

Abstract
Obesity and associated metabolic diseases collectively referred to as the metabolic syndrome increase the risk of skeletal and synovial joint diseases, including osteoarthritis (OA). The relationship between obesity and musculoskeletal diseases is complex, involving biomechanical, dietary, genetic, inflammatory, and metabolic factors. Recent findings illustrate how changes in cellular metabolism and metabolic signaling pathways alter skeletal development, remodeling, and homeostasis, especially in response to biomechanical and inflammatory stressors. Consequently, a better understanding of the energy metabolism of diarthrodial joint cells and tissues, including bone, cartilage, and synovium, may lead to new strategies to treat or prevent synovial joint diseases such as OA. This rationale was the basis of a workshop presented at the 2016 Annual ORS Meeting in Orlando, FL on the emerging role of metabolic signaling in synovial joint remodeling and OA. The topics we covered included (i) the relationship between metabolic syndrome and OA in clinical and pre-clinical studies, (ii) the effect of biomechanical loading on chondrocyte metabolism, (iii) the effect of Wnt signaling on osteoblast carbohydrate and amino acid metabolism with respect to bone anabolism, and (iv) the role of AMP-activated protein kinase in chondrocyte energetic and biomechanical stress responses in the context of cartilage injury, aging, and OA. Although challenges exist for measuring in vivo changes in synovial joint tissue metabolism, the findings presented herein provide multiple lines of evidence to support a central role for disrupted cellular energy metabolism in the pathogenesis of OA.
Keywords: metabolic syndrome, obesity, bone, cartilage, osteoarthritis
Introduction
Osteoarthritis (OA) is the most common cause of disability among musculoskeletal diseases throughout the world [1]. The overall disease burden of OA is two times greater in developed versus underdeveloped regions of the world due in part to the elevated prevalence of obesity [1]. Obesity substantially increases the lifetime risk of developing knee OA, being similar to the effect of joint trauma (Figure 1) [2]. Obesity, however, is not viewed simply as a biomechanical risk factor due to increased weight-bearing because it also increases the risk of OA in the hand [3]. This finding has motivated the search for non-biomechanical factors linking obesity and OA. There is increasing evidence that systemic metabolic dysregulation and inflammation are underlying factors in the pathogenesis of OA, potentially even in the absence of obesity [4]. As a result, there is a growing consensus that the relationship between obesity and OA is complex and multifactorial [5–9].
Figure 1.
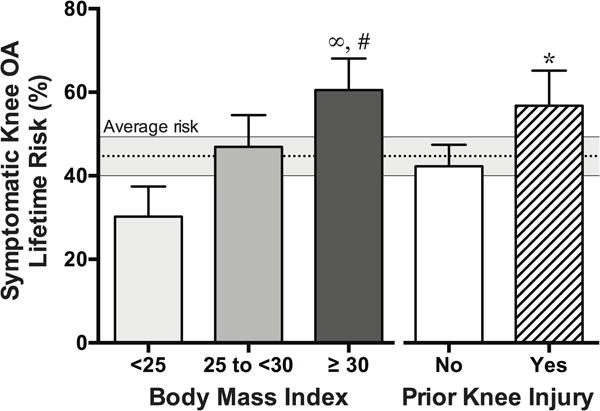
Effect of obesity versus joint injury on the lifetime risk of developing symptomatic knee OA. Obesity, defined as a body mass index ≥ 30, doubles the risk of knee OA compared to normal weight or underweight individuals (body mass index < 25). The effect of obesity on knee OA risk is similar to that for a prior joint injury (60.5% versus 56.8%, respectively). ∞P<0.0001, comparison with normal weight and obese. #P=0.01, comparison with overweight and obese. *P=0.002, comparison of prior joint injury (no and yes). Figure produced from data reported in [2].
A number of excellent recent reviews have discussed the role of metabolic diseases in driving a proposed metabolic OA subtype [10–16]. Many potential links exist between factors involved in metabolic dysfunction (e.g., excess adiposity, insulin resistance, dyslipidemia, and hypertension) and the development of OA, including the production of pro-inflammatory cytokines and adipokines [e.g., 7, 14, 15]. However, the clustering of these metabolic conditions, defined as the Metabolic Syndrome, increases OA risk more than any single condition alone and suggests that a common set of underlying factors contribute to metabolic associated OA [10–16]. In addition to inflammation, prior reviews describe the contribution of vascular dysfunction [12, 16], oxidative stress [10, 13, 16], mitochondrial dysfunction [13], miRNAs [12, 15], and metabolites such as Vitamin D [12, 15] to OA pathogenesis.
An additional phenomenon that has received less attention is metabolic inflexibility. Over-nutrition and insufficient physical activity contribute to a state of metabolic dysregulation characterized by a breakdown of hormonal and metabolite sensing feedback pathways that regulate the delivery and utilization of nutrients (e.g., glucose and lipids) throughout the body. The resulting metabolic inflexibility occurs both systemically and at the cellular level and is considered a key driving force of numerous metabolic pathologies, including type 2 diabetes and cardiovascular disease [17, 18]. The effect of metabolic inflexibility on synovial joint function and OA is poorly understood. This Perspectives Article focuses on emerging areas of research related to aspects of metabolic flexibility that are revealing how changes in cellular energy metabolism, metabolic substrate utilization, and metabolic signaling pathways alter skeletal development and remodeling in response to biomechanical and inflammatory stressors.
This Perspectives Article is based on topics that were discussed at a workshop presented at the 2016 Annual Meeting of the Orthopaedic Research Society in Orlando, FL. These topics include: 1) an update on metabolic stress in OA, focusing in particular on sexually dimorphic pathways, 2) energy metabolism in joint tissues at rest and in response to mechanical loading, 3) glucose and glutamine metabolism in osteoblasts, and 4) AMP-activated protein kinase (AMPK) and metabolism in cartilage and OA. We hope that this overview will stimulate further interest in understanding the metabolic basis of synovial joint health and disease. The development of powerful new techniques for metabolic profiling (i.e., metabolomics) has emerged as a critical factor linking gene expression and proteostasis to cellular function [19]. The impact of this metabolic renaissance is evident in multiple related areas of investigation, including epigenetics, regenerative medicine, immunology, and experimental therapeutics [20–23].
Update on Metabolic Stress in Osteoarthritis
Aging populations and obesogenic environments are increasing the prevalence and comorbidity of metabolic and musculoskeletal diseases [24, 25]. In the United States, approximately 35% of all adults and 50% of adults aged 60 years and older are estimated to have the metabolic syndrome [26]. Based on data from the NHANES III cohort, approximately half of US adults with OA also have metabolic syndrome [27]. Additional cross-sectional studies from other populations also show an association between metabolic syndrome and knee and hand OA [28–31], albeit not always independent of obesity [9, 32]. Several prospective studies, however, confirm that metabolic syndrome increases the risk of knee OA incidence and severity independent of obesity status [33–35]. These findings raise the question: how do components of the metabolic syndrome impact the biology and function of different tissues in synovial joints to promote the pathogenesis of OA?
The metabolic syndrome was originally identified and defined as a group of metabolic conditions that collectively increase the risk of heart disease, diabetes, and stroke. Although several definitions of the metabolic syndrome exist, here we provide the definition set forth by the National Cholesterol Education Program Adult Treatment Panel III and updated by the American Heart Association, where metabolic syndrome is diagnosed as the presence of three or more of the metabolic conditions shown in Figure 2A [26]. Of these conditions, the most prevalent condition in US adults with metabolic syndrome is an elevated waist circumference (93%), followed by elevated triglycerides (85%), low HDL cholesterol (60%), high fasting blood glucose (54%), and high blood pressure (48%) (Figure 2B) [36]. As with knee OA, the overall prevalence of metabolic syndrome in adults in the United States is greater in women than men (37% versus 33%) [26]. Women with metabolic syndrome have a significantly higher prevalence of elevated waist circumference and low HDL cholesterol [36]. However, not all conditions are more prevalent in women. For example, high blood pressure is just as prevalent in women and men with metabolic syndrome, and the prevalence of high triglycerides and high fasting blood glucose are lower in women than men with metabolic syndrome [36].
Figure 2.
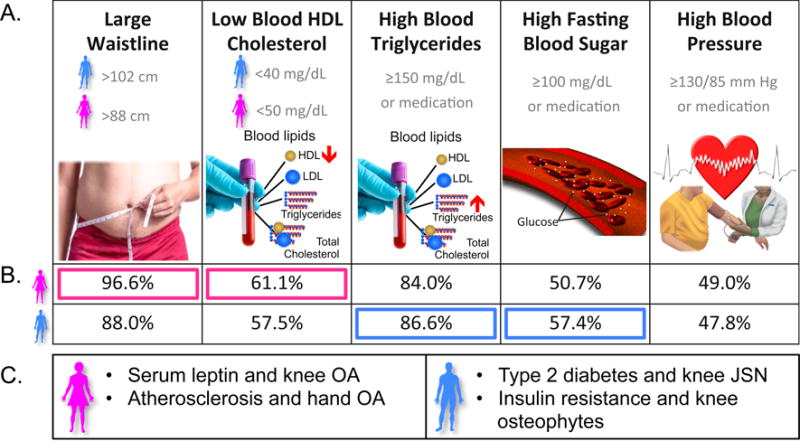
Association of metabolic syndrome components and OA pathology in men and women. A. Metabolic syndrome components as defined by the National Cholesterol Education Program Adult Treatment Panel III and updated by the American Heart Association. Metabolic syndrome is diagnosed as ≥ 3 of these conditions. B. Prevalence of individual metabolic syndrome components in men and women diagnosed with metabolic syndrome. Pink boxes indicated significantly higher prevalence in women, and blue boxes indicated higher prevalence in men. Data from [36]. C. Sex-biases in OA pathology and metabolic syndrome related factors (pink = female, blue = male). References for these relationships are provided in the text.
These sex-specific differences may provide some mechanistic insight into the link between metabolic syndrome and OA. In general, these metabolic syndrome components are thought to promote OA by increasing joint inflammation, impairing bone remodeling, and suppressing cartilage anabolic activity in favor of catabolism. One area that has received considerable attention is the relationship between adiposity (e.g., waist circumference) and the production of systemic and local mediators of inflammation, including adipokines and cytokines [37, 38]. Of particular note is the role of leptin, an adipokine that has been shown to stimulate catabolic pathways in chondrocytes [39, 40]. In both cross-sectional and longitudinal studies, leptin is associated with an increase in knee OA severity and progression in women but not men (Figure 2C) [41–44]. Similarly, a sex-dependent bias was also observed for the relationship between atherosclerosis and OA (Figure 2C). In a longitudinal population-based study, Hoeven and colleagues showed that atherosclerosis was independently associated with baseline knee and hand OA prevalence and with the progression of hand OA in women only [45]. A number of pre-clinical animal studies have shown that elevated adiposity and blood lipids, including hypercholesterolemia and related conditions, increase joint pathology in aging, post-traumatic, and collagenase-induced OA models (e.g., [46–49]). Most obesity-related pre-clinical studies, however, use animals from a single sex. Consequently, the sex-dependent basis for these clinical associations is not known and warrants further investigation.
The sex-bias linking metabolic syndrome components to OA does not only favor women. Recent studies indicate that insulin resistance and hygerglycemia are associated primarily with OA development in men (Figure 2C). For example, HOMA-IR scores, which are derived from a model-based estimate of insulin resistance and β-cell function using fasting plasma insulin and glucose concentrations, were positively associated with osteophyte-defined knee OA in obese men but not women [43]. In addition, male, but not female, patients with symptomatic knee OA and type 2 diabetes had significantly greater joint space narrowing than patients without type 2 diabetes, even when adjusting for age, obesity, hypertension, and dyslipidemia [50]. A recent study by Hamada and colleagues showed that diabetes and synovial insulin resistance increase OA pathology by stimulating synovial inflammation, and in particular tumor necrosis factor production [51]. Whether or not these findings underlie the sex-dependent clinical observations is not known.
As with other obesity-related diseases, a better understanding of the tissue and cell-specific effects of metabolic stressors on OA pathogenesis is expected to lead to the identification of novel disease biomarkers and the development of metabolic-based therapies to prevent or slow the progression of OA. A number of labs are tackling this challenge by investigating the contribution of particular components of the metabolic syndrome to the function of specific joint tissues. Ultimately, though, a systems-based approach that integrates the pro-catabolic effects of metabolic stressors and altered biomechanics will likely be needed to comprehensively understand the role of obesity in OA-pathogenesis and to develop new targeted therapies (e.g., [52]). The following sections address this need by providing new insight into how biomechanical loads, developmental signaling pathways, and cellular metabolic sensors regulate cartilage and bone metabolic and structural homeostasis.
Energy Metabolism in Joint Tissues at Rest and in Response to Mechanical Loading
Joints transmit loads generated during locomotion, which can be in excess of 3 times body weight [53]. For example, contact pressures can reach 2,600 PSI during stair climbing [54]. These loads result in deformation of the soft tissues within the joints. Studies of joint loading in humans and animal models show that typical compressive soft tissue strains are approximately 10% [55, 56]. Higher strains from injury or joint trauma may result in tissue damage and cellular dysfunction. When joint tissues experience mechanical wear and damage, additional biochemical energy is required to repair and remodel the damaged tissue. Frequently, this energy is stored by the nucleotide adenosine triphosphate (ATP). While mechanical energy is dissipated when soft tissues are compressed, it is unknown if this affects the cellular energy landscape. This section describes the changes in energy metabolism at rest and in response to mechanical loading using recent studies of chondrocytes as a model system.
Glucose is metabolized to produce ATP [57], which is the primary energy source for many cellular processes [58]. Glucose is the major source of energy for chondrocytes [59]. Glucose metabolism involves glycolysis, the pentose phosphate pathway (PPP), and the tricarboxylic acid (TCA) cycle (Figure 3), which generates many of the precursors to amino acids. Glycolysis occurs independent of oxygen whereas oxygen is required to complete the TCA cycle and generate ATP using oxidative phosphorylation via the electron transport chain (ETC). The ETC combines molecular oxygen with protons producing water during ATP synthesis. Cells from highly vascularized tissues (e.g., bone, synovium, ligament, and tendon) have rapid access to oxygen, which supports high rates of ATP generation using the TCA cycle and the ETC. In contrast, cells from avascular tissues, such as cartilage and portions of the meniscus, rely primarily on glycolysis to rapidly generate ATP [60, 61]. Interestingly though, there is evidence that TCA cycle activity and mitochondrial respiration positively regulate glycolysis in bovine cartilage explants [62].
Figure 3.
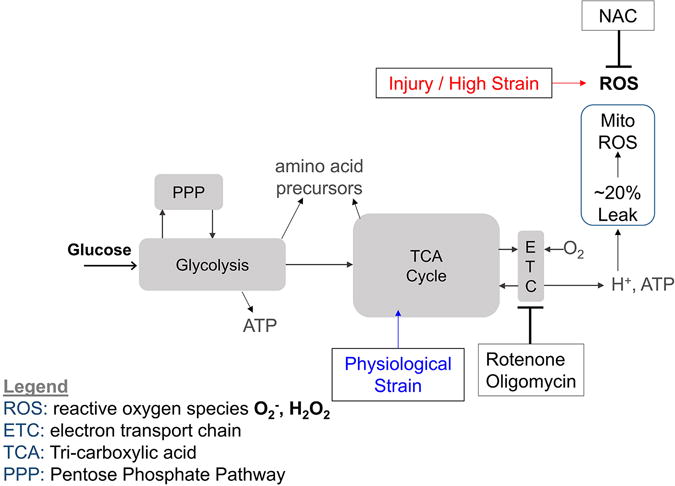
Model of mechanically-induced changes in chondrocyte energy metabolism. As a source of energy, glucose metabolism begins with glycolysis. Glycolysis can route energy to the pentose phosphate pathway (PPP) to make 5-membered sugars including nucleic acids. In the presence of oxygen, the tri-carboxylic acid (TCA) cycle can increase the yield of ATP compared with glycolysis alone. Glucose is also be used to make amino acid precursors for protein synthesis. In both cartilage explants and 3D cultured chondrocytes, physiological loading (blue) appears to stimulate the TCA cycle, while high-magnitude injurious loading (red) increases oxidative stress [62, 68, 69]. Studies have used small molecule inhibitors, including rotenone, oligomycin, and N-acetylcystein (NAC) to modulate metabolic and oxidative processes.
Because soft tissues undergo most of the deformation in the joint during loading, we focused on studies in cartilage and chondrocytes. In chondrocytes from healthy cartilage, glycolysis is enhanced by phosphorylation of the enzyme PFK-2, which is mediated by activation of the serine-threonine kinase, AMP-activated protein kinase (AMPKα) [63]. However, OA and aged chondrocytes exhibit diminished activated AMPKα [64, 65]. Furthermore, OA and aged cartilage exhibit decreased mitochondrial protein expression and biogenesis compared to healthy cartilage [66], suggesting that impaired mitochondrial respiration may limit metabolic resources required to repair and remodel damaged joint tissue. For example, healthy bovine chondrocytes survive a nutrient stress in part by upregulating mitochondrial respiration and reducing the rate of reactive nitrogen and oxygen species production [67]. Conversely, in response to hypoxia, chondrocytes alter their metabolism by increasing glucose uptake and expression of the glucose transporter GLUT1, consistent with an adaptive response to changes in the in vivo environment [59].
Studies of physiological compression in agarose-encapsulated SW1353 chondrocytes show that the TCA cycle is activated and increases production of the protein precursor pyruvate throughout 15–30 minutes of compression [68]. In contrast, primary human OA chondrocytes in the same system exhibit a transient increase in TCA activity that diminishes after 30 minutes. Principal components analysis of 28 glucose derivatives indicates that unloaded controls cluster separately from samples subjected to either 15 or 30 minutes of compression in primary OA chondrocytes [69]. In contrast to physiological loading, injurious loading of cartilage explants increases oxidative stress similar to observations in OA lesions [62].
While these studies focus on glucose metabolism, chondrocytes may also derive energy from metabolism of lipids and other sources such as galactose or glutamine. Because these pathways involve a variety of metabolic intermediates over time, measurements of multiple metabolites are useful for understanding pathway-wide behavior. By performing flux balance analysis on targeted metabolomic data using a stoichiometric model of energy metabolism [70], studies can comprehensively describe changes in cellular metabolism [71, 72].
Glucose and Glutamine Metabolism in Osteoblasts
It is increasingly clear that OA development often couples with changes in the subchondral bone compartment. Understanding how metabolic stress affects OA requires better knowledge about cellular metabolism in osteoblasts. Although the general metabolic pathways are shared among mammalian cells, specific cell types such as osteoblasts may utilize the pathways differently to fulfill their unique biological functions. Studies in the 1960s already revealed that cultured bone slices or osteoblasts utilize glucose briskly to produce lactate as the main end product [73–75]. However, interest in this area of research waned during the past several decades. As a result, the energy and carbon source for osteoblasts remains poorly defined [76]. Moreover, it is not known how cellular metabolic pathways are reprogrammed when mesenchymal progenitors transition to mature osteoblasts.
Recent studies have explored the potential effect of Wnts and PTH, both known bone anabolic signals, on cellular metabolism during osteoblast differentiation. The results indicate that both signals stimulate glycolysis but suppress glucose metabolism in the mitochondria, a process known as aerobic glycolysis or Warburg effect originally associated with cancer cells. In particular, Wnt signaling acutely increases the protein levels of a number of glycolytic enzymes in a manner independent of β-catenin-mediated transcription but dependent on mTORC2 and Akt activity [77]. Such changes result in a rapid increase in glycolysis as well as the production of lactate while simultaneously suppressing glucose metabolism via the TCA cycle [77, 78]. The energy requirement during osteoblast differentiation in response to Wnt signaling is fulfilled partly by increased glutamine metabolism in the TCA cycle [79]. Remarkably, the increased utilization of glutamine notably reduces the intracellular levels of glutamine, thus activating the Gcn2-eIF2α-Atf4 stress pathway to increase the protein synthesis capacity that is integral to osteoblast identity [79]. In contrast to Wnt’s direct reprogramming of cellular metabolism, PTH stimulates aerobic glycolysis secondarily through the induction of Igf signaling [80]. Importantly, with either Wnt or PTH, reversing the increased glycolysis suppresses the bone anabolic effects of the signal [77, 80]. These studies therefore identify stimulation of aerobic glycolysis as a shared mechanism between two major bone anabolic signals. Overall, dysregulation of cellular metabolism in osteoblasts may contribute to the pathology of OA in response to systemic metabolic stress, such as insulin resistance. Targeting metabolic pathways may be a viable therapeutic approach for delaying the occurrence or impeding the progression of joint diseases.
AMPK and Metabolism in Cartilage and Osteoarthritis
Effective regulation of cellular energy metabolism is critical for tissue homeostasis [22, 81, 82]. Emerging evidence indicates a critical interplay between cellular energy metabolism and inflammation [22, 81, 82]. Low-grade systemic inflammation resulting from metabolic disturbances, as well as low-grade local joint inflammation (i.e., synovitis), is implicated in progressive cartilage degeneration and OA progression [14, 83]. Recent findings indicate that AMPK, a master regulator of energy balance and metabolism, plays an important role in cartilage inflammation and tissue homeostasis.
AMPK is an evolutionary conserved serine/threonine protein kinase existing in essentially all eukaryotic cells. AMPK is a heterotrimeric complex consisting of a catalytic α-subunit and two regulatory β- and γ-subunits. Each subunit has 2–3 isoforms encoded by different genes [84, 85]. Importantly, phosphorylation of a conserved threonine within the catalytic domain (Thr172) of the α subunit is crucial for AMPK activity [84, 85]. Articular chondrocytes express α1, α2, β1, β2 and γ1 isoforms of AMPK subunits, and α1 appears to be the predominantly expressed and functionally active AMPK α isoform [64]. AMPK is activated in response to a metabolic stress resulting in an increase in the cellular AMP:ATP ratio, which can be caused by either a net increase in ATP consumption (e.g. exercise/muscle contraction) or decrease in ATP production (e.g. hypoxia). In this manner, AMPK enables cells to adjust to changes in energy demand. AMPK can be activated by pharmacological compounds such as the nucleotide mimetic AICAR (5-aminoimidazole-4- caboxamide 1-b-D-ribofuranoside) and A-769662, which is a selective and direct AMPK activator through binding to the β1 subunit [81, 84]. Some drugs already in the clinic for arthritis and other conditions have also been shown to activate AMPK (e.g., sodium salicylate, methotrexate, metformin, and the natural plant product berberine) [86, 87].
Phosphorylation of AMPKα Thr172 is constitutively present in normal articular chondrocytes/cartilage; however, it is decreased in human knee OA chondrocytes/cartilage, trauma-induced mouse knee OA cartilage, and aged mouse knee cartilage [64, 65]. In vitro studies demonstrated that inflammatory cytokines IL-1β and TNFα, as well as biomechanical injury, can cause de-phosphorylation of AMPKα in normal articular chondrocytes. This is correlated with increased catabolic responses [64, 65]. In addition, chondrocytes deficient in both AMPKα1 and AMPKα2 (achieved via siRNA transfection) significantly enhance catabolic responses to IL-1β and TNFα [64]. Moreover, AMPK pharmacological activators are able to inhibit catabolic responses to either inflammatory cytokines or biomechanical injury [64, 65]. These results indicate that chondrocytes with reduced AMPK activity can disturb the cartilage homeostatic balance by increasing catabolic activity. Recent in vivo studies reveal that AMPKα1 knockout (KO) mice develop spontaneous OA development (unpublished observation). Activation of AMPK by berberine limits both surgery-induced and aging-related spontaneous OA development in mice, indicated by significantly less proteoglycan loss and cartilage degradation [88]. Taken together, these data suggest that maintaining articular chondrocyte AMPK activity is critical to cartilage matrix homeostasis.
Regulation of AMPK activity in articular chondrocytes
Liver protein kinase B1 (LKB1) is the primary upstream kinase of AMPKα in chondrocytes [65]. Notably, phosphorylation of AMPKα Thr172 is nearly completely inhibited in LKB1 knockdown chondrocytes, resulting in significantly enhanced catabolic responses to IL-1β and TNFα [65]. Phosphorylation of LKB1, similar to phosphorylation of AMPKα, is reduced in human knee OA chondrocytes, mouse knee OA cartilage, aged mouse knee cartilage, and chondrocytes challenged with mechanical injury [65]. This suggests that dysregulation of LKB1 in aged and OA cartilage may contribute to suppression of AMPK activity. Chondrocyte AMPK activity can also be negatively regulated through de-phosphorylation by protein phosphatase PP2Cα, as suggested by increased phosphorylation of AMPKα Thr172 in PP2Cα knockdown chondrocytes (unpublished observation). Chondrocytes treated with IL-1β and TNFα have increased expression of PP2Cα, which could be, at least in part, responsible for de-phosphorylated AMPKα Thr172 by these cytokines (unpublished observation). AMPK activation also increases the expression and activity of another energy sensor in articular chondrocytes, SIRT1, which is a NAD+-dependent protein deacetylase [66]. Several studies have demonstrated that dysregulation of SIRT1 leads to OA development and progression. Interestingly, SIRT1 can deacetylate LKB1, forming a positive feedback loop to potentiate AMPK activity and function [89].
AMPK downstream signaling in articular chondrocyte
Mitochondrial dysfunction is well documented in OA chondrocytes and has been associated with abnormalities in articular chondrocyte function and reduced viability that contribute to cartilage degeneration in OA [90–92]. These abnormalities include impaired chondrocyte anabolic and growth responses, excesses oxidative stress, increased apoptosis, and cartilage matrix calcification. In addition, mitochondrial dysfunction has also been linked to enhanced responsiveness to pro-inflammatory cytokines and increased matrix catabolism [90–92]. Mitochondrial biogenesis is important for maintenance of mitochondrial function. Recent findings show that expression of peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α), a transcription co-activator and master regulator of mitochondrial biogenesis and function, is decreased in both mouse knee OA cartilage and in aged mouse knee cartilage [66]. In addition, mitochondrial biogenesis capacity is significantly reduced in advanced human knee OA chondrocytes, as indicated by deceased mitochondrial DNA content and mass. This is associated with reduced mitochondrial respiration and intracellular ATP level [66]. These functional changes are correlated with a concomitant reduction in phosphorylation of AMPKα, reduced expression of SIRT1 and PGC-1α, and increased acetylation of PGC-1α. Importantly, pharmacologic activation of AMPK reverses impaired mitochondrial biogenesis and function in advanced human knee OA chondrocytes through activation of SIRT1 and PGC-1α [66].
Activation of AMPK in articular chondrocytes also increases expression of FoXO3a, a transcription factor that belongs to the forkhead box O (FoXO) family [93]. FoXO3a is a direct transcriptional regulator of PGC-1α, and PGC-1α itself can augment the transcriptional activity of FoXO3a. Not surprisingly, reduced expression of FoXO3a is found in both mouse knee OA cartilage and in aged mouse knee cartilage. Mitochondria dysfunction leads to elevated levels of reactive oxygen species (ROS), and ROS can modulate redox-sensitive NF-κB and other signaling pathways that increase chondrocyte catabolic activity [93]. Pharmacologic activation of AMPK in articular chondrocytes limits excessive oxidative stress by up-regulating expression of antioxidant enzymes SOD2 and catalase [93]. AMPK activation also inhibits inflammatory cytokine-induced NF-κB signaling and catabolic responses through PGC-1α and FoXO3a [93].
An additional cellular process associated with metabolic dysfunction and activated in human knee OA chondrocytes is a stress response termed the unfolded protein response (UPR). UPR can result from a compromise in the function of the endoplasmic reticulum (ER), where all secretory and integral membrane proteins are folded and post-translationally modified [94, 95]. Cellular stress such as perturbations in energy stores, redox state, and metabolic and inflammatory challenges cause ER stress, leading to accumulation of misfolded proteins [94, 95]. UPR protects cells from stress and contributes to cellular homeostasis re-establishment. However, when ER stress is too severe or chronic, or the UPR is unable to resolve the protein-folding defects, cells are led to apoptosis through induction of C/EBP homologous protein (CHOP) [94, 95]. Several factors implicated in OA pathogenesis upregulate expression CHOP in articular chondrocytes, including biomechanical injury, IL-1β, nitric oxide, and advanced glycation end products [96–100]. CHOP potentiates the capacity of IL-1β to induce catabolic responses, superoxide generation and apoptosis in chondrocytes by inhibiting AMPK activity [96]. Pharmacologic AMPK activation blunts CHOP expression and catabolic responses induced by IL-1β and biomechanical injury in chondrocytes [96], suggesting a role of AMPK in alleviating ER stress.
Another OA-related process involved in cellular proteostasis and metabolism is autophagy. Autophagy is a cellular housekeeping and protein quality control mechanism, which can remove damaged or defective proteins and organelles including damaged mitochondria [89, 98]. Chondrocyte autophagy is a constitutive homeostatic mechanism in articular cartilage. It is reduced in human knee OA, mouse knee OA, and aged mouse knee cartilage [101]. Suppressed autophagy is observed in cartilage ex vivo in response to mechanical injury [102]. Inhibition of autophagy in chondrocytes exacerbated IL-1β–induced OA-like gene expression changes and apoptotic signals; whereas, activation of autophagy inhibited these responses, possibly through modulation of ROS in chondrocytes in vitro [103]. AMPK can promote chondrocyte autophagy through inhibition of mTOR [104]. The later steps of autophagy in autophagosome formation can be enhanced by SIRT1 through deacetylating several autophagy-related proteins [89]. Collectively, AMPK exerts its chondroprotective effect through its downstream targets that increase cellular stress resistance by 1) preserving mitochondrial function that inhibits oxidative stress and inflammation, and 2) protecting chondrocyte proteostasis mechanisms that limit ER stress and maintain autophagy.
The increased incidence of OA with aging and metabolic syndrome strongly supports the concept that AMPK dysfunction is involved in OA pathogenesis. Responsiveness of AMPK activation declines during the aging process, and low-grade inflammation present in aging tissues is thought to be at least in part responsible for suppressing AMPK signaling [89]. Nutritional factors can affect AMPK signaling. Caloric restriction is known to stimulate AMPK activity. However, nutritional overload impairs AMPK activity, which induces insulin resistance in many tissues [89]. Dysregulation of AMPK is linked to obesity and diabetes and a variety of other age-related diseases [89, 105]. Reduced capacity of AMPK activation in cartilage and likely other articular joint tissues could promote OA by limiting energy availability for cellular maintenance, triggering cell stress pathways, altering cell metabolism, and ultimately compromising cell survival, tissue integrity and function. Since sustained AMPK activity in articular chondrocytes appears to be critical for maintaining cartilage homeostatic balance, targeted activation of AMPK could be an attractive and novel therapeutic strategy for OA (Figure 4).
Figure 4.
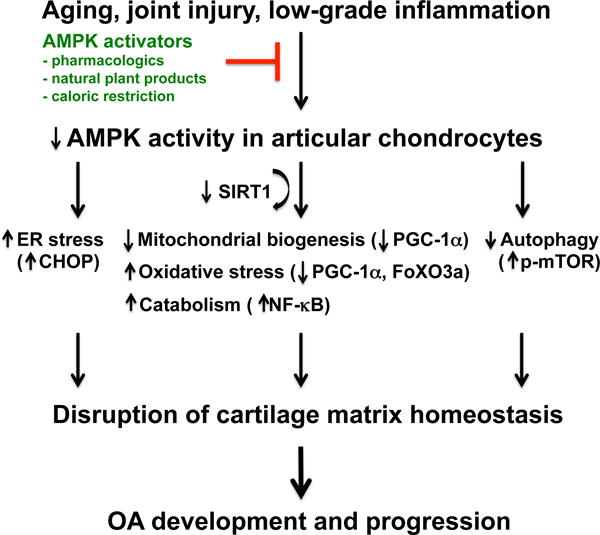
AMPK as a potential therapeutic target for OA. Aging, joint injury, and low-grade inflammation impair AMPK activity in articular cartilage, causing dysregulation of AMPK signaling in articular chondrocytes. This would result in mitochondrial dysfunction (e.g., reduced capacity of mitochondrial biogenesis), increased oxidative stress and inflammation-mediated matrix catabolism, and poor cellular quality control (e.g., increased ER stress and decreased autophagy). All of which can compromise cell survival and cartilage tissue integrity, ultimately leading to OA development and progression. However, targeted activation of AMPK (potentially achieved by pharmacological approach, natural plant products or dietary restriction) could be an attractive therapeutic strategy for OA.
Conclusions
OA is a multifactorial disease with both complex causes and diverse phenotypes. Obesity is a key driver of both altered joint biomechanics and catabolic processes within the joint. Metabolic syndrome affects upwards of 50% of the US population and patients with metabolic syndrome have increased incidence of OA. OA in metabolic syndrome patients displays interesting differences between genders, and further research into the sexual dimorphism of OA within the context of metabolic syndrome may yield important advances in understanding the etiology of OA. Cartilage is an important tissue that deteriorates in OA, and the low oxygen environment of cartilage suggests that mitochondrial respiration is negligible in chondrocytes. However, the historical view that chondrocyte metabolism is primarily glycolytic and that mitochondrial function has minimal impact on chondrocyte function is rapidly changing. Chondrocytes clearly modulate their glycolytic and oxidative central metabolism in response to both physiological and injurious mechanical loads, suggesting that mitochondrial dysfunction may contribute to OA pathogenesis.
Diabetic patients have compromised skeletal health demonstrating the importance of glucose regulation to bone metabolism. However, the carbon sources for osteoblasts remain poorly defined. Exogenous PTH and Wnt agonists are known to stimulate bone anabolism, and both signals increase osteoblast glycolysis. Glucose is primarily metabolized via anaerobic glycolysis, which appears to be a common feature of bone anabolic signals. Inhibiting either Wnt- or PTH-induced glucose metabolism decreases anabolic changes. Wnt also stimulates glutamine metabolism, raising the possibility that impaired TCA cycle function in the mitochondria may also compromise bone anabolism.
Cellular energy sensors are critical mediators of cellular homeostasis. In chondrocytes, AMPK is a master regulator of energy metabolism that becomes impaired in OA cartilage. Both mechanical injury and pro-inflammatory cytokines can decrease the phosphorylation of AMPK, and siRNA knockdown of AMPK enhances cytokine-driven catabolic responses. Pharmacological activators of AMPK inhibit catabolic responses to both injury and pro-inflammatory cytokines. In animal models of OA, berberine, a natural activator of AMPK, decreases cartilage degradation and proteoglycan loss. Restoring AMPK-mediated cellular responses that promote chondrocyte energy homeostasis is an emerging area for therapeutic targeting in OA. Overall, the findings described in this Perspectives Article provide strong evidence that dysregulated metabolism drives musculoskeletal disease.
Many questions remain about the nature of metabolic dysfunction in the onset and progression of musculoskeletal diseases such as OA. Some key questions include:
What are the types and sources of carbon used by the various synovial joint tissues for energy metabolism (e.g., glucose, amino acids, fatty acids), and how do they change during conditions that increase the risk of disease (e.g., aging, obesity, joint injury)?
What are the primary determinants and regulators of metabolic energy usage and nutrient transport in joint tissues?
Is there a central hormonal or metabolic factor driving shared metabolic pathology in all joint tissues, or does each tissue develop a unique cellular metabolic pathology with a distinct temporal onset?
For example, basic studies of how healthy and OA cells modulate energy metabolism with mechanical loading may yield insight into the pathways linking biomechanics, metabolism, and inflammation. By understanding how energy metabolism and mechanotransduction are dysregulated in OA and other joint diseases, future studies may yield novel strategies for improving joint health by a combination of targeted physical and pharmacological interventions.
Acknowledgments
This work was supported by grants from the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases grants R01 AR060456 and AR055923 to FL, R01AR1067966 to RLB, R03AR066828 to TMG, and National Institute on Aging grant R01AG049058 to TMG), the Department Veterans Affairs (1I01BX002234 to RLB), the Arthritis Foundation (6045 to RLB), and the National Science Foundation (1342420 and 1554708 to RKJ). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Author Contributions:
All authors were involved in drafting the article and revising it critically for important intellectual content, and all authors approved the final version to be published.
RKJ has financial interests in Beartooth Biotech that has licensed a stoichiometric model of energy metabolism. FL, RLB, and TMG have no conflicts to disclose.
References
- 1.Brooks PM. The burden of musculoskeletal disease–a global perspective. Clin Rheumatol. 2006;25(6):778–781. doi: 10.1007/s10067-006-0240-3. [DOI] [PubMed] [Google Scholar]
- 2.Murphy L, Schwartz TA, Helmick CG, Renner JB, Tudor G, Koch G, Dragomir A, Kalsbeek WD, Luta G, Jordan JM. Lifetime risk of symptomatic knee osteoarthritis. Arthritis Rheum. 2008;59(9):1207–1213. doi: 10.1002/art.24021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yusuf E, Nelissen RG, Ioan-Facsinay A, Stojanovic-Susulic V, DeGroot J, van Osch G, Middeldorp S, Huizinga TW, Kloppenburg M. Association between weight or body mass index and hand osteoarthritis: a systematic review. Ann Rheum Dis. 2010;69(4):761–765. doi: 10.1136/ard.2008.106930. [DOI] [PubMed] [Google Scholar]
- 4.Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!) Osteoarthritis Cartilage. 2013;21(1):16–21. doi: 10.1016/j.joca.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Issa RI, Griffin TM. Pathobiology of obesity and osteoarthritis: integrating biomechanics and inflammation. Pathobiol Aging Age Relat Dis. 2012;2(2012) doi: 10.3402/pba.v2i0.17470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Messier SP. Obesity and osteoarthritis: disease genesis and nonpharmacologic weight management. Med Clin North Am. 2009;93(1):145–159. xi–xii. doi: 10.1016/j.mcna.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Rai MF, Sandell LJ. Inflammatory mediators: tracing links between obesity and osteoarthritis. Crit Rev Eukaryot Gene Expr. 2011;21(2):131–142. doi: 10.1615/critreveukargeneexpr.v21.i2.30. [DOI] [PubMed] [Google Scholar]
- 8.Thijssen E, van Caam A, van der Kraan PM. Obesity and osteoarthritis, more than just wear and tear: pivotal roles for inflamed adipose tissue and dyslipidaemia in obesity-induced osteoarthritis. Rheumatology (Oxford) 2015;54(4):588–600. doi: 10.1093/rheumatology/keu464. [DOI] [PubMed] [Google Scholar]
- 9.Visser AW, de Mutsert R, le Cessie S, den Heijer M, Rosendaal FR, Kloppenburg M. The relative contribution of mechanical stress and systemic processes in different types of osteoarthritis: the NEO study. Annals of the Rheumatic Diseases. 2015;74(10):1842–1847. doi: 10.1136/annrheumdis-2013-205012. [DOI] [PubMed] [Google Scholar]
- 10.Courties A, Gualillo O, Berenbaum F, Sellam J. Metabolic stress-induced joint inflammation and osteoarthritis. Osteoarthritis Cartilage. 2015;23(11):1955–1965. doi: 10.1016/j.joca.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 11.Hawker GA, Stanaitis I. Osteoarthritis year in review 2014: clinical. Osteoarthritis Cartilage. 2014;22(12):1953–1957. doi: 10.1016/j.joca.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 12.Katz JD, Agrawal S, Velasquez M. Getting to the heart of the matter: osteoarthritis takes its place as part of the metabolic syndrome. Curr Opin Rheumatol. 2010;22(5):512–519. doi: 10.1097/BOR.0b013e32833bfb4b. [DOI] [PubMed] [Google Scholar]
- 13.Le Clanche S, Bonnefont-Rousselot D, Sari-Ali E, Rannou F, Borderie D. Inter-relations between osteoarthritis and metabolic syndrome: A common link? Biochimie. 2016;121:238–252. doi: 10.1016/j.biochi.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Sellam J, Berenbaum F. Is osteoarthritis a metabolic disease? Joint Bone Spine. 2013;80(6):568–573. doi: 10.1016/j.jbspin.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Hunter D, Xu J, Ding C. Metabolic triggered inflammation in osteoarthritis. Osteoarthritis Cartilage. 2015;23(1):22–30. doi: 10.1016/j.joca.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Zhuo Q, Yang W, Chen J, Wang Y. Metabolic syndrome meets osteoarthritis. Nat Rev Rheumatol. 2012;8(12):729–737. doi: 10.1038/nrrheum.2012.135. [DOI] [PubMed] [Google Scholar]
- 17.Griffin TM, Humphries KM, Kinter M, Lim HY, Szweda LI. Nutrient sensing and utilization: Getting to the heart of metabolic flexibility. Biochimie. 2016;124:74–83. doi: 10.1016/j.biochi.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159(6):1253–1262. doi: 10.1016/j.cell.2014.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016 doi: 10.1038/nrm.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153(1):56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013;123(3):980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493(7432):346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 23.Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140(12):2535–2547. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Losina E, Weinstein AM, Reichmann WM, Burbine SA, Solomon DH, Daigle ME, Rome BN, Chen SP, Hunter DJ, Suter LG, et al. Lifetime risk and age at diagnosis of symptomatic knee osteoarthritis in the US. Arthritis Care Res (Hoboken) 2013;65(5):703–711. doi: 10.1002/acr.21898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muthuri SG, Hui M, Doherty M, Zhang W. What if we prevent obesity? Risk reduction in knee osteoarthritis estimated through a meta-analysis of observational studies. Arthritis Care Res (Hoboken) 2011;63(7):982–990. doi: 10.1002/acr.20464. [DOI] [PubMed] [Google Scholar]
- 26.Aguilar M, Bhuket T, Torres S, Liu B, Wong RJ. Prevalence of the metabolic syndrome in the United States, 2003–2012. JAMA. 2015;313(19):1973–1974. doi: 10.1001/jama.2015.4260. [DOI] [PubMed] [Google Scholar]
- 27.Puenpatom RA, Victor TW. Increased prevalence of metabolic syndrome in individuals with osteoarthritis: an analysis of NHANES III data. Postgraduate medicine. 2009;121(6):9–20. doi: 10.3810/pgm.2009.11.2073. [DOI] [PubMed] [Google Scholar]
- 28.Calvet J, Orellana C, Larrosa M, Navarro N, Chillaron JJ, Pedro-Botet J, Galisteo C, Garcia-Manrique M, Gratacos J. High prevalence of cardiovascular co-morbidities in patients with symptomatic knee or hand osteoarthritis. Scandinavian journal of rheumatology. 2015:1–4. doi: 10.3109/03009742.2015.1054875. [DOI] [PubMed] [Google Scholar]
- 29.Lee S, Kim TN, Kim SH, Kim YG, Lee CK, Moon HB, Koh EM, Yoo B. Obesity, metabolic abnormality, and knee osteoarthritis: a cross-sectional study in Korean women. Modern rheumatology / the Japan Rheumatism Association. 2015;25(2):292–297. doi: 10.3109/14397595.2014.939393. [DOI] [PubMed] [Google Scholar]
- 30.Shin D. Association between metabolic syndrome, radiographic knee osteoarthritis, and intensity of knee pain: results of a national survey. The Journal of clinical endocrinology and metabolism. 2014;99(9):3177–3183. doi: 10.1210/jc.2014-1043. [DOI] [PubMed] [Google Scholar]
- 31.Yoshimura N, Muraki S, Oka H, Kawaguchi H, Nakamura K, Akune T. Association of Knee Osteoarthritis with the Accumulation of Metabolic Risk Factors Such as Overweight, Hypertension, Dyslipidemia, and Impaired Glucose Tolerance in Japanese Men and Women: The ROAD Study. The Journal of rheumatology. 2011;38(5):921–930. doi: 10.3899/jrheum.100569. [DOI] [PubMed] [Google Scholar]
- 32.Engström G, Gerhardsson de Verdier M, Rollof J, Nilsson PM, Lohmander LS. C-reactive protein, metabolic syndrome and incidence of severe hip and knee osteoarthritis. A population-based cohort study. Osteoarthritis and Cartilage. 2009;17(2):168–173. doi: 10.1016/j.joca.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Monira Hussain S, Wang Y, Cicuttini FM, Simpson JA, Giles GG, Graves S, Wluka AE. Incidence of total knee and hip replacement for osteoarthritis in relation to the metabolic syndrome and its components: A prospective cohort study. Seminars in Arthritis and Rheumatism. 2014;43(4):429–436. doi: 10.1016/j.semarthrit.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 34.Sowers M, Karvonen-Gutierrez CA, Palmieri-Smith R, Jacobson JA, Jiang Y, Ashton-Miller JA. Knee osteoarthritis in obese women with cardiometabolic clustering. Arthritis Rheum. 2009;61(10):1328–1336. doi: 10.1002/art.24739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshimura N, Muraki S, Oka H, Tanaka S, Kawaguchi H, Nakamura K, Akune T. Accumulation of metabolic risk factors such as overweight, hypertension, dyslipidaemia, and impaired glucose tolerance raises the risk of occurrence and progression of knee osteoarthritis: a 3-year follow-up of the ROAD study. Osteoarthritis and Cartilage. 2012;20(11):1217–1226. doi: 10.1016/j.joca.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Wong RJ. Trends in Prevalence of the Metabolic Syndrome–Reply. JAMA. 2015;314(9):950–951. doi: 10.1001/jama.2015.8628. [DOI] [PubMed] [Google Scholar]
- 37.Kluzek S, Arden NK, Newton J. Adipokines as potential prognostic biomarkers in patients with acute knee injury. Biomarkers. 2015;20(8):519–525. doi: 10.3109/1354750X.2014.948914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann E, Junker S, Schett G, Frommer K, Muller-Ladner U. Adipokines in bone disease. Nat Rev Rheumatol. 2016;12(5):296–302. doi: 10.1038/nrrheum.2016.49. [DOI] [PubMed] [Google Scholar]
- 39.Dumond H, Presle N, Terlain B, Mainard D, Loeuille D, Netter P, Pottie P. Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum. 2003;48(11):3118–3129. doi: 10.1002/art.11303. [DOI] [PubMed] [Google Scholar]
- 40.Hui W, Litherland GJ, Elias MS, Kitson GI, Cawston TE, Rowan AD, Young DA. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. Ann Rheum Dis. 2012;71(3):455–462. doi: 10.1136/annrheumdis-2011-200372. [DOI] [PubMed] [Google Scholar]
- 41.Fowler-Brown A, Kim DH, Shi L, Marcantonio E, Wee CC, Shmerling RH, Leveille S. The mediating effect of leptin on the relationship between body weight and knee osteoarthritis in older adults. Arthritis Rheumatol. 2015;67(1):169–175. doi: 10.1002/art.38913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karvonen-Gutierrez CA, Harlow SD, Jacobson J, Mancuso P, Jiang Y. The relationship between longitudinal serum leptin measures and measures of magnetic resonance imaging-assessed knee joint damage in a population of mid-life women. Ann Rheum Dis. 2014;73(5):883–889. doi: 10.1136/annrheumdis-2012-202685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karvonen-Gutierrez CA, Sowers MR, Heeringa SG. Sex dimorphism in the association of cardiometabolic characteristics and osteophytes-defined radiographic knee osteoarthritis among obese and non-obese adults: NHANES III. Osteoarthritis Cartilage. 2012;20(7):614–621. doi: 10.1016/j.joca.2012.02.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martel-Pelletier J, Raynauld JP, Dorais M, Abram F, Pelletier JP. The levels of the adipokines adipsin and leptin are associated with knee osteoarthritis progression as assessed by MRI and incidence of total knee replacement in symptomatic osteoarthritis patients: a post hoc analysis. Rheumatology (Oxford) 2016;55(4):680–688. doi: 10.1093/rheumatology/kev408. [DOI] [PubMed] [Google Scholar]
- 45.Hoeven TA, Kavousi M, Clockaerts S, Kerkhof HJ, van Meurs JB, Franco O, Hofman A, Bindels P, Witteman J, Bierma-Zeinstra S. Association of atherosclerosis with presence and progression of osteoarthritis: the Rotterdam Study. Ann Rheum Dis. 2013;72(5):646–651. doi: 10.1136/annrheumdis-2011-201178. [DOI] [PubMed] [Google Scholar]
- 46.de Munter W, van der Kraan PM, van den Berg WB, van Lent PL. High systemic levels of low-density lipoprotein cholesterol: fuel to the flames in inflammatory osteoarthritis? Rheumatology (Oxford) 2016;55(1):16–24. doi: 10.1093/rheumatology/kev270. [DOI] [PubMed] [Google Scholar]
- 47.Gierman LM, Kuhnast S, Koudijs A, Pieterman EJ, Kloppenburg M, van Osch GJ, Stojanovic-Susulic V, Huizinga TW, Princen HM, Zuurmond AM. Osteoarthritis development is induced by increased dietary cholesterol and can be inhibited by atorvastatin in APOE*3Leiden. CETP mice–a translational model for atherosclerosis. Ann Rheum Dis. 2014;73(5):921–927. doi: 10.1136/annrheumdis-2013-203248. [DOI] [PubMed] [Google Scholar]
- 48.Griffin TM, Fermor B, Huebner JL, Kraus VB, Rodriguiz RM, Wetsel WC, Cao L, Setton LA, Guilak F. Diet-induced obesity differentially regulates behavioral, biomechanical, and molecular risk factors for osteoarthritis in mice. Arthritis Res Ther. 2010;12(4):R130. doi: 10.1186/ar3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu CL, Jain D, McNeill JN, Little D, Anderson JA, Huebner JL, Kraus VB, Rodriguiz RM, Wetsel WC, Guilak F. Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann Rheum Dis. 2015;74(11):2076–2083. doi: 10.1136/annrheumdis-2014-205601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eymard F, Parsons C, Edwards MH, Petit-Dop F, Reginster JY, Bruyere O, Richette P, Cooper C, Chevalier X. Diabetes is a risk factor for knee osteoarthritis progression. Osteoarthritis Cartilage. 2015;23(6):851–859. doi: 10.1016/j.joca.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 51.Hamada D, Maynard R, Schott E, Drinkwater CJ, Ketz JP, Kates SL, Jonason JH, Hilton MJ, Zuscik MJ, Mooney RA. Suppressive Effects of Insulin on Tumor Necrosis Factor-Dependent Early Osteoarthritic Changes Associated With Obesity and Type 2 Diabetes Mellitus. Arthritis Rheumatol. 2016;68(6):1392–1402. doi: 10.1002/art.39561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chu CR, Andriacchi TP. Dance between biology, mechanics, and structure: A systems-based approach to developing osteoarthritis prevention strategies. J Orthop Res. 2015;33(7):939–947. doi: 10.1002/jor.22817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller RH, Edwards WB, Deluzio KJ. Energy expended and knee joint load accumulated when walking, running, or standing for the same amount of time. Gait Posture. 2015;41(1):326–328. doi: 10.1016/j.gaitpost.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 54.Morrell KC, Hodge WA, Krebs DE, Mann RW. Corroboration of in vivo cartilage pressures with implications for synovial joint tribology and osteoarthritis causation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(41):14819–14824. doi: 10.1073/pnas.0507117102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sutter EG, Widmyer MR, Utturkar GM, Spritzer CE, Garrett WE, Jr, DeFrate LE. In vivo measurement of localized tibiofemoral cartilage strains in response to dynamic activity. Am J Sports Med. 2015;43(2):370–376. doi: 10.1177/0363546514559821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hosseini A, Van de Velde SK, Kozanek M, Gill TJ, Grodzinsky AJ, Rubash HE, Li G. In-vivo time-dependent articular cartilage contact behavior of the tibiofemoral joint. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society. 2010;18(7):909–916. doi: 10.1016/j.joca.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nelson DL, Cox MM. Lehniger: Principles of Biochemistry. W.H. Freeman and Company; 2008. [Google Scholar]
- 58.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the cell. 4th. Garland Science; 2002. [Google Scholar]
- 59.Peansukmanee S, Vaughan-Thomas A, Carter SD, Clegg PD, Taylor S, Redmond C, Mobasheri A. Effects of hypoxia on glucose transport in primary equine chondrocytes in vitro and evidence of reduced GLUT1 gene expression in pathologic cartilage in vivo. J Orthop Res. 2009;27(4):529–535. doi: 10.1002/jor.20772. [DOI] [PubMed] [Google Scholar]
- 60.Kiaer T, Gronlund J, Sorensen KH. Subchondral pO2, pCO2, pressure, pH, and lactate in human osteoarthritis of the hip. Clin Orthop Relat Res. 1988;(229):149–155. [PubMed] [Google Scholar]
- 61.Najafipour H, Ferrell WR. Comparison of synovial PO2 and sympathetic vasoconstrictor responses in normal and acutely inflamed rabbit knee joints. Exp Physiol. 1995;80(2):209–220. doi: 10.1113/expphysiol.1995.sp003841. [DOI] [PubMed] [Google Scholar]
- 62.Coleman MC, Ramakrishnan PS, Brouillette MJ, Martin JA. Injurious Loading of Articular Cartilage Compromises Chondrocyte Respiratory Function. Arthritis Rheumatol. 2016;68(3):662–671. doi: 10.1002/art.39460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10(20):1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 64.Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum. 2011;63(7):1928–1937. doi: 10.1002/art.30333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petursson F, Husa M, June R, Lotz M, Terkeltaub R, Liu-Bryan R. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res Ther. 2013;15(4):R77. doi: 10.1186/ar4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu-Bryan R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol. 2015;67(8):2141–2153. doi: 10.1002/art.39182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lane RS, Fu Y, Matsuzaki S, Kinter M, Humphries KM, Griffin TM. Mitochondrial respiration and redox coupling in articular chondrocytes. Arthritis Res Ther. 2015;17:54. doi: 10.1186/s13075-015-0566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jutila AA, Zignego DL, Hwang BK, Hilmer JK, Hamerly T, Minor CA, Walk ST, June RK. Candidate mediators of chondrocyte mechanotransduction via targeted and untargeted metabolomic measurements. Arch Biochem Biophys. 2014;545:116–123. doi: 10.1016/j.abb.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zignego DL, Hilmer JK, June RK. Mechanotransduction in primary human osteoarthritic chondrocytes is mediated by metabolism of energy, lipids, and amino acids. J Biomech. 2015;48(16):4253–4261. doi: 10.1016/j.jbiomech.2015.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stephanopoulos GN, Aristidou AA, Nielsen J. Metabolic Engineering: Principles and Methodologies. Academic Press; 1998. [Google Scholar]
- 71.Minor CA, Salinas D, Carlson R, June RK. Annual Meeting of the ORS: 2015. Las Vegas, NV: 2015. A Stoichiometric Matrix Model of the Biochemical Network of Central Energy Metabolism in Human Cells for Understanding Chondrocyte Mechanotransduction. [Google Scholar]
- 72.Salinas D, Minor CA, Carlson RP, Mumey B, June RK. Annual Meeting of the ORS: 2015. Las Vegas, NV: 2015. Flux Calculations Based on Metabolomic Data for Human Chondrocyte Central Energy Metabolism in Response to Applied Compression. [Google Scholar]
- 73.Borle AB, Nichols N, Nichols G., Jr Metabolic studies of bone in vitro. I. Normal bone. The Journal of biological chemistry. 1960;235:1206–1210. [PubMed] [Google Scholar]
- 74.Cohn DV, Forscher BK. Aerobic metabolism of glucose by bone. The Journal of biological chemistry. 1962;237:615–618. [PubMed] [Google Scholar]
- 75.Peck WA, Birge SJ, Jr, Fedak SA. Bone Cells: Biochemical and Biological Studies after Enzymatic Isolation. Science. 1964;146(3650):1476–1477. doi: 10.1126/science.146.3650.1476. [DOI] [PubMed] [Google Scholar]
- 76.Esen E, Long F. Aerobic glycolysis in osteoblasts. Curr Osteoporos Rep. 2014;12(4):433–438. doi: 10.1007/s11914-014-0235-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell metabolism. 2013;17(5):745–755. doi: 10.1016/j.cmet.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Karner CM, Esen E, Chen J, Hsu FF, Turk J, Long F. Wnt Protein Signaling Reduces Nuclear Acetyl-CoA Levels to Suppress Gene Expression during Osteoblast Differentiation. The Journal of biological chemistry. 2016;291(25):13028–13039. doi: 10.1074/jbc.M115.708578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karner CM, Esen E, Okunade AL, Patterson BW, Long F. Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. The Journal of clinical investigation. 2015;125(2):551–562. doi: 10.1172/JCI78470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Esen E, Lee SY, Wice BM, Long F. PTH Promotes Bone Anabolism by Stimulating Aerobic Glycolysis Via IGF Signaling. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2015 doi: 10.1002/jbmr.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fullerton MD, Steinberg GR, Schertzer JD. Immunometabolism of AMPK in insulin resistance and atherosclerosis. Mol Cell Endocrinol. 2013;366(2):224–234. doi: 10.1016/j.mce.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 82.Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, Vachharajani VT, Yoza BK, McCall CE. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol. 2012;92(3):499–507. doi: 10.1189/jlb.0212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51(2):249–257. doi: 10.1016/j.bone.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89(3):1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 85.Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci. 2008;65(23):3737–3755. doi: 10.1007/s00018-008-8244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hardie DG, Ross FA, Hawley SA. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol. 2012;19(10):1222–1236. doi: 10.1016/j.chembiol.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hwang JT, Kwon DY, Yoon SH. AMP-activated protein kinase: a potential target for the diseases prevention by natural occurring polyphenols. N Biotechnol. 2009;26(1–2):17–22. doi: 10.1016/j.nbt.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 88.Liu-Bryan R, Zho X, Wang Y, Lee HS, Akasdi A, Terkeltaub R. Activation of AMP-activated Protein Kinase (AMPK) by Berberine Limits Both Surgical Knee Instability-induced And Aging-related Osteoarthritis in Mice. Arthritis and Rheumatology. 2016;66(S10) [Google Scholar]
- 89.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11(2):230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 90.Blanco FJ, Lopez-Armada MJ, Maneiro E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion. 2004;4(5–6):715–728. doi: 10.1016/j.mito.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 91.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7(3):161–169. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 92.Vaamonde-Garcia C, Riveiro-Naveira RR, Valcarcel-Ares MN, Hermida-Carballo L, Blanco FJ, Lopez-Armada MJ. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012;64(9):2927–2936. doi: 10.1002/art.34508. [DOI] [PubMed] [Google Scholar]
- 93.Zhao X, Petursson F, Viollet B, Lotz M, Terkeltaub R, Liu-Bryan R. Peroxisome proliferator-activated receptor gamma coactivator 1alpha and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheumatol. 2014;66(11):3073–3082. doi: 10.1002/art.38791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dufey E, Sepulveda D, Rojas-Rivera D, Hetz C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. American journal of physiology Cell physiology. 2014;307(7):C582–594. doi: 10.1152/ajpcell.00258.2014. [DOI] [PubMed] [Google Scholar]
- 95.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66(2 Suppl 1):S102–109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 96.Husa M, Petursson F, Lotz M, Terkeltaub R, Liu-Bryan R. C/EBP homologous protein drives pro-catabolic responses in chondrocytes. Arthritis Res Ther. 2013;15(6):R218. doi: 10.1186/ar4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Oliver BL, Cronin CG, Zhang-Benoit Y, Goldring MB, Tanzer ML. Divergent stress responses to IL-1beta, nitric oxide, and tunicamycin by chondrocytes. Journal of cellular physiology. 2005;204(1):45–50. doi: 10.1002/jcp.20261. [DOI] [PubMed] [Google Scholar]
- 98.Takada K, Hirose J, Senba K, Yamabe S, Oike Y, Gotoh T, Mizuta H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. International journal of experimental pathology. 2011;92(4):232–242. doi: 10.1111/j.1365-2613.2010.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takada K, Hirose J, Yamabe S, Uehara Y, Mizuta H. Endoplasmic reticulum stress mediates nitric oxide-induced chondrocyte apoptosis. Biomedical reports. 2013;1(2):315–319. doi: 10.3892/br.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamabe S, Hirose J, Uehara Y, Okada T, Okamoto N, Oka K, Taniwaki T, Mizuta H. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. The FEBS journal. 2013;280(7):1617–1629. doi: 10.1111/febs.12170. [DOI] [PubMed] [Google Scholar]
- 101.Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carames B, Taniguchi N, Seino D, Blanco FJ, D’Lima D, Lotz M. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 2012;64(4):1182–1192. doi: 10.1002/art.33444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sasaki H, Takayama K, Matsushita T, Ishida K, Kubo S, Matsumoto T, Fujita N, Oka S, Kurosaka M, Kuroda R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012;64(6):1920–1928. doi: 10.1002/art.34323. [DOI] [PubMed] [Google Scholar]
- 104.Bohensky J, Leshinsky S, Srinivas V, Shapiro IM. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatric nephrology. 2010;25(4):633–642. doi: 10.1007/s00467-009-1310-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]