

Abstract
Previous studies have reported that lipoxin A4 (LXA4) may exert a renoprotective effect on ischemia/reperfusion injury in various animal models. The underlying mechanism of LXA4-induced renoprotection during ischemia/reperfusion injury remains to be elucidated. The present study investigated LXA4-induced protection on renal tubular cells subjected to hypoxia/reoxygenation (H/R) injury, and determined the effects of peroxisome proliferator-activated receptor-γ (PPARγ) and heme oxygenase-1 (HO-1) on LXA4 treatment. HK-2 human tubular epithelial cells exposed to H/R injury were pretreated with LXA4, signal molecule inhibitors or the HO-1 inhibitor zinc protoporphyrin-IX, or were transfected with PPARγ small interfering RNA (siRNA) or nuclear factor E2-related factor 2 (Nrf2) siRNA. The protein and mRNA expression levels of PPARγ and HO-1 were analyzed using western blotting and reverse transcription-quantitative polymerase chain reaction. Binding activity of Nrf2 to the HO-1 E1 enhancer was determined using chromatin immunoprecipitation. Nrf2 binding to the HO-1 antioxidant responsive element (ARE) was assessed using electrophoretic mobility shift assay. Preincubation of cells with LXA4 exposed to H/R injury led to a decreased production of inducible nitrogen oxide synthase, malondialdehyde, γ-glutamyl transpeptidase, leucine aminopeptidase and N-acetyl-β-glucosaminidase. In addition, LXA4 pretreatment increased cell viability, protein and mRNA expression levels of PPARγ and HO-1 and PPARγ and HO-1 promoter activity. SB20358 is a p38 mitogen-activated protein kinase (p38 MAPK) pathway inhibitor, which reduced LXA4-induced PPARγ expression levels. LXA4 treatment upregulated p38 MAPK activation, Nrf2 nuclear translocation and increased binding activity of Nrf2 to HO-1 ARE and E1 enhancer in cells exposed to H/R injury. Transfection of the cells with PPARγ siRNA reduced the LXA4-induced Nrf2 translocation. Transfection of the cells with PPARγ siRNA or Nrf2 siRNA also reduced the LXA4-induced increase in HO-1 expression. In conclusion, LXA4-induced protection of renal tubular cells against H/R injury was associated with the induction of PPARγ and HO-1, via activation of the p38 MAPK pathway, as well as Nrf2 nuclear translocation and binding to HO-1 ARE and E1 enhancer. Therefore, LXA4-induced renoprotection is associated with activation of the p38 MAPK/PPARγ/Nrf2-ARE/HO-1 pathway.
Keywords: lipoxin A4, hypoxia/reoxygenation, heme oxygenase-1, peroxisome proliferator-activated receptor-γ, renal tubular cells, nuclear factor E2-related factor 2
Introduction
Ischemia/reperfusion (I/R)-induced acute kidney injury is a major challenge during circulatory arrest, ischemic stroke, renal and cardiovascular surgery, and kidney transplantation, and may lead to delayed graft function or acute renal failure (1). A previous study attributed renal I/R injury to an inflammatory process characterized by neutrophil infiltration into the ischemic kidney and the generation of proinflammatory cytokines and reactive oxygen species (ROS), which are produced during reperfusion (2). ROS may induce the production of heme oxygenase-1 (HO-1), an important component of the cellular defense mechanism, which acts against ROS-induced I/R tissue injury, including renal I/R injury (3,4). The renoprotective potency of HO-1 may be attributed to the byproducts of the HO-1 enzymatic reaction, including carbon monoxide and bilirubin (3,4). A HO-1 agonist may reduce oxidative stress and inducible nitrogen oxide synthase (iNOS) activity in renal I/R injury (5). The severity of tubular injury following I/R exposure may also be attenuated by inhibiting the cytotoxicity of inflammatory infiltrates (3,4). Furthermore, peroxisome proliferator-activated receptor-γ (PPARγ) may provide renoprotection during I/R injury. It has previously been demonstrated that PPARγ agonists may provide a potential beneficial effect on renal function (6). PPARγ agonists attenuated renal I/R injury through antioxidant and anti-inflammatory effects (7), inhibited renal oxidative stress in diabetic rabbits and rats (6), and diminished podocyte injury triggered by oxygen/glucose deprivation-reoxygenation (8). Furthermore, the renoprotective mechanisms of several drugs on renal I/R injury are associated with upregulation of PPARγ expression (9–11).
Lipoxin A4 (LXA4) is an eicosanoid, which acts as a ‘breaking signal’ in the inflammatory process, by promoting the reduction of inflammation via inhibition of neutrophil infiltration and activation, reducing the response of various cells stimulated by pathogens and proinflammatory cytokines, and the production of proinflammatory cytokines and toxic compounds, including ROS (12,13). Lipoxin analogs may induce renoprotection following I/R injury via regulation of chemokine and cytokine production, and neutrophil recruitment (14). In a previous study, treatment of murine renal I/R injury with a lipoxin analog altered the induction of various pathogenic mediators, including growth factors, cytokines, proteases and adhesion molecules, thus suggesting that a lipoxin analog may have a renoprotective role in the pathophysiology of renal I/R injury (15). At present, it is unclear whether PPARγ or HO-1 contribute to lipoxin-induced renoprotection against I/R injury. In addition to its anti-inflammatory effects, lipoxin-induced production of PPARγ and HO-1 may also contribute to renoprotection following I/R injury. Previous studies have revealed that LXA4 and aspirin-triggered LXA4 upregulated the expression of HO-1 in lung tissues, and endothelial and corneal epithelial cells (16–18). Our previous studies also revealed that LXA4-triggered HO-1 inhibited hypoxia/reoxygenation (H/R) injury-induced cardiomyocyte lesions (19,20). In addition, LXA4 may contribute to neuroprotection as a PPARγ agonist in cerebral ischemia (21). Treatment of adult neutrophils with LXA4 led to increased PPARγ expression levels (22). It remains to be elucidated whether LXA4 treatment increases PPARγ or HO-1 expression in renal tubular epithelial cells, and whether LXA4-induced PPARγ or HO-1 expression may participate in renoprotection following I/R injury.
Previous studies have revealed that the signaling pathways associated with HO-1 induction include phosphatidyinositol-3-kinase (PI3K)/Akt, mitogen-activated protein kinase (MAPK) pathways, nuclear factor-E2-related factor 2 (Nrf2) and antioxidant responsive element (ARE) in the HO-1 gene promoter. The transcription factor Nrf2 interacts with ARE, and is crucial for HO-1 transcriptional activation (19,23–26). The signaling molecules that trigger HO-1 gene expression are activated in a cell-specific and inducer-specific manner. Statins may stimulate protein kinase G to increase the expression of extracellular signal-regulated kinase (ERK) and p38 MAPK pathways, subsequently activating HO-1 gene induction (24). Tyrosine kinase inhibitors, but not inhibitors of the ERK and p38 MAPK pathways, may reduce the induction of HO-1 by cadmium chloride, hemin and sodium arsenite in human HeLa cells (25). Furthermore, nitric oxide may increase HO-1 expression through the formation of the Nrf2/ARE complex, independent of the MAPK or PI3K/Akt pathways in smooth muscle cells (26). Our previous study demonstrated that the protective effects of LXA4-induced HO-1 expression against H/R injury in cardiomyocytes were mediated by activation of the p38 MAPK pathway, nuclear translocation of Nrf2, and Nrf2 binding to the HO-1/ARE complex and the E1 enhancer (19). However, the signaling pathways that contribute to LXA4-induced HO-1 expression in renal cells remain to be elucidated. In a previous study, LXA4 was revealed to stimulate activation of p38 MAPK and ERK; however, not PI3K in human renal mesangial cells (27). In our previous studies, LXA4 promoted the phosphorylation of ERK, but not PI3K/Akt, in renal mesangial cells and lung fibroblasts (28,29). In addition, the phosphorylation of ERK and p38 MAPK, but not PI3K, was increased in endothelial cells (30). Our previous study did not detect LXA4-induced activation of ERK and PI3K/Akt in renal tubular epithelial cells (31). In addition, it has previously been revealed that LXA4 may inhibit lipopolysaccharide-triggered ROS generation via the Nrf2 pathway in human umbilical vein cells (32). PPARγ has also been suggested to regulate Nrf2 (33) and HO-1 (34) expression, and nitric oxide activates PPARγ via the p38 MAPK signaling pathway (35). Therefore, the present study aimed to determine whether p38 MAPK, ERK, PPARγ and Nrf2/ARE contribute to LXA4-induced HO-1 expression in renal tubular epithelial cells. Human renal tubular epithelial cells were used and cellular injury was triggered by incubation in low-glucose medium and H/R injury, which mimicked in vivo renal I/R injury (8).
Materials and methods
Reagents
Fetal calf serum (FCS) and Dulbecco's modified Eagle's medium (DMEM) were obtained from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Enzyme-linked immunosorbent assay (ELISA) kits for N-acetyl-β-glucosaminidase (NAG; CSB-E09450), γ-glutamyl transpeptidase (γ-GT; CSB-EL009394) and leucine aminopeptidase (LAP; CSB-E13840) levels were purchased from Cusabio Biotech Co., Ltd. (Wuhan, China). HO-1 ELISA kit (EKS-800) was obtained from Assays Designs; Enzo Life Science (Farmingdale, NY, USA). The chromatin immunoprecipitation (ChIP) assay kit (17–295) was purchased from EMD Millipore (Billerica, MD, USA). Superoxide dismutase (SOD) activity assay kit (A001-3), iNOS assay kit (A014-1) and malondialdehyde (MDA) assay kit (A003-4) were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Prime Script™ RT reagent kit and SYBR Premix Ex Taq™ were purchased from Takara Bio, Inc. (Otsu, Japan). TRIzol reagents were obtained from Thermo Fisher Scientific, Inc. PPARγ transcription factor assay kit was obtained from Abcam (Cambridge, UK). LXA4, SB203580 (an inhibitor of p38 MAPK phosphorylation) and LY294002 (an inhibitor of the phosphotransferase activity of PI3K), were purchased from Calbiochem (San Diego, CA, USA). Rabbit anti-human HO-1 (sc-10789), PPARγ (sc-7196), Nrf2 (sc-13032), total Akt 1/2/3 (sc-8312) and serine 473-phosphorylated Akt 1/2/3 (p-Akt; sc-7985) antibodies, PPARγ-specific small interfering RNA (siRNA; 5′-GAACAUCGAGUGUCGAAUATT−3′), Nrf2-specific siRNA (5′-CGCUCAGAACUGUAGGAAAAGGAAGAG-3′) and control siRNA (non-specific siRNA) were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Rabbit anti-human total ERK1/2 (BS3628) and threonine 202/tyrosine 204-diphosphorylated ERK1/2 (p-ERK1/2; BS5016) antibodies were purchased from Bioworld Technology, Inc. (St. Louis Park, MN, USA). Rabbit anti-human total p38 MAPK (2307), threonine 180/tyrosine 204-diphosphorylated p38 MAPK (p-p38 MAPK; 4511), α/β-tubulin (2148), β-actin (4967), GAPDH (5174) antibodies, biotin-conjugated anti-rabbit immunoglobulin G (IgG) (14708) and horseradish peroxidase-conjugated goat anti-rabbit IgG (7074) were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). HO-1 activity assay kit was purchased from GenMed Scientifics, Inc. (Arlington, MA, USA). A gel shift assay kit, and total and nuclear protein extraction kit were obtained from Active Motif (Carlsbad, CA, USA). PD98059, an inhibitor of ERK1/2 phosphorylation, zinc protoporphyrin-IX (ZnPP-IX; a specific inhibitor of HO-1 activity), L-glutamine, insulin, sodium pyruvate, CdCl2, trypsin, EDTA, Triton X-100, bovine serum albumin and pioglitazone (PPARγ agonist) were purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). Lipofectamine 2000 reagents were obtained from Invitrogen; Thermo Fisher Scientific, Inc. Chemiluminescent horseradish peroxidase substrate was purchased from Merck Millipore. Protein extraction kit and bicinchoninic acid (BCA) protein assay kit were obtained from KeyGen Biotech Co., Ltd. (Nanjing, China). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). Enhanced chemiluminescence (ECL) reagent system was obtained from Amersham; GE Healthcare (Little Chalfont, UK). Hoechst staining kit was purchased from Beyotime Institute of Biotechnology (Shanghai, China).
Cell culture
HK-2 human proximal tubular epithelial cells were obtained from Type Culture Collection of the Chinese Academy of Sciences (Wuhan, China), originated from American Type Culture Collection (Manassas, VA, USA; no. CRL-2190). The cell monolayers were incubated in DMEM containing 1,000 mg/l glucose, 5 µg/ml insulin, 10% FCS, 4 mmol/l L-glutamine, 110 mg/l sodium pyruvate, 100 µg/ml streptomycin and 100 U/ml penicillin in a 5% CO2 incubator at 37°C. Following digestion with 0.01% EDTA and 0.25% trypsin, 8×105 cells were seeded in 50 ml plastic culture bottles and allowed to reach 60–70% sub-confluence. Cellular H/R injury was induced by H/R treatment. Briefly, the cells were incubated in low-glucose DMEM in a modular incubator chamber (BioSpherix, Parish, NY, USA) with 1% O2, 5% CO2 and 94% N2 for 24 h (hypoxia for 24 h), then cultured in an atmosphere containing 21% O2, 5% CO2 and 74% N2 for 6 h (reoxygenation for 6 h). The H/R injury was induced following pretreatment with 10 nM LXA4 for 12 h with or without co-incubation with 30 µM SB203580, 10 µM LY294002 or 40 µM PD98059 for 30 min, 10 µM CdCl2 for 1 h, or 10 µM pioglitazone or 20 µM ZnPP-IX for 12 h.
siRNA transfection
The cells were cultured in 6-well plates (1×105 cells/well) for 24 h and subsequently transfected with PPARγ-siRNA, Nrf2-siRNA or nonspecific siRNA (control siRNA) using Lipofectamine 2000 reagents according to the manufacturer's protocol. The cells were preincubated with or without LXA4 for 12 h, then subjected to H/R injury. The mRNA and protein expression levels of PPARγ and HO-1 in the transfected cells were quantified using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blotting. Nrf2 protein expression was determined using western blotting. The efficiency of Nrf2 and PPARγ-specific siRNA transfection was determined using non-specific siRNA.
Cell viability assay and iNOS, SOD, MDA, LAP, NAG and γ-GT quantification
Cell viability was assessed using the CCK-8 assay. HK-2 cells were cultured in 96-well plates and allowed to reach 40–50% sub-confluence. Subsequently, the cells were treated with various concentrations of LXA4 (0, 0.1, 1 and 10 nM) for 6, 12 and 24 h; subsequently, the cells were exposed to H/R injury. CCK-8 solution (10 µl) was added to each well and the incubation at 37°C was continued for an additional 1 h. Cell viability was quantified at A450 nm using a spectrophotometer in three wells of each group. The iNOS and SOD activity, and MDA levels in whole cell lysates were determined using the assay kits, according to the manufacturer's protocols. The LAP, NAG and γ-GT levels in cellular supernatants were determined using ELISA kits according to the manufacturer's protocols.
RT-qPCR analysis
RT-qPCR was conducted as previously described (30). Briefly, total RNA was isolated using TRIzol. The RNA was reverse transcribed to cDNA using the PrimeScrpt™ RT reagent kit according to the manufacturer's protocol. qPCR was performed using heat-activated SYBR Premix EX Taq DNA polymerase in a TaqMan ABI 5700 Sequence Detection system (Applied Biosystems; Thermo Fisher Scientific, Inc.). GAPDH served as internal control. The following sets of primers were selected using Primer Premier version 5.0 software analysis: HO-1, sense 5′-CAACCCTGCTTGCGTCCTA-3′, antisense 5′-ACCGTTCCTCCCTCCAACTA-3′; PPARγ, sense 5′-GGTCTCGATGTTGGCGCTAT-3′, antisense 5′-CCCCTCACGAAGCAGACTTT-3′; and GAPDH, sense 5′-ACCACAGTCCATGCCATCAC-3′ and antisense 5′-TCCACCACCTGTTGCTGTA-3′. Identical amplification conditions were applied for all reactions: 95°C for 2 min for hot start and template denaturation prior to PCR cycling, which consisted of three stages: 30 sec at 95°C for denaturation, 30 sec at 59°C for annealing, 30 sec at 72°C for extension, and an additional 20 sec at 72°C for fluorescent signal acquisition. A total of 30 cycles were conducted. Subsequently, the Cq values were calculated for target genes in the samples and the results were analyzed (36).
Western blot analysis
The cellular total and nuclear proteins of the lysates were extracted using protein extraction kits according to the manufacturer's protocol. Protein levels were determined using the BCA protein assay kit according to the manufacturer's protocol. Subsequently, 30 µg protein was separated by 10% SDS-polyacrylamide gel electrophoresis, and transferred onto polyvinylidene difluoride membranes with an electroblotting apparatus. The membranes were incubated with the primary antibodies against HO-1, PPARγ, Nrf2, p38 MAPK, p-p38 MAPK, p-EKR, ERK, Akt or p-Akt at 1:200 dilution, and tubulin or β-actin at 1:1,000 dilution overnight at 4°C prior to washing with TBS containing 0.1% Tween-20. Subsequently, the membranes were exposed to the horseradish peroxidase-conjugated secondary antibodies at 1:2,000 dilution for 1 h at 37°C. Finally, the membranes were incubated with an ECL reagent system and exposed to Kodak Biomax films (Eastman Kodak, Rochester, NY, USA). Semi-quantitative analysis was performed using GelDoc-It2 imaging system (UVP, LLC, Upland, CA, USA).
Quantification of HO-1 activity and levels
HO-1 activity was assessed using the HO-1 activity kit according to the manufacturer's protocol. The cellular HO-1 activity of the lysates was presented as pmol/mg/h. The cellular HO-1 concentration (ng/mg) in the lysates was determined using the ELISA kit according to the manufacturer's protocol.
Immunofluorescence assay
Intracellular Nrf2 localizations were quantified using an immunofluorescence assay. The cells (1×105/ml) were incubated for 30 min at 37°C on glass coverslips, and subsequently fixed with 4% paraformaldehyde, washed and permeabilized with 0.1% Triton X-100, and blocked with 2% bovine serum albumin in PBS at room temperature for 30 min. Subsequently, the cells were exposed to the Nrf2 antibodies (1:100 dilution) at room temperature for 1 h, and were washed and incubated for 30 min at 37°C with biotin-conjugated anti-rabbit IgG at 1:500 dilution. The cells were then incubated with fluorescein isothiocyanate-conjugated streptavidin at room temperature for 1 h. Hoechst stain for DNA was performed using the Hoechst staining kit, according to the manufacturer's protocol. Coverslips were mounted on the slides, and images of the labeled cells were observed under a fluorescence microscope (Axiovert 200 M; Carl Zeiss AG, Oberkochen, Germany).
HO-1 promoter analysis
The sequences for dominant-negative Nrf2 mutant (dnNrf2) that has had its transactivation domain deleted, for mutant E1 enhancer (M739) that had its three ARE core sequences mutated, and for the wild-type E1 enhancer coupled to a minimum HO-1 promoter (E1), were synthesized as previously described (20). The plasmids expressing dnNrf2 (pEF-F2/Nrf2), 1 µg/ml pCMVβ-galactosidase, an empty vector and the 1 µg/ml promoter/luciferase constructs were co-transfected into the HK-2 cells using Lipofectamine 2000 reagent according to the manufacturer's protocol. The cells were incubated for an additional 24 h and subsequently treated with 10 µM CdCl2 for 1 h or 10 nM LXA4 for 12 h. The luciferase activity of the reporter enzyme was assessed using a TD-20/20 Turner Designs luminometer and quantified at 560 nm using a spectrophotometer.
PPARγ transcriptional activity assay
The transcriptional activity of PPARγ was determined using the PPARγ transcription factor assay kit according to the manufacturer's protocol. The nuclear extracts were extracted from the cells using the aforementioned kit and were incubated for 15 min at 37°C in wells coated with specific PPAR response element oligonucleotide sequences, then exposed to the primary anti-PPARγ polyclonal antibody. Subsequently, the horseradish peroxidase-conjugated secondary antibody and the 3,39,5,59-tetramethylbenzidine substrate were added and the absorbance was quantified at 450 nm using a spectrophotometer.
Electrophoretic mobility shift assay (EMSA)
The cellular nuclear protein was extracted using a nuclear protein extraction kit according to the manufacturer's protocol. EMSA was performed using a gel shift assay kit according to the manufacturer's protocol. Briefly, the nuclear extracts containing 30 µg protein were pretreated with gel shift binding buffer for 10 min, and then exposed to the double-stranded, biotin-labeled oligonucleotide probe of ARE (3 µg) for 20 min. The oligonucleotide pairs of ARE were 5′-TTTATGCTGTGTCATGGTT-3′ and 5′-AACCATGACACAGCATAAA-3′. The resulting nuclear protein-DNA complexes were merged in 4% non-denaturing polyacrylamide gels and electrophoresis was performed at 220 V for 2 h. Subsequently, the active bands in the gels were visualized on X-ray films. The antibody supershift assay was performed using 1 µg Nrf2 antibody added to the reaction mixture and incubated for 3 h at 4°C prior to addition of the probe. To determine the reaction specificity, competition assays were conducted with 100-fold excess of unlabeled consensus oligonucleotide pairs of ARE, which were added to the binding reaction mixture 10 min prior to the addition of the labeled probes.
ChIP assay
ChIP assays were conducted using the ChIP assay kit according to the manufacturer's protocol. Briefly, the cells were lysed in SDS-lysis buffer and subsequently sonicated. The DNA and proteins were cross-linked with formaldehyde. Sheared chromatin was immunocleared using protein agarose-A. The portions of the precleared chromatin were stored and labeled as ‘input DNA’. The remaining chromatin was immunoprecipated with Nrf2 antibodies, IgG was used as a control. The protein-DNA complexes were eluted using elution buffer from the antibodies. The formaldehyde cross-links were reversed by exposure to NaCl and heating at 65°C for 4 h. The DNA was purified and PCR was performed under aforementioned thermocycling conditions using a primer pair that spanned the mouse HO-1 E1 enhancer. The following primers were used: E1 forward, 5′-AAGAGCTCCACCCCCACCCA-3′ and reverse, 5′-GGGCTAGCATGCGAAGTGAG-3′. The PCR products were separated and examined using electrophoresis with 1.5% agarose gel and ethidium bromide.
Statistical analysis
Data are expressed as the mean ± standard deviation. Significant differences were analyzed using one-way analysis of variance followed by least significant difference test using SPSS version 14.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
LXA4 reduces H/R injury
The treatment duration required for LXA4-induced protection against H/R injury was assessed. Pretreatment with LXA4 for 12 h at 10 nM provided maximum protection during H/R injury as indicated by CCK-8 assessment (data not shown). As indicated in Fig. 1A, LXA4 pretreatment led to significantly increased cell viability during H/R injury (P<0.05). Similarly, the H/R injury-induced changes in oxidative and nitrosative stress parameters, such as MDA and iNOS, were significantly reduced by LXA4 pretreatment (Fig. 1B; P<0.05). H/R injury significantly reduced SOD levels, which in turn may increase MDA levels, this was also significantly reversed in the group that received LXA4 pretreatment (P<0.05; Fig. 1C). As presented in Fig. 1C and D the levels of γ-GT, LAP and NAG were significantly higher in the group undergoing H/R injury compared with the control group (P<0.05). LXA4 pretreatment significantly reduced the levels of γ-GT, LAP and NAG during H/R injury compared with the H/R only treatment group (P<0.05). In addition, inhibition of PPARγ expression with PPARγ siRNA and suppression of HO-1 with ZnPP-IX, significantly reduced the cytoprotection of LXA4 on cell viability (P<0.05; Fig. 1A) and the changes of MDA, iNOS, SOD, γ-GT, LAP and NAG levels (Fig. 1B-D).
Figure 1.
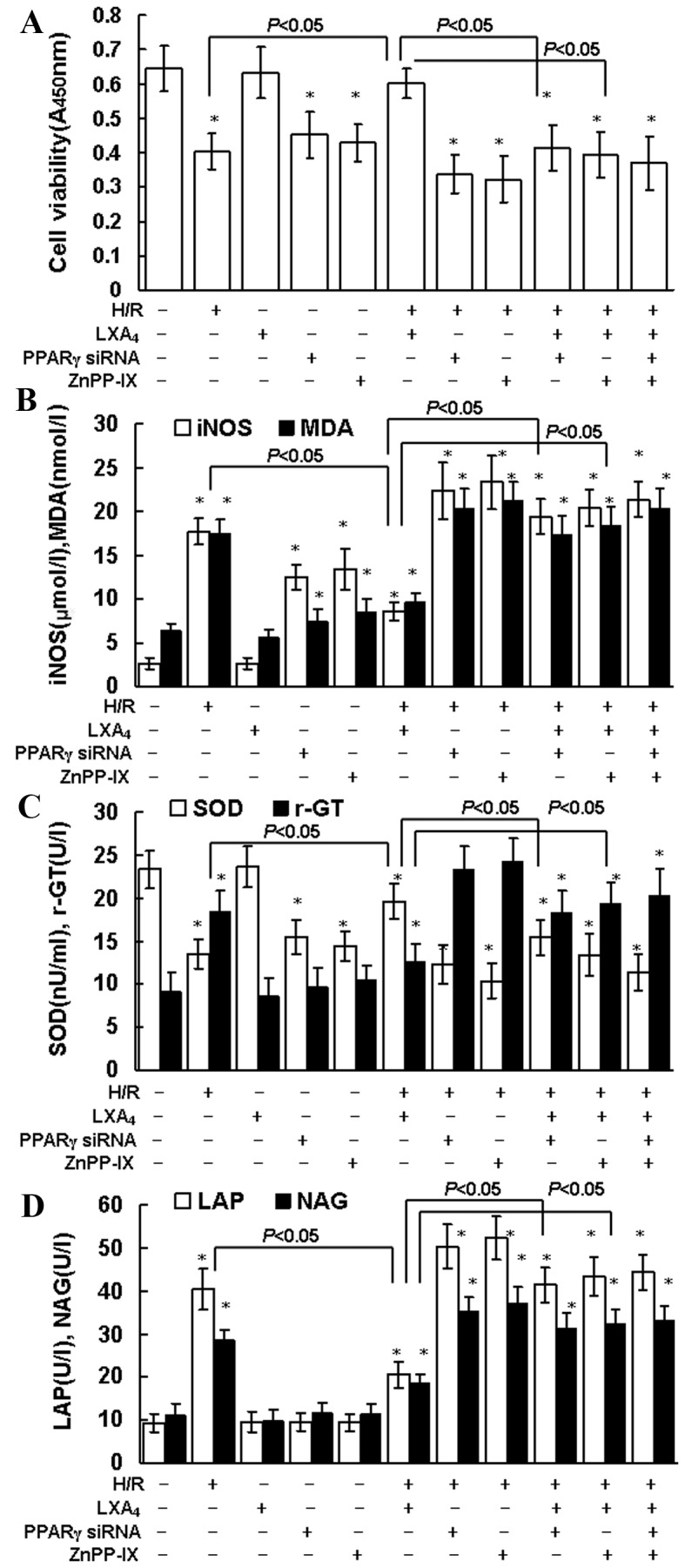
Cell viability, iNOS, MDA, SOD, γ-GT, LAP and NAG production. (A) HK-2 cell viability was determined using Cell Counting Kit-8 assay. (B) iNOS (µmol/l) and MDA (nmol/l) concentration, and (C) SOD and γ-GT activity in whole cell lysates were determined using the assay kits. (D) LAP and NAG concentrations in cellular supernatants were assessed using ELISA kits. The cells were pretreated with 10 nM LXA4 for 12 h, 20 µM ZnPP-IX or PPARγ siRNA transfection for 12 h, and then exposed to H/R. Untreated cells were used as the control group. Data are presented as the mean ± standard deviation of 5 independent experiments. *P<0.05 vs. control group. iNOS, nitric oxide synthase; MDA, malondialdehyde; SOD, superoxide dismutase; γ-GT, γ-glutamyl transpeptidase; LAP, leucine aminopeptidase; NAG, N-acetyl-β-glucosaminidase; LXA4, lipoxin A4; PPARγ, peroxisome proliferator-activated receptor-γ; siRNA, small interfering RNA; H/R, hypoxia/reoxygenation.
LXA4 induces PPARγ expression
As presented in Fig. 2A and B, LXA4 treatment significantly increased PPARγ mRNA and protein expression, and transcriptional activity in groups with or without H/R injury (P<0.05). However, as shown in Fig. 2C, the LXA4- and pioglitazone (PPARγ agonist)-induced PPARγ expression was specifically suppressed by transfection of the cells with PPARγ siRNA.
Figure 2.

Upregulation of PPARγ expression and activity due to LXA4 treatment in HK-2 cells. Cells were pretreated with 10 nM LXA4 for 12 h, PPARγ siRNA or control siRNA transfection, or 10 µM pioglitazone for 12 h, and then exposed to H/R. (A) mRNA expression of PPARγ was determined using quantitative PCR. The amount of PCR products was normalized to GAPDH to determine the relative expression ratio (mRNA expression ratio ×1/10) for each mRNA. For western blot, tubulin protein was used as a loading control. (B) PPARγ transcriptional activity was expressed as 450 nm value of PPARγ/control. (C) Nuclear protein expression levels of PPARγ are presented as PPARγ/tubulin ratio for each sample. Data are presented as the mean ± standard deviation of 5 independent experiments. PPARγ, peroxisome proliferator-activated receptor-γ; siRNA, small interfering RNA; H/R, hypoxia/reoxygenation; LXA4, lipoxin A4; PCR, polymerase chain reaction.
LXA4 induces PPARγ expression via p38 MAPK
The role of ERK, Akt and p38 MAPK inhibition on the induction of PPARγ is presented in Fig. 3. SB203580 (a p38 MAPK pathway inhibitor) significantly reduced LXA4-induced, H/R injury-triggered, and LXA4 pretreatment followed by H/R injury-induced PPARγ expression, conversely the ERK inhibitor PD98059 or PI3K inhibitor LY294002 did not affect PPARγ expression. As indicated in Fig. 3B, LXA4 pretreatment slightly increased p-p38 MAPK expression compared with the control group. However, no significant difference was observed in p-Akt or p-ERK1/2 expression between the control and LXA4 groups (Fig. 3C and D). H/R exposure significantly upregulated p-ERK1/2, p-p38 MAPK and p-Akt expression levels in cells without LXA4 pretreatment (P<0.05; Fig. 3). In addition, LXA4 pretreatment significantly upregulated H/R injury-induced p-p38 MAPK expression levels (P<0.05; Fig. 3B); however, no significant difference was identified for p-ERK1/2 or p-Akt expression levels (Fig. 3C and D).
Figure 3.

Role of p38 MAPK in LXA4-triggered PPARγ expression. HK-2 cells were pretreated with 10 nM LXA4 for 12 h, 40 µM PD98059, 10 µM LY294002 and 30 µM SB203580 for 30 min, and then exposed to H/R injury. (A) mRNA and nuclear protein expression levels of PPARγ were determined using quantitative PCR and western blotting. The amount of PCR products was normalized with GAPDH to determine the relative expression ratio (mRNA expression ratio ×1/10) for each mRNA. PPARγ protein expression was presented as PPARγ/tubulin ratio for each sample. Expression levels of (B) total and p-p38 MAPK, (C) total and p-Akt, (D) and total and p-ERK1/2 protein were assessed using western blotting. Data are presented as the mean ± standard deviation of 5 independent experiments. *P<0.05 vs. control group. PPARγ, peroxisome proliferator-activated receptor-γ; H/R, hypoxia/reoxygenation; LXA4, lipoxin A4; p-p38MAPK, phosphorylated-p38 mitogen-activated protein kinase; p-Akt, phosphorylated-Akt; p-ERK1/2, phosphorylated extracellular-signal regulated kinase; PCR, polymerase chain reaction.
LXA4-triggered Nrf2 translocation is dependent on PPARγ activation
As shown in Fig. 4A, intracellular Nrf2 translocation from cytoplasm to nuclei was triggered by H/R injury and LXA4 pretreatment followed by H/R injury, since Nrf2 protein expression was suppressed in the cytoplasm and enhanced in the nuclei of the cells after H/R exposure and LXA4 pretreatment followed by H/R injury. Pretreatment with LXA4 increased the H/R injury-induced Nrf2 translocation from the cytoplasm to the nucleus (Fig. 4). Fluorescence microscopy confirmed the translocation of Nrf2. As presented in Fig. 4B, Nrf2 protein was only detected in the cytoplasm of cells that were not exposed to H/R and LXA4. In addition, LXA4 pretreatment of H/R-injured cells led to a significantly increased expression of Nrf2 in the nucleus compared with the control group (P<0.05; Fig. 4A). No immunofluorescence was identified in the negative controls using secondary antibodies alone (data not shown). Furthermore, it was determined that Nrf2 translocation was dependent on PPARγ activation since transfection of the cells with PPARγ siRNA completely abolished the Nrf2 translocation induced by H/R injury and LXA4 pretreatment followed by H/R injury. The cytosolic and nuclear Nrf2 expressions in the cells transfected with PPARγ siRNA were the same as the expressions in the control cells without any treatment (Fig. 4A).
Figure 4.
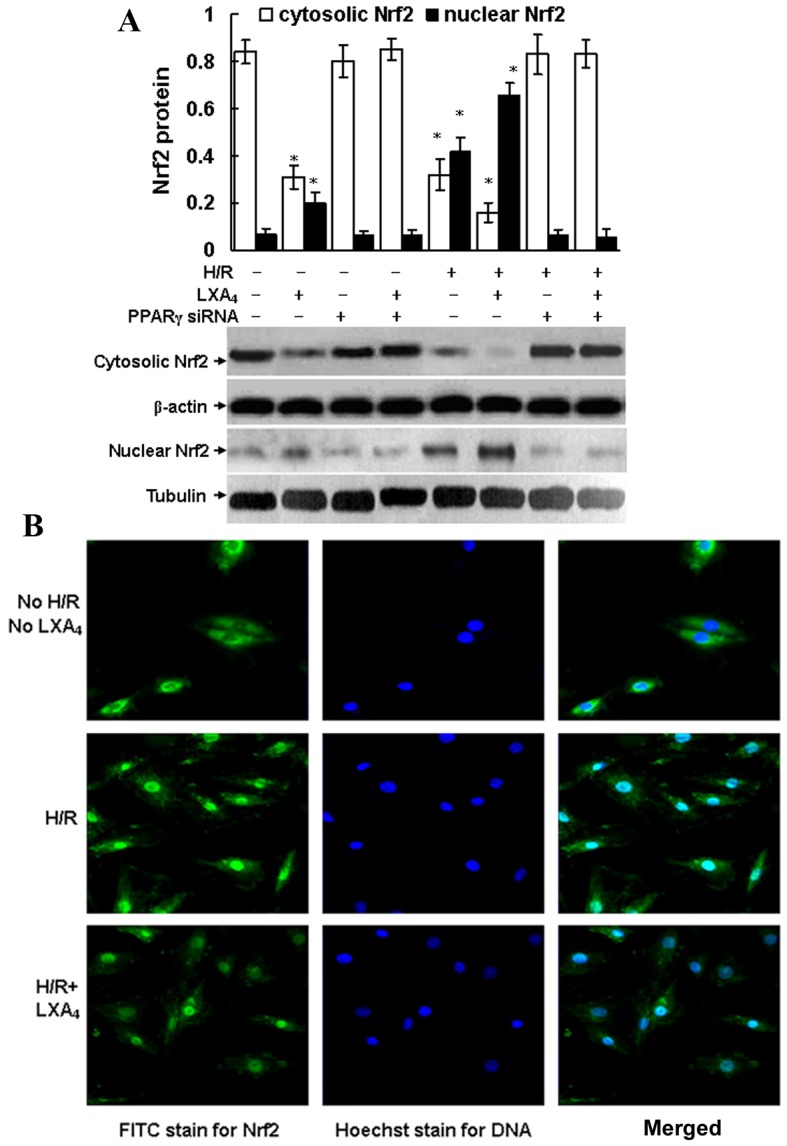
LXA4-stimulated Nrf2 translocation was dependent on PPARγ activation. (A) HK-2 cells were pretreated with 10 nM LXA4 for 12 h and transfected with PPARγ siRNA, and then exposed to H/R. The cytosolic or nuclear Nrf2 proteins were determined using western blotting. Nuclear Nrf2 was expressed as Nrf2/tubulin ratio and the cytosolic Nrf2 was expressed as Nrf2/β-actin ratio for each sample. Data are presented as the mean ± standard deviation of 5 independent experiments. *P<0.05 vs. control group. (B) Nrf2 translocation was assessed using fluorescence microscopy (magnification ×400) and cells were pretreated with LXA4 and exposed to H/R. Nrf2, nuclear factor E2-related factor 2; PPARγ, peroxisome proliferator-activated receptor-γ; siRNA, small interfering RNA; H/R, hypoxia/reoxygenation; LXA4, lipoxin A4; FITC, fluorescein isothiocyanate.
LXA4 pretreatment induces HO-1 expression
As illustrated in Fig. 5A and B, LXA4 significantly increased HO-1 mRNA and protein expression in cells with or without H/R injury (P<0.05). LXA4 pretreatment significantly upregulated HO-1 activity levels in the cells without H/R injury (P<0.05; Fig. 5C). In addition, LXA4 pretreatment significantly upregulated the levels and enzyme activity of HO-1 in a dose-dependent manner in the cells enduring H/R injury (P<0.05; Fig. 5C). Furthermore, it was revealed that the LXA4-induced HO-1 expression levels were dependent of PPARγ and Nrf2 expression, since cells transfected with PPARγ siRNA or Nrf2 siRNA exhibited significantly reduced HO-1 expression levels (P<0.05; Fig. 5A and B).
Figure 5.
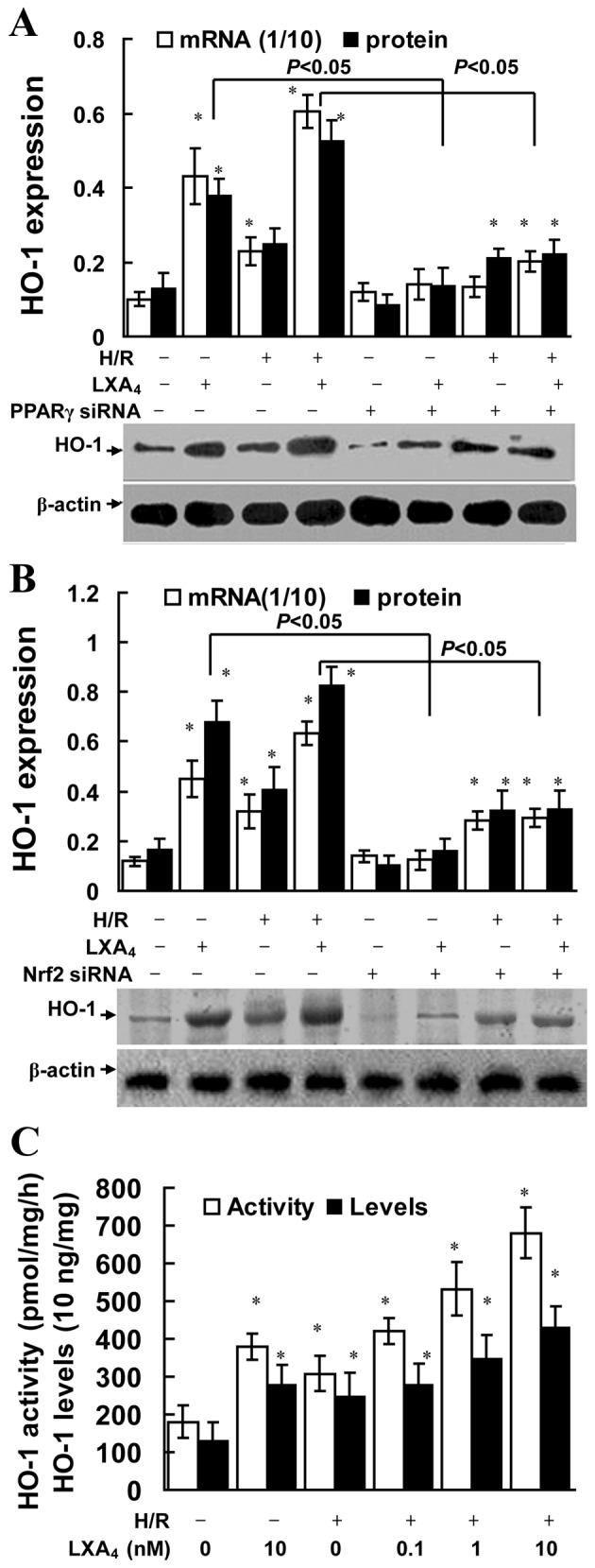
LXA4-induced HO-1 expression was dependent on PPARγ activation and Nrf2 translocation. HK-2 cells were pretreated with 10 nM LXA4 for 12 h and transfected with (A) PPARγ siRNA or (B) Nrf2 siRNA, then exposed to H/R. mRNA and protein expression levels of HO-1 were determined using quantitative PCR and western blotting. The amount of PCR products was normalized to GAPDH to determine the relative expression ratio (mRNA expression ratio ×1/10) for each mRNA Protein expression of HO-1 was presented as HO-1/β-actin ratio for each sample. (C) HK-2 cells were pretreated with LXA4 for 12 h, then exposed to H/R, and HO-1 activity and expression levels were quantified. HO-1 concentration in cell lysates was determined using ELISA. Data are presented as the mean ± standard deviation of 5 independent experiments. *P<0.05 vs. control group. HO-1, heme oxygenase-1; H/R, hypoxia/reoxygenation; LXA4, lipoxin A4; PPARγ, peroxisome proliferator-activated receptor-γ; siRNA, small interfering RNA; Nrf2, nuclear factor E2-related factor 2.
LXA4 stimulates HO-1 gene transcription through formation of the Nrf2/ARE complex
As presented in Fig. 6A, treatment with CdCl2, which is an established activator of the HO-1 promoter, led to a 4.5-fold increase in HO-1 promoter activity. Pretreatment with LXA4 or exposure to H/R injury let to a 4.8 fold increase in HO-1 promoter activity. Pretreatment with LXA4 and exposure to H/R injury may induce a 6.3 fold upregulation in HO-1 promoter activity. Transfection with M739, which induced an ARE mutation and served as a negative control for E1 enhancer, reduced the cellular HO-1 promoter response to LXA4 pretreatment, H/R exposure, and LXA4 pretreatment followed by H/R injury. In addition, transfection with dnNrf2, which blocked formation of the Nrf2/ARE complex, limited the cellular HO-1 promoter response to LXA4 pretreatment, H/R exposure and LXA4 pretreatment followed by H/R injury. EMSA demonstrated the Nrf2/ARE complex formation. As depicted in Fig. 6B, the Nrf2 binding activity was upregulated following the pretreatment with LXA4, H/R exposure and LXA4 pretreatment followed by H/R injury. Nrf2 antibody pretreatment reduced Nrf2 binding activity triggered by LXA4 pretreatment, H/R exposure and LXA4 pretreatment followed by H/R injury and promoted the formation of the Nrf2-anti-Nrf2 complex. Competition assay was performed using the unlabeled oligonucleotide probes in order to determine the specificity of ARE. As indicated in Fig. 6C, the ChIP assay confirmed the findings elucidated from EMSA. The ChIP assay used the Nrf2 antibody and demonstrated the binding of Nrf2 to the E1 enhancer and that this binding was upregulated following pretreatment with LXA4, H/R exposure and LXA4 pretreatment followed by H/R injury.
Figure 6.
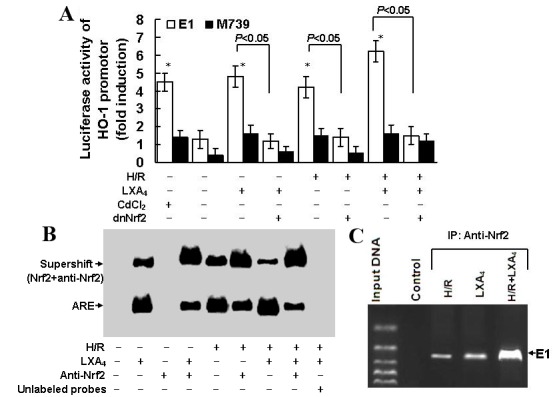
LXA4-induced HO-1 expression was dependent on formation of the Nrf2/ARE complex. (A) HK-2 cells were transfected with M739, E1, dnNrf2, then treated with LXA4 followed by exposure to H/R injury. Fold induction of luciferase activity of the HO-1 promoter was determined using reporter gene transfection assays. Data are presented as the mean ± standard deviation of 5 independent experiments. *P<0.05 vs. untreated control group. (B) Nuclear extracts of the cells were subjected to electrophoretic mobility shift assay with biotin-labeled double-stranded oligonucleotide probe of ARE. Supershift assay was conducted using the Nrf2 antibody. (C) Binding activity of Nrf2 to E1 was assessed using a chromatin immunoprecipitation assay in cells subjected to H/R injury, LXA4 or H/R injury and LXA4. HO-1, heme oxygenase-1; E1, mouse HO-1 promoter construct; M739, mutated mouse HO-1 promoter construct; H/R, hypoxia/reoxygenation; LXA4, lipoxin A4; dnNrf, dominant negative nuclear factor E2-related factor 2; ARE, antioxidant responsive element.
Discussion
A previous study suggested that there is therapeutic potential for the use of lipoxin and its analogs in animal models of renal disease (37). In a previous study, a lipoxin analog reduced neutrophil recruitment in anti-glomerular basement membrane cells in a murine nephritis model (38), whereas LXA4 and benzo-LXA4 have been reported to attenuate experimental renal fibrosis (39). Furthermore, the LXA4-triggered increase of let-7c has been reported to lead to suppression of renal fibrosis (40) and LXA4 protected renal function against rhabdomyolysis-induced acute kidney injury in rats (41). Our previous study also revealed that the LXA4 analog inhibited mesangial proliferation and inflammation in mesangioproliferative nephritis in rats (42). As aforementioned, LXA4 analogs may lead to protection against renal I/R injury (15) and ischemic acute renal failure (14). The present study revealed that LXA4 protected renal tubular epithelial cells against H/R injury. The CCK-8 assay demonstrated that LXA4 provided protection for cells exposed to H/R injury, with regards to cellular viability (Fig. 1A). In addition, H/R injury-induced changes in oxidative and nitrosative stress parameters were reversed by LXA4 pretreatment (Fig. 1B). LXA4 pretreatment also reduced the release of γ-GT, LAP and NAG from tubular epithelial cells subjected to H/R injury (Fig. 1C).
Since in vitro H/R injury mimics in vivo renal I/R injury (8), the present study was conducted in HK-2 cells in order to determine the intracellular mechanism by which LXA4 induced protection against H/R injury. Previous studies have reported that LXA4-induced protection against renal I/R injury may be attributed to the inhibition of neutrophil recruitment, and to the suppression of proinflammatory cytokines and chemokines (14,15). To the best of our knowledge, the present study was the first to reveal that LXA4-induced PPARγ and HO-1 expression was involved in LXA4-induced renoprotection. Initially, LXA4 pretreatment alone or LXA4 pretreatment followed by H/R injury increased the mRNA and protein expression levels, and activities, of PPARγ and HO-1 in HK-2 cells (Figs. 2A and B; 5A and C). In addition, ZnPP-IX treatment and PPARγ siRNA transfection inhibited the LXA4-induced protection on cell viability (Fig. 1A), and reversed the LXA4-modulated oxidative and nitrosative stress parameters (Fig. 1B), and release of γ-GT, LAP and NAG (Fig. 1C and D) in cells exposed to H/R injury. These findings demonstrated that LXA4-induced renoprotection was mediated by PPARγ and HO-1 upregulation in HK-2 cells subjected to H/R injury. These findings are consistent with a previous study, which indicated that LXA4 induced neuroprotection by acting as a PPARγ agonist in cerebral ischemia (21). Furthermore, treatment of adult neutrophils with LXA4 previously resulted in increased PPARγ expression (22), and topical application of LXA4 exerted an anti-inflammatory effect on corneal wound healing through HO-1 upregulation in murine and human corneal epithelial cells (17). In addition, an LXA4 analog ameliorated lipopolysaccharide-evoked acute lung injury in mice via upregulation of HO-1 in lung tissues (18), LXA4-triggered HO-1 has also been revealed to reduce cardiomyocyte injury induced by exposure to H/R (19). The present study demonstrated that LXA4-induced HO-1 expression was dependent on PPARγ expression, since transfection with PPAR-γ siRNA reduced LXA4-induced HO-1 upregulation (Fig. 5A). Similarly, previous studies indicated that HO-1 induction was upregulated by PPARγ in human smooth muscle cells and vascular endothelial cells (34), and cilostazol, an inhibitor of phosphodiesterase type III, protected endothelial cells against tumor necrosis factor-α-induced cytotoxicity through HO-1 induction via a PPARγ-dependent pathway (43).
As aforementioned, signaling transduction pathways that participate in PPARγ and HO-1 production, act in an inducer-specific and cell-specific manner (24–26,35). In the present study, LXA4 alone and LXA4 pretreatment followed by H/R injury induced PPARγ expression, which was partially dependent on p38 MAPK activation, since suppression of p-p38MAPK with SB203580, but not p-ERK or PI3K/Akt suppression, reduced PPARγ production triggered by LXA4 pretreatment, H/R exposure and LXA4 pretreatment followed by H/R injury (Fig. 3A). In addition, LXA4 increased p-p38 MAPK expression; however, no difference in p-Akt or p-ERK1/2 expression was detected in the cells with or without LXA4 treatment (Fig. 3B-D). These findings, to the best of our knowledge, were the first to indicate that p38 MAPK phosphorylation may contribute to PPARγ/HO-1 production triggered by LXA4 pretreatment in renal cells. It has previously been demonstrated that nitric oxide-induced PPARγ activation may occur in a p38 MAPK signaling pathway-dependent manner in human umbilical vein endothelial cells (35). Furthermore, LXA4-induced HO-1 was able to inhibit cardiomyocyte injury following exposure to H/R, via p38 MAPK activation (19).
Previous studies have suggested that Nrf2 acts as regulator of the transcriptional upregulation of HO-1 and of the adaptive response to oxidative stress (44,45). Previous studies have also indicated that Nrf2 may contribute to ARE-triggered transcriptional upregulation of HO-1 and antioxidant gene expression (44,45). LXA4 reduced the permeability of endothelial cells via Nrf2 activation in HO-1 upregulation (32). Therefore, the role of Nrf2/ARE in LXA4-induced renoprotection required further investigation. The present study was, to the best of our knowledge, the first to clarify that Nrf2 translocation and Nrf2/ARE activation were required for LXA4-induced HO-1 expression in renal cells. Initially, LXA4 pretreatment and LXA4 pretreatment followed by H/R injury induced upregulation of HO-1 mRNA and protein levels, which were reduced following transfection with Nrf2-siRNA (Fig. 5B). In addition, LXA4 pretreatment, H/R exposure or LXA4 pretreatment followed by H/R injury all promoted Nrf2 translocation from the cytoplasm to the nucleus (Fig. 4). Immunofluorescence also confirmed that LXA4 resulted in Nrf2 translocation from the cytoplasm into the nuclei following exposure to H/R, since LXA4 treatment led to an increase in nuclear Nrf2 staining (Fig. 4B).
The present study aimed to determine whether LXA4-induced HO-1 expression was associated with transcriptional activation of ARE in the HO-1 promoter by transfecting cells with a HO-1 promoter construct E1 and mutant E1 (M739). Subsequently, LXA4 pretreatment and LXA4 pretreatment followed by H/R injury upregulated HO-1 basal transcription, whereas transfection with M739 reduced the upregulation of HO-1 promoter activity (Fig. 6A). Therefore, it is possible that LXA4 activated HO-1 gene transcription via transcriptional activation of ARE. Furthermore, the Nrf2/ARE binding in the cells following LXA4 pretreatment was assessed using EMSA, which demonstrated that the DNA binding activity of Nrf2 was enhanced by LXA4 pretreatment, H/R exposure and LXA4 pretreatment followed by H/R injury. From the supershift EMSA reactions, it was evident that treatment with anti-Nrf2 reduced migration of the ARE complex, thus suggesting Nrf2 was present in the ARE-nuclear protein complex (Fig. 6B). In addition, ChIP assays indicated that Nrf2 binding to the HO-1 E1 enhancer may be triggered by LXA4 pretreatment, H/R exposure and LXA4 pretreatment followed by H/R injury (Fig. 6C). The present study also demonstrated that LXA4 pretreatment and LXA4 pretreatment followed by H/R injury-induced translocation of Nrf2 were dependent on PPARγ expression levels, since cells transfected with PPARγ siRNA exhibited reduced Nrf2 translocation (Fig. 4A). A previous study demonstrated that PPARγ was able to regulate the expression of Nrf2 in the pathogenesis of intracerebral hemorrhage (33).
In conclusion, to the best of our knowledge, the present study was the first to demonstrate that LXA4 protected tubular epithelial cells against H/R injury via activation of the p38 MAPK/PPARγ/Nrf2-ARE/HO-1 pathway; however, the PI3K/Akt or ERK pathways were not involved. These findings elucidated the underlying mechanisms by which LXA4 induced renoprotection during H/R injury. In conjunction with previous studies on the efficacy of LXA4 and LXA4 analogs in treatment of renal diseases (14,15,38–42), the present study determined that LXA4 may be a novel promising therapeutic agent for ischemic renal diseases.
Acknowledgements
The present study was supported by the National Natural Scientific Grant (grant nos. 81270821 and 81300521) from the Government of China and by the Priority Academic Program Development of Jiangsu Higher Education Institutions (grant no. JX10231801).
References
- 1.Wen X, Murugan R, Peng Z, Kellum JA. Pathophysiology of acute kidney injury: A new perspective. Contrib Nephrol. 2010;165:39–45. doi: 10.1159/000313743. [DOI] [PubMed] [Google Scholar]
- 2.Eltzschig HK, Eckle T. Ischemia and reperfusion-from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chok MK, Ferlicot S, Conti M, Almolki A, Dürrbach A, Loric S, Benoît G, Droupy S, Eschwège P. Renoprotective potency of heme oxygenase-1 induction in rat renal ischemia-reperfusion. Inflamm Allergy Drug Targets. 2009;8:252–259. doi: 10.2174/187152809789352186. [DOI] [PubMed] [Google Scholar]
- 4.Nath KA. Heme oxygenase-1: A provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70:432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 5.Li Volti G, Sorrenti V, Murabito P, Galvano F, Veroux M, Gullo A, Acquaviva R, Stacchiotti A, Bonomini F, Vanella L, Di Giacomo C. Pharmacological induction of heme oxygenase-1 inhibits iNOS and oxidative stress in renal ischemia-reperfusion injury. Transplant Proc. 2007;39:2986–2991. doi: 10.1016/j.transproceed.2007.09.047. [DOI] [PubMed] [Google Scholar]
- 6.Iglesias P, Diez JJ. Peroxisome proliferator-activated receptor gamma agonists in renal disease. Eur J Endocrinol. 2006;154:613–621. doi: 10.1530/eje.1.02134. [DOI] [PubMed] [Google Scholar]
- 7.Reel B, Guzeloglu M, Bagriyanik A, Atmaca S, Aykut K, Albayrak G, Hazan E. The effects of PPAR-γ agonist pioglitazone on renal ischemia/reperfusion injury in rats. J Surg Res. 2013;182:176–184. doi: 10.1016/j.jss.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 8.Miglio G, Rosa AC, Rattazzi L, Grange C, Collino M, Camussi G, Fantozzi R. The subtypes of peroxisome proliferator-activated receptors expressed by human podocytes and their role in decreasing podocyte injury. Br J Pharmacol. 2011;162:111–125. doi: 10.1111/j.1476-5381.2010.01032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ragab D, Abdallah DM, El-Abhar HS. Cilostazol renoprotective effect: Modulation of PPAR-γ, NGAL, KIM-1 and IL-18 underlies its novel effect in a model of ischemia-reperfusion. PLoS One. 2014;9:e95313. doi: 10.1371/journal.pone.0095313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh JP, Singh AP, Bhatti R. Explicit role of peroxisome proliferator-activated receptor gamma in gallic acid-mediated protection against ischemia-reperfusion-induced acute kidney injury in rats. J Surg Res. 2014;187:631–639. doi: 10.1016/j.jss.2013.11.1088. [DOI] [PubMed] [Google Scholar]
- 11.Wu QQ, Wang Y, Senitko M, Meyer C, Wigley WC, Ferguson DA, Grossman E, Chen J, Zhou XJ, Hartono J, et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ and HO-1. Am J Physiol Renal Physiol. 2011;300:F1180–F1192. doi: 10.1152/ajprenal.00353.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: A new pharmacologic genus. Br J Pharmacol. 2008;153(Suppl 1):S200–S215. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Fierro IM. Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: A novel antioxidative mechanism. Thromb Haemost. 2007;97:88–98. [PubMed] [Google Scholar]
- 14.Leonard MO, Nannan K, Burne MJ, Lappin DW, Doran P, Coleman P, Stenson C, Taylor C, Daniels F, Godson C, et al. 15-Epi-15-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J Am Soc Nephrol. 2002;13:1657–1662. doi: 10.1097/01.ASN.0000015795.74094.91. [DOI] [PubMed] [Google Scholar]
- 15.Kieran NE, Doran PP, Connolly SB, Greenan MC, Higgins DF, Leonard M, Godson C, Taylor CT, Henger A, Kretzler M, et al. Modification of the transcriptomic response to renal ischemia/reperfusion injury by lipoxin analog. Kidney Int. 2003;64:480–492. doi: 10.1046/j.1523-1755.2003.00106.x. [DOI] [PubMed] [Google Scholar]
- 16.Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Villela CG, Fierro IM. Novel lipid mediator aspirin-triggered lipoxin A4 induces heme oxygenase-1 in endothelial cells. Am J Physiol Cell Physiol. 2005;289:C557–C563. doi: 10.1152/ajpcell.00045.2005. [DOI] [PubMed] [Google Scholar]
- 17.Biteman B, Hassan IR, Walker E, Leedom AJ, Dunn M, Seta F, Laniado-Schwartzman M, Gronert K. Interdependence of lipoxin A4 and heme-oxygenase in counter-regulating inflammation during corneal wound healing. FASEB J. 2007;21:2257–2266. doi: 10.1096/fj.06-7918com. [DOI] [PubMed] [Google Scholar]
- 18.Jin SW, Zhang L, Lian QQ, Liu D, Wu P, Yao SL, Ye DY. Posttreatment with aspirin-triggered lipoxin A4 analog attenuates lipopolysaccharide-induced acute lung injury in mice: The role of heme oxygenase-1. Anesth Analg. 2007;104:369–377. doi: 10.1213/01.ane.0000252414.00363.c4. [DOI] [PubMed] [Google Scholar]
- 19.Chen XQ, Wu SH, Zhou Y, Tang YR. Lipoxin A4-induced heme oxygenase-1 protects cardiomyocytes against hypoxia/reoxygenation injury via p38 MAPK activation and Nrf2/ARE complex. PLoS One. 2013;8:e67120. doi: 10.1371/journal.pone.0067120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen XQ, Wu SH, Zhou Y, Tang YR. Involvement of K+ channel-dependent pathways in lipoxin A4-induced protective effects on hypoxia/reoxygenation injury of cardiomyocytes. Prostaglandins Leukot Essent Fatty Acids. 2013;88:391–397. doi: 10.1016/j.plefa.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Sobrado M, Pereira MP, Ballesteros I, Hurtado O, Fernández-López D, Pradillo JM, Caso JR, Vivancos J, Nombela F, Serena J, et al. Synthesis of lipoxin A4 by 5-lipoxygenase mediates PPARgamma-dependent, neuroprotective effects of rosiglitazone in experimental stroke. J Neurosci. 2009;29:3875–3884. doi: 10.1523/JNEUROSCI.5529-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinberger B, Quizon C, Vetrano AM, Archer F, Laskin JD, Laskin DL. Mechanisms mediating reduced responsiveness of neonatal neutrophils to lipoxin A4. Pediatr Res. 2008;64:393–398. doi: 10.1203/PDR.0b013e318180e4af. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des. 2003;9:2499–2511. doi: 10.2174/1381612033453730. [DOI] [PubMed] [Google Scholar]
- 24.Chen JC, Huang KC, Lin WW. HMG-CoA reductase inhibitors upregulate heme oxygenase-1 expression in murine RAW264.7 macrophages via ERK, p38 MAPK and protein kinase G pathways. Cell Signal. 2006;18:32–39. doi: 10.1016/j.cellsig.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 25.Masuya Y, Hioki K, Tokunaga R, Taketani S. Involvement of the tyrosine phosphorylation pathway in induction of human heme oxygenase-1 by hemin, sodium arsenite, and cadmium chloride. J Biochem. 1998;124:628–633. doi: 10.1093/oxfordjournals.jbchem.a022158. [DOI] [PubMed] [Google Scholar]
- 26.Liu XM, Peyton KJ, Ensenat D, Wang H, Hannink M, Alam J, Durante W. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc Res. 2007;75:381–389. doi: 10.1016/j.cardiores.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMahon B, Stenson C, McPhillips F, Fanning A, Brady HR, Godson C. Lipoxin A4 antagonizes the mitogenic effects of leukotriene D4 in human renal mesangial cells. Differential activation of MAP kinases through distinct receptors. J Biol Chem. 2000;275:27566–27575. doi: 10.1074/jbc.M001015200. [DOI] [PubMed] [Google Scholar]
- 28.Wu SH, Wu XH, Lu C, Dong L, Chen ZQ. Lipoxin A4 inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am J Respir Cell Mol Biol. 2006;34:65–72. doi: 10.1165/rcmb.2005-0184OC. [DOI] [PubMed] [Google Scholar]
- 29.Wu SH, Wu XH, Lu C, Dong L, Zhou GP, Chen ZQ. Lipoxin A4 inhibits connective tissue growth factor-induced production of chemokines in rat mesangial cells. Kidney Int. 2006;69:248–256. doi: 10.1038/sj.ki.5000025. [DOI] [PubMed] [Google Scholar]
- 30.Wu SH, Liao PY, Dong L, Chen ZQ. Signal pathway involved in inhibition by lipoxin A(4) of production of interleukins in endothelial cells by lipopolysaccharide. Inflamm Res. 2008;57:430–437. doi: 10.1007/s00011-008-7147-1. [DOI] [PubMed] [Google Scholar]
- 31.Wu SH, Zhang YM, Tao HX, Dong L. Lipoxin A(4) inhibits transition of epithelial to mesenchymal cells in proximal tubules. Am J Nephrol. 2010;32:122–136. doi: 10.1159/000315121. [DOI] [PubMed] [Google Scholar]
- 32.Pang H, Yi P, Wu P, Liu Z, Liu Z, Gong J, Hao H, Cai L, Ye D, Huang Y. Effect of lipoxin A4 on lipopolysaccharide-induced endothelial hyperpermeability. Sci World Journal. 2011;11:1056–1067. doi: 10.1100/tsw.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao X, Gonzales N, Aronowski J. Pleiotropic role of PPARγ in intracerebral hemorrhage: An intricate system involving Nrf2, RXR, and NF-κB. CNS Neurosci Ther. 2015;21:357–366. doi: 10.1111/cns.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kronke G, Kadl A, Ikonomu E, Bluml S, Fürnkranz A, Sarembock IJ, Bochkov VN, Exner M, Binder BR, Leitinger N. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2007;27:1276–1282. doi: 10.1161/ATVBAHA.107.142638. [DOI] [PubMed] [Google Scholar]
- 35.Ptasinska A, Wang S, Zhang J, Wesley RA, Danner RL. Nitric oxide activation of peroxisome proliferator-activated receptor gamma through a p38 MAPK signaling pathway. FASEB J. 2007;21:950–961. doi: 10.1096/fj.06-6822com. [DOI] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta deltaC(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Kieran NE, Maderna P, Godson C. Lipoxins: Potential anti-inflammatory, proresolution and antifibrotic mediators in renal disease. Kidney Int. 2004;65:1145–1154. doi: 10.1111/j.1523-1755.2004.00487.x. [DOI] [PubMed] [Google Scholar]
- 38.Ohse T, Ota T, Kieran N, Godson C, Yamada K, Tanaka T, Fujita T, Nangaku M. Modulation of interferon-induced genes by lipoxin analogue in anti-glomerular membrane nephritis. J Am Soc Nephrol. 2004;15:919–927. doi: 10.1097/01.ASN.0000119962.69573.CC. [DOI] [PubMed] [Google Scholar]
- 39.Börgeson E, Docherty NG, Murphy M, Rodgers K, Ryan A, O'Sullivan TP, Guiry PJ, Goldschmeding R, Higgins DF, Godson C. Lipoxin A4 and benzo-lipoxin A4 attenuate experimental renal fibrosis. FASEB J. 2011;25:2967–2979. doi: 10.1096/fj.11-185017. [DOI] [PubMed] [Google Scholar]
- 40.Brennan EP, Nolan KA, Börgeson E, Gough OS, McEvoy CM, Docherty NG, Higgins DF, Murphy M, Sadlier DM, Ali-Shah ST, et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFβR1. J Am Soc Nephrol. 2013;24:627–637. doi: 10.1681/ASN.2012060550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng LL, Zhong L, Lei JR, Tang L, Liu L, Xie SQ, Liao XH. Protective effect of lipoxin A4 against rhabdomyolysis-induced acute kidney injury in rats. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2012;28:907–910. (In Chinese) [PubMed] [Google Scholar]
- 42.Wu SH, Wu XH, Liao PY, Dong L. Signal transduction involved in protective effects of 15(R/S)-methyl-lipoxin A(4) on mesangioproliferative nephritis in rats. Prostaglandins Leukot Essent Fatty Acids. 2007;76:173–180. doi: 10.1016/j.plefa.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Park SY, Bae JU, Hong KW, Kim CD. HO-1 induced by cilostazol protects against TNF-α-associated cytotoxicity via a PPAR-γ-dependent pathway in human endothelial cells. Korean J Physiol Pharmacol. 2011;15:83–88. doi: 10.4196/kjpp.2011.15.2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mann GE, Niehueser-Saran J, Watson A, Gao L, Ishii T, de Winter P, Siow RC. Nrf2/ARE regulated antioxidant gene expression in endothelial and smooth muscle cells in oxidative stress: Implications for atherosclerosis and preeclampsia. Sheng Li Xue Bao. 2007;59:117–127. [PubMed] [Google Scholar]
- 45.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]