

Abstract
Transforming growth factor-β1 (TGF-β1) and collagen type I (Col-1) serve a critical role in the development and progression of hypertrophic scarring (HS). The present study hypothesized that a post-translational mechanism of microRNAs (miR) regulated the expression of TGF-β1 and Col-1 in HS fibroblasts (HSFBs). A collection of 20 HS tissues was compared with corresponding normal tissues from clinical patients, and the expression of miR-185 was measured. Using PicTar, TargetScan and miRBase databases, it was identified that miR-185 may be a regulator of TGF-β1 and Col-1 in humans. Based on these hypotheses, the expression of miR-185, TGF-β1 and Col-1 in HS tissues was investigated. The results demonstrated that the expression of miR-185 was markedly suppressed, and TGF-β1 and Col-1 levels were increased, in HS tissues. The expression levels of endogenous miR-185 negatively correlated with the TGF-β1 and Col-1 mRNA levels (Pearson's correlation coefficient r=−0.674, P<0.01 and r=−0.590, P<0.01, respectively). In vitro, miR-185 can regulate TGF-β1 and Col-1 through the predicted binding sites in its 3′-untranslated region. miR-185 had an effect on cell proliferation and apoptosis, thereby regulating HSFBs growth. In addition, miR-185 gain-of-function decreased TGF-β1 and Col-1 protein expression, and miR-185 loss-of-function increased TGF-β1 and Col-1 protein expression in HSFBs. In conclusion, overexpressed miR-185 could inhibit HSFBs growth, and the underlying mechanism was mediated, at least partly, through the suppression of TGF-β1 and Col-1 expression. However, above all, miR-185 might serve as a potential therapeutic approach for the treatment of HS.
Keywords: microRNA-185, transforming growth factor-β1, collagen type I, hypertrophic scarring, fibroblasts
Introduction
Hypertrophic scarring (HS) is a type of pathological scar that occurs during the healing process of a dermal wound, and is characterized by the excessive development and persistence of myofibroblasts (1). These cells contract the surrounding extracellular matrix resulting in the increased tissue density characteristic of scar tissue, which not only affects the appearance and functions of skin, however can additionally result in psychological trauma for the patient (2). The treatment of pathological scars is difficult due to a lack of effective therapeutic options and frequently involves scar revision surgery, which can induce renewed scar formation (3,4). Therefore, it was important to elucidate the underlying molecular mechanisms in HS as they may serve as novel targets for therapeutic interventions.
The pathophysiological response to wounding in adult tissue is the formation of a scar, which can be grouped temporally into three distinct phases; inflammation, proliferation and remodeling (5,6). During the inflammatory phase of healing, chemotactic and inflammatory factors are induced, which are the prerequisites for subsequent processes, including angiogenesis, re-epithelialization, recruitment, the proliferation of fibroblasts and extracellular matrix (ECM) deposition (7,8). Fibroblasts recruitment and proliferation, the production of ECM, and the inhibition of the production of proteases required to maintain the balance between production and degradation, are influenced predominantly by the fibrogenic growth factors, including insulin-like growth factor, platelet-derived growth factor and transforming growth factor-β (TGF-β) (6). Among these growth factors, TGF-β1 has been widely reported to be associated with HS formation and mediates fibroblast proliferation, collagen production, ECM deposition and myofibroblast differentiation in wound healing (9–12). In keloid fibroblasts, TGF-β1 increases the phosphorylation of Smad2/3 and Smad4 and decreases Smad7, subsequently resulting in the overabundant accumulation of collagen-1 (Col-1) (13). In addition, TGF-β1 can induce collagen production and contraction in HS-derived human skin fibroblasts (HSFBs) (9). These results indicate that TGF-β1 is a key target for the development of novel therapeutic strategies for HS. However, the underlying molecular mechanisms of microRNAs (miRs) via targeting TGF-β1 and the Col-1 contribution to the induction of HS remain to be elucidated.
The present study hypothesized that a post-translational mechanism may exist for TGF-β1 and Col-1 expression and may be regulated by miRs in HSFBs. Using PicTar, TargetScan and miRBase databases, it was indicated that miR-185 is a regulator of TGF-β1 and Col-1 in HSFBs, and its aberrant regulation has been demonstrated in a wide variety of diseases (14,15). miR-185 was analyzed for its potential use as a therapeutic target for developing novel strategies for HS prevention and treatment.
Materials and methods
Patient samples
A total of 20 HS tissues and paired corresponding adjacent normal tissues (NC) were collected from patients who had undergone surgical excision at the Shanghai Ninth People's Hospital (Shanghai Jiao Tong University, School of Medicine, Shanghai, China) between June 2012 and June 2015. The tissue samples were immediately stored at liquid nitrogen until use. Human samples were obtained with written informed consent from all patients. The present study was approved by the Ethics Committee of the Shanghai Jiao Tong University, School of Medicine.
Cell culture
HSFBs were established as described previously (16) and cultured in Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) that contained 10% fetal calf serum (Gibco; Thermo Fisher Scientific, Inc.), 10% L-glutamine, 0.5% penicillin/streptomycin, 10% nonessential amino acids and 10% pyruvate, and were incubated in a 5% CO2 atmosphere at 37°C.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cell proliferation was monitored using an MTT Cell Proliferation/Viability Assay kit (R&D Systems, Inc., Minneapolis, MN, USA), in according to the manufacturer's protocol.
Caspase-3 activity assay
HSFBs lysates were prepared and incubated with anti-caspase 3. Immunocomplexes were incubated with peptide substrate in assay buffer for 2 h at 37°C. Release of p-nitroaniline was measured at 405 nm using an ELISA reader (MD SpectraMax M5; Molecular Devices, Sunnyvale, CA, USA) according to the manufacturer's protocol.
Quantification of apoptosis by flow cytometry
Quantitative assessment of apoptotic cells was also performed by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick endlabeling (TUNEL) method, which examines DNA-strand breaks during apoptosis, using a BD ApoAlert DNA Fragmentation Assay kit (BD Biosciences, Franklin Lakes, NJ, USA). The HSFBs were trypsinized, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton-X-100 in 0.1% sodium citrate. The cells were washed with phosphate buffered saline twice and then incubated with the reaction mixture for 60 min at 37°C. Cells were immediately analyzed using a FACScan flow cytometer and the BD CellQuestTM Pro program (catalog no. 648089, BD Biosciences).
Histological observation and immunohistochemical staining
Formalin-fixed and paraffin-embedded HS tissues were cut into ~4 µm sections, which were then stained with hematoxylin and eosin and visualized under a light microscope.
Paraffin embedded HS tissues were cut into ~4 µm sections and mounted on glass slides and stained by immunoperoxidase. The paraffin sections were heated in an oven at 65°C for 24 h, then dewaxed and rehydrated and rinsed with PBS three times. The washed sections were placed in EDTA buffer for microwave antigen retrieval, were brought to the boil, then kept on a low heat after 10 min. After being allowed to cool, the sections were washed with PBS three times. The sections were added into 3% hydrogen peroxide solution and incubated at room temperature for 10 min, to block endogenous peroxidase, then washed with PBS three times, and sealed with 5% bovine serum albumin (BSA) for 20 min. Following the removal of the BSA, 50 µl diluted primary antibody, α-smooth muscle actin (α-SMA; catalog no. sc-53142; 1:50; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), transforming growth factor-β1 (TGF-β1; catalog no. sc-130348; 1:100; Santa Cruz Biotechnology, Inc.), type 1 collagen (Col-1, catalog no. sc-59772; 1:50; Santa Cruz Biotechnology, Inc.), type 3 collagen (Col-3, catalog. no. ab82354; 1:50; Abcam, Cambridge, UK), was added to each section, which was incubated overnight at 4°C, prior to being washed with PBS three times. Subsequent to the removal of PBS, 50–100 µl of the secondary antibody (goat anti-mouse IgG; catalog no. A0216; 1:5,000; Beyotime Institute of Biotechnology, Nanjing, China) was added to each section, which was then incubated at 4°C for 50 min, prior to being washed three times with PBS. To each section was added 50–100 µl freshly prepared DAB solution (under the microscope to control color). The sections were being washed and then counterstained with hematoxylin, rinsed with tap water, dehydrated and mounted and examined under a light microscope.
Luciferase reporter gene activity assay
The 3′-untranslated region (UTR) of the TGF-β1 and Col-1 gene containing the predicated target sites for miR-185 was obtained by polymerase chain reaction (PCR) amplification. The fragment was inserted into the multiple cloning sites in the pMIR-REPORT luciferase microRNA expression reporter vector (Ambion; Thermo Fisher Scientific, Inc.). HEK-293 cells (Cell Resource Center, Shanghai Institute for Biological Sciences, Shanghai, China) were co-transfected with 0.1 µg of luciferase reporters containing TGF-β1 and Col-1 3′-UTR and miR-329 mimics by Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.). The cell lysates were harvested after 48 h transfection and the luciferase activity measured with a dual luciferase reporter assay kit (catalog no. RG027; Beyotime Institute of Biotechnology) according to manufacturer's protocol.
Transfection of miR-185 mimics and inhibitor
The fluorescein amidite-modified 2′-OMe-oligonucleotides were chemically synthesized and purified by high-performance liquid chromatography (Shanghai GenePharma Co., Ltd., Shanghai, China). The 2′-OMe-miR-185 mimics were composed of RNA duplexes with the following sequence: 5′-UGGAGAGAAAGGCAGUUCCUGA-3′. The sequences of 2′-OMe-miR-185 inhibitor and 2′-Ome-scramble oligonucleotides were as follows: 5′-UCAGGAACUGCCUUUCUCUCCA-3′; and 5′-ACUUUCAGAGGUCUUGACCUAG-3′. Cells were transfected using Lipofectamine2000 (Invitrogen; Thermo Fisher Scientific, Inc.) at a final concentration of 50 nM. At 24 h following transfection, the culture medium was changed. After 48 h, the cells were harvested for analysis.
Reverse transcription-quantitative PCR (RT-qPCR)
RNA extraction was performed according to the TRIzol manufacturer's (Invitrogen; Thermo Fisher Scientific, Inc.) protocol. A reaction mixture (20 µl) containing 4 µg of total RNA was used to reverse transcribe the RNA to cDNA using moloney murine leukemia virus reverse transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.) with oligo dT (15) primers (Fermentas; Thermo Fisher Scientific, Inc., Pittsburgh, PA, USA) as described by the manufacturer's protocol. The miR-185 level was quantified by the mirVana RT-qPCR miR detection kit (Ambion; Thermo Fisher Scientific, Inc.) in conjunction with RT-qPCR with SYBR-Green (Roche Diagnostics GmbH, Mannheim, Germany). Following the cycle reaction, the threshold cycle (Ct) was determined and relative miR-329 level was calculated based on the Ct values and normalized to U6 level in each sample, as described previously (17). PCR with the following primers: TGF-β1, forward 5′-CGAAATCTAGACAAGTTCAAGCAG-3′ and reverse 5′-GAGGTATCGCCAGGAATTGTTG-3′; Col1a1, forward 5′-GAGGGCAACAGCAGGTTCACTTA-3′ and reverse 5′-TCAGCACCACCGATGTCCA-3′; GAPDH, forward 5′-GCACCGTCAAGCTGAGAAC-3′ and reverse 5′-TGGTGAAGACGCCAGTGGA-3′.
Western blotting
HS tissues and HSFBs were homogenized and extracted in NP-40 buffer, followed by 5–10 min boiling and centrifugation (10,000 × g, 4°C, 10 min) to obtain the supernatant. Samples containing 30 µg of protein were separated on 10% SDS-PAGE gel and then transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA). After saturation with 5% (w/v) non-fat dry milk in TBS and 0.1% (w/v) Tween-20 (TBST), the membranes were incubated with the primary antibodies: TGF-β1 (catalog no. sc-130348; 1:1,000; Santa Cruz Biotechnology, Inc.), Col-1 (catalog no. sc-59772; 1:500; Santa Cruz Biotechnology, Inc.) and GAPDH (catalog no. sc-51907; 1:2,000; Santa Cruz Biotechnology, Inc.). The membranes were washed three with TBST and then incubated with the appropriate horseradish peroxidase-conjugated antibody (goat anti-mouse IgG; catalog no. A0216; 1:5,000; Beyotime Institute of Biotechnology) and visualized with chemiluminescence (Thermo Labsystems, Santa Rosa, CA, USA).
Statistical analysis
The data from these experiments were reported as the mean ± standard deviation for each group. All statistical analyses were performed by using PRISM version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Inter-group differences in genes expression between HS and NC were assessed by the Paired-Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
Histological observation
In the lesioned tissues (HS group), the collagen bundles were running parallel to the epidermis in a large part of the dermis. However, the thickness and complexity of the collagen bundles were reduced in the control group compared with the HS group (Fig. 1A). This indicated that a large number of fibroblasts, collagen tissue and blood vessels were induced to increase with the development of HS. To further investigate the induction of fibrogenesis in HS, α-smooth muscle actin (SMA) expression levels in lesioned tissues were measured by immunohistochemical staining. The results demonstrated that α-SMA was significantly increased in the HS group compared with the NC group (Fig. 1B).
Figure 1.
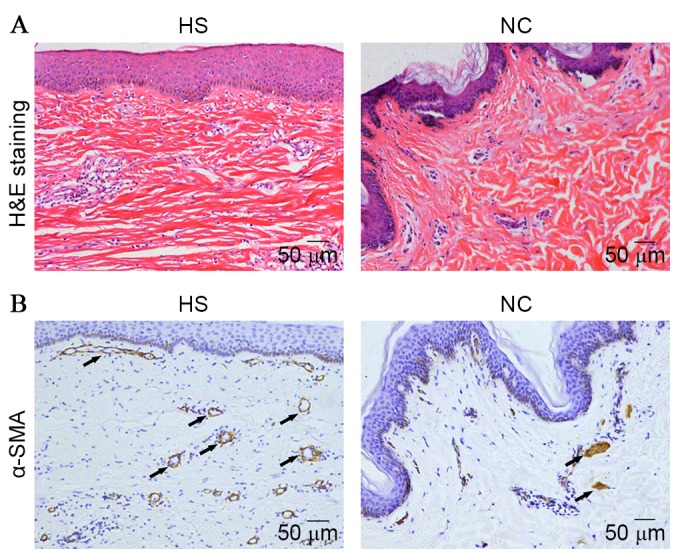
The histological observation on hypertrophic scarring tissues and paired corresponding adjacent normal tissues from clinical patients. (A) H&E staining (magnification, ×100). (B) α-SMA expression measured by immunohistochemical staining (magnification, ×100). H&E, hematoxylin and eosin; α-SMA, α-smooth muscle actin; HS, hypertrophic scarring tissues; NC, corresponding adjacent normal tissues.
miR-185 and TGF-β1 expression in HS
To explore the miR expression profiles in HS tissues, the miR expression between HS tissues and adjacent normal tissues was compared. miR-185 was notably less expressed in HS tissues compared with adjacent normal tissues. In addition, PicTar, TargetScan and miRBase databases suggested that miR-185 was a regulator of TGF-β1. Based on these results, the expression of miR-185 and TGF-β1 in HS tissues was investigated. The investigation led to the conclusion that the expression of miR-185 was markedly suppressed in HS tissues (Fig. 2A) while the expression TGF-β1 was increased; this was confirmed by RT-qPCR (Fig. 2B), western blotting (Fig. 2C) and immunohistochemical staining assay (Fig. 2D and E). To validate the correlation between miR-185 and TGF-β1, the TGF-β1 mRNA and miR-185 expression levels were analyzed in 20 human HS tissue samples using RT-qPCR. As presented in Fig. 2F, the expression levels of endogenous miR-185 negatively correlated with the TGF-β1 mRNA levels (Pearson's correlation coefficient r=−0.674; P<0.01). These data provide further evidence of a functional link between miR-185 and TGF-β1 in HS tissues. In addition, Col-1 was investigated as an additional predicted target of miR-185 in HS tissues. Immunohistochemical staining demonstrated that HS tissues indicated enhanced expression of Col-1 and Col-3 compared with normal adjacent tissues (Fig. 3A). Subsequently, Col-1 mRNA was measured by RT-qPCR, and there was an evident difference between the HS and NC groups (Fig. 3B). The correlation between miR-185 and Col-1 was investigated and the expression levels of endogenous miR-185 negatively correlated with the Col-1 mRNA levels (Pearson's correlation coefficient r=−0.590, P<0.01; Fig. 3C).
Figure 2.

miR-185 expression was examined by RT-qPCR and normalized to U6 expression in 20 HS tissues and paired NC tissues. (A) mRNA and (B and C) protein expression of TGF-β1 were examined by RT-qPCR and western blotting respectively, and normalized to GAPDH expression. (D) TGF-β1 expression was measured by immunohistochemical staining in HS and NC tissues (magnification, ×100), and (E) the histogram represented the mean integral optical density of TGF-β1 in HS and NC tissues. (F) Inverse correlation of miR-185 and TGF-β1 expression in HS tissues. miR, microRNA; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; HS, hypertrophic scarring tissues; NC, corresponding adjacent normal tissues; TGF-β1, transforming growth factor-β1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IOD, integral optical density.
Figure 3.

Col-1 and Col-3 expression were measured by (A) immunohistochemical staining in HS and NC tissues (magnification, ×100). (B) mRNA expression of Col-1 was examined by RT-qPCR. (C) Inverse correlation of miR-185 and Col-1 expression in HS tissues. Col, collagen; HS, hypertrophic scarring tissues; NC, corresponding adjacent normal tissues; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; miR, microRNA.
ΜicroRNA-185 is a regulator of TGF-β1 and Col-1
To determine whether miR-185 takes effect through direct binding to the 3′-UTR of TGF-β1 and Col-1, the alignment predicted by the TargetScan algorithm was evaluated and the potential miR-185 binding sites within the 3′-UTR of TGF-β1 and Col-1 in humans were identified (Fig. 4A). To further determine whether miR-185 regulated TGF-β1 and Col-1 through the predicted binding sites in its 3′-UTR, the 3′-UTR of TGF-β1 and Col-1 containing the potential miR-185 binding site was cloned for use in a firefly luciferase reporter assay. A construct carrying a luciferase gene upstream of the wild-type 3′-UTR or the mutant 3′-UTR (containing mismatches in the seed region of the binding site) was utilized. Cotransfection with this luciferase construct containing wild-type 3′-UTR and miR-185 mimics resulted in a ~60% decline in the luciferase activity compared with the control group. Following the cotransfection of HEK293 cells with this recombination structure containing mutant 3′-UTR of TGF-β1 and Col-1, no significant difference was observed in the reporter activity compared with the control group (Fig. 4B).
Figure 4.

(A) Schematic representation of the putative miR-185 binding site in the TGF-β1 and Col-1 3′-UTR as in TargetScan and (B) luciferase activity assay. miR, microRNA; TGF-β1, transforming growth factor-β1; Col, collagen; UTR, untranslated region.
miR-185 regulates TGF-β1 and Col-1 expression in HSFBs
In HSFBs, the miR-185 mimics or inhibitor was transfected to evaluate the potential effect on cell proliferation and apoptosis of miR-185 in vitro. HSFBs cell viability was measured by MTT assay when cells were transfected with miR-185 mimics or inhibitor for 24, 48 and 72 h. The viability of HSFBs transfected with miR-185 mimics was noticeably lower compared with those of the untreated group. However, miR-185 loss-of-function increased the growth of HSBFs compared with the scramble cells (Fig. 5A). In addition, whether miR-185 mimics or inhibitor induced apoptosis in HSFBs through an apoptotic mechanism was examined. Caspase-3 activity assay and TUNEL staining were measured after HSFBs were transfected with miR-185 mimics or inhibitor for 48 h. The results indicated that the caspase-3 activity in the miR-185 mimics group were noticeably higher compared with the control group. However, HSFBs transfected with miR-185 inhibitor suppressed caspase-3 activity (Fig. 5B). TUNEL staining demonstrated that miR-185 gain-of-function induced cell apoptosis and that miR-185 loss-of-function suppressed cell apoptosis in HSFBs (Fig. 5C). These results suggested that miR-185 exercised an effect on cell proliferation and apoptosis, thereby regulating growth of HSFBs. The protein expression of TGF-β1 and Col-1 was measured by western blotting in HSFBs transfected with miR-185 mimics or inhibitor for 48 h. The results demonstrated that miR-185 gain-of-function decreased TGF-β1 and Col-1 protein expression, and that miR-185 loss-of-function increased TGF-β1 and Col-1 protein expression in HSFBs (Fig. 5D).
Figure 5.

(A) MTT assays were performed on HSFBs transfected with miR-185 mimics or inhibitor for 24, 48 and 72. (B) Caspase-3 activity assay and (C) TUNEL staining were measured after HSFBs were transfected with miR-185 mimics or inhibitor for 48 h. (D) The protein expression of TGF-β1 and Col-1 was measured by western blotting in HSFBs transfected with miR-185 mimics or inhibitor for 48 h. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; HSFBs, hypertrophic scarring tissue fibroblasts; miR, microRNA; TUNEL, triphosphate nick-end labeling; TGF-β1, transforming growth factor-β1; Col-1, collagen-1; OD, optical density; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Discussion
miR-185, located on 22q11.21, has been reported to be dysregulated in several malignant tumors and acts as a tumor suppressor in non-small cell lung cancer (18,19), breast cancer (20), hepatocellular carcinoma (21) and gastric cancer (22). In addition, miR-185 can regulate the expression of lipid metabolism genes and improves insulin sensitivity in mice with non-alcoholic fatty liver disease (23). Notably, miR-185 is involved in dermal wound repair and fibroblast migration (24). These results suggest that miR-185 provide novel insights into the underlying mechanism and therapeutic strategies for damaged tissues.
The precise mechanisms using the regulation of TGF-β1 and Col-1 expression in HS patients remain to be elucidated, however the results obtained in the present study suggest that this induction is provoked as a consequence of miRs dysfunction. The present study examined miR-185 expression levels in HS tissues from clinical patients and the results indicated that miR-185 was markedly underexpressed in HS tissues compared with adjacent normal tissues. In addition, PicTar, TargetScan, and miRBase databases indicated that miR-185 was a regulator of TGF-β1 and Col-1. The predicted alignment was evaluated and the potential miR-185 binding sites within the 3′-UTR of TGF-β1 and Col-1 in humans were identified. A construct carrying a luciferase gene upstream of the wild-type 3′-UTR or the mutant 3′-UTR was utilized and revealed that miR-185 mimics targeting 3′-UTR of TGF-β1 and Col-1 resulted in a lower luciferase activity in HEK293 cells. In addition, the mRNA and protein expression of TGF-β1 and Col-1 were significantly increased in HS tissues compared with adjacent normal tissues. In vitro, miR-185 gain-of-function decreased TGF-β1 and Col-1 protein expression, and miR-185 loss-of-function increased TGF-β1 and Col-1 protein expression in HSFBs. These results suggested that overexpressed miR-185 can inhibit HSFBs growth and that the underlying mechanism was mediated, at least partly, through the suppression of TGF-β1 and Col-1 expression. HS occurs as a result of a pathological wound-healing process, characterized by excess collagen deposition and hyperproliferation of fibroblasts (2,6,8).
Numerous biological actions of TGF-β1 contribute to the normal wound-healing process and have been implicated in a wide variety of fibrotic disorders. Immediately following injury, high levels of TGF-β are released and become involved in collagen production and ECM deposition (5,9). In addition, TGF-β1 can induce collagen production and contraction in hypertrophic scar-derived human skin fibroblasts (9). TGF-β1 has been demonstrated to upregulate the expression of α-SMA and stimulate normal fibroblasts into differentiating into myofibroblasts (25). In the present study, α-SMA expression levels in lesioned tissues were measured by immunohistochemical staining and were markedly increased in the HS group compared with the NC group. It has been previously demonstrated that α-SMA is associated with tissue fibrosis (26). During wound repair and skin regeneration, myofibroblasts, as a specialized subgroup of cells with the features of smooth muscle cells, can induce α-SMA expression (27). In addition, TGF-β1 can induce α-SMA expression in human dermal fibroblasts (28). Notably, TGF-β1-Smad2/3 and TGF-β1-extracellular signal-related kinase/c-Jun n-terminal kinase signaling pathways can promote HS formation through the acceleration of collagen production and ECM deposition (9,29). In the present study, immunohistochemical staining of HS tissues demonstrated enhanced expression of Col-1 and Col-3 compared with NC tissues, simultaneously it was demonstrated that Col-1 mRNA expression was clearly increased in the HS group compared with the NC group.
In conclusion, the data reported in the present study indicated that overexpressed miR-185 can inhibit HSFBs growth and that the underlying mechanism is mediated, at least in part, through the suppression of TGF-β1 and Col-1 expression. However, notably, miR-185 may serve a potential therapeutic strategy for the treatment of HS.
Acknowledgements
The current study was supported by the National Natural Science Foundation of China (grant no. 81272109).
References
- 1.Zhang H, Liu X, Liu Y, Wu Y, Li H, Zhao C, Li H, Meng Q, Li W. Effect of hematoporphyrin monomethyl ether-sonodynamic therapy (HMME-SDT) on hypertrophic scarring. PLoS One. 2014;9:e86003. doi: 10.1371/journal.pone.0086003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crawford J, Nygard K, Gan BS, O'Gorman DB. Periostin induces fibroblast proliferation and myofibroblast persistence in hypertrophic scarring. Exp Dermatol. 2015;24:120–126. doi: 10.1111/exd.12601. [DOI] [PubMed] [Google Scholar]
- 3.Gras C, Ratuszny D, Hadamitzky C, Zhang H, Blasczyk R, Figueiredo C. miR-145 contributes to hypertrophic scarring of the skin by inducing myofibroblast activity. Mol Med. 2015;21:296–304. doi: 10.2119/molmed.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kathju S, Gallo PH, Satish L. Scarless integumentary wound healing in the mammalian fetus: Molecular basis and therapeutic implications. Birth Defects Res C Embryo Today. 2012;96:223–236. doi: 10.1002/bdrc.21015. [DOI] [PubMed] [Google Scholar]
- 5.Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies. Mol Med. 2011;17:113–125. doi: 10.2119/molmed.2009.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niessen FB, Spauwen PH, Schalkwijk J, Kon M. On the nature of hypertrophic scars and keloids: a review. Plast Reconstr Surg. 1999;104:1435–1458. doi: 10.1097/00006534-199910000-00031. [DOI] [PubMed] [Google Scholar]
- 7.Nirodi CS, Devalaraja R, Nanney LB, Arrindell S, Russell S, Trupin J, Richmond A. Chemokine and chemokine receptor expression in keloid and normal fibroblasts. Wound Repair Regen. 2000;8:371–382. doi: 10.1111/j.1524-475X.2000.00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahmani-Neishaboor E, Yau FM, Jalili R, Kilani RT, Ghahary A. Improvement of hypertrophic scarring by using topical anti-fibrogenic/anti-inflammatory factors in a rabbit ear model. Wound Repair Regen. 2010;18:401–408. doi: 10.1111/j.1524-475X.2010.00598.x. [DOI] [PubMed] [Google Scholar]
- 9.Fan C, Dong Y, Xie Y, Su Y, Zhang X, Leavesley D, Upton Z. Shikonin reduces TGF-β1-induced collagen production and contraction in hypertrophic scar-derived human skin fibroblasts. Int J Mol Med. 2015;36:985–991. doi: 10.3892/ijmm.2015.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Jiang L, Li H, Shi H, Luo H, Zhang Y, Yu C, Jin Y. Mesenchymal stem cells prevent hypertrophic scar formation via inflammatory regulation when undergoing apoptosis. J Invest Dermatol. 2014;134:2648–2657. doi: 10.1038/jid.2014.169. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Chu J, Wen CJ, Fu SB, Qian YL, Wo Y, Wang C, Wang DR. Functional characterization of TRAP1-like protein involved in modulating fibrotic processes mediated by TGF-β/Smad signaling in hypertrophic scar fibroblasts. Exp Cell Res. 2015;332:202–211. doi: 10.1016/j.yexcr.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Li W, Ning Y, Liu T, Shao J, Wang Y. Ski diminishes TGF-β1-induced myofibroblast phenotype via up-regulating Meox2 expression. Exp Mol Pathol. 2014;97:542–549. doi: 10.1016/j.yexmp.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Wu CS, Wu PH, Fang AH, Lan CC. FK506 inhibits the enhancing effects of transforming growth factor (TGF)-β1 on collagen expression and TGF-β/Smad signalling in keloid fibroblasts: Implication for new therapeutic approach. Br J Dermatol. 2012;167:532–541. doi: 10.1111/j.1365-2133.2012.11023.x. [DOI] [PubMed] [Google Scholar]
- 14.Tang H, Liu Q, Liu X, Ye F, Xie X, Xie X, Wu M. Plasma miR-185 as a predictive biomarker for prognosis of malignant glioma. J Cancer Res Ther. 2015;11:630–634. doi: 10.4103/0973-1482.146121. [DOI] [PubMed] [Google Scholar]
- 15.Jing R, Chen W, Wang H, Ju S, Cong H, Sun B, Jin Q, Chu S, Xu L, Cui M. Plasma miR-185 is decreased in patients with esophageal squamous cell carcinoma and might suppress tumor migration and invasion by targeting RAGE. Am J Physiol Gastrointest Liver Physiol. 2015;309:G719–G729. doi: 10.1152/ajpgi.00078.2015. [DOI] [PubMed] [Google Scholar]
- 16.Kashiyama K, Mitsutake N, Matsuse M, Ogi T, Saenko VA, Ujifuku K, Utani A, Hirano A, Yamashita S. miR-196a downregulation increases the expression of type I and III collagens in keloid fibroblasts. J Invest Dermatol. 2012;132:1597–1604. doi: 10.1038/jid.2012.22. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi Y, Forrest AR, Maeno E, Hashimoto T, Daub CO, Yasuda J. MiR-107 and MiR-185 can induce cell cycle arrest in human non small cell lung cancer cell lines. PLoS One. 2009;4:e6677. doi: 10.1371/journal.pone.0006677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Ma Y, Hou X, Liu Y, Li K, Xu S, Wang J. miR-185 acts as a tumor suppressor by targeting AKT1 in non-small cell lung cancer cells. Int J Clin Exp Pathol. 2015;8:11854–11862. [PMC free article] [PubMed] [Google Scholar]
- 20.Tang H, Liu P, Yang L, Xie X, Ye F, Wu M, Liu X, Chen B, Zhang L, Xie X. miR-185 suppresses tumor proliferation by directly targeting E2F6 and DNMT1 and indirectly upregulating BRCA1 in triple-negative breast cancer. Mol Cancer Ther. 2014;13:3185–3197. doi: 10.1158/1535-7163.MCT-14-0243. [DOI] [PubMed] [Google Scholar]
- 21.Qadir XV, Han C, Lu D, Zhang J, Wu T. miR-185 inhibits hepatocellular carcinoma growth by targeting the DNMT1/PTEN/Akt pathway. Am J Pathol. 2014;184:2355–2364. doi: 10.1016/j.ajpath.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan Z, Jiang H, Wu Y, Xie L, Dai W, Tang H, Tang S. miR-185 is an independent prognosis factor and suppresses tumor metastasis in gastric cancer. Mol Cell Biochem. 2014;386:223–231. doi: 10.1007/s11010-013-1860-y. [DOI] [PubMed] [Google Scholar]
- 23.Wang XC, Zhan XR, Li XY, Yu JJ, Liu XM. MicroRNA-185 regulates expression of lipid metabolism genes and improves insulin sensitivity in mice with non-alcoholic fatty liver disease. World J Gastroenterol. 2014;20:17914–17923. doi: 10.3748/wjg.v20.i47.17914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang W, Yee AJ. Versican 3′-untranslated region (3′UTR) promotes dermal wound repair and fibroblast migration by regulating miRNA activity. Biochim Biophys Acta. 2014;1843:1373–1385. doi: 10.1016/j.bbamcr.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 25.Varga J, Abraham D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madala SK, Edukulla R, Phatak M, Schmidt S, Davidson C, Acciani TH, Korfhagen TR, Medvedovic M, Lecras TD, Wagner K, Hardie WD. Dual targeting of MEK and PI3K pathways attenuates established and progressive pulmonary fibrosis. PLoS One. 2014;9:e86536. doi: 10.1371/journal.pone.0086536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng L, Hui Q, Tang L, Zheng L, Jin Z, Yu B, Wang Z, Lin P, Yu W, Li H, et al. TAT-mediated acidic fibroblast growth factor delivery to the dermis improves wound healing of deep skin tissue in rat. PLoS One. 2015;10:e0135291. doi: 10.1371/journal.pone.0135291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldberg MT, Han YP, Yan C, Shaw MC, Garner WL. TNF-alpha suppresses alpha-smooth muscle actin expression in human dermal fibroblasts: An implication for abnormal wound healing. J Invest Dermatol. 2007;127:2645–2655. doi: 10.1038/sj.jid.5700890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He T, Bai X, Yang L, Fan L, Li Y, Su L, Gao J, Han S, Hu D. Loureirin B inhibits hypertrophic scar formation via inhibition of the TGF-β1-ERK/JNK pathway. Cell Physiol Biochem. 2015;37:666–676. doi: 10.1159/000430385. [DOI] [PubMed] [Google Scholar]