

Abstract
Stable and isolable alkanethiolate-stabilized Pt nanoparticles (PtNP) were synthesized using the two-phase thiosulfate method with sodium S-alkylthiosulfate as ligand precursor. The mechanistic formation of octanethiolate-capped PtNP (Pt-SC8) from both sodium S-octylthiosulfate and 1-octanethiol ligands was investigated by using 1H NMR and UV–vis spectroscopies, which revealed the formation of different Pt complexes as the reaction intermediates. The synthesis using S-octylthiosulfate ligand precursor produced Pt-SC8 in higher yields than that using 1-octanethiol ligand. The obtained nanoparticles were characterized by 1H NMR, UV–vis spectroscopy, infrared spectroscopy (IR), thermogravimetric analysis, and transmission electron microscopy (TEM). The results obtained from 1H NMR, IR, and UV–vis spectroscopy were consistent with the formation of stable and pure alkanethiolate-capped PtNP. TEM images of PtNP confirmed their small average core size (∼1.5 nm) and high monodispersity. The partially poisoned PtNP with thiolate monolayer ligands were further investigated for the hydrogenation of various alkynes to understand the organic ligands-induced geometric and electronic surface properties of colloidal Pt nanoparticle catalysts. The high catalytic activity of activated terminal alkynes, but the significantly low activity of internal alkynes and unactivated terminal alkynes, were observed under the mild reaction conditions (room temperature and atmospheric pressure). These results indicated that the presence of alkanethiolate ligands could decrease the coordination activity of PtNP surface especially for the bulkier and unactivated substrates.
Keywords: platinum, nanoparticles, catalysis, unsupported, colloidal, alkylthiosulfate
Introduction
Research on metal nanoparticles has been popular for more than two decades due to their unique properties such as catalytic, electronic, and optical properties1−4 that were led to applications ranging from catalysis5−9 to electronic devices.10−12 The biggest obstacle of using metal nanoparticles for such applications has been their tendency to aggregate over time, which led to a deterioration in their overall activity. The stability of metal nanoparticles against aggregation and oxidation could be adequately enhanced by using various organic ligands. Different organic compounds containing reactive head groups such as disulfide,13−15 ammonium,16−18 thiosulfate,19−21 and amine22−24 have been used as protecting ligands. It was found that the type of ligands and applied synthetic conditions could systematically alter the chemical and physical properties of metal nanoparticles in addition to their size and ligand–metal ratio.
The synthesis of alkanethiolate-protected Au nanoparticles (AuNP) was popularized after the development of the two-phase Brust–Schiffrin method in 1994.25 The AuNP produced by this method was highly stable and could be easily isolated.26,27 In addition to their potential applications described above, this thiolate-stabilized AuNP has received growing interests from biomedical communities in targeting cancer and drug delivery.28 In a typical two-phase Brust–Schiffrin reaction, the metal source and the phase transfer agent, tetra-n-octylammonium bromide (TOAB), were mixed until the metal ions were transferred to organic phase. To the organic layer, thiol ligand was added to reduce Au(III) to Au(I), and the subsequent addition of reducing agent, NaBH4, reduced Au(I)-SR to form AuNP stabilized by thiolate. The synthesis of monodisperse nanoparticles using the same method could be further extended to different metals such as Ag,29,30 Pd,4,31 and Pt.32
The drawback of thiolate-stabilized nanoparticles synthesized using the Brust–Schiffrin method was that the thiol ligand binds strongly to the metal site and forms densely packed monolayer, which in turn inhibits the catalytic property.3,12 Thiolate-capped metal nanoparticles with lower ligand density were recently starting to gain more interests due to their potential as chemo-, regio-, and stero-selective catalysts.21,33 The previous studies from our group have shown that nanoparticles synthesized from S-alkylthiosulfate ligand show better catalytic activity compared to those generated from alkanethiol ligand.34−38 Alkanethiolate-protected Pd nanoparticles (PdNP) generated from S-alkylthiosulfate selectively catalyzed the isomerization of allylic alcohols and alkenes over the hydrogenation process.21,34 In addition to PdNP, magnetic iridium nanoparticles (IrNP) have recently been synthesized using S-alkylthiosulfate, and the mechanism for the formation of nanoparticles was investigated with a focus on the difference between the activities of S-alkylthiosulfate and alkanethiol ligands.39
With the success in both PdNP catalysis and IrNP synthesis, the target of this research was to synthesize alkanethiolate ligand-stabilized Pt nanoparticles (PtNP), which can further expand the scope for selective catalysts for various organic reactions. Platinum nanoparticles have been widely studied for their applications in heterogeneous catalysis,23,24 electrocatalysis,40,41 fuel cells,40 and hydrogen storage materials.4 PtNP has been synthesized using thiol ligand precursors by various research groups with low thiol/Pt reaction condition, but with a relatively low yield.32,42,43 Cliffel group has also synthesized different functionalized PtNP that are soluble in organic or aqueous media, but their catalytic activity was fairly low.44 The current research tries to expand the versatility of the thiosulfate protocol to PtNP synthesis, discover the catalytic utility of PtNP, and further enhance the understanding on the mechanism of nanoparticle core growth and surface ligand capping process of PtNP. The formation of PtNP from both S-alkylthiosulfate and alkanethiol ligands was systematically studied, and the catalytic activities of this PtNP were investigated as a potential catalyst for hydrogenation of alkynes. In particular, understanding the influence of alkanethiolate ligands on the geometric and electronic surface properties of unsupported PtNP was a focal point of the present catalysis studies.
Experimental Section
Chemicals
All reagents were used as received from the following suppliers. 1-Bromooctane (C8H17Br), 1-bromododecane (C12H24Br), sodium thiosulfate pentahydrate (Na2O3S2·5H2O), and hydrogen hexachloroplatinate (IV) hydrate (H2PtCl6) were obtained from Sigma-Aldrich. Tetra-n-octylammonium bromide (TOAB), 1-octanethiol, sodium borohydride (NaBH4), potassium tetrachloroplatinate(II) (K2PtCl4), phenylacetylene, diphenylacetylene, tert-butyl propiolate, 3,3-dimethyl-1-butyne, methyl propiolate, 3-butyn-2-one, 2-methyl-3-butyn-2-ol, 1-pentyne, and dimethyl acetylenedicarboxylate (DMAD) were obtained from Acros. Ethanol, methanol, acetone, dichloromethane (DCM), and toluene (tol) were obtained from Fisher Scientific. Deuterium oxide (D2O), chloroform-d (CDCl3), dichloromethane-d2 (CD2Cl2), methanol-d4 (CD3OD), and toluene-d8 were purchased from Cambridge Isotope Laboratories, Inc.
Synthesis of Sodium S-Octylthiosulfate
The synthesis followed the previously published procedure from our group.38 1-Bromooctane (25 mmol) was dissolved in 50 mL of ethanol, and Na2S2O3·5H2O (25 mmol) was dissolved in 50 mL of water. Two solutions were mixed in 250 mL round-bottom flask, which was then connected to the reflux condenser. After the solution mixture was refluxed for 3 h, the reaction flask was cooled to room temperature, and the resulting solvents were removed by using rotary evaporator. The white solid product was isolated, dissolved in hot ethanol, and recrystallized to form crystalline solid. 1H NMR (400 MHz, D2O): triplet (δ 3.1 ppm, α-CH2–S), quintet (δ 1.7 ppm, β-CH2CH2–S), broad peak (δ 1.3 ppm, −CH2−), and another triplet (δ 0.9 ppm, CH3−). More characterization results are available in Supporting Information and the previous publication.38
Synthesis of Sodium S-Dodecylthiosulfate
The synthetic method was the same as the synthesis of sodium S-octylthiosulfate, except that 1-bromodocdecane was used as the substrate. 1H NMR (400 MHz, CD3OD): triplet (δ 3.1 ppm, α-CH2–S), quintet (δ 1.8 ppm, β-CH2CH2–S), broad peak (δ 1.3 ppm, −CH2−), and another triplet (δ 0.9 ppm, CH3−). More characterization results are available in the previous publication.35
Synthesis of Pt Nanoparticles Using Sodium S-Octylthiosulfate
Hydrogen hexachloroplatinate(IV) hydrate (H2PtCl6; 0.4 mmol) was dissolved in 12 mL of nanopure water, and TOAB (2.0 mmol) was dissolved in 25 mL of toluene. Two solutions were mixed and stirred for ∼15 min. After the phase transfer, the aqueous layer was separated and discarded by using the separatory funnel. The synthesized sodium S-octylthiosulfate ligand (0.8 mmol) was dissolved in 10 mL of 25% methanol. The ligand and TOAB (2.0 mmol) were added to the separated organic layer, and the reaction mixture was stirred for 15 min. NaBH4 (8.0 mmol) was dissolved in 7 mL of nanopure water before it was added to the vigorously stirring reaction flask within 10 s. The reaction mixture first turned dark orange and then black, which indicated the formation of Pt nanoparticles. The reaction was stirred for additional 3 h, and then the aqueous layer was removed by separatory funnel. The solvents from the organic layer were removed by rotary evaporator. The crude Pt nanoparticles were washed with methanol and ethanol for several times before they were dried under vacuum.
Synthesis of Pt Nanoparticles Using 1-Octanethiol
The synthetic method was the same as the synthesis of Pt nanoparticles using sodium S-octylthiosulfate, except that 1-octanethiol (0.2 mmol) was used instead of sodium S-octylthiosulfate ligand. 1-Octanethiol ligand is readily soluble in toluene; therefore, the ligand was directly added to the reaction mixture instead of dissolving it in 25% methanol.
Synthesis of Pt Nanoparticles Using Deuterated Solvents
H2PtCl6 (0.02 mmol) was dissolved in 0.6 mL of D2O, and TOAB (0.1 mmol) was dissolved in 1.25 mL of tolune-d8. Two solutions were vigorously stirred for ∼15 min to complete the phase transfer of PtCl62–. The 1H NMR spectra of both organic layer (toluene-d8) and aqueous layer (D2O) were obtained after the two layers were separated. The thiosulfate ligand (0.04 mmol) dissolved in 0.4 mL of 25% methanol-d4, and TOAB (0.1 mmol) was added to the organic layer, which was stirred for additional 15 min. The 1H NMR spectra of organic layer (toluene-d8) and aqueous layer (D2O) were again obtained. NaBH4 (0.4 mmol) was dissolved in 0.35 mL of D2O, and the solution was added rapidly to the stirred reaction mixture. The reaction was continuously stirred for 3 h, and the 1H NMR spectra of organic layer (toluene-d8) and aqueous layer (D2O) were obtained. The solvents from the organic layer were removed, and the crude PtNP was washed with methanol and ethanol several times.
Characterization of Nanoparticles
1H NMR spectra were obtained by using Bruker Fourier 400 MHz, and UV–vis spectra were obtained by using the Shimadzu UV-2450 UV-spectrometer. Infrared spectra were obtained by using Bruker Alpha FTIR spectrometer. The NMR samples were prepared as follows: the sodium S-octylthiosulfate ligand was prepared in D2O, sodium S-dodecylthiosulfate ligand was prepared in CD3OD, and the Pt nanoparticles were prepared in CDCl3. For the UV–vis measurement of Pt nanoparticles, dichloromethane (CH2Cl2) solvent was used. Thermogravimetric analysis (TGA) was obtained using TA Instruments SDT Q600. Transmission electron microscopy (TEM) images were obtained using JEOL 1200 EX II electron microscope.
Catalysis Experiments
The catalysis experiments were performed by dissolving 5 mol % of the PtNP, and 0.25 mmol of substrate in 2 mL of CDCl3. The solution mixture was purged with hydrogen gas for ∼10 min. The reaction mixture was stoppered, para-filmed, and stirred for 24 h. The resulting products were analyzed by 1H NMR.
Results and Discussion
Synthesis of Pt Nanoparticles
PtNP was synthesized from dihydrogen hexachloroplatinate(IV) (H2PtCl6·6H2O) by the two-phase thiosulfate method using S-alkylthiosulfate ligand precursor (Bunte salts) as shown in Scheme 1. Synthetic conditions such as the mole equivalents and chain length of thiosulfate ligand and the solvent media of the reaction were systematically varied to observe the influence on the yield, core size, and surface composition of PtNP.35 The reaction condition and nanoparticle yields are summarized in Table 1. The resulting black powder of PtNP was easily soluble in organic solvents such as chloroform and tetrahydrofuran (THF). This indicated that the synthesized PtNP is stable and prevented from aggregation. The mole ratio of thiosulfate ligand to metal salts was varied from 2:1 to 4:1 for the nanoparticle synthesis. As the mole equivalents of S-octylthiosulfate ligand was increased from 2 to 4, the yield of the octanethiolate-capped PtNP (Pt-SC8) almost doubled (entries 1 and 2). Further increase in particle yield was, however, not observed when the additional amount of S-octylthiosulfate was used. Pt-SC8 was also synthesized in dichloromethane (CH2Cl2) instead of toluene (entries 3 and 4). Employing CH2Cl2 solvent resulted in higher Pt-SC8 yield (≥32 mg) compared to that in toluene solvent (≤21 mg). The overall Pt % recovery could be increased from 10% to 29% by changing the reaction condition as mentioned above. The results indicated that the surface passivation activity of alkylthiosulfate might be higher in CH2Cl2 than in toluene.
Scheme 1. Reaction Mechanism of PtNP Using Thiosulfate Ligand.

Table 1. Synthesis of PtNP on Various Conditions and Their Core Size and Composition.
| entry | PtNP | equiv ligand per Pt | sol | yield (mg) | Pt % recovery | TEM (diameter, nm) | TGA (wt %Pt) | No. of Pt atoms/No. of ligandsa | surface ligand density No. of ligands/surface Pta |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Pt-SC8b | 2 | tol | 11 | 10 | 1.58 ± 0.64 | 78 | ∼Pt140(SC8)53 | ∼0.55 |
| 2 | Pt-SC8 | 4 | tol | 21 | 19 | 1.24 ± 0.36 | 73 | ∼Pt79(SC8)39 | ∼0.65 |
| 3 | Pt-SC8 | 2 | DCM | 33 | 29 | 1.51 ± 0.62 | 70 | ∼Pt116(SC8)67 | ∼0.86 |
| 4 | Pt-SC8 | 4 | DCM | 32 | 29 | 1.48 ± 0.52 | 71 | ∼Pt116(SC8)64 | ∼0.82 |
| 5 | Pt-SC12 | 2 | tol | 30 | 20 | 1.45 ± 0.42 | 68 | ∼Pt116(SC12)53 | ∼0.68 |
| 6 | Pt-SC12 | 4 | tol | 34 | 18 | 1.40 ± 0.39 | 66 | ∼Pt116(SC12)58 | ∼0.74 |
These calculated values involve some errors, because the calculation is based on the assumed average molecular weight of somewhat polydisperse nanoparticles. Therefore, the obtained values must be viewed as rough estimates and just as guidelines for the comparison of various PtNPs.
Stable Pt-SC8 was not formed when 2 equiv of 1-octanethiol was used instead of the same amount of S-octylthiosulfate.
When the chain length of the ligand was increased from S-octylthiosulfate to S-dodecylthiosulfate, the yield of PtNP also increased. This result suggested that the longer chain length of surface ligand might provide an enhanced protection of PtNP surface and a better colloidal stability. For dodecanethiolate-capped PtNP (Pt-SC12), however, the mole equivalents of the ligand had no significant effect on the resulting yield of PtNP.
Thiolate-capped Pt nanoparticles were also synthesized by using 1-octanethiol as the ligand precursor. These nanoparticles are abbreviated as PtSH-SC8 to be distinguished from the Pt-SC8 generated from S-octylthiosulfate. When the mole ratio of ligand to metal was fixed at 2:1 for 1-octanethiol ligand, stable PtSH-SC8 was not formed. Chen and Kimura also reported no Pt nanoparticle formation when the thiol ligand to metal ratio was greater than 1.14.42 There have been reports describing the reason for unsuccessful results to be the formation of metal thiolate complexes through decomposition of nanoparticles by excess thiols.43,45 When the reduced mole ratio (1:2 ratio of ligand to metal) was used to lessen the decomposition problem, the Brust–Schiffrin reaction still only produced a small amount (∼2 mg) of PtSH-SC8 suggesting the method’s inefficiency for the PtNP synthesis. Because of the presence of Pt(II) complexes in the product (vide infra) even after the use of reduced mole ratio, the extensive washing of the isolated PtNP was insufficient in obtaining pure PtSH-SC8.
Effect on the oxidation state of platinum sources for the synthesis of PtNP was studied by comparing another platinum complex: potassium tetrachloroplatinate(II) (K2PtCl4). Ulman et al. have studied the formation of alkanethiol-capped Pt nanoparticles using both H2PtCl6·6H2O and K2PtCl4 as the platinum source.41 They found that PtNP was formed by using the one-phase synthesis with the addition of strong reducing agent (lithium triethylborohydride) in THF to the solution containing chloroplatinic acid. However, PtNP was only formed with a relatively low yield after heating the reaction mixture to ∼35 °C for the synthesis using K2PtCl4.41 Our attempt to use K2PtCl4 for the thiosulfate method also resulted in only 1–2 mg of stable PtNP. The yield was much lower than that (11–21 mg) obtained from the reaction using chloroplatinic acid as the Pt source as shown in Table 1. These results were consistent with the findings of Ulman group indicating that the nanoparticle formation by the liquid-phase reduction of platinum complex occurs more favorably with the use of H2PtCl6·6H2O, the platinum source with higher oxidation state.
Characterization of Pt Nanoparticles
The 1H NMR spectrum of the Pt-SC8 in CDCl3 is shown in Figure 1a. The two broad signals are seen at ∼0.90 and ∼1.33 ppm for methyl (−CH3) and methylene (−CH2) groups, respectively. The small broad peak at 1.64 ppm is due to the presence of β-CH2 from S, which only appears for small cluster-like alkanethiolate-capped metal nanoparticles.39,46 The absence of the peaks for α-CH2S groups confirms that the ligand is chemisorbed on the metal surface forming an alkanethiolate monolayer. The 1H NMR spectrum of PtSH-SC8 generated from 1-octanethiol was similar to that of S-octylthiosulfate-derived PtNP showing the two broad peaks for −CH2– and −CH3– groups at 0.90 and 1.3 ppm (Figure S1). The IR spectra for the ligand precursor and the PtNP are shown in Figures S2 and S3, respectively. The absence of strong S=O stretching bands at ∼1210 and ∼1040 cm–1 in the IR spectrum of Pt-SC8 generated from S-octylthiosulfate indicated the absence of any unbound thiosulfate ligands and the elimination of sulfite moiety (most likely as SO3) from thiosulfate groups. The UV–vis spectra were also obtained for the Pt-SC8 using dichloromethane as the solvent. As shown in the Figure 1b, the UV–vis spectrum of Pt-SC8 synthesized from thiosulfate ligand shows no absorbance peak corresponding to nanoparticles from 3 to 20 nm in diameter and is almost identical with the spectra reported for small Pt nanoparticles stabilized with thiol ligands.44 In addition, any other absorbance corresponding to oxidized platinum species was absent. This indicated that Pt(II) complexes, which are formed by the oxidized decomposition of Pt nanoparticles, were not present during and after the two-phase thiosulfate reaction.
Figure 1.

(a) 1H NMR spectrum and (b) UV–vis spectrum of Pt-SC8 generated from sodium S-octylthiosulfate.
TEM image and core-size histogram of Pt-SC8 synthesized from sodium S-octylthiosulfate under the standard condition (Table 1, entry 1) are shown in Figure 2. On the basis of the image, the nanoparticles are monodisperse, spherical, and free of any aggregate formation. TGA data of Pt-SC8 in Table 1 (entry 1) showed that the organic content of the nanoparticles is ∼22 wt %. By using both the TEM and TGA results, the theoretical number of Pt atoms present in the particle and the average number of ligand on the surface of Pt can be roughly estimated.47 For the Pt-SC8 nanoparticle (Table 1, entry 1), the estimated ligand surface coverage was ∼0.55 ligand/surface Pt, and the average molecular formula was determined to be ∼Pt140(SC8)53. Since these estimated values are based on the model for Au nanoparticles with the truncated octahedron structure, they are only presented here to provide some rough insights regarding the relative changes in surface ligand densities for different PtNP.
Figure 2.

TEM image and size distribution histogram of Pt-SC8 (Table 1, entry 1) generated from sodium S-octylthiosulfate.
The analyzed core diameters, TGA data, and estimated surface ligand density are also summarized in Table 1 for all other PtNP. As the amount of S-octylthiosulfate ligand increased, the average core diameter of the Pt-SC8 decreased slightly from 1.58 ± 0.64 to 1.24 ± 0.36 nm (Table 1, entry 2). The previous studies have found that the average core size of the nanoparticle could be controlled by changing the mole ratio of ligand to metal.43 The results also showed the organic content of the nanoparticle slightly increases as the result of increases in surface area to volume ratio. The estimated results indicated that the surface ligand density of Pt-SC8 increases slightly from ∼0.55 to ∼0.65, too. The core diameter of the synthesized Pt-SC8 in CH2Cl2 were mostly consistent with that of particles synthesized in toluene with small and monodisperse particle sizes (Table 1, entry 3). The organic weight content and estimated surface ligand density for Pt-SC8 generated in CH2Cl2 was a little higher than those of Pt-SC8 prepared in toluene (Table 1, entries 3 and 4), which was likely the reason for the higher yield of isolated nanoparticles in CH2Cl2. When sodium S-dodecylthiosulfate was used as a ligand precursor, the average core sizes of Pt-SC12 were also close to those of Pt-SC8 synthesized under the same condition using toluene as a solvent (Table 1, entries 5 and 6). As the mole equivalents of the ligand increased, the relative organic content and surface ligand density of Pt-SC12 also slightly increased.
Mechanistic Studies
To follow the reaction species present throughout the process of synthesizing Pt nanoparticles for both S-octylthiosulfate and 1-octanethiol, the reactions were run using deuterated solvents and scaled down enough to be monitored by 1H NMR. The first 1H NMR spectrum (Figure 3a) was obtained for the organic layer of [TOA+]2[PtCl62–]. The spectrum showed the α-CH2–N signal at δ 3.52 ppm, β-CH2CH2–N signal at δ 1.39 ppm, and CH3– signal at δ 1.01 ppm. In addition, the UV–vis spectrum of the organic layer of [TOA+]2[PtCl62–] showed a strong absorption band at ∼270 nm. The second spectrum (Figure 3b) was obtained after 4 equiv of thiol ligand was added to the organic layer of [TOA+]2[PtCl62–] after the phase transfer to observe the reaction between PtCl62– with 1-octanethiol. In theory, 1 equiv of 1-octanethiol would be used for the reduction of Pt4+ to Pt3+, and another equivalent of 1-octanethiol would be used for the reduction of Pt3+ to Pt2+. The other 2 equiv of thiols would be used for the formation of complex with Pt2+ resulting in Pt(SR)2. The signals corresponding to 1,1-dioctyldisulfide and 1-octanethiol (or 1-octanethiolate) were observed at δ 2.54 ppm (triplet) and δ 2.20 ppm (quartet + triplet). If the reaction took place as expected, the relative intensity of the signal integrations for 1,1-dioctyldisulfide and 1-octanethiolate was postulated to be 2 to 2 with the formation of Pt(SR)2 complex. However, the relative intensity of the signals corresponding to these two species was not determined to be 2:2 ratios in 1H NMR spectrum (Figure 3b). The UV–vis spectrum of the reaction mixture still showed a strong absorption band at ∼270 nm. These results indicated that 1-octanethiol was not able to fully reduce Pt ions and ligated to form Pt(SR)2. The slight upfield shift of the α-CH2–N signal at δ 3.52 ppm to δ 3.42 ppm indicated the intercalation/interaction of 1-octanthiol ligand to/with [TOA+]2[PtCl62–].
Figure 3.

1H NMR spectra of (a) organic (toluene-d8) layer of TOAB (2.0 mmol) after mixing with the aqueous (D2O) layer of H2PtCl6 (0.4 mmol). (b) 1-Octanethiol (1.6 mmol) added to (a). (c) NaBH4 (4.0 mmol) added to (b). (d) S-Octylthiosulfate (0.8 mmol) added to (a). (e) Pt-SC8 in CDCl3 isolated after the reduction by NaBH4 of the solution (d). The peak at δ 2.10 ppm corresponds to the toluene solvent peak.
The spectrum (Figure 3c) was obtained after the reducing agent NaBH4 was added to the reaction mixture (b). A quartet centered at δ −0.03 ppm could be observed due to the presence of excess unreacted NaBH4 (data not shown). After the addition of NaBH4, all the octanethiolate ligands were to be converted to disulfide after the complete reduction of Pt complexes. However, the NMR data showed that Pt(II)-octanethiolate (2.20 ppm, triplet) was still present in the reaction mixture (c), suggesting an incomplete reduction by NaBH4 or a fast decomposition of PtNP to Pt(SR)2 species. Moreover, the UV–vis spectrum of the reaction mixture after NaBH4 addition showed a strong absorption band at ∼270 nm, confirming the presence of Pt complexes, without a signature feature for nanoparticles with an exponential decay of spectrum (Figure 4). This indicated that instead of the metal being completely reduced from Pt2+ to Pt0 by NaBH4 and stabilized by the adsorption of thiolate ligand, other side reactions such as the formation of other Pt complexes such as Pt(OH)62– or Pt(OH)42– or thiol-induced decomposition of PtNP would take place.48,49 Similar results for metal complex formation were also observed from the attempted synthesis of alkanethiolate-capped iridium nanoparticles using the Brust–Schiffrin method.38
Figure 4.

UV–vis spectrum of resulting nanoparticle solution after NaBH4 reduction in the presence of 1-octanethiol ligand.
The reaction pathway for the synthesis of Pt-SC8 using the thiosulfate protocol with sodium S-octylthiosulfate was compared to that with 1-octanethiol ligand. After 2 equiv of sodium S-octylthiosulfate ligand was added to the organic layer of [TOA+]2[PtCl62–] (a), the α-CH2–S signal and β-CH2CH2–S signal from S-octylthiosulfate appeared at ∂ 3.31 ppm and ∂ 1.94 ppm, respectively, in the spectrum (Figure 3d). The signals corresponding to disulfide or thiolate in addition to platinum thiolate complex were not observed for the reaction run using thiosulfates. The slight upfield shift of the α-CH2–N peak observed for 1-octanthiol also took place for the thiosulfate indicating the same intercalation/interaction of S-octylthiosulfate to/with [TOA+]2[PtCl62–]. The 1H NMR spectrum of the D2O layer showed no other signals except for H2O. This indicated that TOA was able to transfer the thiosulfate ligand from aqueous layer to organic layer completely.
The 1H NMR spectrum of purified PtNP (Figure 3e) generated by the reduction of complex (d) showed three broad signals corresponding to the −CH2– and −CH3 groups as explained in the previous section. The absence of the peaks for α-CH2S groups corresponding to thiol, disulfide, thiosulfate, and platinum thiolate after isolation confirmed the chemisorption of ligands as thiolate on the Pt surface forming a monolayer.
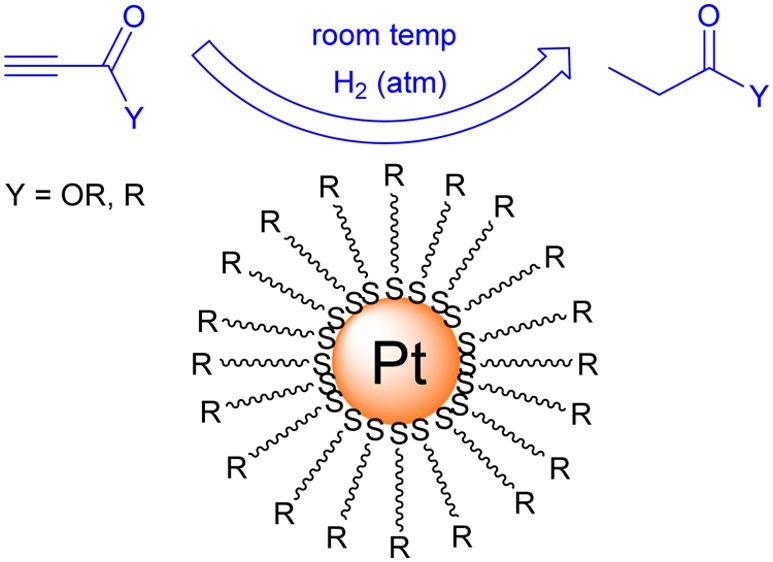
Catalytic Reactions
Catalytic reactions of alkynes were performed in CDCl3 by using 5 mol % of purified PtNP (Scheme 2), and the reaction was monitored by 1H NMR (Figure 5). First, the kinetic study of hydrogenation of methyl propiolate was performed using Pt-SC12 (Table 1, entry 5), and the 1H NMR spectra were obtained after 3, 6, 12, and 24 h reactions for kinetic information (Table 2). The spectrum obtained at 3 h showed that the substrate is converted to 49% semihydrogenation product and 51% full-hydrogenated product. At 6 h, the reaction was very close to completion toward full hydrogenation. The 1H NMR spectra obtained after 12 h reactions confirmed that the signals for starting material were not present from the spectra of the reaction mixture (Figure 5). In addition, the signals for semihydrogenation products were not observed. This suggested that the triple bond of methyl propiolate could be fully hydrogenated by Pt-SC12 to form methyl propanoate. The calculated initial turnover frequency (TOF) was determined to be 30.9/active site·hour.
Scheme 2. Catalysis Reaction of Methyl Propiolate by PtNP.

Figure 5.

1H NMR spectra (CDCl3) of methyl propiolate (bottom) and the reaction solution after the hydrogenation of methyl propiolate using 5 mol % of Pt-SC12 (top).
Table 2. Kinetic Study of Hydrogenation of Methyl Propiolate Using Pt-SC12.
| time (h) | substrate | semihydrogenation | full hydrogenation |
|---|---|---|---|
| 0 | 100 | 0 | 0 |
| 3 | 0 | 49 | 51 |
| 6 | 0 | 0 | 100 |
| 12 | 0 | 0 | 100 |
The catalysis reactions of methyl propiolate were then tested using various Pt-SC8 and Pt-SC12 nanoparticles, and the 1H NMR spectra were obtained after 24 h of reaction. All three Pt-SC8 (Table 1, entries 1, 2, and 4) tested for the same reaction produced the identical catalysis results. Even though an increase in the surface ligand density of the nanoparticle could hinder the reactivity of PtNP, the catalytic activity of Pt-SC8 (entry 4) was as efficient as other Pt-SC8 (entries 1 and 2) for the hydrogenation of methyl propiolate. The results showed that the effect of surface ligand density does not play a major role in the hydrogenation reaction of activated terminal alkyne. The PtSH-SC8, however, was not very active for the hydrogenation of even activated terminal alkynes resulting in low yields (<20% conversion). One of the main reasons was that the PtSH-SC8 nanoparticles were not pure even after extensive washing. The UV–vis spectrum of the purified nanoparticles showed that there was an absorbance peak resulting from the Pt complexes. This caused the inefficiency of synthesized PtSH-SC8 for catalysis reactions. Both batches of Pt-SC12 (Table 1, entries 5 and 6) were also able to fully hydrogenate methyl propiolate to the corresponding methyl propanoate. This indicated that the slightly longer chain length of surface ligands does not lower the activity of PtNP.
Recycling study was performed for the hydrogenation of methyl propiolate by using Pt-SC8 (Table 1, entry 2). The reaction was performed at room temperature and pressure for 24 h after purging with hydrogen gas. After the reaction was completed, the nanoparticles were washed with methanol and dried in vacuum for several days before being reused for additional catalysis cycles. The nanoparticles were still efficient in the third cycle resulting in 100% hydrogenation of methyl propiolate. TEM images of the recycled particles showed they were stable and monodisperse (Figure 6). The UV–vis spectrum of the resulting nanoparticles showed no absorption band corresponding to Pt(II) species (Figure 7).
Figure 6.

TEM image and size distribution histogram of recycled Pt-SC8 nanoparticles.
Figure 7.

UV–vis spectra of recycled Pt-SC8 nanoparticles.
Hydrogenation of various alkynes was tested by using Pt-SC8 (entry 2), and the results are summarized in Table 3. The alkynes that are activated by the C=O functional group such as methyl propiolate (1) and 3-butyn-2-one (2) were fully hydrogenated to their corresponding alkanes with high selectivity. For tert-butyl propiolate (3), Pt-SC8 was not able to fully hydrogenate the alkyne but still produced the full hydrogenation product in high yield (∼95%) with only ∼5% of semihydrogenation product. This indicated that the tert-butyl group of this substrate has a slight steric influence on the reactivity of Pt-SC8. For both phenylacetylene (4) and 2-methyl-3-butyn-2-ol (5), Pt-SC8 produced the full-hydrogenation product in ∼70% yield and the semihydrogenation product in ∼30% yield. Both phenyl and hydroxyl functional groups are known to facilitate the binding of substrate to the surface of metal nanoparticles via the coordination of p orbitals.50 The catalytic reactions of terminal alkynes by Pt-SC12 (1–3, 5) produced similar results as Pt-SC8. For alkynes without any activating group such as the reactions by 3,3-dimethyl-1-butyne (6) and 1-pentyne (7), the catalytic activity of Pt-SC8 was quite low leaving significant amount of substrates to remain unreacted. Compared to 1-pentyne, 3,3-dimethyl-1-butyne resulted in even lower substrate consumption (<20%). This was again due to the steric influence by the tert-butyl group in 3,3-dimethyl-1-butyne. For internal alkynes such as dimethyl acetylenedicarboxylate (DMAD, 8) and diphenylacetylene (9), Pt-SC8 was not active enough to hydrogenate the alkynes to the full-hydrogenation products (only trace amount). The results confirmed the catalytic activity of terminal alkynes is much higher than that of internal alkynes with activating groups.
Table 3. Hydrogenation of Alkynes by Pt-SC8 (entry 2).
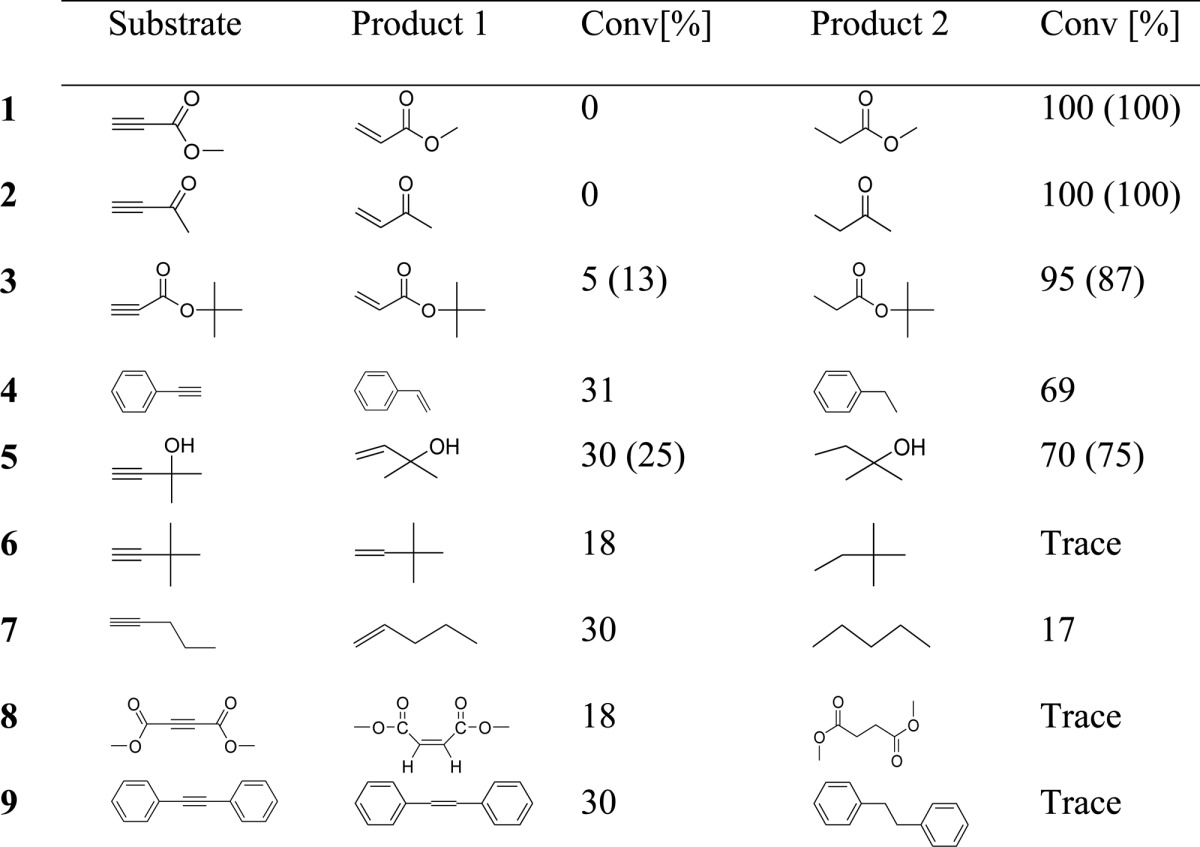
The yields in parentheses are for Pt-SC12 nanoparticles.
The yields are obtained after 24 h of reaction.
The overall catalysis results clearly showed that the partial poisoning by alkanethiolate surface ligands clearly influences the geometric and electronic surface properties of colloidal PtNP. The complete full-hydrogenation products were only obtained from the catalytic reactions of terminal alkynes with a conjugated carbonyl group. Unactivated alkynes without conjugation and bulkier alkynes including internal alkynes would only result in low substrate conversions, whereas only trace amount of full hydrogenation products were obtained. There are many other metal catalysts that show an excellent selectivity for semihydrogenation of alkynes in the literature.51−55 To our knowledge, however, this study is one of the limited examples showing the unique substrate selectivity of nanoparticle-based catalysts for the hydrogenation of different alkynes. For example, Pd nanoparticles supported on nitrogen-doped carbon nanofibers has shown different activities and selectivities between activated internal and terminal alkynes as shown in Table 4.55 In comparison, most other ligand-capped nanoparticle catalysts such as tetra(ethylene glycol)-stabilized Pd nanoparticles supported on titania showed strong activities for internal alkyne hydrogenation.56 This unique selectivity of alkanethiolate-capped PtNP clearly implies the importance of developing a new synthetic protocol that allows the systematic partial poisoning of nanoparticle surfaces.
Table 4. Comparison of Different Metal Catalysts for the Hydrogenation of Terminal and Internal Alkynes.

Pd nanoparticles supported on nitrogen-doped, herringbone-type carbon nanofibers.
Tetra(ethylene glycol)-stabilized Pd nanoparticles supported on titania.
Conclusion
Alkanethiolate-capped PtNP was successfully synthesized from both sodium S-octylthiosulfate and sodium S-dodecylthiosulfate ligand precursors. The structure of these PtNP was confirmed by 1H NMR, UV–vis, TGA, and TEM analyses. The systematic modification of synthetic conditions for the thiosulfate protocol resulted in increasing the yield and controlling the core size and ligand density. Comparison between alkanethiosulfate ligand precursor and alkanethiol ligand confirmed the efficiency of synthesizing PtNP using the thiosulfate protocol. The synthesized PtNP exhibited the high activity for the hydrogenation of activated terminal alkynes. Lower activity for unactivated terminal alkynes and internal alkynes suggested the potential of PtNP as a chemoselective hydrogenation reagent. This selectivity was due to the presence of thiolate poisoning ligands covering active sites of PtNP surfaces, which blocks the hydrogenation of less-reactive alkynes. We plan to explore this partially poisoned PtNP for selective hydrogenation of various alkynes and alkenes with different functional groups.
Acknowledgments
This research was supported by the W. M. Keck Foundation and the National Institute of General Medical Science (SC3GM089562). The NMR instrumentation was provided by the National Science Foundation (MRI CHE-1337559).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.7b02765.
1H NMR spectrum of octanethiol-capped Pt nanoparticles, IR spectra of dodecylthiosullfate ligand and platinum nanoparticles. (PDF)
National Institute of General Medical Science and W. M. Keck Foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Sanchez C.; Julián B.; Belleville P.; Popall M. Applications of Hybrid Organic-inorganic Nanocomposites. J. Mater. Chem. 2005, 15, 3559–3592. 10.1039/b509097k. [DOI] [Google Scholar]
- Burda C.; Chen X.; Narayanan R.; El-Sayed M. A. Chemistry and Properties of Nanocrystals of Different Shapes. Chem. Rev. 2005, 105, 1025–1102. 10.1021/cr030063a. [DOI] [PubMed] [Google Scholar]
- Isaacs S. R.; Cutler E. C.; Park J.; Lee T. R.; Shon Y. Synthesis of Tetraoctylammonium-protected Gold Nanoparticles with Improved Stability. Langmuir 2005, 21, 5689–5692. 10.1021/la050656b. [DOI] [PubMed] [Google Scholar]
- Corthey G.; Rubert A. A.; Picone A. L.; Casillas G.; Giovanetti L. J.; Ramallo-López J. M.; Zelaya E.; Benitez G. A.; Requejo F. G.; José-Yacamán M.; Salvarezza R. C.; Fonticelli M. H. New Insights into the Chemistry of Thiolate-protected Palladium Nanoparticles. J. Phys. Chem. C 2012, 116, 9830–9837. 10.1021/jp301531n. [DOI] [Google Scholar]
- Dhakshinamoorthy A.; Garcia H. Catalysis by Metal Nanoparticles Embedded on Metal-organic Frameworks. Chem. Soc. Rev. 2012, 41, 5262–5284. 10.1039/c2cs35047e. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Cui X.; Shi F.; Deng Y. Nano-gold Catalysis in Fine Chemical Synthesis. Chem. Rev. 2012, 112, 2467–2505. 10.1021/cr200260m. [DOI] [PubMed] [Google Scholar]
- Stratakis M.; Garcia H. Catalysis by Supported Gold Nanoparticles: Beyond Aerobic Oxidative Processes. Chem. Rev. 2012, 112, 4469–4506. 10.1021/cr3000785. [DOI] [PubMed] [Google Scholar]
- Scholten J. D.; Leal B. C.; Dupont J. Transition Metal Nanoparticle Catalysis in Ionic Liquids. ACS Catal. 2012, 2, 184–200. 10.1021/cs200525e. [DOI] [Google Scholar]
- Mikami Y.; Dhakshinamoorthy A.; Alvaro M.; García H. Catalytic Activity of Unsupported Gold Nanoparticles. Catal. Sci. Technol. 2013, 3, 58–69. 10.1039/C2CY20068F. [DOI] [Google Scholar]
- Kane J.; Ong J.; Saraf R. F. Chemistry, Physics, and Engineering of Electrically Percolating Arrays of Nanoparticles: A Mini Review. J. Mater. Chem. 2011, 21, 16846–16858. 10.1039/c1jm12005k. [DOI] [Google Scholar]
- Karmakar S.; Kumar S.; Rinaldi R.; Maruccio G. Nano-electronics and Spintronics with Nanoparticles. J. Phys. Conf. Ser. 2011, 292, 012002. 10.1088/1742-6596/292/1/012002. [DOI] [Google Scholar]
- Moreno M.; Kissell L. N.; Jasinski J. B.; Zamborini F. P. Selectivity and Reactivity of Alkylamine- and Alkanethiolate-stabilized Pd and PdAg Nanoparticles for Hydrogenation and Isomerization of Allyl Alcohol. ACS Catal. 2012, 2, 2602–2613. 10.1021/cs300361y. [DOI] [Google Scholar]
- Zaluzhna O.; Li Y.; Allison T. C.; Tong Y. J. Inverse-micelle-encapsulated Water-enabled Bond Breaking of Dialkyl Diselenide/Disulfide: A Critical Step for Synthesizing High-quality Gold Nanoparticles. J. Am. Chem. Soc. 2012, 134, 17991–17996. 10.1021/ja3068758. [DOI] [PubMed] [Google Scholar]
- Oh E.; Susumu K.; Goswami R.; Mattoussi H. One-phase Synthesis of Water-soluble Gold Nanoparticles with Control Over Size and Surface Functionalities. Langmuir 2010, 26, 7604–7613. 10.1021/la904438s. [DOI] [PubMed] [Google Scholar]
- Volkert A. A.; Subramaniam V.; Ivanov M. R.; Goodman A. M.; Haes A. J. Salted-mediated Self-assembly of Thioctic Acid on Gold Nanoparticles. ACS Nano 2011, 5, 4570–4580. 10.1021/nn200276a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shon Y.-S.; Chuc S.; Voundi P. Stability of Tetraoctylammonium Bromide-capped Gold Nanoparticles: Effects of Anion Treatments. Colloids Surf., A 2009, 352, 12–17. 10.1016/j.colsurfa.2009.09.037. [DOI] [Google Scholar]
- Calò V.; Nacci A.; Monopoli A.; Cotugno P. Heck Reactions with Palladium Nanoparticles in Ionic Liquids: Coupling of Aryl Chlorides with Deactivated Olefins. Angew. Chem., Int. Ed. 2009, 48, 6101–6103. 10.1002/anie.200902337. [DOI] [PubMed] [Google Scholar]
- Ibañez F. J.; Zamborini F. P. Reactivity of Hydrogen with Solid-state Films of Alkylamine- and Tetraoctylammonium Bromide-stabilized Pd, PdAg, and PdAu Nanoparticles for Sensing and Catalysis Applications. J. Am. Chem. Soc. 2008, 130, 622–633. 10.1021/ja076000h. [DOI] [PubMed] [Google Scholar]
- Mari A.; Imperatori P.; Marchegiani G.; Pilloni L.; Mezzi A.; Kaciulis S.; Cannas C.; Meneghini C.; Mobilio S.; Suber L. High Yield Synthesis of Pure Alkanethiolate-capped Silver Nanoparticles. Langmuir 2010, 26, 15561–15566. 10.1021/la102062p. [DOI] [PubMed] [Google Scholar]
- Lohse S. E.; Dahl J. A.; Hutchison J. E. Direct Synthesis of Large Water-soluble Functionalized Gold Nanoparticles Using Bunte Salts as Ligand Precursors. Langmuir 2010, 26, 7504–7511. 10.1021/la904306a. [DOI] [PubMed] [Google Scholar]
- Sadeghmoghaddam E.; Lam C.; Choi D.; Shon Y.-S. Synthesis and Catalytic Properties of Alkanethiolate-capped Pd Nanoparticles Generated from Sodium S-dodecylthiosulfate. J. Mater. Chem. 2011, 21, 307–312. 10.1039/C0JM01761B. [DOI] [Google Scholar]
- Mazumder V.; Sun S. Oleylamine-mediated Synthesis of Pd Nanoparticles for Catalytic Formic Acid Oxidation. J. Am. Chem. Soc. 2009, 131, 4588–4589. 10.1021/ja9004915. [DOI] [PubMed] [Google Scholar]
- Kwon S. G.; Krylova G.; Sumer A.; Schwartz M. M.; Bunel E. E.; Marshall C. L.; Chattopadhyay S.; Lee B.; Jellinek J.; Shevchenko E. V. Capping Ligands as Selectivity Switchers in Hydrogenation Reactions. Nano Lett. 2012, 12, 5382–5388. 10.1021/nl3027636. [DOI] [PubMed] [Google Scholar]
- Li D.; Wang C.; Tripkovic D.; Sun S.; Markovic N. M.; Stamenkovic V. R. Surface Removal for Colloidal Nanoparticles from Solution Synthesis: The Effect on Catalytic Performance. ACS Catal. 2012, 2, 1358–1362. 10.1021/cs300219j. [DOI] [Google Scholar]
- Brust M.; Walker M.; Bethell D.; Schiffrin D. J.; Whyman R. Synthesis of Thiol-derivatised Gold Nanoparticles in a Two-phase Liquid-liquid System. J. Chem. Soc., Chem. Commun. 1994, 0, 801–802. 10.1039/C39940000801. [DOI] [Google Scholar]
- Love J. C.; Estroff L. A.; Kriebel J. K.; Nuzzo R. G.; Whitesides G. M. Self-assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology. Chem. Rev. 2005, 105, 1103–1169. 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- Sardar R.; Funston A. M.; Mulvaney P.; Murray R. W. Gold Nanoparticles: Past, Present, and Future. Langmuir 2009, 25, 13840–13851. 10.1021/la9019475. [DOI] [PubMed] [Google Scholar]
- Saha K.; Agasti S. S.; Kim C.; Li X.; Rotello V. M. Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev. 2012, 112, 2739–2779. 10.1021/cr2001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakr O. M.; Amendola V.; Aikens C. M.; Wenseleers W.; Li R.; Dal Negro L.; Schatz G. C.; Stellacci F. Silver Nanoparticles with Broad Multiband Linear Optical Absorption. Angew. Chem., Int. Ed. 2009, 48, 5921–5926. 10.1002/anie.200900298. [DOI] [PubMed] [Google Scholar]
- Stewart A.; Zheng S.; McCourt M. R.; Bell S. E. J. Controlling Assembly of Mixed Thiol Monolayers on Silver Nanoparticles to Tune Their Surface Properties. ACS Nano 2012, 6, 3718–3726. 10.1021/nn300629z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Klabunde K. J.; Sorensen C. M. From Monodisperse Sulfurized Palladium Nanoparticles to Tiara Pd(II) Thiolate Clusters: Influence of Thiol Ligand on Thermal Treatment of a Palladium(II)-amine System. J. Phys. Chem. C 2007, 111, 18143–18147. 10.1021/jp0765751. [DOI] [Google Scholar]
- Dablemont C.; Lang P.; Mangeney C.; Piquemal J.-Y.; Petkov V.; Herbst F.; Viau G. FTIR and XPS Study of Pt Nanoparticle Functionalization and Interaction with Alumina. Langmuir 2008, 24, 5832–5841. 10.1021/la7028643. [DOI] [PubMed] [Google Scholar]
- Molnár Á.; Sárkány A.; Varga M. Hydrogenation of Carbon-carbon Multiple Bonds: Chemo-, Regio- and Stero-selectivity. J. Mol. Catal. A: Chem. 2001, 173, 185–221. 10.1016/S1381-1169(01)00150-9. [DOI] [Google Scholar]
- Sadeghmoghaddam E.; Gaieb K.; Shon Y.-S. Catalytic Isomerization of Allyl Alcohols to Carbonyl Compounds Using Poisoned Pd Nanoparticles. Appl. Catal., A 2011, 405, 137–141. 10.1016/j.apcata.2011.07.038. [DOI] [Google Scholar]
- Gavia D.; Shon Y.-S. Controlling Surface Ligand Density and Core Size of Alkanethiolate-capped Pd Nanoparticles and Their Effects on Catalysis. Langmuir 2012, 28, 14502–14508. 10.1021/la302653u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghmoghaddam E.; Gu H.; Shon Y.-S. Pd Nanoparticle-catalyzed Isomerization vs Hydrogenation of Allyl Alcohol: Solvent-dependent Regioselectivity. ACS Catal. 2012, 2, 1838–1845. 10.1021/cs300270d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavia D. J.; Maung M. S.; Shon Y.-S. Water-soluble Pd Nanoparticles Synthesized from ω-carboxyl-S-alkanethiosulfate Ligand Precursors as Unimolecular Micelle Catalysts. ACS Appl. Mater. Interfaces 2013, 5, 12432–12440. 10.1021/am4035043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. S.; Shon Y.-S. Mechanistic Interpretation of Selective Catalytic Hydrogenation and Isomerization of Alkenes and Dienes by Ligand Deactivated Pd Nanoparticles. Nanoscale 2015, 7, 17786–17790. 10.1039/C5NR05090A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavia D. J.; Do Y.; Gu J.; Shon Y.-S. Mechanistic Insights into the Formation of Dodecanethiolate-stabilized Magnetic Iridium Nanoparticles: Thiosulfate vs Thiol ligands. J. Phys. Chem. C 2014, 118, 14548–14554. 10.1021/jp504239x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolini E. Carbon Supports for Low-temperature Fuel Cell Catalysts. Appl. Catal., B 2009, 88, 1–24. 10.1016/j.apcatb.2008.09.030. [DOI] [Google Scholar]
- Hu J.; Lu X.; Foord J. S. Nanodiamond Pretreatment for the Modification of Diamond Electrodes by Platinum Nanoparticles. Electrochem. Commun. 2010, 12, 676–679. 10.1016/j.elecom.2010.03.004. [DOI] [Google Scholar]
- Yee C.; Scotti M.; Ulman A.; White H.; Rafailovich M.; Sokolov J. One-phase Synthesis of Thiol-functionalized Platinum Nanoparticles. Langmuir 1999, 15, 4314–4316. 10.1021/la9814283. [DOI] [Google Scholar]
- Chen S.; Kimura K. Synthesis of Thiolate-stabilized Platinum Nanoparticles in Protolytic Solvents as Isolable Colloids. J. Phys. Chem. B 2001, 105, 5397–5403. 10.1021/jp0037798. [DOI] [Google Scholar]
- Eklund S. E.; Cliffel D. E. Synthesis and Catalytic Properties of Soluble Platinum Nanoparticles Protected by a Thiol Monolayer. Langmuir 2004, 20, 6012–6018. 10.1021/la049787n. [DOI] [PubMed] [Google Scholar]
- Zamborini F. P.; Gross S. M.; Murray R. W. Synthesis, Characterization, Reactivity, and Electrochemistry of Palladium Monolayer Protected Clusters. Langmuir 2001, 17, 481–488. 10.1021/la0010525. [DOI] [Google Scholar]
- Qian H.; Zhu M.; Andersen U. N.; Jin R. Facile, Large-scale Synthesis of Dodecanethiol-stabilized Au38 Clusters. J. Phys. Chem. A 2009, 113, 4281–4284. 10.1021/jp810893w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler M. J.; Wingate J. E.; Zhong C.-J.; Harris J. E.; Vachet R. W.; Clark M. R.; Londono J. D.; Green S. J.; Stokes J. J.; Wignall G. D.; Glish G. L.; Porter M. D.; Evans N. D.; Murray R. W. Alkanethiolate Gold Cluster Molecules with Core Diameters from 1.5 to 5.2 nm: Core and Monolayer Properties as a Function of Core Size. Langmuir 1998, 14, 17–30. 10.1021/la970588w. [DOI] [Google Scholar]
- Castro E. G.; Salvatierra R. V.; Schreiner W. H.; Oliveira M. M.; Zarbin A. J. G. Dodecanethiol-stabilized Platinum Nanoparticles Obtained by a Two-phase Methods: Synthesis, Characterization, Mechanism of Formation, and Electrocatalytic Properties. Chem. Mater. 2010, 22, 360–370. 10.1021/cm902748k. [DOI] [Google Scholar]
- Spieker W. A.; Liu J.; Miller J. T.; Kropf A. J.; Regalbuto J. R. An EXAFS Study of the Co-ordination Chemistry of Hydrogen Hexachloroplatinate(IV): 1. Speciation in Aqueous Solution. Appl. Catal., A 2002, 232, 219–235. 10.1016/S0926-860X(02)00116-3. [DOI] [Google Scholar]
- Astruc D.Organometallic Chemistry and Catalysis ;Springer-Verlag: Berlin, Germany, 2007. [Google Scholar]
- Yun S.; Lee S.; Yook S.; Patel H. A.; Yavuz C. T.; Choi M. Cross-Linked “Poisonous” Polymer: Thermochemically Stable Catalyst Support for Tuning Chemoselectivity. ACS Catal. 2016, 6, 2435–2442. 10.1021/acscatal.5b02613. [DOI] [Google Scholar]
- McKenna F.-M.; Wells R. P. K.; Anderson J. A. Enhanced Selectivity in Acetylene Hydrogenation by Ligand Modified Pd/TiO2 Catalysts. Chem. Commun. 2011, 47, 2351–2353. 10.1039/C0CC01742F. [DOI] [PubMed] [Google Scholar]
- Fedorov A.; Liu H.-J.; Lo H.-K.; Copéret C. Silica-supported Cu Nanoparticle Catalysts for Alkyne Semihydrogenation: Effect of Ligands on Rates and Selectivity. J. Am. Chem. Soc. 2016, 138, 16502–16507. 10.1021/jacs.6b10817. [DOI] [PubMed] [Google Scholar]
- Long W.; Brunelli N. A.; Didas S. A.; Ping E. W.; Jones C. W. Aminopolymer-silica Composite-supported Pd Catalysts for Selective Hydrogenation of Alkynes. ACS Catal. 2013, 3, 1700–1708. 10.1021/cs3007395. [DOI] [Google Scholar]
- Lee Y.; Motoyama Y.; Tsuji K.; Yoon S.-H.; Mochida I.; Nagashima H. (Z)-Selective Partial Hydrogenation of Internal Alkynes by Using Palladium Nanoparticles Supported on Nitrogen-doped Carbon Nanofiber. ChemCatChem 2012, 4, 778–781. 10.1002/cctc.201200058. [DOI] [Google Scholar]
- Kim N.; Kwon M. S.; Park C. M.; Park J. One-pot Synthesis of Recyclable Palladium Catalysts for Hydrogenations and Carbon-carbon Coupling Reactions. Tetrahedron Lett. 2004, 45, 7057–7059. 10.1016/j.tetlet.2004.07.126. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.