

Abstract
Previous studies performed in cell lines have shown that the heat shock protein, DNAJB6, protects against the proteotoxic effects of mutant huntingtin (mut-Htt) via direct interaction with mut-Htt. However, these studies were performed primarily using in vitro models and cell lines. We report that when expressed in primary neurons, DNAJB6 induces cell death. Neurotoxicity is observed with both the DNAJB6a isoform, which is strictly nuclear, and the DNAJB6b isoform, which is predominantly cytoplasmic, suggesting that neurotoxicity is mediated in the nucleus. However, when co-expressed in primary neurons with mut-Htt, DNAJB6 protects against mut-Htt neurotoxicity. This suggests that the contrasting effect of DNAJB6 on neuronal viability depends on the presence or absence of proteotoxic stress. Neurotoxicity of DNAJB6 cannot be prevented by inhibition of glycogen synthase kinase 3 beta (GSK3β) or c-Jun N-terminal kinase (JNK) but is prevented by pharmacological inhibition of cyclin-dependent kinases (CDKs). Expression of dominant-negative forms of CDK2 or CDK4, or of p21CIP1, the physiological inhibitor of CDKs, also inhibits DNAJB6 neurotoxicity. DNAJB6 neurotoxicity can also be inhibited by histone deacetylase-4 (HDAC4), which interacts with DNAJB6 and which has previously been described to inhibit cell cycle progression. These results conclude that neurotoxicity resulting from elevated DNAJB6 is cell cycle dependent.
Keywords: DNAJB6, Heat shock proteins, Neuronal death, Neuronal survival, Neurodegeneration, Huntington’s disease
Heat shock proteins (HSPs) are molecular chaperones that were first identified by their increased synthesis following heat shock [1, 2]. Production of HSPs is also increased in response to the accumulation of misfolded proteins [1–4]. A critical function of HSPs is to refold such misfolded proteins or stimulate their clearance through degradation. Deficiencies in managing protein quality control leads to the accumulation of misfolded proteins and protein aggregates within cells, threatening their viability. Indeed, several adult-onset neurodegenerative diseases are caused by the accumulation of misfolded proteins and protein aggregates. It is believed that reduced effectiveness of HSPs is a contributor to these age-related neurodegenerative diseases [5, 6]. A number of recent studies have shown that overexpression of specific HSPs or administration of pharmacological agents that stimulate HSP production protect against neuronal loss in experimental models of neurodegenerative diseases [5, 6].
Among the best studied of the HSPs in the context of neurodegenerative diseases are members of the HSP70 and HSP90 families [3, 5, 6]. Recent studies indicate that members of the HSP40 family also play a key role in protection against proteotoxic neurodegeneration [7–10]. In humans, the HSP40 family, also referred to as the DNAJ family, has over 40 members and is subdivided into three subfamilies based on the presence or absence of certain domains—the DNAJA, DNAJB, and DNAJC subfamilies [9, 11]. All DNAJ proteins contain a J-domain at the N-terminus through which they interact with HSP70 proteins, stimulating their ATPase activity and determining substrate specificity of HSP70 proteins [12, 13]. Members of the DNAJA subfamily of proteins also contain a glycine/phenylalanine-rich and a C-terminal cysteine-rich C-domain [12, 13]. Members of the DNAJB subfamily possess the glycine/phenylalanine-rich domain but lack the C-domain. DNAJC proteins lack both the glycine/phenylalanine-rich domain and the C-domain [12, 13].
Among the DNAJ family of proteins, members of the DNAJB subfamily may play a particularly important role in protecting against neurotoxicity caused by protein aggregation. Evidence for this comes from cell culture models of Huntington’s disease (HD), a genetic neurodegenerative disorder caused by a polyglutamine expansion of the huntingtin protein (polyQ-Htt). In a study utilizing cell line models in which many members of the HSP70, HSP110, and DNAJ families were tested for their ability to suppress aggregation of polyQ-Htt, two members of the DNAJB subfamily, DNAJB6 and DNAJB8, were found to be efficacious [7]. In addition to inhibiting aggregation, overexpression of DNAJB6 and DNAJB8 reduced cytotoxicity by polyQ-Htt expression [7]. In vitro analyses showed that the inhibition of polyQ aggregation is ATP independent and due to direct interaction between polyQ and DNAJB6 [9, 11]. DNAJB6 also inhibits Aβ42 fibril formation in vitro [14]. In a separate study DNAJB6 has been reported to be present within the core of Lewy bodies in the substantia nigra (SN) and cortex of Parkinson’s disease (PD) patients [15]. Moreover, the level of DNAJB6 is highly upregulated in glial cells within the SN of the PD brain [15]. Whether this upregulation in astrocytes is part of a protective response is currently unclear. Recent studies have linked mutations in DNAJB6 to limb girdle muscular dystrophy (LGMD), a progressive myopathy with degeneration of skeletal muscle [16]. Additionally, mutations in DNAJB6 have been reported to cause frontotemporal dementia with LGMD [17].
Although DNAJB6 is likely to play beneficial roles in neurons and skeletal muscle, particularly in the presence of protein aggregation, a systematic analysis of the normal role of DNAJB6 and DNAJB8 in primary neurons has not been conducted. Moreover, whether DNAJB6 and DNAJB8 can protect neurons when death is not due to accumulation of protein aggregates has not been investigated. Using cultured cerebellar granule neurons (CGNs) and embryonic cortical neurons we have investigated the role of DNAJB6. We report that in sharp contrast to what was previously observed in cell line models and proteinopathic stress, DNAJB6 has a toxic effect when overexpressed in primary neurons. The neurotoxic effect of DNAJB6 can be reduced by treatment with cell cycle inhibitors as well as by the expression of molecules known to suppress cell cycle progression, such as p21CIP1 and histone deacetylase-4 (HDAC4). Given the large body of evidence linking abortive cell cycle progression to neuronal death, our results suggest that DNAJB6 expression promotes neuronal death by stimulating abortive re-entry into the cell cycle.
Materials and Methods
Materials
Unless specified, all cell culture reagents were purchased from Life Technologies (Grand Island, NY) and all chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO). Poly-L-lysine for neuronal cultures was purchased from Trevigen (Gaithersburg, MD). Antibodies used in this paper are as follows: Flag (cat. no. F1804, Sigma-Aldrich), α-tubulin (cat. no. T9026, Sigma-Aldrich), β-tubulin (cat. no. D71G9, Cell Signaling, (Danvers, MA)), ERK1/2 (cat. no. 9102, Cell Signaling), c-Jun (L70B11 cat. no. 2315, Cell Signaling), GFP (B-2 cat. no. sc-9996; FL cat. no. sc-8334, Santa Cruz Biotechnology (Dallas, TX)), HA (Y-11 cat. no. sc-805, Santa Cruz), V5 (cat. no. MCA1360GA, AbD Serotec (Raleigh, NC)), DNAJB6 (B-5 cat. no. sc-365574, Santa Cruz; cat. no. AV52499, Sigma-Aldrich), HDAC4 (cat. no. 2072, Cell Signaling), Sirt2 (A-5 cat. no. sc-28298, Santa Cruz), HDAC1 (10E2 cat. no. sc-81598, Santa Cruz), HSF1 (E-4 cat. no. sc-17757, Santa Cruz), HDAC3 (H-99 cat. no. sc-11417, Santa Cruz), and IgG (3E8 cat. no. sc-69786, Santa Cruz). Primary antibodies were used at dilutions from 0.1 to 2.0 ng/μL for Western blot analysis and 1.0 to 5.0 ng/μL for immunocytochemistry in 5 % bovine serum albumin (BSA). Secondary antibodies for Western blotting (Thermo Fisher Scientific, Waltham, MA) were used at dilutions from 0.1 to 0.5 ng/μL. DyLight™ 594- and AlexaFluor® 488-conjugated secondary antibodies for immunocytochemistry were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA) and used at a concentration of 2.0 ng/μL.
Plasmids
Unless specified, plasmids were purchased from Addgene (Cambridge, MA). Plasmids for DNAJB6 expression (cat. no. 19502, 19501, 19529, 19528) were donated by Dr. Harm Kampinga [18] (University of Groningen, Groningen, The Netherlands). Plasmid for expression of Flag-p21CIP1 (cat. no. 16240) was donated by Dr. Mien-Chie Hung [19] (University of Texas MD Anderson Cancer Center, Houston, TX). Plasmid for expression of Flag-Sirt2 (cat. no. 13813) was donated by Dr. Eric Verdin [20]. (University of California, San Francisco, CA). Plasmids for expression of dominant-negative cyclin-dependent kinase (CDK) mutants (cat. no. 1889, 1882, 1877) were donated by Dr. Sander van den Heuvel [21] (Utrecht University, Utrecht, The Netherlands). Plasmid for expression of eGFP-HSP70 (cat. no. 15215) was donated by Dr. Lois Greene [22] (McGill University, Montreal, Canada). Plasmids for expression of eGFP-Htt were a kind gift from Dr. Troy Littleton (Massachusetts Institute of Technology, Cambridge, MA). Plasmid for expression of Flag-HDAC4 was generated by our lab [23].
Neuronal Cultures, Treatments, and Transfection
Cerebellar granule neurons (CGNs) were harvested from 7- to 8-day-old Wistar rats as described previously [24] and plated in basal medium Eagle (BME) supplemented with 10 % FBS, 25 mM KCl, 2 mM L-glutamine, and 0.2 % gentamycin [24]. Unless specified, all transfections were performed 5 days later using the calcium phosphate method as described previously, which typically produces a transfection efficiency in primary cells of 1–2 % [25–27]. For co-transfections of two plasmids not involving small hairpin (shRNA) constructs, cells were transfected at a ratio of 6:6 μg. Eight hours after transfection, CGNs were treated with high-potassium (HK; BME supplemented with 25 mM KCl) or low-potassium (LK; BME without supplemented KCl) medium or inhibitors for 24 h prior to fixing with paraformaldehyde. Viability of transfected cells was determined through immunocytochemistry and 4′,6-diamidino-2-phenylindole (DAPI) staining [25–28]. Treatment of CGNs for Western blotting or RT-PCR analysis was performed 7 days after harvest.
Cortical neurons (CNs) were harvested from 17- to 18-day-old embryos of Wistar rats using neurobasal medium with B27 supplement as previously described [29, 30]. Transfections were performed 5 days later using the calcium phosphate method and treated with 1 mM homocysteic acid (HCA; pH 7.5) for 15–16 h prior to fixing with paraformaldehyde [29, 30]. Viability of transfected cells was determined through immunocytochemistry and DAPI staining as previously described [29, 30].
Culturing of Cerebellar Glial Cells and Kidney Fibroblasts
Cerebellar glial cells were harvested from 7- to 8-day-old Wistar rats under the same protocol as CGNs. Once the single cell suspension was obtained after dissection, the cell pellet was resuspended in BME supplemented with 10 % FBS, 25 mM KCl, 2 mM L-glutamine, and 0.2 % gentamycin and plated without poly-L-lysine. Glial cells were passaged twice to remove neuronal cells and transfected using Lipofectamine 2000 (Life Technologies), which routinely produces a transfection efficiency of ~50 % in glial cells and kidney fibroblasts. Cells were transfected for 24 h prior to fixing with paraformaldehyde. Viability of transfected cells was determined through immunocytochemistry and DAPI staining. Kidneys were dissected from 7- to 8-day-old Wistar rats and placed in Dulbecco’s modified Eagle’s medium (DMEM), diced, then trypsinized for 8–10 min. After treatment with trypsin inhibitor and centrifugation at 500×g for 1 min, the cell pellet was resuspended in DMEM and broken up with a Pasteur pipette. The supernatant was centrifuged at 500×g for 1 min, then the cell pellet was resuspended and plated in DMEM supplemented with 10 % FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin. Primary fibroblasts were passaged twice before transfection using Lipofectamine 2000 for 24 h prior to fixing with paraformaldehyde. Viability of transfected cells was determined through immunocytochemistry and DAPI staining as described above for neurons.
RNA Preparation and RT-PCR
RNA was extracted from cell cultures using TRIzol® Reagent (Life Technologies). Complementary DNA (cDNA) was prepared using the Verso cDNA Synthesis Kit (Thermo Fisher Scientific). PCR was performed with GoTaq Green Master Mix (Promega, Madison, WI). The primers used for PCR are as follows:
DNAJB6a-Rat-FP: 5′-CAGGCTTTACTCCATTCG-3′
DNAJB6a-Rat-RP: 5′-TTCATCTTCCCAGTTGCT-3′
18S-FP: 5′-GCTACCACATCCAGGGAAGG-3′
18S-RP: 5′-GGCCTCGAAAGAGTCCTGTA-3′
Actin-FP: 5′-AGGACTCCTATGTGGGTGACGA-3′
Actin-RP: 5′-CGTTGCCAATAGTGATGACCTG-3′
c-Jun-FP: 5′-GATGGAAACGACCTTCTACG-3′
c-Jun-RP: 5′-GTTGAAGTTGCTGAGGTTGG-3′
Western Blotting
Primary neuron and cell line cultures were lysed in 1× cell lysis buffer (20 mM Tris–HCl at pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 % Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin, and a protease inhibitor cocktail tablet from Roche (Mannheim, Germany)). Rat brain tissue samples were lysed in 1× RIPA buffer (Cell Signaling) containing a protease inhibitor cocktail tablet. The lysates were stored at −80 °C overnight before being analyzed by Western blotting as described previously.
Cell Line and Transfection
HEK293T human embryonic kidney (cat. no. CRL-3216) cells were purchased from ATCC (Manassas, VA) and cultured in DMEM (supplemented with L-glutamine, 110 mg/L sodium pyruvate, 4.5 g/L D-glucose, and 10 % fetal bovine serum (FBS)). NIH/3 T3 mouse embryonic kidney cell lines (cat. no. CRL-1658) were purchased from ATCC and cultured in DMEM (supplemented with L-glutamine, 110 mg/L sodium pyruvate, 4.5 g/L D-glucose, and 10 % newborn calf serum (NCS)). HT22 mouse hippocampal cells were a kind gift from Dr. Rajiv Ratan (Burke Medical Research Institute, NY) and maintained in DMEM (supplemented with L-glutamine, 4.5 g/L D-glucose, and 10 % FBS). As needed, media was supplemented with 0.5 % penicillin-streptomycin solution (cat. no. P4333, Sigma-Aldrich). For viability experiments, HEK293T, HT22, and NIH/3T3 cell lines were transfected at ~25 % confluency using Lipofectamine 2000, which routinely produces a transfection efficiency of 30–50 % in cell lines. Cells were transfected for 24 h prior to fixing with paraformaldehyde. For co-transfections of two plasmids not involving shRNA constructs, cells were transfected at a ratio of 6:6 μg. Viability of transfected cells was determined through immunocytochemistry and DAPI staining. For immunoprecipitation, cells were plated and transfected at 60 % confluency using Lipofectamine 2000 for 48 h. For shRNA knockdown, HT22 cells were transfected at an initial confluence of 40–50 %, incubated for 72 h after transfection, and replenished with media daily until RNA and whole cell lysates were collected for RT-PCR and Western blotting.
shRNA-Mediated Suppression
For the knockdown of DNAJB6, five shRNA constructs were obtained from Sigma-Aldrich (TRCN0000008558, TRCN0000008559, TRCN0000008560, TRCN0000008561, TRCN0000321351, denoted as sh1, sh2, sh3, sh4, and sh5, respectively). pLKO.1-TRC, a plasmid encoding a non-hairpin 18-bp insert, was purchased from Addgene and used as a negative control. shRNAs were co-transfected with GFP into CGNs at a ratio of 13:2 μg for 48 h before treatment with HK medium and LK medium as described previously. Viability of transfected CGNs was determined through immunocytochemistry and DAPI staining.
Immunoprecipitation
Cell lysates were collected as described above. An aliquot of the whole cell lysate (15 %) was mixed with 6× sodium dodecyl sulfate (SDS) loading buffer and prepared for analysis through Western blotting. The remainder of the lysate was incubated 1 h with Protein A/G agarose beads (cat. no. sc-2003, Santa Cruz) while rocking gently at 4 °C, spun down once at 4000 rpm for 2 min, then immunoprecipitated overnight at 4 °C with suitable antibodies bound to Protein A/G agarose beads. Afterwards, the immunoprecipitated lysates were spun down at 4000 rpm for 2 min and washed with 1× cell lysis buffer three times, mixed with 3× SDS loading buffer, incubated at 95° for 5 min, and analyzed through Western blotting. For immunoprecipitation of endogenous protein from CGNs, CGNs were treated with HK or LK medium for 6 h prior to lysis. An aliquot of the whole cell lysate (100 μg) was mixed with 6× SDS loading buffer and prepared for analysis through Western blotting. Nine hundred micrograms of lysate was immunoprecipitated as described above using 5 μg of appropriate antibody.
Statistical Analysis
GraphPad Prism 5 for Windows (GraphPad Software, San Diego, CA) was used to generate the graphs in this paper. Two-tailed Student’s t test was used to perform statistical analysis, and results are shown as mean± standard deviation. p values of <0.05 were considered as statistically significant. One-way ANOVA with Bonferroni’s multiple comparison post-test was used to compare multiple data sets. Asterisks are used to indicate statistical significance: *p<0.05, **p<0.01, and ***p<0.001, respectively. Unless specified otherwise, all experiments were performed in duplicate and repeated in triplicate. At least 200 cells were counted in each experiment.
Results
Elevated Expression of DNAJB6 Promotes Neuronal Death
In humans, DNAJB6 is expressed as two isoforms due to alternative splicing [31]. While the longer isoform, DNAJB6a, contains a functional nuclear localization sequence, the shorter isoform, DNAJB6b, is present in both the nucleus and cytosol [7, 31, 32]. Whereas human DNAJB6b was shown to suppress polyQ-Htt aggregation and toxicity, DNAJB6a was effective only when polyQ-Htt localization was targeted to the nucleus using a targeting sequence [7], suggesting that neuroprotection against polyQ-Htt involved nuclear mechanisms. Much of the work on DNAJB6 in HD models has been done using cell lines in which proteotoxicity was induced by overexpression of polyQ-containing proteins. To examine the effect of forced DNAJB6 expression in primary neurons and in a paradigm in which neuronal death is not due to proteotoxicity, we used CGNs. Cultured CGNs undergo apoptosis when switched from medium containing depolarizing levels of potassium (HK) to medium containing lower, non-depolarizing levels of potassium (LK) [24, 33]. Surprisingly, in view of its protective effect against polyQ-Htt in cell lines, overexpression of either isoform of DNAJB6 induced apoptosis in otherwise healthy neurons (Fig. 1a, b). The extent of neuronal death was not increased in LK (Fig. 1a, b). Similar results were observed using V5-tagged DNAJB6 instead of the GFP-tagged form shown in Fig. 1a, although V5-tagged DNAJB6 was slightly more toxic in LK-treated cells than the GFP-tagged form (Fig. 1c). As described previously in cell lines [7], ectopically expressed DNAJB6a is strictly nuclear in primary neurons while DNAJB6b is predominantly cytosolic but also present in the nucleus. This result suggests that DNAJB6 neurotoxicity is likely to be mediated by effects in the nucleus. To examine whether the toxic effect of DNAJB6 was restricted to CGNs or a more general effect, we used embryonic cortical neurons. Cultures of cortical neurons die when treated with homocysteic acid (HCA), which induces oxidative stress [34, 35]. As observed in CGNs, viability of otherwise healthy neurons was dramatically reduced by DNAJB6 overexpression (Fig. 1d, e). Again, the extent of death did not increase further in the presence of HCA.
Fig. 1.

Expression of DNAJB6 induces apoptosis in neuronal cell cultures. Primary neuronal cultures were transfected with plasmids encoding eGFP, eGFP-tagged DNAJB6 (eGFP-DNAJB6), or V5-tagged DNAJB6 (V5-DNAJB6) isoforms as described in materials and methods. a Viability of CGNs transfected with eGFP or eGFP-DNAJB6 isoforms. b Fluorescent visualization of CGNs transfected with eGFP-DNAJB6 isoforms and treated with either HK or LK medium. c Viability of CGNs transfected with eGFP or V5-DNAJB6 isoforms. d Viability of cortical neurons transfected with eGFP or eGFP-DNAJB6 isoforms and left untreated (UN) or treated with HCA for 15–16 h. e Fluorescent visualization of cortical neurons transfected with eGFP-DNAJB6 isoforms. For all experiments, N=3
HSP40 proteins work in conjunction with HSP70 to refold protein substrates [13]. To examine whether the toxic effect of DNAJB6 is due to an imbalance between the levels of these two types of chaperones, we examined the effect of overexpressing HSP70 on DNAJB6 toxicity. As shown in Fig. S1, elevating HSP70 has no effect on the level of DNAJB6 toxicity. The failure of HSP70 to protect against DNAJB6 overexpression suggests that death is not induced by an imbalance of HSP40/HSP70 levels.
Neurotoxic Effect of DNAJB6 Is Cell-Type Selective
To examine if other primary cell types were also sensitive to overexpressed DNAJB6, we overexpressed DNAJB6 in cerebellar glial cells and in kidney fibroblasts cultured from rats. DNAJB6 induces apoptosis in cerebellar glial cells but has no toxic effect in kidney fibroblasts (Fig. 2a–d), suggesting that the toxic effects of DNAJB6 are restricted to neural tissue. In contrast to its effect in primary neurons, the overexpression of DNAJB6 displayed no toxicity in HT22 cells, a cell line derived from the mouse hippocampus (Fig. 2e). Similarly, overexpression of DNAJB6 had no effect on the viability of HEK293T and NIH/3T3 cultures, two non-neuronal cell lines (Fig. 2f, g). These results suggest that transformed cells, including cells of neural origin, are insensitive to the toxic effects of DNAJB6 overexpression.
Fig. 2.

Expression of DNAJB6 has varying effects in primary cerebellar glials, kidney fibroblasts, and immortalized cell lines. Primary cerebellar glials, kidney fibroblasts (KFs), and cell lines were transfected with eGFP or eGFP-DNAJB6 isoforms with Lipofectamine. a Viability of transfected cerebellar glials. b Fluorescent visualization of cerebellar glials transfected with eGFP-DNAJB6 isoforms. c Viability of transfected KFs. d Fluorescent visualization of KFs transfected with eGFP-DNAJB6 isoforms. e Viability of transfected HT22 mouse hippocampal cells. f Viability of transfected HEK293T human embryonic kidney cells. g Viability of transfected NIH/3T3 mouse embryonic fibroblast cells. For all experiments, N=3
The Effect of DNAJB6 on Neuronal Viability Depends on the Absence or Presence of Proteotoxic Stress
Because our results obtained from primary neurons distinctly contrasted with the neuroprotective effects of DNAJB6 previously reported, we proceeded to confirm the results of Hageman et al. [7]. As reported, the expression of DNAJB6a or DNAJB6b was protective against polyQ-Htt toxicity and aggregation in HEK293T cells (Fig. 3a–c). To examine whether the neurotoxic effect of DNAJB6 in neurons depended on the presence of proteinopathic stress, we co-expressed DNAJB6 with polyQ-Htt in CGNs. While DNAJB6 has a toxic effect on normally healthy CGNs and cortical neurons, when CGNs were challenged with mut-Htt, it protected against the toxic effect of mut-Htt (Fig. 3d, e). This result suggests that whether DNAJB6 is protective or toxic, at least in part, depends on the presence of proteinopathic stress.
Fig. 3.

Expression of DNAJB6 protects against mutant-Htt proteotoxicity. HEK293T cells and CGNs were co-transfected with eGFP or eGFP-tagged wild-type or mutant huntingtin (Htt-Q15 and Htt-Q138, respectively), as well as V5-DNAJB6 isoforms. a Quantification of cell death of co-transfected HEK293T cells. b Quantification of presence of huntingtin aggregates in co-transfected HEK293T cells. c Fluorescent visualization of co-transfected HEK293T cells. d Viability of co-transfected CGNs. e Fluorescent visualization of co-transfected CGNs. For all experiments, N=3
DNAJB6 Is Expressed in Post-mitotic Neurons but Its Expression Does Not Change During Neuronal Death
Although overexpression of DNAJB6 kills CGNs, whether DNAJB6 is normally expressed in neurons and whether its expression is elevated during apoptosis was unclear. To address this issue we used both CGNs and cortical neurons. While two DNAJB6 isoforms have been reported in humans, only expression of DNAJB6a protein was clearly observed in CGNs, despite the fact that the epitope recognized by the antibody in DNAJB6a is also present in DNAJB6b. A similar absence of DNAJB6b was also found with another antibody (from Sigma-Aldrich). Expression levels of the DNAJB6 messenger RNAs (mRNAs) or protein was not changed by LK treatment. (Fig. 4a, b). DNAJB6 is also expressed in cortical neurons. As observed in CGNs, the expression of DNAJB6 does not change in cortical neurons induced to die by treatment with HCA (Fig. 5a). Although a quantitative comparison of DNAJB6 in CGNs and cortical neurons cannot be made, based on the relative signal intensity in the Western blots and the higher exposure times required to detect DNAJB6 in cortical neurons, we infer that expression of DNAJB6 is significantly lower in cortical neurons. A study of whole brain lysates from embryonic, postnatal, and adult rats reveals that DNAJB6a protein expression increases through development with much higher levels observed in adulthood (Fig. 5b), while DNAJB6b expression remains relatively low. In adult rats, analysis of lysates shows that DNAJB6 protein is expressed at high levels in the cerebellum, hippocampus, and olfactory bulb, but at relatively low levels in the cortex and striatum (Fig. 5c).
Fig. 4.

DNAJB6 is expressed in post-mitotic neurons. a RT-PCR analysis of DNAJB6 expression in CGNs. CGN cultures were treated with HK or LK medium for 3, 6, and 9 h or left untreated (UN). RT-PCR analysis was performed using primers specific for DNAJB6. Actin was used as a loading control, and c-Jun was used as a marker for induction of apoptosis. Bottom, densitometric analysis from three separate experiments. b Western blot analysis of DNAJB6 expression in CGNs. CGN cultures were treated with HK or LK medium for 3, 6, and 9 h or left untreated (UN). DNAJB6 expression was analyzed through Western blotting. ERK1/2 was used as a loading control, and c-Jun was used as a marker for induction of apoptosis. Bottom, densitometric analysis from three separate experiments
Fig. 5.
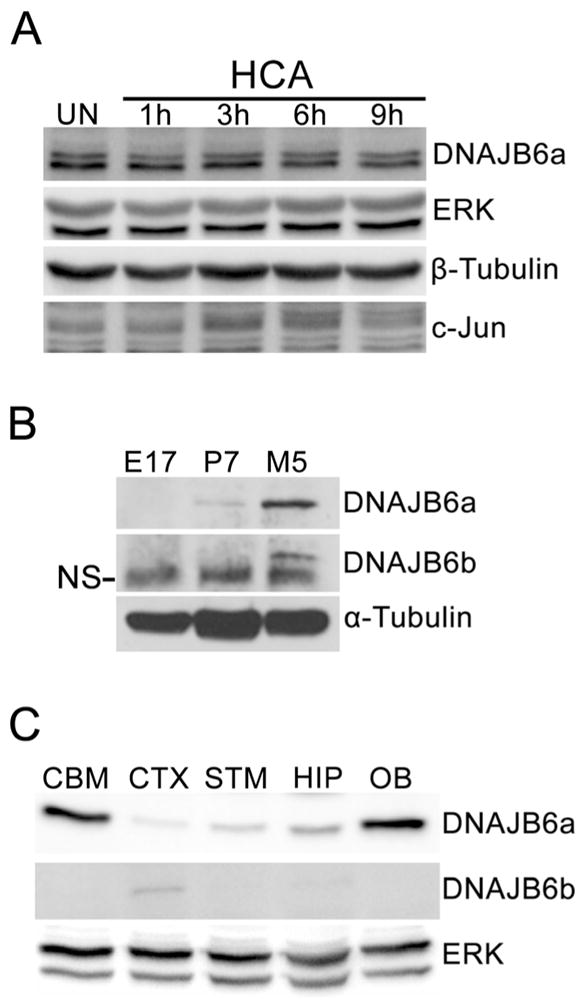
Expression of DNAJB6 in cortical neurons and rat brain. a Western blot analysis of DNAJB6 expression in cortical neurons (CNs). Embryonic CN cultures were treated with 1 mM HCA for 1, 3, 6, and 9 h or left untreated (UN). DNAJB6 expression was analyzed through Western blotting. ERK1/2 and β-tubulin were used as loading controls, and c-Jun was used as a marker for induction of apoptosis. b Western blot analysis of DNAJB6 expression in whole brain samples at different ages. Whole brains were harvested from embryonic (E17), postnatal (P7), and adult (M5) rats and homogenized. DNAJB6 expression was analyzed through Western blotting. α-tubulin was used as a loading control. NS non-specific band. c Western blot analysis of DNAJB6 expression in adult rat brain regions. Brain regions were harvested from adult rats and homogenized. DNAJB6 expression was analyzed through Western blotting. CBM cerebellum, CTX cortex, STM striatum, HIP hippocampus, OB olfactory bulb. ERK was used as a loading control. For all experiments, N=3
Suppression of DNAJB6 with shRNA Protects Against Neuronal Death
Although DNAJB6 overexpression is sufficient to induce apoptosis in otherwise healthy neurons, it was unclear whether DNAJB6 expression was necessary for the progression of apoptosis in LK-treated neurons. To elucidate this, we used shRNA-encoding plasmids to suppress the expression of DNAJB6. We tested the efficacy of five shRNA constructs through transfection in HT22 cells. RT-PCR shows that shRNA constructs sh1 and sh2 were effective in reducing endogenous DNAJB6 mRNA, while shRNA constructs sh3 and sh4 were ineffective in suppressing DNAJB6 expression (Fig. 6a). shRNA construct sh5 was also effective in reducing endogenous DNAJB6 mRNA, but to a lesser extent than the other constructs. These results were confirmed through analysis of protein lysates from HT22 cells transfected with these shRNA constructs (Fig. 6b). As expected, in CGNs, sh1 and sh2 were effective in protecting cells from LK-induced apoptosis, while the other constructs had no significant effect (Fig. 6c). This suggests that while DNAJB6 may be protective against proteinopathic stress, it is required for the progression of apoptosis in CGNs.
Fig. 6.

Suppression of DNAJB6 expression protects neurons. a RT-PCR analysis of shRNA knockdown. HT22 cells were left untransfected (UT) or transfected with either a control plasmid (pLKO.1) or one of five plasmids encoding DNAJB6-specific shRNAs (sh1–5). Expression of DNAJB6 was analyzed through RT-PCR with DNAJB6-specific primers. 18S rRNAwas used as a loading control. Bottom, results of densitometric analysis from three separate experiments. b Western blot analysis of shRNA knockdown. HT22 cells were left untransfected (UT) or transfected with either a control plasmid (pLKO.1) or sh1–5. ERK1/2 and β-tubulin were used as loading controls. c Viability of CGNs transfected with DNAJB6-specific shRNA. CGNs were transfected with eGFP only (UN) or co-transfected with eGFP and a control plasmid (pLKO.1) or eGFP and sh1–5 at a ratio of 1:6.5 for 48 h prior to treatment with HK or LK medium. For all experiments, N=3
DNAJB6 Induces Apoptosis Through Activation of CDKs
Signaling kinases including GSK-3β, c-Jun N-terminal kinase (JNK), and CDKs have been shown to play a critical role in the regulation of apoptosis in neurons [36]. To clarify whether any of these signaling pathways are involved in the toxicity of elevated DNAJB6, we examined whether DNAJB6-induced apoptosis was suppressed when these kinases were pharmacologically inhibited. Inhibition of GSK-3β or JNK fails to protect DNAJB6-transfected neurons from apoptosis. However, treatment with two CDK inhibitors, roscovitine and HSB13 [37], completely abrogates DNAJB6-induced death (Fig. 7a). This suggests that elevated DNAJB6 induces cell cycle progression in neurons. A number of studies have described that abortive cell cycle re-entry induces neuronal death [38–40]. To take into account the possibility that pharmaceutical inhibitors could have effects on molecules other than their primary targets, we examined the effect of expressing p21CIP1, an endogenous inhibitor of various CDKs, on DNAJB6 neurotoxicity. As shown in Fig. 7b, co-expression of p21CIP1 inhibits the toxicity of elevated DNAJB6. To further confirm this result, we co-expressed dominant-negative mutants of CDKs with DNAJB6. As seen with pharmaceutical inhibition and p21CIP1 co-expression, expression of dominant-negative forms of CDK2 and CDK4 also protect against DNAJB6-induced apoptosis (Fig. 7c). Surprisingly, expression of dominant-negative CDK1 failed to protect. While CDK2 acts at the late G1 and S phases and CDK4 acts at the G1 phase, CDK1 is activated at G2 and M phases of the cell cycle. These results suggest that DNAJB6 induces apoptosis through a CDK-dependent mechanism resulting in cell cycle activation and that this toxicity is mediated at the G1 or S phase.
Fig. 7.

Inhibition of CDKs protects against DNAJB6-induced death. a Viability of CGNs treated with inhibitors of apoptotic pathways. CGNs were transfected with eGFP or eGFP-DNAJB6 isoforms and then treated with either HK medium or HK medium supplemented with inhibitors for GSK-3β (SB216), JNK (SP600), or CDKs (HSB-13 and Rosco). SB216 SB216763 (5 μM), SP600 SP600125 (10 μM), HSB-13 (25 μM), Rosco Roscovitine (50 μM). b Viability of CGNs co-transfected with FLAG-p21CIP1 (F-P21-CIP) and eGFP or eGFP-DNAJB6 isoforms (JB6a and JB6b). pLKO.1 was used as a control plasmid. c Viability of CGNs co-transfected with HA-tagged dominant-negative mutants of CDKs (DN-CDK1, DN-CDK2, and DN-CDK4) and eGFP or eGFP-DNAJB6 isoforms. For all experiments, N=3
Effect of DNAJB6-Interacting Partners on Neurotoxicity
DNAJB6 has been shown to interact with Sirt2, a member of the sirtuin subfamily of HDACs, and HDAC4, a classical HDAC [7]. We and others have found that Sirt2 contributes to the promotion of neuronal death, raising the possibility that DNAJB6 and Sirt2 cooperate in inducing neurotoxicity [28, 41–43]. We confirmed that both isoforms of DNAJB6 interact with Sirt2 as well as HDAC4 when these two proteins are co-expressed in HEK293T cells (Fig. 8c). More interaction is observed with DNAJB6b that DNAJB6a. This is likely due to the fact that in HEK293 cells we find both HDAC4 and Sirt2 to be predominantly cytoplasmic (data not shown). Cytoplasmic localization of these two proteins has also been described by other groups [22, 44–46]. We examined whether these interactions also occurred in primary neurons. Using CGNs, we observed interaction with HDAC4 (Fig. 8d) but were unable to detect interaction between endogenous DNAJB6 and Sirt2 (data not shown). It is unclear whether this lack of interaction is due to antibody limitations or is related to the abundance of these proteins in CGNs. We also detected a lack of interaction between HDAC1, HSF1 (Fig. 8d), and c-Jun (data not shown). When co-expressed, Sirt2 does not exacerbate toxicity induced by DNAJB6 (Fig. 8a), suggesting that Sirt2 and DNAJB6 could function in the same cell death signaling pathway. Another protein found to interact with DNAJB6 is HDAC4 [7]. A previous study from our lab has shown that HDAC4 protects neurons from death and does so by inhibiting CDK1 and cell cycle progression [23]. Neuroprotection by HDAC4 has also been described by other labs [47]. As previously reported, we found that both isoforms of DNAJB6 interact with HDAC4 when co-expressed in HEK293T cells (Fig. 8c) and at endogenous levels in CGNs (Fig. 8d). Interestingly, co-expression of HDAC4 inhibits the ability of DNAJB6 to induce neuronal death (Fig. 8b).
Fig. 8.

Co-expression of interacting partners protects against DNAJB6 neurotoxicity. a Viability of CGNs co-transfected with Flag-tagged Sirt2 (F-Sirt2) and eGFP or eGFP-DNAJB6 isoforms (JB6a and JB6b). pLKO.1 was used as a control plasmid. b Viability of CGNs co-transfected with Flag-tagged HDAC4 (F-HDAC4) and eGFP or eGFP-DNAJB6 isoforms (JB6a and JB6b). pLKO.1 was used as a control plasmid. c Western blot analysis of co-immunoprecipitation of GFP-tagged DNAJB6 isoforms with Flag-tagged Sirt2 and HDAC4. Lysates from co-transfected HEK293Tcells were immunoprecipitated (IP) with Mouse anti-IgG (IgG) or Mouse anti-Flag (FLAG) antibody; 15 % of lysates was used to verify overexpression of transfected plasmids (Input). Both membranes were probed with GFP and Flag antibodies to evaluate pulldown and expression. d Western blot analysis of co-immunoprecipitation of endogenous DNAJB6 with HDAC4, HDAC1, and HSF1. Lysates from HK- and LK-treated CGNs were immunoprecipitated (IP) with Mouse anti-IgG (IgG) or Mouse anti-DNAJB6 (JB6) antibody; One hundred micrograms of lysate was used to verify expression of respective proteins (Input). For viability experiments, N=3
Discussion
Although the nature of the mutation and neuropathology of neurodegenerative disease varies between each disorder, a common theme in neurodegeneration is the aggregation of misfolded proteins in apoptotic neurons. As it has been noted that the adult age of onset in most patients of neurodegenerative disease coincides with the dysfunction of the heat shock response, it has been proposed that restoration of the heat shock response may be a viable route to the treatment of neurodegenerative disease. DNAJB6, a member of the HSP40 family of heat shock proteins, has been reported as an effective suppressor of the proteinopathic aggregation of polyQ-Htt in cell lines [7]. Other studies conducted using in vitro systems has shown that DNAJB6 can interact with polyQ-Htt and Aβ42 [11, 14]. These studies notwithstanding, the effectiveness of DNAJB6 to regulate neuronal survival was not previously studied in neurons. We report that, in contrast to its previously reported protective effects in immortalized cell lines, elevated DNAJB6 induces apoptosis in post-mitotic CGNs and cortical neurons. In contrast, overexpression in cell lines has no effect. Interestingly, when DNAJB6 is co-expressed in neurons with mut-Htt, it inhibits mut-Htt neurotoxicity. This suggests that the effect of elevated levels DNAJB6 on viability is cell-specific and whether it protects or kills neurons, depends on the presence of proteotoxic stress. Based on its opposing roles on neuronal viability we suggest that elevating expression of DNAJB6 as a therapeutic approach may not be without dangers.
DNAJB6 is expressed as two isoforms in humans. While DNAJB6a is nuclear, DNAJB6b is present in the nucleus although predominantly present in the cytoplasm [7, 31, 32]. Forced expression of these two isoforms produce an almost identical effect on neuronal viability either in the absence or presence of proteinopathic stress. This suggests that the effect of DNAJB6b on neuronal viability is likely to be mediated in the nucleus.
The toxic effect of DNAJB6 in neurons under normal conditions can be prevented by inhibiting CDKs either pharmacologically or by co-expression of dominant-negative forms of CDK2 and CDK4. Additionally, co-expression of the physiological CDK inhibitor, p21CIP1 also inhibits DNAJB6 neurotoxicity. Taken together, these results indicate that elevated DNAJB6 promotes death of neurons by a cell cycle-dependent mechanism. Aberrant activation of cell cycle machinery has been implicated in cell death in a variety of in vitro and in vivo paradigms of neurodegeneration [6, 31, 36, 37]. Indeed, expression of molecules that inhibit cell cycle progression or treatment of neurons with pharmacological inhibitors of the cell cycle protects neurons against death induced by a variety of apoptotic stimuli [38–40]. As previously described, DNAJB6 interacts with HDAC4 [7]. We previously reported that HDAC4 is a neuroprotective protein acting by inhibiting cell cycle progression [23]. We find that HDAC4 inhibits DNAJB6 neurotoxicity suggesting that HDAC4 acts by sequestering DNAJB6 thus preventing it from cell cycle activation. Sirt2 is another protein known to interact with DNAJB6 [7]. Previous studies have reported that Sirt2 can be neurotoxic [28, 41]. Indeed, inhibition of Sirt2 can prevent neuronal death in both tissue culture and in vivo models of neurodegenerative disease [28, 40, 41]. Although we confirmed interaction between Sirt2 and DNAJB6 when the two proteins are overexpressed, co-expression of Sirt2 did not enhance neurotoxicity by DNAJB6. Although it is not known how Sirt2 mediated neuronal death, our results suggest that both Sirt2 and DNAJB6 may be part of the same cell cycle-dependent signaling pathway.
We found that DNAJB6 also induces apoptosis in cerebellar glia. This suggests that, in addition to neurons, sensitivity to elevated levels of DNAJB6 may extend to other neural types in the CNS. Although this has not been explored in this study, it may be possible that co-expression of mut-Htt with DNAJB6 in glia inhibits its toxicity, similar to the effect seen in CGNs. In knock-in models in which mut-Htt is selectively expressed in astrocytes and oligodendrocytes, glial dysfunction, subsequent impaired glutamate uptake, and myelin breakdown is sufficient to induce neurodegeneration accompanied by an HD-like phenotype [48, 49]. It would be interesting to see whether DNAJB6 reverses these effects through its interaction with mut-Htt.
In summary, despite the general impression that elevated levels of neuroprotective heat shock proteins such as DNAJB6 could be a therapeutic strategy for proteinopathic neurodegenerative diseases, our results suggest that DNAJB6 can have detrimental effects on healthy neurons. Our results indicate that the neurotoxic effect of elevated DNAJB6 is cell cycle dependent. Recent studies have shown that mutation in DNAJB6 can cause LGMD as well as frontotemporal dementia with LGMD8 [16, 17], indicating that normal level of DNAJB6 may protect against muscle and neuronal atrophy and degeneration. Taken together with our results, this suggests that while normal levels of DNAJB6 is beneficial, elevated levels in the absence of proteinopathic stress has a detrimental effect on neuronal survival and serves as a cautionary note on elevating HSPs as a therapeutic approach for neurodegenerative diseases.
Supplementary Material
Acknowledgments
We thank Lulu Wang for technical assistance. We thank Jason A. Pfister, Jade M. Franklin, and Tanzeen Yusuff for reading the manuscript and providing commentary. This research was supported by NIH grant NS040408 to S.R.D.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s12035-015-9452-3) contains supplementary material, which is available to authorized users.
Conflict of Interest The authors declare that they have no conflict of interest.
References
- 1.Lindquist S. The heat-shock response. Annu Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- 2.Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 3.Fujimoto M, Nakai A. The heat shock factor family and adaptation to proteotoxic stress. FEBS J. 2010;277:4112–4125. doi: 10.1111/j.1742-4658.2010.07827.x. [DOI] [PubMed] [Google Scholar]
- 4.Bjork JK, Sistonen L. Regulation of the members of the mammalian heat shock factor family. FEBS J. 2010;277:4126–4139. doi: 10.1111/j.1742-4658.2010.07828.x. [DOI] [PubMed] [Google Scholar]
- 5.Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron. 2011;69:22–32. doi: 10.1016/j.neuron.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Gestwicki JE, Garza D. Protein quality control in neurode-generative disease. Prog Mol Biol Transl Sci. 2012;107:327–353. doi: 10.1016/B978-0-12-385883-2.00003-5. [DOI] [PubMed] [Google Scholar]
- 7.Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HM, Lubsen NH, et al. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell. 2010;37:355–369. doi: 10.1016/j.molcel.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Fayazi Z, Ghosh S, Marion S, Bao X, Shero M, Kazemi-Esfarjani P. A Drosophila ortholog of the human MRJ modulates polyglutamine toxicity and aggregation. Neurobiol Dis. 2006;24:226–244. doi: 10.1016/j.nbd.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 9.Gillis J, Schipper-Krom S, Juenemann K, Gruber A, Coolen S, van den Nieuwendijk R, van Veen H, Overkleeft H, et al. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J Biol Chem. 2013;288:17225–17237. doi: 10.1074/jbc.M112.421685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zijlstra MP, Rujano MA, Van Waarde MA, Vis E, Brunt ER, Kampinga HH. Levels of DNAJB family members (HSP40) correlate with disease onset in patients with spinocerebellar ataxia type 3. Eur J Neurosci. 2010;32:760–770. doi: 10.1111/j.1460-9568.2010.07352.x. [DOI] [PubMed] [Google Scholar]
- 11.Mansson C, Kakkar V, Monsellier E, Sourigues Y, Harmark J, Kampinga HH, Melki R, Emanuelsson C. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones. 2014;19:227–239. doi: 10.1007/s12192-013-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitra A, Shevde LA, Samant RS. Multi-faceted role of HSP40 in cancer. Clin Exp Metastasis. 2009;26:559–567. doi: 10.1007/s10585-009-9255-x. [DOI] [PubMed] [Google Scholar]
- 13.Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63:2560–2570. doi: 10.1007/s00018-006-6192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Månsson C, Arosio P, Hussein R, Kampinga HH, Hashem RM, Boelens WC, Dobson CM, Knowles TP, et al. Interaction of the molecular chaperone DNAJB6 with growing amyloid-beta 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J Biol Chem. 2014;289:31066–31076. doi: 10.1074/jbc.M114.595124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durrenberger PF, Filiou MD, Moran LB, Michael GJ, Novoselov S, Cheetham ME, Clark P, Pearce RK, et al. DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in parkinsonian astrocytes. J Neurosci Res. 2009;87:238–245. doi: 10.1002/jnr.21819. [DOI] [PubMed] [Google Scholar]
- 16.Couthouis J, Raphael AR, Siskind C, Findlay AR, Buenrostro JD, Greenleaf WJ, Vogel H, Day JW, et al. Exome sequencing identifies a DNAJB6 mutation in a family with dominantly-inherited limb-girdle muscular dystrophy. Neuromuscul Disord. 2014;24:431–435. doi: 10.1016/j.nmd.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yabe I, Tanino M, Yaguchi H, Takiyama A, Cai H, Kanno H, Takahashi I, Hayashi YK, et al. Pathology of frontotemporal dementia with limb girdle muscular dystrophy caused by a DNAJB6 mutation. Clin Neurol Neurosurg. 2014;127:10–12. doi: 10.1016/j.clineuro.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Hageman J, Kampinga HH. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones. 2009;14:1–21. doi: 10.1007/s12192-008-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 20.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 21.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 22.Zeng XC, Bhasin S, Wu X, Lee JG, Maffi S, Nichols CJ, Lee KJ, Taylor JP, et al. Hsp70 dynamics in vivo: effect of heat shock and protein aggregation. J Cell Sci. 2004;117:4991–5000. doi: 10.1242/jcs.01373. [DOI] [PubMed] [Google Scholar]
- 23.Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dastidar SG, Landrieu PM, D’Mello SR. FoxG1 promotes the survival of postmitotic neurons. J Neurosci. 2011;31:402–413. doi: 10.1523/JNEUROSCI.2897-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardai FH, Verma P, Smith C, Rawat V, Wang L, D’Mello SR. Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J Neurosci. 2013;33:11833–11838. doi: 10.1523/JNEUROSCI.5831-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma C, D’Mello SR. Neuroprotection by histone deacetylase-7 (HDAC7) occurs by inhibition of c-jun expression through a deacetylase-independent mechanism. J Biol Chem. 2011;286:4819–4828. doi: 10.1074/jbc.M110.146860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfister JA, Ma C, Morrison BE, D’Mello SR. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma P, Pfister JA, Mallick S, D’Mello SR. HSF1 protects neurons through a novel trimerization- and HSP-independent mechanism. J Neurosci. 2014;34:1599–1612. doi: 10.1523/JNEUROSCI.3039-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mallick S, D’Mello SR. JAZ (Znf346), a SIRT1-interacting protein, protects neurons by stimulating p21 (WAF/CIP1) protein expression. J Biol Chem. 2014;289:35409–35420. doi: 10.1074/jbc.M114.597575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanai R, Mashima K. Characterization of two isoforms of a human DnaJ homologue, HSJ2. Mol Biol Rep. 2003;30:149–153. doi: 10.1023/a:1024916223616. [DOI] [PubMed] [Google Scholar]
- 32.Mitra A, Fillmore RA, Metge BJ, Rajesh M, Xi Y, King J, Ju J, Pannell L, et al. Large isoform of MRJ (DNAJB6) reduces malignant activity of breast cancer. Breast Cancer Res. 2008;10:R22. doi: 10.1186/bcr1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 36.D’Mello SR, Chin PC. Treating neurodegenerative conditions through the understanding of neuronal apoptosis. Curr Drug Targets CNS Neurol Disord. 2005;4:3–23. doi: 10.2174/1568007053005118. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Ankati H, Akubathini SK, Balderamos M, Storey CA, Patel AV, Price V, Kretzschmar D, et al. Identification of novel 1,4-benzoxazine compounds that are protective in tissue culture and in vivo models of neurodegeneration. J Neurosci Res. 2010;88:1970–1984. doi: 10.1002/jnr.22352. [DOI] [PubMed] [Google Scholar]
- 38.Greene LA, Liu DX, Troy CM, Biswas SC. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta. 2007;1772:392–401. doi: 10.1016/j.bbadis.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci. 2007;8:368–378. doi: 10.1038/nrn2124. [DOI] [PubMed] [Google Scholar]
- 40.Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Memo M. Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci. 2001;24:25–31. doi: 10.1016/s0166-2236(00)01663-5. [DOI] [PubMed] [Google Scholar]
- 41.Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 42.Chopra V, Quinti L, Kim J, Vollor L, Narayanan KL, Edgerly C, Cipicchio PM, Lauver MA, et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012;2:1492–1497. doi: 10.1016/j.celrep.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Oliveira RM, Sarkander J, Kazantsev AG, Outeiro TF. SIRT2 as a Therapeutic Target for Age-Related Disorders. Front Pharmacol. 2012;3:82. doi: 10.3389/fphar.2012.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walkinshaw DR, Weist R, Xiao L, Yan K, Kim GW, Yang XJ. Dephosphorylation at a conserved SP motif governs cAMP sensitivity and nuclear localization of class IIa histone deacetylases. J Biol Chem. 2013;288:5591–5605. doi: 10.1074/jbc.M112.445668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang B, Wei W, Wang G, Gaertig MA, Feng Y, Wang W, Li XJ, Li S. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron. 2015;85:1212–1226. doi: 10.1016/j.neuron.2015.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. 2014;17(5):694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.