

Abstract
The present study aimed to analyze RNA-seq data of kidney renal clear cell carcinoma (KIRC) to identify prognostic genes. RNA-seq data were downloaded from The Cancer Genome Atlas. Feature genes with a coefficient of variation (CV) >0.5 were selected using the genefilter package in R. Gene co-expression networks were constructed with the WGCNA package. Cox regression analysis was performed using the survive package. Furthermore, a functional enrichment analysis was conducted using Database for Annotation, Visualization and Integrated Discovery tools. A total of 533 KIRC samples were collected, from which 6,758 feature genes with a CV >0.5 were obtained for further analysis. The KIRC samples were divided into two sets: The training set (n=319 samples) and the validation set (n=214 samples). Subsequently, gene co-expression networks were constructed for the two sets. A total of 12 modules were identified, and the green module was significantly associated with survival time. Genes from the green module were revealed to be implicated in the cell cycle and p53 signaling pathway. In addition, a total of 11 hub genes were revealed, and 10 of them (CCNA2, CDC20, CDCA8, GTSE1, KIF23, KIF2C, KIF4A, MELK, TOP2A and TPX2) were validated as possessing prognostic value, as determined by conducting a survival analysis on another gene expression dataset. In conclusion, a total of 10 prognostic genes were identified in KIRC. These findings may help to advance the understanding of this disease, and may also provide potential biomarkers for therapeutic development.
Keywords: kidney renal clear cell carcinoma, gene co-expression network, survival analysis, hub genes
Introduction
Kidney renal clear cell carcinoma (KIRC) is the eighth most common type of cancer, which accounts for the majority of malignant kidney tumors (1). KIRC is known to be associated with radiotherapy and chemotherapy resistance (2), and the 2-year survival rate of patients with metastatic KIRC is <20% (3,4). Early diagnosis and surgical resection may result in a good prognosis; therefore, further investigations regarding the genomic alterations and underlying molecular mechanisms of KIRC are essential for improvements in early diagnosis and treatment.
Certain advances have been made in unveiling the complicated molecular mechanisms underlying KIRC, since numerous relevant pathways have been implicated in its pathogenesis. Components of the mammalian target of rapamycin pathway have been reported to be significantly associated with the pathological features and survival of KIRC (5). Frequent mutations in genes encoding ubiquitin-mediated proteolysis pathway components have also been observed in KIRC (6). The Sonic hedgehog signaling pathway (7) and MYC pathway (8) are also activated in KIRC and serve a role in tumor growth. Furthermore, numerous biomarkers have been identified, including cluster of differentiation 70 (8), succinate dehydrogenase B (8) and transforming growth factor beta 1 (9). Nevertheless, further studies are required to identify novel prognostic genes and provide potential therapeutic targets.
Previous studies have focused on the identification of differentially expressed genes, which may serve roles in the pathogenesis of KIRC (10,11). The present study performed a gene co-expression network analysis and a survival analysis on RNA-seq data in order to screen out prognostic genes in KIRC. These findings may help improve understanding regarding the pathogenesis of KIRC, and also provide potential markers for prognosis and treatment.
Materials and methods
Gene expression data
RNA-seq (Illumina RNASeqV2, Level 3; Illumina, San Diego, CA, USA) rsem.gene.results data of KIRC were downloaded from The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/) on September 25, 2015, including 533 KIRC samples. Clinical information, including status, follow-up time and time of death, was also collected.
Screening of feature genes
Raw data were normalized and filtered using the TCGAbiolinks package in R (version 3.2.2, http://www.r-project.org/). Genes with an average expression level <0.25 in all samples were excluded from the subsequent analyses. Feature genes with a coefficient of variation (CV) >0.5 in all samples were selected using the genefilter package in R.
Construction of a gene co-expression network
The KIRC samples were divided into two sets: The training set (n=319 samples) and the validation set (n=214 samples), with a ratio of 3:2 using the caTools package in R.
Gene co-expression networks were constructed using the weighted gene co-expression network analysis (WGCNA) (12) package in R. Adjacency coefficient (aij) was calculated as follows:
Where xi and xj are vectors of expression value for genes i and j; cor represents Pearson's correlation coefficient of the two vectors; and aij is adjacency coefficient, which is acquired via exponential transform of Sij.
The WGCNA method takes topological properties into consideration in order to identify modules from a gene co-expression network. Therefore, this method not only considers the relationship between two connected nodes, but also takes associated genes into account. Weighting coefficient (Wij) is calculated from aij as follows:
Where u represents common genes linked gene I and gene j together; aiu, the connection coefficient of gene i and gene u; and auj, the connection coefficient of gene u and gene j. Wij considers overlapping between neighbor genes of genes i and j. Modules were identified via hierarchical clustering of weighting coefficient matrix W.
Survival analysis
A univariate Cox regression analysis was performed using the survive package in R.
Functional enrichment analysis
Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed using DAVID (Database for Annotation, Visualization and Integration Discovery; http://david.abcc.ncifcrf.gov/) (13).
Validation of the hub genes
A KIRC gene expression dataset (accession no. E-GEOD-22541) was downloaded from ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) to validate the reliability of the 11 hub genes. Cases were divided into two groups (high and low) based upon the expression levels of certain hub genes, using the gene average expression level in all samples as the cut-off. The samples in which the gene expression level were higher than average expreesion level were defined as high exp; the other samples were defined as low exp, correspondingly. Survival analysis was performed using the Kaplan-Meier method.
Results
Feature genes
A total of 533 KIRC samples were collected from TCGA. After pretreatment, 13,742 genes were selected according to the threshold (average expression level >0.25 in all samples). Finally, 6,758 feature genes with a CV >0.5 were acquired for further analysis.
Gene co-expression network
The training set included 319 samples and the validation set contained 214 samples. The training set was used to construct a gene co-expression network, whereas the validation set was used to examine the stability and accuracy of the network. The soft threshold was set as 5 to construct the network (Fig. 1).
Figure 1.

(A) Scale-free fit R2 vs. various soft thresholds. The red line indicates an R2 of 0.85. (B) Mean Connectivity vs. different soft threshold β.
When the soft threshold was set as 5, both training set and validation set networks obeyed power-law distribution, exhibiting scale-free characteristics (Fig. 2). The correlation coefficient between the two networks was 0.75, when the soft threshold was 5.
Figure 2.

Distribution of genes in terms of degree (soft threshold, 5). (A) Training set; (B) Validation set; X-axis indicates degree k; Y-axis indicates percentage of genes with degree k. (C) Correlation between the training dataset and validation dataset co-expression networks. The x-axis indicates degree k in the training dataset; y-axis indicates degree k in the validation dataset.
Survival-related modules
A total of 12 modules were revealed using the cuttreeStaticColor function from WGCNA package (cutHeight=0.93; minSize=50) (Fig. 3). A Cox regression analysis was performed for each gene in both datasets and a P-value was obtained. Hub genes may serve critical roles in disease; therefore, degree (k) was also calculated for each gene. The correlation between k and -log10(p) was subsequently determined. Survival-associated genes were significantly over-represented in the green module (Fig. 4).
Figure 3.

Results of a cluster analysis, and 12 modules identified from the gene expression networks. (A) Training set; (B) validation set. Gray represents no module.
Figure 4.

Enrichment of survival-associated genes in each module. (A) Training set; (B) validation set. X-axis indicates modules; Y-axis indicates significance of enrichment.
Biological functions of the green module
Significantly over-represented GO biological process terms (Table I) and KEGG pathways (Table II) were identified for genes from the green module. The cell cycle and p53 signaling pathway were revealed to be closely associated with KIRC.
Table I.
Top 10 GO biological process terms of genes from the green module.
| No. | Biological process | Count | P-value |
|---|---|---|---|
| GO:0007049 | Cell cycle | 98 | 1.85E-74 |
| GO:0022403 | Cell cycle phase | 79 | 3.66E-72 |
| GO:0000279 | M phase | 73 | 4.21E-71 |
| GO:0022402 | Cell cycle process | 83 | 1.43E-66 |
| GO:0000278 | Mitotic cell cycle | 68 | 7.52E-60 |
| GO:0000280 | Nuclear division | 56 | 8.53E-57 |
| GO:0007067 | Mitotic nuclear division | 56 | 8.53E-57 |
| GO:0000087 | M phase of mitotic cell cycle | 56 | 2.56E-56 |
| GO:0048285 | Organelle fission | 56 | 9.80E-56 |
| GO:0051301 | Cell division | 54 | 1.95E-46 |
GO, gene ontology.
Table II.
Significantly over-represented Kyoto Encyclopedia of Genes and Genomes pathways of genes from the green module.
| No. | Pathway | Count | P-value |
|---|---|---|---|
| hsa04110 | Cell cycle | 25 | 2.63E-23 |
| hsa04114 | Oocyte meiosis | 13 | 7.35E-09 |
| hsa04914 | Progesterone-mediated oocyte maturation | 11 | 8.09E-08 |
| hsa04115 | p53 signaling pathway | 7 | 1.86E-04 |
| hsa03440 | Homologous recombination | 5 | 3.73E-04 |
Hub genes in the green module
A total of 202 genes were included in the green module. Genes with P<0.01 in the Cox regression analysis of the training and validation sets were selected. The intramodular degree (kWithin) was then calculated for each gene. The top 20 genes in the training and validation sets were subsequently obtained. The overlapping genes were regarded as hub genes. A total of 11 hub genes were identified (Table III): Cyclin A2 (CCNA2), cyclin B2 (CCNB2), cell division cycle 20 (CDC20), cell division cycle associated 8 (CDCA8), G2 and S-phase expressed 1 (GTSE1), kinesin family member 23 (KIF23), kinesin family member 2C (KIF2C), kinesin family member 4A (KIF4A), maternal embryonic leucine zipper kinase (MELK), topoisomerase II alpha (TOP2A) and TPX2 microtubule-associated (TPX2).
Table III.
Summary of the 11 hub genes.
| P-value | k Total | k Within | ||||
|---|---|---|---|---|---|---|
| Gene | T set | V set | T set | V set | T set | V set |
| CCNA2 | 2.29E-06 | 8.15E-11 | 85.594 | 57.123 | 68.745 | 48.839 |
| CCNB2 | 9.08E-07 | 1.89E-08 | 94.399 | 68.515 | 72.728 | 55.378 |
| CDC20 | 6.17E-08 | 1.27E-08 | 93.507 | 60.032 | 74.198 | 50.181 |
| CDCA8 | 2.76E-05 | 5.21E-08 | 89.649 | 64.707 | 73.107 | 52.065 |
| GTSE1 | 1.88E-06 | 1.30E-08 | 93.828 | 63.922 | 73.780 | 53.611 |
| KIF23 | 3.21E-08 | 1.07E-08 | 91.183 | 60.441 | 69.097 | 48.626 |
| KIF2C | 3.00E-07 | 8.09E-08 | 88.153 | 64.374 | 70.517 | 54.608 |
| KIF4A | 1.14E-04 | 4.07E-08 | 92.184 | 63.336 | 69.749 | 51.397 |
| MELK | 9.74E-07 | 2.37E-07 | 85.264 | 60.536 | 69.125 | 52.317 |
| TOP2A | 3.88E-08 | 1.72E-08 | 88.265 | 61.531 | 72.680 | 53.977 |
| TPX2 | 7.24E-07 | 1.40E-08 | 88.309 | 68.001 | 71.906 | 57.164 |
T set, training set; V set, validation set; CCNA2, cyclin A2; CCNB2, cyclin B2; CDC20, cell division cycle 20; CDCA8, cell division cycle associated 8; GTSE1, G2 and S-phase expressed 1; KIF23, kinesin family member 23; KIF2C, kinesin family member 2C; KIF4A, kinesin family member 4A; MELK, maternal embryonic leucine zipper kinase; TOP2A, topoisomerase II alpha; TPX2, TPX2 microtubule-associated.
Validation of the hub genes
With the exception of CCNB2, the other 10 hub genes exhibited good prognostic effects in the validation dataset E-GEOD 22541. The Kaplan-Meier survival curve of CCNA2 is presented in Fig. 5.
Figure 5.
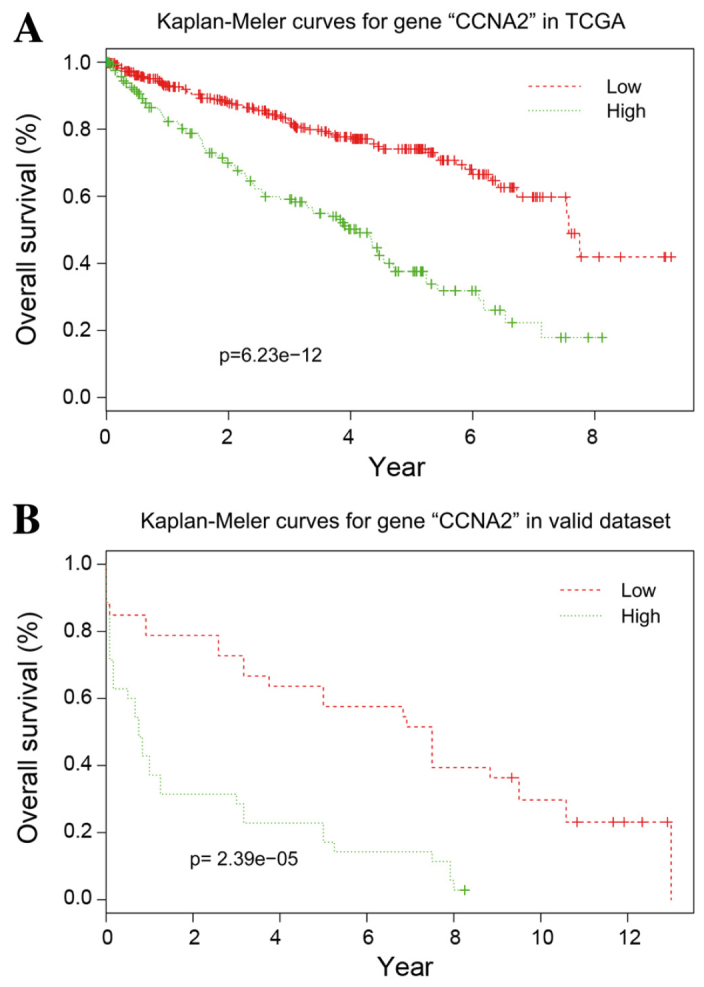
Kaplan-Meier survival curves of CCNA2. Based on gene expression data from (A) TCGA and the (B) E-GEOD-22541 dataset. CCNA2, cyclin A2; TCGA, the Cancer Genome Atlas.
Discussion
In the present study, a total of 533 KIRC samples were collected from TCGA and 6,758 feature genes were revealed, based upon which gene co-expression networks were constructed. A total of 12 modules were identified; however, only one module (green) was significantly associated with survival time. The green module included 202 genes, which were implicated in the cell cycle and p53 signaling pathway. Finally, a total of 11 hub genes were revealed by network analysis combined with survival analysis; 10 of which were validated using another gene expression dataset.
The majority of the validated hub genes were involved in the cell cycle, including CCNA2, CDC20 and CDCA8. CDC20 acts as a regulatory protein at numerous points in the cell cycle. It is negatively regulated by p53 and may be considered a good potential therapeutic target (14). Increased TOP2A expression is associated with more aggressive pathological features and an increased risk of cancer-specific mortality among patients undergoing surgery for localized KIRC (15). Chen et al indicated that TOP2A is a prognostic marker in advanced renal cell carcinoma (16). Furthermore, overexpression of TOP2A has been reported in other types of cancer (17,18) and is considered a therapeutic target (19). The results of the present study indicated that it may also be a therapeutic target in KIRC. GTSE1 accumulates in the nucleus and binds to p53, resulting in its translocation out of the nucleus and suppression of its apoptosis-inducing ability. In addition, GTSE1 suppresses apoptotic signaling and confers cisplatin resistance in gastric cancer cells (20). Overexpression of GTSE1 has previously been observed in KIRC (21) and may therefore exert a similar function in KIRC.
Several prognostic genes have been implicated in various types of cancer; however, their roles in KIRC require further research. Kinesins are a family of molecular motor proteins that travel along microtubule tracks in order to fulfill their numerous roles in intracellular transport and cell division (22). Several kinesins that are involved in mitosis have emerged as potential targets for cancer drug development (23). Three kinesins (KIF23, KIF2C and KIF4A) were identified as prognostic genes in KIRC in the present study. Previous studies have indicated their roles in lung cancer (24), colorectal cancer (25) and oral cancer (26). MELK, which is a highly conserved serine/threonine kinase, is a regulator in cell cycle control and cancer (27,28). Dysregulated expression of MELK is associated with a poor prognosis in breast cancer (29). In addition, a MELK inhibitor has been reported to have potential as a novel molecular targeted therapy, which targets human cancer stem cells (30). TPX2 is associated with various types of cancer, including esophageal squamous cell carcinoma (31), bladder carcinoma (32) and cervical carcinoma (33). In addition, it contributes to the growth and metastasis of hepatocellular carcinoma (34). Further studies regarding these genes may provide novel insights into the pathogenesis of KIRC and provide potential prognostic markers.
In conclusion, the present study identified 11 critical genes associated with KIRC. The prognostic value of 10 genes was validated using another gene expression dataset, which provides important evidence regarding the pathogenesis of KIRC. Further studies are required to better define their roles in KIRC.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Linehan WM. Genetic basis of kidney cancer: Role of genomics for the development of disease-based therapeutics. Genome Res. 2012;22:2089–2100. doi: 10.1101/gr.131110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mickisch GH. Principles of nephrectomy for malignant disease. BJU Int. 2002;89:488–495. doi: 10.1046/j.1464-410X.2002.02654.x. [DOI] [PubMed] [Google Scholar]
- 4.Janzen NK, Kim HL, Figlin RA, Belldegrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am. 2003;30:843–852. doi: 10.1016/S0094-0143(03)00056-9. [DOI] [PubMed] [Google Scholar]
- 5.Robb VA, Magdalena K, Klein-Szanto AJ, Henske EP. Activation of the mTOR signaling pathway in renal clear cell carcinoma. J Urol. 2007;177:346–352. doi: 10.1016/j.juro.2006.08.076. [DOI] [PubMed] [Google Scholar]
- 6.Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, Jia W, Li Z, He M, Sun L, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet. 2012;44:17–19. doi: 10.1038/ng.1014. [DOI] [PubMed] [Google Scholar]
- 7.Dormoy V, Danilin S, Lindner V, Thomas L, Rothhut S, Coquard C, Helwig JJ, Jacqmin D, Lang H, Massfelder T. The sonic hedgehog signaling pathway is reactivated in human renal cell carcinoma and plays orchestral role in tumor growth. Mol Cancer. 2009;8:123. doi: 10.1186/1476-4598-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang SW, Chang WH, Su YC, Chen YC, Lai YH, Wu PT, Hsu CI, Lin WC, Lai MK, Lin JY. MYC pathway is activated in clear cell renal cell carcinoma and essential for proliferation of clear cell renal cell carcinoma cells. Cancer Lett. 2009;273:35–43. doi: 10.1016/j.canlet.2008.07.038. [DOI] [PubMed] [Google Scholar]
- 9.Lebdai S, Verhoest G, Parikh H, Jacquet SF, Bensalah K, Chautard D, Leclercq N Rioux, Azzouzi AR, Bigot P. Identification and validation of TGFBI as a promising prognosis marker of clear cell renal cell carcinoma. Urol Oncol. 2015;33:69.e11–e18. doi: 10.1016/j.urolonc.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Wang J, Sun G. Identification of key genes and pathways in renal cell carcinoma through expression profiling data. Kidney Blood Press Res. 2015;40:288–297. doi: 10.1159/000368504. [DOI] [PubMed] [Google Scholar]
- 11.Valletti A, Gigante M, Palumbo O, Carella M, Divella C, Sbisà E, Tullo A, Picardi E, D'Erchia AM, Battaglia M, et al. Genome-wide analysis of differentially expressed genes and splicing isoforms in clear cell renal cell carcinoma. PLoS One. 2013;8:e78452. doi: 10.1371/journal.pone.0078452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-9-r60. [DOI] [PubMed] [Google Scholar]
- 14.Kidokoro T, Tanikawa C, Furukawa Y, Katagiri T, Nakamura Y, Matsuda K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene. 2008;27:1562–1571. doi: 10.1038/sj.onc.1210799. [DOI] [PubMed] [Google Scholar]
- 15.Gardner FP, Joseph RW, Serie D, Hilton TW, Parasramka M, Eckel-Passow J, Cheville J, Bradley C. Association of topoisomerase II expression and cancer-specific death in patients with surgically resected clear cell renal cell carcinoma. J Clin Oncol. 2013;31(Suppl 6) doi: 10.1200/jco.2013.31.6_suppl.446. abstr 446. [DOI] [Google Scholar]
- 16.Chen D, Maruschke M, Riesenberg R, Zimmermann W, Stief CG, Buchner A. MP29-03 TET3, hells, TOP2A and ATAD2 are novel independent prognostic markers in advanced renal cell carcinoma. J Urol. 2014;191:e305. doi: 10.1016/j.juro.2014.02.745. [DOI] [Google Scholar]
- 17.Wong N, Yeo W, Wong WL, Wong NL, Chan KY, Mo FK, Koh J, Chan SL, Chan AT, Lai PB, et al. TOP2A overexpression in hepatocellular carcinoma correlates with early age onset, shorter patients survival and chemoresistance. Int J Cancer. 2009;124:644–652. doi: 10.1002/ijc.23968. [DOI] [PubMed] [Google Scholar]
- 18.Lan J, Huang HY, Lee SW, Chen TJ, Tai HC, Hsu HP, Chang KY, Li CF. TOP2A overexpression as a poor prognostic factor in patients with nasopharyngeal carcinoma. Tumour Biol. 2014;35:179–187. doi: 10.1007/s13277-013-1022-6. [DOI] [PubMed] [Google Scholar]
- 19.Jain M, Zhang L, He M, Zhang YQ, Shen M, Kebebew E. TOP2A is overexpressed and a therapeutic target for adrenocortical carcinoma. Endocr Relat Cancer. 2013;20:361–370. doi: 10.1530/ERC-12-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subhash VV, Tan SH, Tan WL, Yeo MS, Xie C, Wong FY, Kiat ZY, Lim R, Yong WP. GTSE1 expression represses apoptotic signaling and confers cisplatin resistance in gastric cancer cells. BMC Cancer. 2015;15:550. doi: 10.1186/s12885-015-1550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q, Su PF, Zhao S, Shyr Y. Transcriptome-wide signatures of tumor stage in kidney renal clear cell carcinoma: Connecting copy number variation, methylation and transcription factor activity. Genome Med. 2014;6:117. doi: 10.1186/s13073-014-0117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rath O, Kozielski F. Kinesins and cancer. Nature Rev Cancer. 2012;12:527–539. doi: 10.1038/nrc3310. [DOI] [PubMed] [Google Scholar]
- 23.Sakowicz R, Finer JT, Beraud C, Crompton A, Lewis E, Fritsch A, Lee Y, Mak J, Moody R, Turincio R, et al. Antitumor activity of a kinesin inhibitor. Cancer Res. 2004;64:3276–3280. doi: 10.1158/0008-5472.CAN-03-3839. [DOI] [PubMed] [Google Scholar]
- 24.Taniwaki M, Takano A, Ishikawa N, Yasui W, Inai K, Nishimura H, Tsuchiya E, Kohno N, Nakamura Y, Daigo Y. Activation of KIF4A as a prognostic biomarker and therapeutic target for lung cancer. Clin Cancer Res. 2007;13:6624–6631. doi: 10.1158/1078-0432.CCR-07-1328. [DOI] [PubMed] [Google Scholar]
- 25.Gnjatic S, Cao Y, Reichelt U, Yekebas EF, Nölker C, Marx AH, Erbersdobler A, Nishikawa H, Hildebrandt Y, Bartels K, et al. NY-CO-58/KIF2C is overexpressed in a variety of solid tumors and induces frequent T cell responses in patients with colorectal cancer. Int J Cancer. 2010;127:381–393. doi: 10.1002/ijc.25058. [DOI] [PubMed] [Google Scholar]
- 26.Minakawa Y, Kasamatsu A, Koike H, Higo M, Nakashima D, Kouzu Y, Sakamoto Y, Ogawara K, Shiiba M, Tanzawa H, Uzawa K. Kinesin family member 4A: A potential predictor for progression of human oral cancer. PLoS One. 2013;8:e85951. doi: 10.1371/journal.pone.0085951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang P, Zhang D. Maternal embryonic leucine zipper kinase (MELK): A novel regulator in cell cycle control, embryonic development, and cancer. Int J Mol Sci. 2013;14:21551–21560. doi: 10.3390/ijms141121551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ganguly R, Mohyeldin A, Thiel J, Kornblum HI, Beullens M, Nakano I. MELK-a conserved kinase: Functions, signaling, cancer, and controversy. Clin Transl Med. 2015;4:11. doi: 10.1186/s40169-014-0045-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pickard MR, Green AR, Ellis IO, Caldas C, Hedge VL, Mourtada-Maarabouni M, Williams GT. Dysregulated expression of Fau and MELK is associated with poor prognosis in breast cancer. Breast Cancer Res. 2009;11:R60. doi: 10.1186/bcr2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung S, Nakamura Y. MELK inhibitor, novel molecular targeted therapeutics for human cancer stem cells. Cell Cycle. 2013;12:1655–1656. doi: 10.4161/cc.24988. [DOI] [PubMed] [Google Scholar]
- 31.Hsu PK, Chen HY, Yeh YC, Yen CC, Wu YC, Hsu CP, Hsu WH, Chou TY. TPX2 expression is associated with cell proliferation and patient outcome in esophageal squamous cell carcinoma. J Gastroenterol. 2014;49:1231–1240. doi: 10.1007/s00535-013-0870-6. [DOI] [PubMed] [Google Scholar]
- 32.Yan L, Li S, Xu C, Zhao X, Hao B, Li H, Qiao B. Target protein for Xklp2 (TPX2), a microtubule-related protein, contributes to malignant phenotype in bladder carcinoma. Tumor Biol. 2013;34:4089–4100. doi: 10.1007/s13277-013-1000-z. [DOI] [PubMed] [Google Scholar]
- 33.Jiang P, Shen K, Wang X, Song H, Yue Y, Liu T. TPX2 regulates tumor growth in human cervical carcinoma cells. Mol Med Rep. 2014;9:2347–2351. doi: 10.3892/mmr.2014.2106. [DOI] [PubMed] [Google Scholar]
- 34.Huang Y, Guo W, Kan H. TPX2 Is a prognostic marker and contributes to growth and metastasis of human hepatocellular carcinoma. Int J Mol Sci. 2014;15:18148–18161. doi: 10.3390/ijms151018148. [DOI] [PMC free article] [PubMed] [Google Scholar]