate the protective effect of fluoxetine against MA-induced chronic pulmonary inflammation and to evaluate the potential role of nuclear factor erythroid 2-related factor 2 (Nrf2)-mediated antioxidative stress. Wistar rats were divided into control, MA and two fluoxetine-treated groups. Rats in the MA and the two fluoxetine-treated groups were treated daily with intraperitoneal injection of 10 mg/kg MA twice daily. Rats in the two fluoxetine-treated groups were injected intragastrically with fluoxetine (2 and 10 mg/kg) once daily, respectively. After 5 weeks, the rats were euthanized and hematoxylin and eosin staining, immunohistochemistry, western blot analysis and redox assay were performed. It was demonstrated that chronic exposure to MA can induce pulmonary inflammation in rats, with the symptoms of inflammatory cell infiltration, crowded lung parenchyma, thickened septum and a reduced number of alveolar sacs. Fluoxetine attenuated pulmonary inflammation and the expression of interleukin-6 and tumor necrosis factor-α in rat lungs. Fluoxetine inhibited MA-induced increases in the expression levels of serotonin transporter (SERT) and p-p38 mitogen-activated protein kinase (MAPK), and reversed the MA-induced decrease in nuclear Nrf2 and human heme oxygenase-1 in lungs. Fluoxetine at 10 mg/kg significantly reversed the reduced glutathione (GSH) level, the ratio of GSH/oxidized glutathione, and the reactive oxygen species level in rat lungs from the MA group. These findings suggested that fluoxetine, a SERT inhibitor, has a protective effect against MA-induced lung inflammation by suppressing oxidative stress through the SERT/p38 MAPK/Nrf2 pathway in rats.
Keywords: methamphetamine, fluoxetine, serotonin transporter, nuclear factor erythroid 2-related factor 2, human heme oxygenase-1, reduced glutathione, serotonin, oxidative stress, inflammation
Introduction
Methamphetamine (MA) abuse is a growing health problem worldwide, and there has been an increase in the number of medical complications and fatalities associated with the toxicity of MA (1,2). A previous study (3) reported that the widest distribution and the highest uptake of MA in the human body occurred in the lungs, and that this may render the lungs vulnerable to infection, pulmonary hypertension and pulmonary edema (3,4). The long-lasting pulmonary toxic effects of MA have placed an increased burden on healthcare costs (5). Therefore, it is particularly important to investigate the mechanism of pulmonary toxicity for drug targets. A previous study (6) demonstrated that serotonin is possibly associated with MA-induced pulmonary toxicity. Pulmonary toxicity is pathologically characterized by parenchymal damage, recruitment of inflammatory cells and a progression of the inflammatory processes (7). Inflammatory changes in lung tissue are the key to the pulmonary toxicity. However, problems remain about the exact pathogenesis of MA-induced chronic pulmonary inflammation through serotonin.
The neurotransmitter serotonin (5-hydroxytryptamine; 5-HT) is implicated in increasing inflammatory reactions of the skin, lungs and gastrointestinal tract (8). An etiological agent of chronic inflammation is deregulation of the tissue macrophage polarization balance (9). The modulation of the phenotypic and functional polarization of macrophages is by 5-HT, which regulates the inflammation and the tissue restoration via a large set of receptors (5-HTR1-7) and/or a transporter (9). Lung macrophages express the serotonin transporter (SERT) and 5-HT receptors 2a, and 2b (10). A previous study (11) reported that the inhibition of 5-HTR2a and 5-HTR2b had no effect on efferocytosis, although inhibiting SERT prevented 5-HT-impaired efferocytosis. A previous epidemiological study (12) suggested that MA abuse significantly increased the risk of developing pulmonary arterial hypertension (PAH). The disease severity and susceptibility to PAH may be associated with the increased SERT activity (13). Pulmonary vascular remodeling and pulmonary inflammation in pulmonary hypertension were associated with a SERT-induced rapid activation of extracellular signal-regulated kinase (ERK) 1/2 (14,15). The phosphorylation and subsequent activation of ERK, p38 mitogen activated protein kinases (p38 MAPK) and Akt, and the production of reactive oxygen species (ROS) appeared to be a common mechanism of proliferation and inflammation (16,17). A previous study (18) reported that MAPK signaling is involved in the regulation of nuclear factor erythroid 2-related factor 2 (Nrf2).
Nrf2 is a basic leucine zipper redox-sensitive transcriptional factor that serves a central role in the transcriptional regulation of antioxidant and/or detoxifying genes (19). Nuclear localization of Nrf2 activation efficiently protects cells from ROS-induced damage in vivo and in vitro by inducing the expression of numerous detoxifying enzymes and antioxidant proteins (20). As Nrf2 is a transcription factor with potent antioxidant effects against cell death caused by ROS-induced damage, targeting Nrf2 may serve an essential role in the protection against various inflammatory diseases (20). However, the role of Nrf2 in MA-induced pulmonary inflammation and the protective mechanism of fluoxetine against MA-induced oxidative stress and pulmonary inflammation remain to be elucidated. Therefore, the present study was designed to further evaluate the potential role of Nrf2 and to investigate if fluoxetine can ameliorate MA-induced oxidative stress and pulmonary inflammation throughthep38 MAPK/Nrfr2 pathway in rats.
Materials and methods
Drugs
MA was obtained from the China Criminal Police University (Shenyang, Liaoning, China). The purity of the MA was identified as 97% by a Bio-Rad REMEDi HS system (Bio-Rad, Milan, Italy) and by liquid chromatography-mass spectrometry-mass spectrometry (Shimadzu Corporation, Kyoto, Japan). MA was dissolved in 0.9% sterile saline and prepared as 4 mg/ml for drug administration.
Animal and experimental paradigm
A total of 30 male Wistar rats (180±10 g) were obtained from the Animal Resource Center, China Medical University (Shenyang, Liaoning, China; certificate number: Liaoning 034) and divided into four groups: i) Control; ii) MA; iii) MA plus fluoxetine 2 mg/kg (MA+F2); and iv) MA plus fluoxetine 10 mg/kg (MA+F10). Rats in the MA and the two fluoxetine-treated groups were also treated daily with intraperitoneal injection of 10 mg/kg MA (China Criminal Police University) twice daily for 5 weeks. Rats in the control group received the respective vehicles only. Rats in the MA+F2 and MA+F10 groups were treated with intragastric fluoxetine (Cadila Pharmaceuticals, Ankleshwar, India) at 2 or 10 mg/kg once daily for 5 weeks, respectively. All the rats were housed in a controlled humidity (50–70%) and temperature (18–22°C), and were given access to food and water ad libitum in an alternating 12 h light/dark cycle over a period of 5 weeks. All experimental protocols for the present study were approved by the Institutional Animal Care and Use Committee of China Medical University.
Morphological analysis by hematoxylin and eosin (H&E) staining
A total of 3% sodium pentobarbital (45 mg/kg) was used to euthanize the rats. The right lower lung tissues were dissected, fixed with paraformaldehyde and embedded in paraffin wax. Sections (5 µm) were stained with H&E for observation and analysis under light microscopy. The inflammatory changes of rat lung parenchyma were evaluated by the thickness of alveolar septum and the destructive index (DI; three randomly selected sites were analyzed in each section; magnification, ×200).
DI, a measure of alveolar septal damage and emphysema, has been proposed as a sensitive index of lung destruction that closely reflects functional abnormalities (21). DI represents the percentage of destroyed space as a fraction of the total alveolar and duct space (21). The quantification of this destruction can add greatly to the microscopic analysis of changes due to pulmonary inflammation.
Immunohistochemistry
After processing the tissue and embedding in paraffin wax, 5 µm thick sections were stained by immunohistochemical (IHC) procedures using Ultrasensitive TM SP kit (Maxin-Bio Co., Fuzhou, China) and DAB Staining kit (Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China). IHC staining followed a basic indirect protocol using a citrate antigen retrieval method. A primary rabbit anti-interleukin-6 (IL-6; cat. no. bs 0379R, Beijing Biosynthesis Biotechnology Co., Ltd., Beijing, China) was diluted at 1:50 and incubated overnight at 4°C. For the negative control, the primary antibody was replaced by 0.01 M PBS in the incubation step. A biotin-labeled secondary antibody from SP kit (cat. no. KIT-9706; Maxin-Bio Co.) was incubated for 10 min at room temperature to detect the primary antibody.
The positive expression of IL-6 was examined using light microscopy. At least six visual fields of lung tissue were examined on each slide. For the convenience of understanding and statistical processes, the quantity of protein in the rat lung was analyzed and calculated as optical density average by microscope with a digital camera and MetaMorph software version 7.7 (Molecular Devices, LLC, Sunnyvale, CA, USA).
Western blot analysis
Nuclear and cytoplasmic fractions were extracted using a Nuclear and Cytoplasmic Protein Extraction kit (Beyotime Institute of Biotechnology, Shanghai, China). The protein concentrations were determined using a BCA protein assay kit (Beyotime Institute of Biotechnology) prior to storage at −80°C. Electrophoresis was performed with 10% SDS-polyacrylamide gel using 80 µg total protein in each lane. Following electrophoresis, the protein was transferred onto a PVDF membrane using a semi-dry transfer unit (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and the membranes were incubated for 1 h in blocking buffer (5% non-fat dry milk, PBS and 0.1% Tween). The membranes were probed with primary mouse monoclonal anti-β-actin (1:2,000; cat. no. sc-130300; Santa Cruz Biotechnology, Inc., Dallas, USA) and α-tubulin 1:2,000; cat. no. 66031; ProteinTech Group, Inc., Chicago, IL, USA) and rabbit polyclonal anti-SERT (1:200; cat. no. bs 1893R), IL-6 (1:200; cat. no. bs 0379R), tumor necrosis factor-α (TNF-α; 1:200; cat. no. bs 0078R), human heme oxygenase-1 (HO-1; 1:400; cat. no. bs 2075R) (Biosynthesis Biotechnology Co. Ltd.), anti-Nrf2 (1:600; cat. no. 16396-1-AP; ProteinTech Group, Inc.), p38 (1:600; cat. no. ZS-7149), and phosphorylated (p)-p38 (1:600; cat. no. ZS-101759; Zhongshan Golden Bridge Biotechnology Co., Ltd.) overnight at 4°C. Following washing with PBS-0.1% Tween, the membranes were incubated in the presence of goat anti-mouse secondary antibody (cat. no. ZB-2305; Zhongshan Golden Bridge Biotechnology Co., Ltd.) for β-actin and α-tubulin at a dilution of 1:4,000 for 2 h, and goat anti-rabbit secondary antibody (cat. no. SA00001-2; ProteinTech Group, Inc.) for other proteins at a dilution of 1:2,000 for 2 h at room temperature, followed by enhanced chemiluminescence (Pierce™ ECL Western Blotting Substrate; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The relative protein expression was quantified by densitometry using Molecular Dynamics Image Quant software (GE Healthcare Life Sciences, Chalfont, UK). The result of the expression of nuclear Nrf2 was represented by the relative yield against α-tubulin, and the other protein expression levels were represented by the relative yield against β-actin.
Reduced glutathione (GSH) and oxidized glutathione (GSSG) assay
Lung tissues were homogenized with 10 ml ice-cold lysis buffer (50 mM phosphate buffer containing 1 mM EDTA/g). Following centrifugation at 10,000 × g for 15 min at 4°C, the supernatant was removed, deproteinated and stored at −20°C until further analyses. Total glutathione and oxidized glutathione levels were determined using a GSH and GSSG Assay kit (Beyotime Institute of Biotechnology), according to the manufacturer's protocol.
ELISA
Samples of the rat lung tissue in each group were homogenized using a Polytron homogenizer (Kinematical, Lucerne, Switzerland) to extract protein. The homogenate was centrifuged at 15,000 × g for 30 min at 4°C and the supernatant was collected and stored at −80°C for ELISA. ROS concentration in the lung tissues was measured by the rat ROS ELISA kits (R&D Systems, Inc., Minneapolis, MN, USA), according to the manufacturer's protocol. The absorbance was measured at 450 nm and the corresponding concentration was determined from the standard curve.
Statistical analysis
All data are presented as the mean ± standard deviation. Statistical analysis was performed by one-way analysis of variance with SPSS software, version 22.0 (IBM SPSS, Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of fluoxetine on the inflammatory changes induced by MA in rat lungs
Representative H&E sections from the different groups indicated that compared with the control group (Fig. 1A) lung injury was markedly induced by 10 mg/kg MA (Fig. 1B) and dose-dependently attenuated by fluoxetine (Fig. 1C and D).
Figure 1.
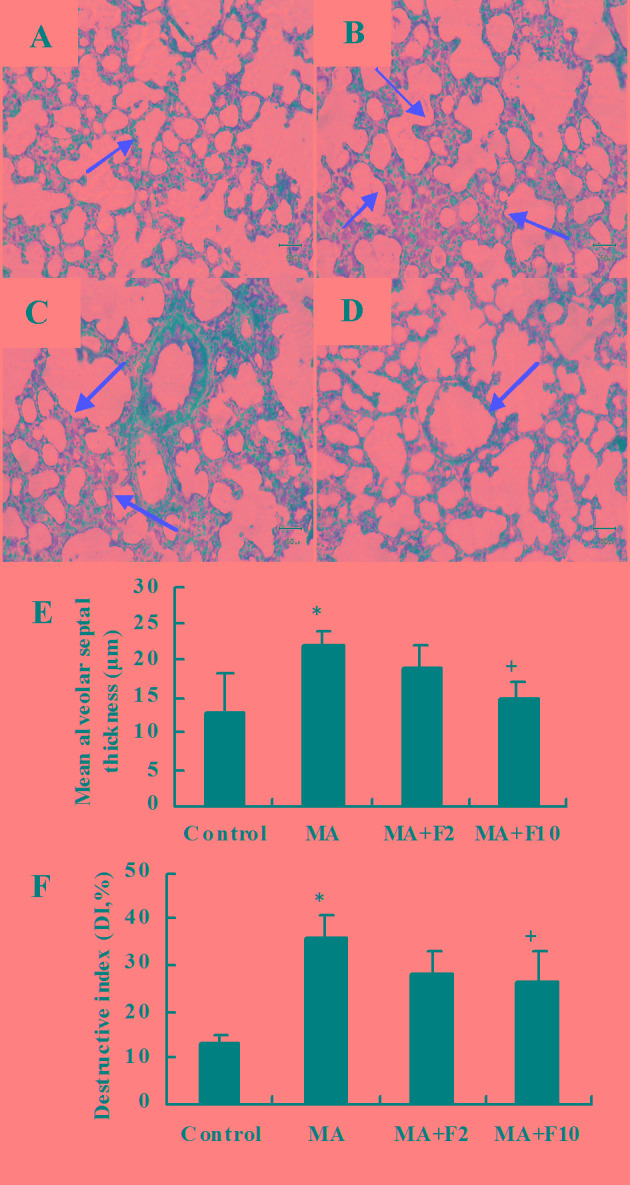
Effect of fluoxetine on the inflammatory changes induced by MA in rat lungs. (A) Control group, (B) MA group, (C) MA+F2 group and (D) MA+F10 group (magnification, ×200). The changes in alveolar septum thickness were marked by red arrows. Compared with (A) the control group, in the (B) MA group the inflammatory injury was apparent in the rat lungs; inflammatory cell infiltration, alveoli fusion, a reduced number of alveolar sacs and an increased thickness of the alveolar septum. (C and D) All inflammatory changes were dose-dependently attenuated by fluoxetine. (E) The mean alveolar septal thickness and (F) destructive index (%) were determined. The data are expressed as the mean ± standard deviation (n=6; *P<0.05 vs. the control group; +P<0.05 vs. the MA group). MA, methamphetamine; F2, fluoxetine at 2 mg/kg; F10, fluoxetine at 10 mg/kg.
Under the microscope, the rat lung tissue was infiltrated by inflammatory cells in the MA group, the lung parenchyma was more compact and the septum thickened (P=0.003, MA vs. control; Fig. 1E). The DI was significantly increased in the MA group compared with the control group (P=0.007, MA vs. control; Fig. 1F). The inflammatory damage to the lung tissues was partly ameliorated following the administration of fluoxetine at 10 mg/kg (P=0.028 vs. MA).
Immunohistochemical analysis of IL-6 protein expression in pulmonary arteries
Sections were stained with rabbit anti-IL-6 (brown) and counterstained with hematoxylin (blue) in the different groups. Compared with control group, IL-6 protein expression in the lungs in the rats of the MA group was significantly increased (P=0.003), and administration of fluoxetine dose-dependently decreased IL-6 protein expression. IL-6 expression in the rats of MA+F10 group was significantly decreased, compared with the MA group (P=0.0174; Fig. 2).
Figure 2.

IL-6 protein expression illustrated by immunohistochemical analysis. (A) Control group, (B) MA group, (C) MA+F2 group, (D) MA+F10 group (magnification, ×400) and (E) IL-6 (optical density). Tissue sections (5 µm) from the different groups were stained with rabbit anti-IL-6 (brown) and counterstained with hematoxylin (blue). The data are expressed as the mean ± standard deviation (n=6; *P<0.05, **P<0.01 vs. the control group; +P<0.05 vs. the MA group). IL-6, interleukin-6; MA, methamphetamine; F2, fluoxetine at 2 mg/kg; F10, fluoxetine at 10 mg/kg.
Western blot analysis of IL-6 and TNF-α expression in rat lungs
Results from western blot analysis demonstrated that compared with the control group, IL-6 protein expression was significantly increased in the MA and MA+F2 groups (0.84±0.20 vs. 0.40±0.10, 0.67±0.10 vs. 0.40±0.10; P=0.004). IL-6 expression was decreased following administration of fluoxetine at 10 mg/kg. Compared with the MA group, relative IL-6 protein level was significantly decreased in the MA+F10 group (0.43±0.10 vs. 0.84±0.20, P=0.006; Fig. 3A).
Figure 3.
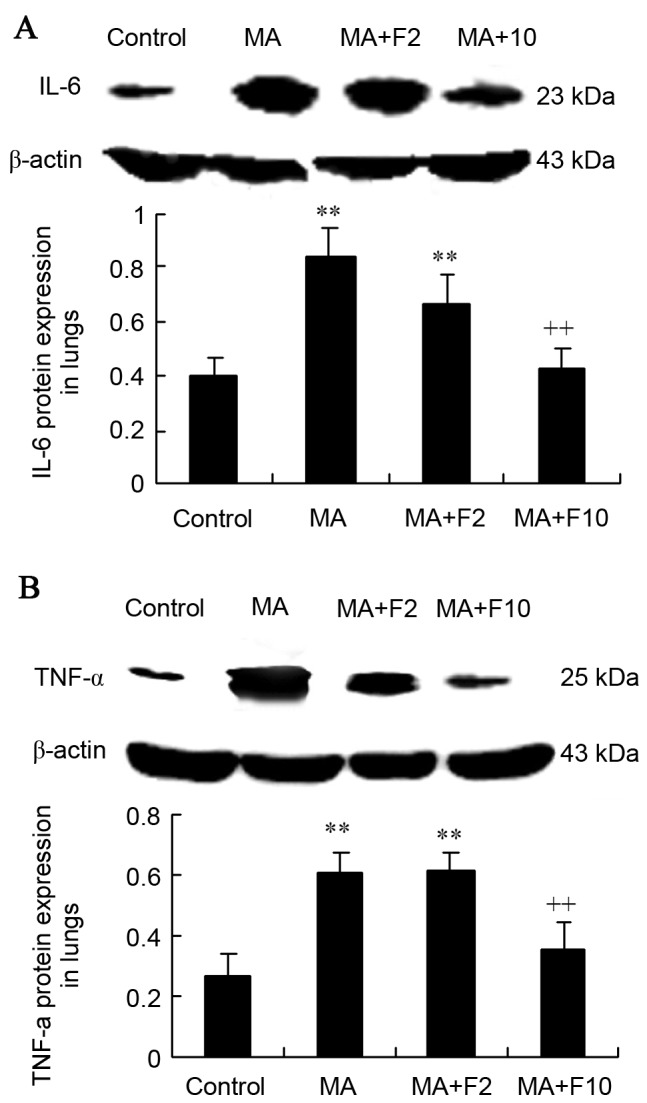
IL-6 and TNF-α protein expression in rat lungs in the different groups. (A) IL-6 and (B) TNF-α protein expression in lungs was assessed and quantified. The data are expressed as the mean ± standard deviation (n=6; **P<0.01 vs. the control group; ++P<0.01 vs. the MA group). IL-6, interleukin-6; MA, methamphetamine; TNF-α, tumor necrosis factor-α; F2, fluoxetine at 2 mg/kg; F10, fluoxetine at 10 mg/kg.
Western blot analysis was performed to further demonstrate that TNF-α protein expression was significantly increased in the MA group compared with the control group (0.60±0.07 vs. 0.26±0.08; P=0.003). Compared with the MA group, relative TNF-α protein expression in the lungs was not markedly changed in the MA+F2 group, but was significantly decreased in the MA+F10 group (0.35±0.09 vs. 0.60±0.07, P=0.032; Fig. 3B).
SERT and p38 MAPK protein expression in lungs
Results from western blot analysis also demonstrated that SERT expression was significantly upregulated in lungs in the MA and MA+F2 groups, compared with the control group (0.89±0.15 vs. 0.34±0.08, P=0.005; 0.76±0.20 vs. 0.34±0.08, P=0.017), and that it was downregulated in the MA+F10 group (0.57±0.14 vs. 0.89±0.18, P=0.034), compared with the MA group (Fig. 4A).
Figure 4.
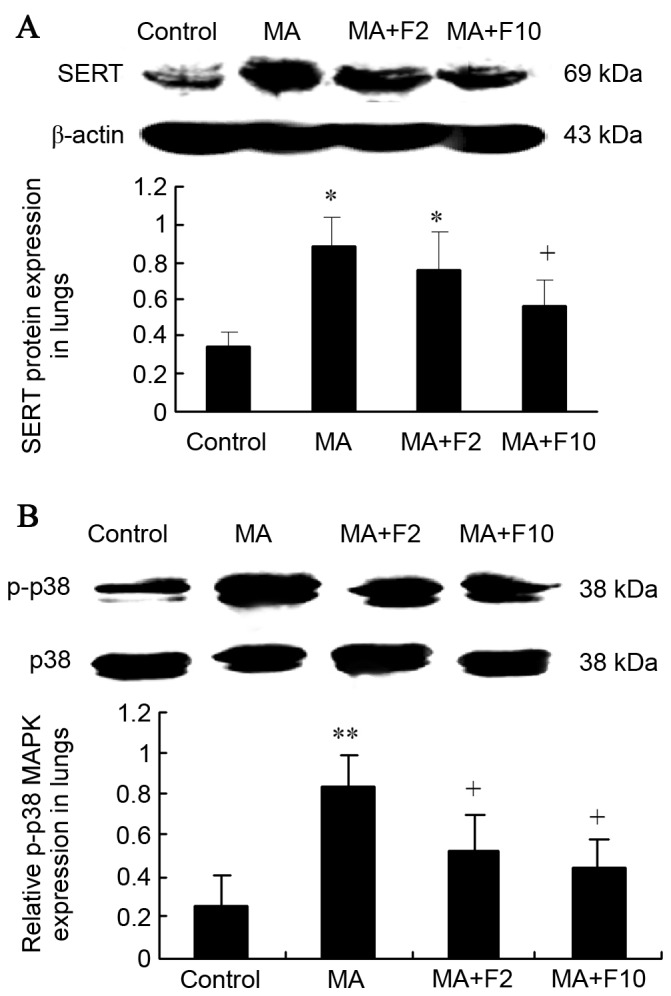
SERT protein expression and P-38 MAPK phosphorylation in rat lungs in the different groups. (A) SERT protein expression and (B) the relative p-p38 MAPK expression in lungs were determined and quantified. The data are expressed as the mean ± standard deviation (n=6; *P<0.05, **P<0.01 vs. the control group; +P<0.05 vs. the MA group). SERT, serotonin transporter; MAPK, mitogen activated protein kinases; MA, methamphetamine; F2, fluoxetine at 2 mg/kg; F10, fluoxetine at 10 mg/kg.
Western blot analysis was performed to further demonstrate that in the M10 group p38 MAPK was phosphorylated compared with the control group (0.84±0.14 vs. 0.25±0.06; P=0.003). In addition, p-p38 MAPK expression was decreased following administration of fluoxetine. Compared with the M10 group, the relative p-p38 MAPK protein level in lungs was markedly decreased in the M10+F2 group (0.52±0.18 vs. 0.84±0.14; P=0.034) and in the M10+F10 group (0.44±0.13 vs. 0.84±0.14, P=0.028; Fig. 4B).
Nrf2/HO-1 expression in the lungs from the different groups
Results from western blot analysis also demonstrated that nuclear Nrf2 expression was significantly decreased in the lungs in the MA and MA+F2 groups, compared with the control group (0.21±0.11 vs. 0.61±0.12, 0.22±0.09 vs. 0.61±0.12, respectively; P=0.004) and that it was increased in the MA+F10 group (0.48±0.19 vs. 0.21±0.0.11, P=0.032), compared with the MA group (Fig. 5A).
Figure 5.
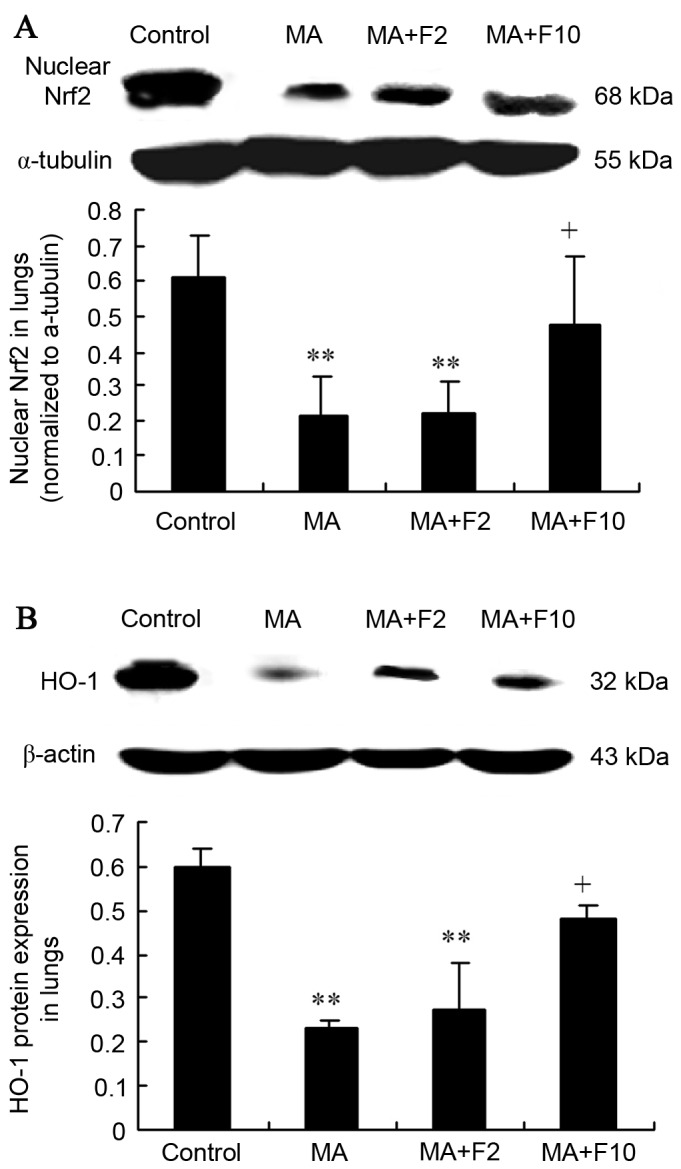
Nrf2 and HO-1 expression in lungs in different groups. (A) Nuclear Nrf2 in lungs was determined and quantified. (B) HO-1 protein expression in lungs was determined and quantified. The data are expressed as the mean ± standard deviation (n=6; **P<0.01 vs. the control group; +P<0.05 vs. the MA group). Nrf2, nuclear factor erythroid 2-related factor 2; HO-1, human heme oxygenase-1; MA, methamphetamine; F2, fluoxetine at 2 mg/kg; F10, fluoxetine at 10 mg/kg.
Western blot analysis further demonstrated that the HO-1 expression was significantly downregulated in the lungs of the MA and MA+F2 groups, compared with the control group (0.23±0.02 vs. 0.60±0.04, P=0.004; 0.27±0.11 vs. 0.60±0.04; P=0.005). However, it was markedly upregulated in the MA+F10 group, compared with the MA group (0.48±0.03 vs. 0.23±0.02, P=0.036; Fig. 5B).
Effect of fluoxetine and MA on GSH and GSSG in lungs
In the present study, MA markedly downregulated the GSH level and upregulated the GSSG level in rat lungs, compared with the control group (2.2±0.7 vs. 4.1±1.0, P=0.035; 0.5±0.18 vs. 0.2±0.1, P=0.027; Fig. 6A and B). However, both were markedly reversed in the MA+F10 group compared with the MA group.
Figure 6.
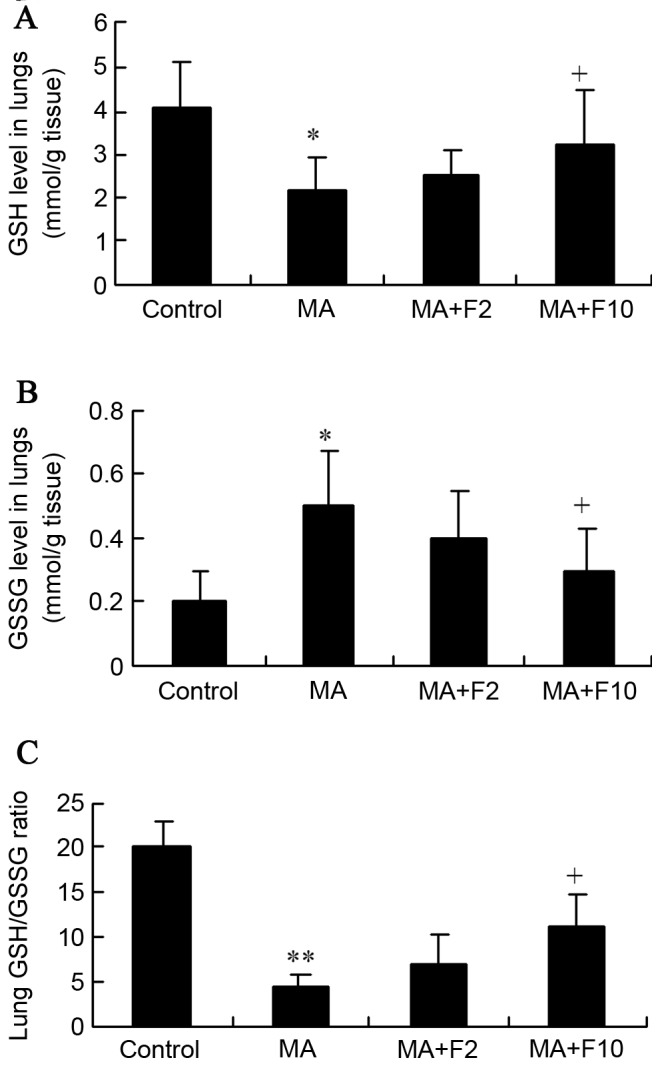
Effect of fluoxetine and MA on GSH and GSSG in lungs. (A) GSH and (B) GSSG levels in the lungs were assessed (mM/g tissue). (C) The lung GSH/GSSG ratio was determined. The data are expressed as the mean ± standard deviation (n=6; *P<0.05, **P<0.01 vs. the control group; +P<0.05 vs. the MA group). MA, methamphetamine; GSH, reduced glutathione; GSSG, oxidized glutathione; F2, fluoxetine at 2 mg/kg; F10, fluoxetine at 10 mg/kg.
Together with the decreased GSH level, the GSH/GSSG ratios were significantly reduced by MA, as is presented in Fig. 6C (4.4±1.4 vs. 20±3.0, P=0.001). However, the ratio was markedly reversed from 4.4±1.4 in the MA group to 11±3.8 in the MA+F10 group. These findings supported the hypothesis that the protective effect of fluoxetine was possibly performed by suppressing the Nrf2-mediated antioxidative stress in the lungs.
Effect of fluoxetine and MA on lung ROS level
The results of the ELISA analysis demonstrated that ROS in the lungs were at a low level in the control group, (control: 1,608.4±364.6 U/mg protein). However, in the MA and MA+F2 groups, lung ROS levels were markedly enhanced to 4,938.8±265.1 and 4,256.2±470.1 U/mg protein (P<0.0001, vs. control). The ROS level in lungs was significantly decreased in the MA+F10 group (2,271.4±334.5 vs. 4,938.8±265.1 U/mg protein, P=0.015 vs. MCT; Fig. 7).
Figure 7.
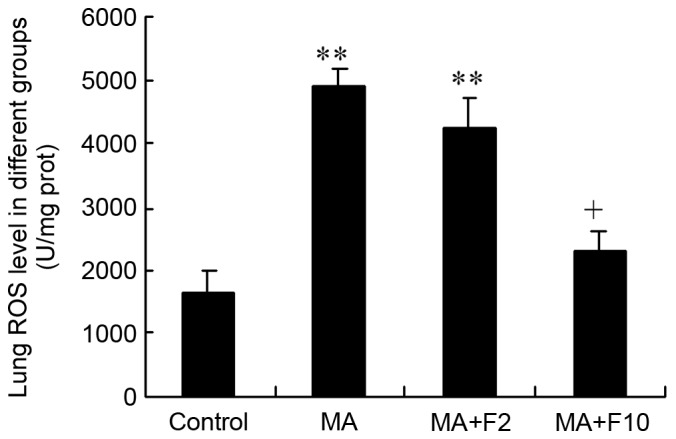
Lung ROS level in different groups (U/mg protein). The data are expressed as the mean ± standard deviation (n=6; **P<0.01 vs. the control group; +P<0.05 vs. the MA group). ROS, reactive oxygen species; MA, methamphetamine; F2, fluoxetine at 2 mg/kg; F10, fluoxetine at 10 mg/kg.
Discussion
Results from the present study demonstrated that chronic use of MA caused rat pulmonary inflammation; inflammatory cell infiltration, crowded lung parenchyma, thickened septum and increased DI. Fluoxetine attenuated the inflammatory changes and the expression of the inflammatory factors IL-6 and TNF-α in rat lungs. Fluoxetine also inhibited MA-induced increases in the expression of SERT and p-p38 MAPK, and reversed the MA-induced decrease in nuclear Nrf2 and HO-1 in lungs. Additionally, fluoxetine at 10 mg/kg significantly reversed the increases in GSH level, GSH/GSSG ratio and the ROS level in rat lungs from the MA group. These findings suggested that fluoxetine, a SERT inhibitor, has a protective effect against MA-induced lung inflammation by suppressing oxidative stress through the SERT/p38 MAPK/Nrf2 pathway in rats.
Pulmonary toxicity is pathologically characterized by parenchymal damage, the recruitment of inflammatory cells and the progression of the inflammatory process (7). Inflammatory changes in lung tissue are the key to pulmonary toxicity. Endogenous IL-6 serves a critical role in the inflammatory response to injury in the lungs of mice (22). TNF-α is involved in lung matrix fragmentation, macrophage activation and endothelial cell apoptosis (23). Thus, both IL-6 and TNF-α are the classical markers of pulmonary inflammation (24). It was demonstrated in the present study that the expression of IL-6 protein was significantly increased in MA-induced lung inflammation in rats, that fluoxetine dose-dependently inhibited the expression of IL-6 and TNF-α, and that fluoxetine significantly attenuated the inflammatory cell infiltration and reduced the inflammation of the alveoli with thickened septum; this all suggested the protective effect of fluoxetine on MA-induced lung inflammation.
Fluoxetine inhibited the SERT-mediated reuptake of cytoplasmic 5-HT in the lungs (25). 5-HT, released from enterochromaffin cells, can act on various innate and adaptive immune cells, and, under inflammatory conditions, increased 5-HT production can promote local inflammation (26). In a previous study (6), it was identified that MA induced the increasedconcentration of 5-HT, which indicated that the serotonin mechanism is involved in MA-induced pulmonary toxicity. MA, as a substrate for SERT, was transported into cells and subsequently inhibited the metabolism of 5-HT by inhibiting monoamine oxidase A (6,27). The activation of SERT can promote 5-HT-impaired efferocytosis (11). SERT is the predominant protein responsible for the uptake and release of serotonin by transporting serotonin in either direction (28). Accordingly, SERT contributes to the modulation of extracellular and intracellular 5-HT concentration (29). In the present study, it was demonstrated that fluoxetine at 10 mg/kg attenuated MA-induced pulmonary inflammation and inhibited MA-induced upregulation of SERT expression in the lung. These findings suggested that SERT may serve an important role in MA-induced pulmonary inflammation.
SERT-induced rapid activation of MAPK is associated with 5-HT-induced pulmonary inflammation (30). The p38 MAPK signaling pathways are involved in the regulation of the Nrf2-mediated cytoprotective effect (31). The present study demonstrated that MA induced the upregulation of SERT and the phosphorylation of p38 MAPK in the lungs. In addition, MA inhibited the expression of Nrf2 in nucleoprotein. Therefore, it is hypothesized that Nrf2 expression is possibly regulated by the activation of SERT/p38 MAPK signaling in MA-induced pulmonary inflammation.
Nrf2 is a transcription factor that controls the expression of a variety of antioxidant and detoxification genes (32). Nrf2 is an important protein involved in the transcriptional upregulation of numerous target genes in phase II drug metabolizing enzymes, including γ-glutamylcysteine synthetase and HO-1 (32). Oxidant/antioxidant balance may serve an important role in a number of the processes of inflammation and fibrosis (33). GSH is an abundant endogenous antioxidant and a critical regulator of oxidative stress (34,35). The GSH level and the ratio of GSH to GSSG (GSH/GSSG) serve a role in the Nrf2-mediated antioxidative stress (36). ROS activation in oxidative stress is considered to be the key factor in inflammatory amplification (37). The present study demonstrated that MA significantly inhibited the protein expression levels of Nrf2 and HO-1 in rat lungs, and that the GSH level and the ratio of GSH/GSSG were decreased in the MA group, accompanied by increasing ROS level in the lungs. The above indices of antioxidative stress from MA were significantly reversed by fluoxetine at 10 mg/kg. Taken together, it is suggested that fluoxetine may alleviate pulmonary inflammation, and may be associated with the inhibition of oxidative stress including upregulation of the endogenous antioxidative factor GSH and the downregulation of ROS levels in the lungs by the activation of the Nrf2/HO-1 pathway.
In conclusion, long-term administration of MA induces chronic pulmonary inflammation. Fluoxetine, as a SERT inhibitor, alleviated the MA-induced lung inflammation in rats. The potential protective mechanisms of fluoxetine may be associated with suppressing oxidative stress through the SERT/p38 MAPK/Nrf2 pathways in rats.
Acknowledgements
The present study was funded by the National Natural Science Foundation of China (grant no. 81503058) and the Natural Science Foundation of Liaoning Province (grant no. 2014021,065).
Glossary
Abbreviations
- MA
methamphetamine
- 5-HT
serotonin (5-hydroxytryptamine)
- SERT
serotonin transporter
- PAH
pulmonary arterial hypertension
- ERK1/2
extracellular signal-regulated kinase 1/2
- p38 MAPK
p38 mitogen activated protein kinases
- ROS
reactive oxygen species
- Nrf2
nuclear factor erythroid 2-related factor 2
- HO-1
human heme oxygenase-1
- IL-6
interleukin-6
- TNF-α
tumor necrosis factor-α
- GSH
reduced glutathione
- GSSG
oxidized glutathione
References
- 1.Rawson RA. Current research on the epidemiology, medical and psychiatric effects, and treatment of methamphetamine use. J Food Drug Anal. 2013;21:S77–S81. doi: 10.1016/j.jfda.2013.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albertson TE, Derlet RW, Van Hoozen BE. Methamphetamine and the expanding complications of amphetamines. West J Med. 1999;170:214–219. [PMC free article] [PubMed] [Google Scholar]
- 3.Volkow ND, Fowler JS, Wang GJ, Shumay E, Telang F, Thanos PK, Alexoff D. Distribution and pharmacokinetics of methamphetamine in the human body: Clinical implications. PLoS One. 2010;5:e15269. doi: 10.1371/journal.pone.0015269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peerzada H, Gandhi JA, Guimaraes AJ, Nosanchuk JD, Martinez LR. Methamphetamine administration modifies leukocyte proliferation and cytokine production in murine tissues. Immunobiology. 2013;218:1063–1068. doi: 10.1016/j.imbio.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma J, Wan J, Meng J, Banerjee S, Ramakrishnan S, Roy S. Methamphetamine induces autophagy as a pro-survival response against apoptotic endothelial cell death through the Kappa opioid receptor. Cell Death Dis. 2014;5:e1099. doi: 10.1038/cddis.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Liu M, Wang HM, Bai Y, Zhang XH, Sun YX, Wang HL. Involvement of serotonin mechanism in methamphetamine-induced chronic pulmonary toxicity in rats. Hum Exp Toxicol. 2013;32:736–746. doi: 10.1177/0960327112468174. [DOI] [PubMed] [Google Scholar]
- 7.Hollinger MA. Drug-induced lung toxicity. Int J Toxicol. 1993;12:31–47. doi: 10.3109/10915819309140620. [DOI] [Google Scholar]
- 8.Kushnir-Sukhov NM, Gilfillan AM, Coleman JW, Brown JM, Bruening S, Toth M, Metcalfe DD. 5-hydroxytryptamine induces mast cell adhesion and migration. J Immunol. 2006;177:6422–6432. doi: 10.4049/jimmunol.177.9.6422. [DOI] [PubMed] [Google Scholar]
- 9.de Las Casas-Engel M and Corbí AL, corp-author. Serotonin modulation of macrophage polarization: Inflammation and beyond. Adv Exp Med Biol. 2014;824:89–115. doi: 10.1007/978-3-319-07320-0_9. [DOI] [PubMed] [Google Scholar]
- 10.Mann DA, Oakley F. Serotonin paracrine signaling in tissue fibrosis. Biochim Biophys Acta. 2013;1832:905–910. doi: 10.1016/j.bbadis.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka T, Doe JM, Horstmann SA, Ahmad S, Ahmad A, Min SJ, Reynolds PR, Suram S, Gaydos J, Burnham EL, Vandivier RW. Neuroendocrine signaling via the serotonin transporter regulates clearance of apoptotic cells. J Biol Chem. 2014;289:10466–10475. doi: 10.1074/jbc.M113.482299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M, Wang Y, Wang HM, Bai Y, Zhang XH, Sun YX, Wang HL. Fluoxetine attenuates chronic methamphetamine-induced pulmonary arterial remodelling: Possible involvement of serotonin transporter and serotonin 1B receptor. Basic Clin Pharmacol Toxicol. 2013;112:77–82. doi: 10.1111/j.1742-7843.2012.00933.x. [DOI] [PubMed] [Google Scholar]
- 13.Dempsie Y, MacLean MR. Pulmonary hypertension: Therapeutic targets within the serotonin system. Br J Pharmacol. 2008;155:455–462. doi: 10.1038/bjp.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dizeyi N, Hedlund P, Bjartell A, Tinzl M, Austild-Taskén K, Abrahamsson PA. Serotonin activates MAP kinase and PI3K/Akt signaling pathways in prostate cancer cell lines. Urol Oncol. 2011;29:436–445. doi: 10.1016/j.urolonc.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Han DD, Wang HM, Liu M, Zhang XH, Wang HL. Downregulation of osteopontin is associated with fluoxetine amelioration of monocrotaline-induced pulmonary inflammation and vascular remodelling. Clin Exp Pharmacol Physiol. 2011;38:365–372. doi: 10.1111/j.1440-1681.2011.05516.x. [DOI] [PubMed] [Google Scholar]
- 17.Singh M, Foster CR, Dalal S, Singh K. Osteopontin: Role in extracellular matrix deposition and myocardial remodeling post-MI. J Mol. Cell Cardiol. 2010;48:538–543. doi: 10.1016/j.yjmcc.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sahu BD, Mahesh Kumar J, Sistla R. Baicalein, a bioflavonoid, prevents cisplatin-induced acute kidney injury by up-regulating antioxidant defenses and down-regulating the MAPKs and NF-κB pathways. PLoS One. 2015;10:e0134139. doi: 10.1371/journal.pone.0134139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pandurangan AK, Mohebali N, Norhaizan ME, Looi CY. Gallic acid attenuates dextran sulfate sodium-induced experimental colitis in BALB/c mice. Drug Des Devel Ther. 2015;9:3923–3934. doi: 10.2147/DDDT.S86345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryu J, Kwon MJ, Nam TJ. Nrf2 and NF-κB signaling pathways contribute to porphyra-334-mediated inhibition of UVA-induced inflammation in skin fibroblasts. Mar Drugs. 2015;13:4721–4732. doi: 10.3390/md13084721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saetta M, Shiner RJ, Angus GE, Kim WD, Wang NS, King M, Ghezzo H, Cosio MG. Destructive index: A measurement of lung parenchymal destruction in smokers. Am Rev Respir Dis. 1985;131:764–769. doi: 10.1164/arrd.1985.131.5.764. [DOI] [PubMed] [Google Scholar]
- 22.Rincon M, Irvin CG. Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci. 2012;8:1281–1290. doi: 10.7150/ijbs.4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lockett AD, Kimani S, Ddungu G, Wrenger S, Tuder RM, Janciauskiene SM, Petrache I. α1-Antitrypsin modulates lung endothelial cell inflammatory responses to TNF-α. Am J Respir Cell Mol Biol. 2013;49:143–150. doi: 10.1165/rcmb.2012-0515OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stromps J, Fuchs P, Demir E, Grieb G, Reuber K, Pallua N. Intraalveolar TNF-α in combined burn and inhalation injury compared with intraalveolar interleukin-6. J Burn Care Res. 2015;36:e55–e61. doi: 10.1097/BCR.0000000000000108. [DOI] [PubMed] [Google Scholar]
- 25.Rothman RB, Baumann MH. Therapeutic and adverse actions of serotonin transporter substrates. Pharmacol Ther. 2002;95:73–88. doi: 10.1016/S0163-7258(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 26.Cloëz-Tayarani I, Changeux JP. Nicotine and serotonin in immune regulation and inflammatory processes: A perspective. J Leukoc Biol. 2007;81:599–606. doi: 10.1189/jlb.0906544. [DOI] [PubMed] [Google Scholar]
- 27.Wells SM, Buford MC, Porter VM, Brunell HL, Bunderson-Schelvan M, Nevin AB, Cardozo-Pelaez F, Holian A. Role of the serotonergic system in reduced pulmonary function after exposure to methamphetamine. Am J Respir Cell Mol Biol. 2010;42:537–544. doi: 10.1165/rcmb.2009-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tavoulari S, Forrest LR, Rudnick G. Fluoxetine (Prozac) binding to serotonin transporter is modulated by chloride and conformational changes. J Neurosci. 2009;29:9635–9643. doi: 10.1523/JNEUROSCI.0440-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose'Meyer R. A review of the serotonin transporter and prenatal cortisol in the development of autism spectrum disorders. Mol Autism. 2013;4:37. doi: 10.1186/2040-2392-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai Y, Wang HM, Liu M, Wang Y, Lian GC, Zhang XH, Kang J, Wang HL. 4-Chloro-DL-phenylalanine protects against monocrotaline-induced pulmonary vascular remodeling and lung inflammation. Int J Mol Med. 2014;33:373–382. doi: 10.3892/ijmm.2013.1591. [DOI] [PubMed] [Google Scholar]
- 31.Chen HH, Wang TC, Lee YC, Shen PT, Chang JY, Yeh TK, Huang CH, Chang HH, Cheng SY, Lin CY, et al. Novel Nrf2/ARE activator, trans-coniferylaldehyde, induces a HO-1-mediated defense mechanism through a dual p38α/MAPKAPK-2 and PK-N3 signaling pathway. Chem Res Toxicol. 2015;28:1681–1692. doi: 10.1021/acs.chemrestox.5b00085. [DOI] [PubMed] [Google Scholar]
- 32.Ishikawa T. Genetic polymorphism in the NRF2 gene as a prognosis marker for cancer chemotherapy. Front Genet. 2014;5:383. doi: 10.3389/fgene.2014.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni S, Wang D, Qiu X, Pang L, Song Z, Guo K. Bone marrow mesenchymal stem cells protect against bleomycin-induced pulmonary fibrosis in rat by activating Nrf2 signaling. Int J Clin Exp Pathol. 2015;8:7752–7761. [PMC free article] [PubMed] [Google Scholar]
- 34.Richie JP, Jr, Nichenametla S, Neidig W, Calcagnotto A, Haley JS, Schell TD, Muscat JE. Randomized controlled trial of oral glutathione supplementation on body stores of glutathione. Eur J Nutr. 2015;54:251–263. doi: 10.1007/s00394-014-0706-z. [DOI] [PubMed] [Google Scholar]
- 35.Sinha-Hikim I, Shen R, Lee WN Paul, Crum A, Vaziri ND, Norris KC. Effects of a novel cystine-based glutathione precursor on oxidative stress in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2010;299:C638–C642. doi: 10.1152/ajpcell.00434.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin T, Yin Y, Yu Q, Yang Q. Bursopentin (BP5) protects dendritic cells from lipopolysaccharide-induced oxidative stress for immunosuppression. PLoS One. 2015;10:e0117477. doi: 10.1371/journal.pone.0117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang AL, Niu Q, Shi N, Wang J, Jia XF, Lian HF, Liu Z, Liu CX. Glutamine ameliorates intestinal ischemia-reperfusion Injury in rats by activating the Nrf2/Are signaling pathway. Int J Clin Exp Pathol. 2015;8:7896–7904. [PMC free article] [PubMed] [Google Scholar]