

Abstract
Three new ulapualides (3–5) were isolated from egg masses of the nudibranch Hexabranchus sanguineus. The structures of 3–5 were deduced by analyses of physical and spectroscopic data in comparisons with ulapualides A (1) and B (2). Ulapualide C demonstrated submicromolar cytotoxicity against select NCI cell lines (768-0, DU-145, MDA-MB-231, and A549) with the most potent activity against MDA-MB-231 (IC50 0.58 μM). Ulapualides A (1) and B (2) were two to four-fold more potent than 3.
Graphical Abstract
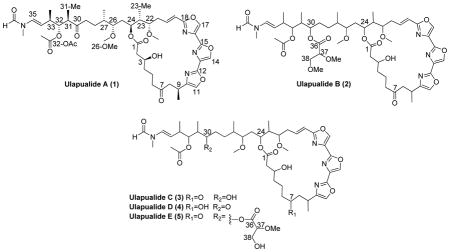
Originally isolated in 1986 from egg masses of the nudibranch Hexabranchus sanguineus, the ulapualides1 derive their Hawaiian name from the flowerlike appearance of the Spanish dancer’s eggs underwater (ula=red and pua=flower). The ulapualides contain a characteristic tris-oxazole moiety and a long aliphatic tail similar to the mycalolides2 and kabiramides.3 These compounds exhibit strong cytotoxicity which is proposed to protect the egg masses from predation. The cytotoxicity arises from the conserved aliphatic tail region that irreversibly binds to G-actin and behaves as a molecular mimic of the actin severing protein gelsolin.4 Taking advantage of this strong affinity allowed co-crystallization with G-actin that provided the absolute configuration of ulapualide A (1) via X-ray crystallography.5,6
As part of our ongoing search for BACE1 inhibitors, preliminary data suggested the ulapualides in crude extracts bound to that enzyme. After purification, however, ulapualides A (1) and B (2) showed no significant inhibition of BACE1 when tested at concentrations up to 30 μM. Further chemical investigation of the extract led to the isolation and identification of three new analogs, ulapualides C-E (3–5), described within.
RESULTS AND DISCUSSION
Two egg masses from the west shore of Oahu (Electric Beach) were extracted with 1:1 MeOH:CH2Cl2 overnight three times. The resultant extract was dry loaded on C8 silica gel and subjected to a stepped gradient of MeOH:H2O. The 75% MeOH fraction was separated by reversed-phase HPLC leading to the isolation of ulapualides A and B (1–2), along with ulapualide C (3) and a mixture of two other analogs. Subjecting this mixture to further HPLC using a different eluent afforded ulapualides D (4) and E (5). 1H and 13C NMR data were collected on all isolated compounds (Tables 1 and 2, respectively), but insufficient sample was available for direct detection of 13C NMR for 4 and 5. Consequently, 13C chemical shifts were obtained from indirect HSQC and HMBC experiments for these two compounds.7
Table 1.
1H NMR Chemical Shifts of Ulapualides C-E (3–5) in CDCl3
| position | Ulapualide C (3) | Ulapualide D (4) | Ulapualide E (5) |
|---|---|---|---|
|
|
|
|
|
| δH (J in Hz)a [minor]b | δH (J in Hz)a [minor]b | δH (J in Hz)a [minor]b | |
| 2 | 2.45, m | 2.49, m | 2.46, m |
| 3 | 4.23, septet (4.1) | 4.17, m | 4.23, septet (4.1) |
| 4 | 1.65, m 1.55, m |
1.72, m | 1.72, m 1.60, m |
| 5 | 1.95, m 1.79, m |
1.25, m | 1.96, m 1.76, m |
| 6 | 2.55, m | 1.76, m 1.41, m |
2.57, m |
| 7 | 4.07, br t (8.4) | ||
| 8 | 3.10, m 2.51, dd (4.9, 16.3) |
1.76, m 1.71, m |
3.01, m 2.51, dd (15.5, 3.7) |
| 9 | 3.41, m | 2.98, m | 3.42, m |
| 9-Me | 1.31, d (7.0) | 1.32, d (7.0) | 1.32, d (6.9) |
| 11 | 7.41, d (1.0) | 7.40, d (1.3) | 7.40, s |
| 14 | 8.06, s | 8.08, s | 8.06, s |
| 17 | 8.06, s | 8.06, s | 8.06, s |
| 19 | 6.38, d (16.0) | 6.31, d (15.7) | 6.39, d (16.5) |
| 20 | 7.05, dt (16.0, 7.4) | 7.09, ddd (15.3, 8.9, 6.0) | 7.01, m |
| 21 | 2.66, ddt (14.4, 4.6, 1.8) 2.44, m |
2.70, m 2.36, dd (14.3, 8.2) |
2.65, br d (14.5) 2.42, m |
| 22 | 3.41, m | 3.66, t (6.1) | 3.40, m |
| 22-OMe | 3.37, s | 3.38, s | 3.37, s |
| 23 | 1.85, m | 1.61, t (7.1) | 1.81, m |
| 23-Me | 0.91, d (7.0) | 0.88, d (6.9) | 0.87, d (6.9) |
| 24 | 5.24, t (8.2) | 5.26, t (9.6) | 5.23, m |
| 25 | 1.54, m | 1.69, m 1.41, m |
1.49, m |
| 26 | 2.95, br d (9.4) | 2.98, m | 2.93, dt (8.8, 3.3) |
| 26-OMe | 3.30, s | 3.31, s | 3.28, s |
| 27 | 1.71, m | 1.70, m | 1.72, m |
| 27-Me | 0.832, d (6.7) [0.828, d] | 0.792, d (6.7) [0.788, d] | 0.83, d (6.9) [0.82, d] |
| 28 | 1.65, m 0.90, m |
1.70, m 1.25, m |
0.94, m |
| 29 | 1.25, br s | 2.44, m | 1.57, m |
| 30 | 3.44, m | 5.10, t (7.2) | |
| 31 | 1.54, m | 2.77, dd (8.8, 7.3) | 1.81, m |
| 31-Me | 0.85, d (7.0) [0.84, d] | 1.06, d (7.1) [1.05, d] | 0.954, d (6.9) [0.947, d] |
| 32 | 4.80, td (9.8, 2.9) | 5.12, dd (8.9, 3.9) [5.13 dd] | 4.75 dd (10.4, 2.4) |
| 32-OAc | 2.16, s [2.15, s] | 2.00, s [1.99, s] | 2.11, s [2.10, s] |
| 33 | 2.56, m | 2.48, m | 2.53, m |
| 33-Me | 1.05, d (6.8) [1.04, d] | 1.04, d (7.1) [1.03, d] | 1.00, d (5.9) [0.99, d] |
| 34 | 5.00, dd (14.1, 9.4) | 4.98, td (13.6, 9.4) | 4.96, dt (14., 9.6) |
| 35 | 6.50, d (14.3) | 6.49, d (14.0) | 6.50, d (14.9) |
| 35-NMe | 3.02, s [3.06, s] | 3.02, s [3.07, s] | 3.02, s [3.06, s] |
| 35-NCHO | 8.29, s [8.07, s] | 8.28, s [8.07, s] | 8.29, s [8.07, s] |
| 37 | 3.81, t (4.8) | ||
| 37-OMe | 3.45, s | ||
| 38 | 3.86, br s |
500 MHz.
Proton signals in brackets are for the minor rotamer.
Table 2.
13C NMR Chemical Shifts of Ulapualides A-E (1–5) in CDCl3
| position | 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
||||||
| δCa,b | type | δCa,b | type | δCa,b | type | δCa,c | type | δCa,c | type | |
| 1 | 172.5 | C | 172.5 | C | 172.5 | C | 172.6 | C | 172.4 | C |
| 2 | 43.0 | CH2 | 43.0 | CH2 | 43.0 | CH2 | 43.4 | CH2 | 42.8 | CH2 |
| 3 | 68.1 | CH | 68.1 | CH | 68.0 | CH | 69.0 | CH | 68.3 | CH |
| 4 | 37.1 | CH2 | 37.1 | CH2 | 37.1 | CH2 | 37.5 | CH2 | 37.0 | CH2 |
| 5 | 20.8 | CH2 | 20.7 | CH2 | 20.8 | CH2 | 29.8 | CH2 | 20.5 | CH2 |
| 6 | 43.8 | CH2 | 43.7 | CH2 | 43.8 | CH2 | 36.8 | CH2 | 43.7 | CH2 |
| 7 | 210.5 | C | 210.4 | C | 210.5 | C | 71.2 | CH | 210.0 | C |
| 8 | 47.8 | CH2 | 47.7 | CH2 | 47.7 | CH2 | 45.4 | CH2 | 47.8 | CH2 |
| 9 | 27.4 | CH | 27.3 | CH | 27.3 | CH | 31.1 | CH | 27.4 | CH |
| 9-Me | 19.6 | CH3 | 19.6 | CH3 | 19.7 | CH3 | 19.3 | CH3 | 19.4 | CH3 |
| 10 | 146.3 | C | 146.3 | C | 146.3 | C | 146.8 | C | 146.1 | C |
| 11 | 133.5 | CH | 133.5 | CH | 133.5 | CH | 133.8 | CH | 133.2 | CH |
| 12 | 154.1 | C | 154.1 | C | 154.1 | C | 154.4 | C | 153.8 | C |
| 13 | 130.1 | C | 130.0 | C | 129.8 | C | NDd | NDd | ||
| 14 | 137.3 | CH | 137.3 | CH | 137.3 | CH | 137.0 | CH | NDd | |
| 15 | 156.1 | C | 156.0 | C | 156.1 | C | 156.2 | C | 155.8 | C |
| 16 | 131.5 | C | 131.4 | C | 131.5 | C | NDd | NDd | ||
| 17 | 137.7 | CH | 137.7 | CH | 137.7 | CH | 137.3 | CH | NDd | |
| 18 | 162.7 | C | 162.7 | C | 162.7 | C | 162.9 | C | 162.4 | C |
| 19 | 116.9 | CH | 116.8 | CH | 116.8 | CH | 115.9 | CH | 116.9 | CH |
| 20 | 139.9 | CH | 140.0 | CH | 140.1 | CH | 141.4 | CH | 139.7 | CH |
| 21 | 33.8 | CH2 | 33.9 | CH2 | 34.0 | CH2 | 35.0 | CH2 | 33.4 | CH2 |
| 22 | 79.9 | CH | 79.9 | CH | 79.9 | CH | 78.3 | CH | 79.6 | CH |
| 22-OMe | 57.7 | CH3 | 57.7 | CH3 | 57.8 | CH3 | 58.1 | CH3 | 57.6 | CH3 |
| 23 | 40.3 | CH | 40.3 | CH | 40.4 | CH | 41.0 | CH | 40.1 | CH |
| 23-Me | 9.0 | CH3 | 8.9 | CH3 | 9.0 | CH3 | 9.0 | CH3 | 9.0 | CH3 |
| 24 | 72.8 | CH | 72.9 | CH | 72.9 | CH | 72.6 | CH | 72.6 | CH |
| 25 | 31.9 | CH2 | 31.8 | CH2 | 32.1 | CH2 | 33.6 | CH2 | 31.6 | CH2 |
| 26 | 81.7 | CH | 81.6 | CH | 81.1 | CH | 81.6 | CH | 81.4 | CH |
| 26-OMe | 58.0 | CH3 | 57.9 | CH3 | 58.1 | CH3 | 58.0 | CH3 | 57.8 | CH3 |
| 27 | 34.4 | CH | 34.7 | CH | 35.5 | CH | 34.5 | CH | 34.4 | CH |
| 27-Me | 15.4 | CH3 | 15.5 | CH3 | 15.4 | CH3 | 15.7 | CH3 | 15.6 | CH3 |
| 28 | 25.0 | CH2 | 27.0 | CH2 | 28.4 | CH2 | 25.2 | CH2 | 26.6 | CH2 |
| 29 | 39.8 | CH2 | 30.4 | CH2 | 29.7 | CH2 | 39.8 | CH2 | 30.3 | CH2 |
| 30 | 211.6 | C | 73.03 | CH | 70.0 | CH | 211.6 | C | 72.7 | CH |
| 31 | 48.6 | CH | 37.5 | CH | 39.7 | CH | 48.6 | CH | 37.2 | CH |
| 31-Me | 13.3 | CH3 | 9.7 | CH3 | 8.5 | CH3 | 13.6 | CH3 | 9.1 | CH3 |
| 32 | 77.2 | CH | 79.8 | CH | 79.3 | CH | 77.2 | CH | 76.4 | CH |
| 32-OAc | 170.0 | C | 170.6 | C | 172.5 | C | 170.0 | C | 171.1 | C |
| 20.8 | CH3 | 20.9 | CH3 | 20.9 | CH3 | 20.9 | CH3 | 20.8 | CH3 | |
| 33 | 36.8 | CH | 36.9 | CH | 36.5 | CH | 36.9 | CH | 36.7 | CH |
| 33-Me | 18.8 | CH3 | 19.3 | CH3 | 19.4 | CH3 | 19.0 | CH3 | 19.4 | CH3 |
| 34 | 110.4 | CH | 110.2 | CH | 110.0 | CH | 110.5 | CH | 109.5 | CH |
| 35 | 129.4 | CH | 129.4 | CH | 129.4 | CH | 129.4 | CH | 129.4 | CH |
| 35-NMe | 27.5 | CH3 | 27.5 | CH3 | 27.5 | CH3 | 27.5 | CH3 | 27.5 | CH3 |
| 35-NCHO | 162.1 | CH | 162.1 | CH | 162.1 | CH | 162.1 | CH | 161.9 | CH |
| 36 | 170.2 | C | 170.3 | C | ||||||
| 37 | 80.6 | CH | 81.4 | CH | ||||||
| 37-OMe | 58.6 | CH3 | 58.2 | CH3 | ||||||
| 38 | 72.98 | CH2 | 62.9 | CH2 | ||||||
| 38-OMe | 59.3 | CH3 | ||||||||
Carbon spectra recorded at 125 MHz.
Resonances for minor rotamers can be found in the supporting information.
Carbon resonances extrapolated from HSQC and HMBC experiments.
Not detected.
All new compounds provided 1H NMR spectra consistent with tris-oxazole macrolides. These characteristic signals included the presence of a minor rotamer in an approximate 2:1 ratio evidenced by doubling of the signals for the N-methyl group (δH 3.02 and 3.06; 35-NMe) and formamide proton (δH 8.29 and 8.07; 35-NCHO). This doubling arises from the slow interconversion of the s-cis- and s-trans-rotamers of tertiary amide and has been documented in molecules containing similar tail regions including tolytoxin,8 scytophycins,9 luminaolide,10 and rhizapodin.11 Additionally, for compounds 3–5, the deshielded 1H NMR signals for H-11, H-14, and H-17 (Table 1) were consistent with the three contiguous 2,4-disubstituted oxazole rings. Co-isolation of 1–2 from this egg mass supported the conclusion that 3–5 were ulapualide analogs. In addition, 1–5 contained the same UV chromophore with a maximum at 249 nm and produced a consistent pattern of ESI-MS cations including [M+H-H2O]+, [M+H]+, [M+NH4]+, and [M+Na]+ where the [M+H]+ were the least abundant.
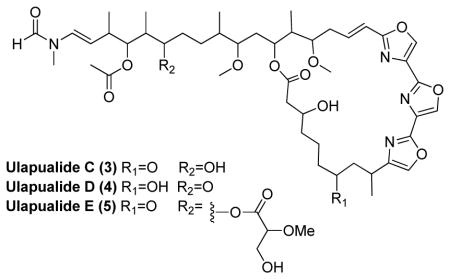
High resolution mass spectrometry of 3 produced a protonated molecule at m/z 883.4700 [M+H]+. This datum is consistent with a molecular formula of C46H66N4O13 for the parent molecule. Comparison to the 1H NMR spectrum of 1 indicated 3 contained two more protons, while the 13C NMR spectrum of 3 contained an additional sp3 oxygenated carbon δC 70.0 (C-30; 3) at the expense of a keto group at δC 211.6 (C-30; 1). HMBC correlations from the methyl group at position 31 to C-30 confirm the keto group on the aliphatic tail of 1 was reduced rather than the one in the macrolide ring.
High resolution mass spectrometry of 4 provided a protonated molecule at m/z 883.4662 [M+H]+ as well. Comparison between the NMR data of 4 and 1 showed an additional oxygenated sp3 carbon δC 71.2 (C-7; 4) and a corresponding proton signal at δH 4.07 (H-7; 4) again at the expense of a carbonyl resonance δC 210.5 (C-7; 1). COSY correlations from the methyl group at position 9 to H-9 and from H2-8 to both H-9 and H-7 support the conclusion that C-7 was a secondary alcohol in 4.
High resolution mass spectrometry of 5 gave a protonated molecule at m/z 985.4976 [M+H]+ consistent with the molecular formula (C50H72N4O16) 14 amu smaller than 2. Comparison of the 13C and 1H NMR spectra of 5 with 2 indicated that 5 contained one less methoxy group. Taken together, the data suggested 5 contained a hydroxy rather than methoxy group. HMBC correlations from the existing OMe groups in 5 pinpoint the placement of the methoxy groups and through deduction that C-38 was a primary alcohol.
The relative and absolute configurations of 3–5 could not be determined due to the limited amount of material available. In mass limited cases, configurational assignment based on carbon chemical shift comparisons with known analogs is a common tactic. In this case, synthetic studies have demonstrated that this approach is unreliable for distinguishing ulapualide diasteromers.12 However, all of the tris-oxazole compounds characterized by X-ray crystallography have the same absolute configuration (with the omission of position 9),13 this suggests that those stereocenters are conserved via a shared biogenesis. In addition, the two other stereocenters in 5 are most likely 30R,37R consistent with 2 and the structurally-related mycalolides.14
With the exception of the kabiramides, also isolated from nudibranch eggs, all other tris-oxazole containing molecules were isolated from sponges suggesting that these molecules may be sequestered through predation. The support for this hypothesis includes observed predation of Halichondria sp. by Hexabranchus sanguineus, isolation of dihydrohalichondramide from both species, and sequestration of dihydrohalichondramide in the mantle and the digestive system/gonad of the nudibranch.15 That similar tris-oxazole compounds have been isolated from sponges of dissimilar phylogeny suggests the ultimate source may be microbial as was found with the bryostatins, metabolites originally isolated from the bryozoan Bugula neritina16 and later discovered to be a product of the symbiont Endobugula sertula.17 The symbiont E. sertula is vertically transmitted from bryozoan adult to embryo18 and provides chemical defenses for the otherwise unprotected larvae.
Compound 3 could be an artifact as hydrolysis of the O-methylated glycerate in 2 would yield the free alcohol 3. That significant amounts of 3 can be observed via LCMS in the original MeOH:CH2Cl2 extract, before buffers were used suggests this is not the case. Furthermore, treatment of 2 in a 2:1 mixture of MeOH:H2O with 0.1% formic acid for seven days yielded no hydrolysis product detectable by LCMS.
Compounds 1–3 were tested against select cancer cell lines within the NCI 60-cell panel. These included human renal cell adenocarcinoma (768-0), prostate carcinoma (DU-145), mammary gland adenocarcinoma (MDA-MB-231) and lung carcinoma (A549) cell lines. Samples were tested in triplicate in two independent trials. For the second independent experiment, the concentration was determined through colorimetry using a standard curve made from the more abundant 2. As expected, low to sub-micromolar IC50 values were observed for all samples (Table 3). The activities of 1–2 were similar in all cell-lines tested, while 3 was two to four times less potent. Removal of methyl or methoxy groups from the tail region or the introduction of charged groups is predicted to decrease binding affinity to G-actin which is dominated by hydrophobic interactions in the cleft between actin subdomains 1 and 3.5 The observed decrease in activity could be attributed to replacement of the modified glycerate or keto group with a more polar hydroxy group.
Table 3.
IC50 Values of Ulapualides A-C (1–3) Against Select NCI Cell Lines (μM)
| 768-0 | DU-145 | MDA-MB-231 | A549 | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| Trial 1 | Trial 2 | Trial 1 | Trial 2 | Trial 1 | Trial 2 | Trial 1 | Trial 2 | |
| Ulapualide A (1) | 0.28 | 0.26 | 0.26 | 0.25 | 0.25 | 0.23 | 0.28 | 0.29 |
| Ulapualide B (2) | 0.26 | 0.66 | 0.28 | 0.34 | 0.29 | 0.29 | 0.33 | 0.26 |
| Ulapualide C (3) | 1.1 | 1.4 | 0.76 | 0.85 | 0.58 | 0.76 | 0.68 | 0.60 |
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were measured on a Jasco DIP-370 Digital Polarimeter at the sodium line (589 nm). UV absorbances were measured on a Varian Cary 50 Bio UV-Vis Spectrophotometer. IR spectroscopy was measured as a thin film on a CaF2 disk using a Shimadzu IRAffinity-1 FTIR. 1H, 13C NMR and 2D NMR experiments on the natural products were carried out on Varian Unity Inova 500 MHz spectrometer. NMR spectra were referenced to the appropriate residual solvent signal (δH 7.26, δC 77.0 for CDCl3). The HSQC experiments were optimized for 1JC,H = 140 Hz and HMBC experiments for 3JC,H = 7 Hz. High-resolution mass spectrometric data were obtained on a Agilent 6210 TOF LC/MS using the ESI source in positive mode.
Collection and Isolation
The egg masses of Hexabranchus sanguineus were collected via SCUBA off the west shore of O’ahu at Electric Beach (21°21′14.3″N 158°07′49.7″W) at a depth of 20 feet on September 23rd, 2011. The two egg masses collected were bright pink and were immediately extracted with 1:1 MeOH:CH2Cl2 three times overnight to yield 2.7 g of extract. The combined extract was then dry loaded on C8 silica gel and was subjected to a solid phase extraction procedure consisting of five steps of increasing MeOH:H2O content (0%, 25%, 50%, 75%, 100%, and an isopropanol wash). The 75% MeOH fraction was subjected to reversed-phase HPLC on a Phenomenex column (Luna C8; 250x10 mm, 5 μ) using a flow rate of 2.8 mL/min and a concentration gradient of 50%-70% (CH3CN:H2O) over 30 min. Detection was by UV, using a PDA and monitoring at 249 nm. This afforded pure compounds ulapualide B (tR = 24 min, 8 mg, 0.32% yield), ulapualide A (tR = 21 min, 6 mg, 0.24% yield; [α]22D-39, c 0.20, MeOH), ulapualide C (3, tR = 18.5 min, 1.6 mg, 0.064% yield) and a mixture of ulapualides D and E. This mixture was further purified using a C18 column (Luna; 100x4.6 mm, 5 μ) at a flow of 1.0 mL/min and using an isocratic system of 65% MeOH-H2O to yield ulapualide D (4, tR =20 min, 0.5 mg, 0.02% yield) and ulapualide E (5, tR =24 min, 0.5 mg, 0.02% yield). Purity was assessed at 210 nm and determined to be 95.3%, 94.5%, and 95.6% for 1, 2 and 3 respectively, prior to biological testing.
Ulapualide C (3)
white amorphous powder; [α]22D-17 (c 0.32, MeOH); UV (MeOH) λmax (log ε) 249 (4.5), 237 (4.6), 202 (4.5); IR νmax 3600-3200 (br), 3162, 2964, 2934, 1720, 1691, 1655, 1597, 1458, 1241, 1086, 916 cm-1. Tables 1 and 2 for NMR data; HRESI-TOFMS m/z 883.4700 [M+H]+ (calcd for C46H67N4O13, 883.4705).
Ulapualide D (4)
white amorphous powder; [α]22D-10 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 249 (4), 213 (5), 202 (5); Tables 1 and 2 for NMR data; HRESI-TOFMS m/z 883.4662 [M+H]+ (calcd for C46H67N4O13, 883.4705).
Ulapualide E (5)
white amorphous powder; [α]22D-23 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 249 (4), 213 (5), 202 (5); Tables 1 and 2 for NMR data; HRESI-TOFMS m/z 985.4976 [M+H]+ (calcd for C50H73N4O16, 985.5022).
Cytotoxicity Assay of Ulapualide A, B, and C
Human renal cell adenocarcinoma (768-0), prostate carcinoma (DU-145), mammary gland adenocarcinoma (MDA-MB-231) and lung carcinoma (A549) cell lines were maintained in RPME 1640 Medium (Gibco, REF: 11875-093) supplemented with 10% premium fetal bovine serum (Atlanta biological, Cat. No.: S11150) and 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco, REF: 15140-122). The day before treatment, cancer cells were seeded with 4,000 cells per well into a 96-well tissue culture plate. Twenty hours post seeding, the serial diluted compounds were added to the cells for the cytotoxicity assay, and co-incubated at 37 °C with 5% CO2 for 72 h. Then the medium with compounds were replaced with 1 × dye binding solution prepared according to the manufacturer’s instruction (CyQuant NF Cell Proliferation Assay Kit [C35006, Invitrogen]) and incubated at 37 °C with 5% CO2 for 60 minutes. After that, cell viability data were collected with a PerkinElmer Multimode Plate Reader according to the manufacturer’s instruction. IC50 curves were generated using GraphPad Prism 5.
Supplementary Material
Acknowledgments
We would like to thank C. Hughes and D. Hana of UCSD along with M. Hamann and C. Geny at the Medical School of South Carolina for assistance with NMR data acquisition. This work was funded by grants from the National Institute on Aging (5R01AG039468-03). Funds for the upgrades of the NMR instrumentation were provided by the CRIF program of the National Science Foundation (CH E9974921) and the Elsa Pardee Foundation. The purchase of the Agilent LC-MS was funded by grant W911NF-04-1-0344 from the Department of Defense.
Footnotes
Dedicated to Professor Phil Crews, of University of California, Santa Cruz, for his pioneering work on bioactive natural products
Supporting Information. Copies of the 1H, 13C, and 2D NMR spectroscopic data for all new compounds associated with this article and a photo of the producing organism are available free of charge on the ACS Publications website at DOI:
References and Notes
- 1.Roesener J, Scheuer P. J Am Chem Soc. 1986;108:846–847. [Google Scholar]
- 2.Fusetani N, Yasumuro K, Matsunaga S, Hashimoto K. Tetrahedron Lett. 1989;30:2809–2812. [Google Scholar]
- 3.Matsunaga S, Fusetani N, Hashimoto K. J Org Chem. 1989;54:1360–1363. [Google Scholar]
- 4.Tanaka J, Choi J, Bai J, Yan Y, Klenchin VA, Rayment I, Marriott G. Proc Nat Acad Sci. 2003;100:13851–13856. doi: 10.1073/pnas.2233339100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klenchin VA, Allingham J, King R, Tanaka J, Marriott G, Rayment I. Nat Struct Bio. 2003;10:1058–1063. doi: 10.1038/nsb1006. [DOI] [PubMed] [Google Scholar]
- 6.Allingham JS, Tanaka J, Marriott G, Rayment I. Org Lett. 2004;6:597–599. doi: 10.1021/ol036458y. [DOI] [PubMed] [Google Scholar]
- 7.Attempts at collecting 13C data on a 600 MHz NMR with cryoprobe and 850 MHz with TCI probe were unsuccessful.
- 8.Carmeli S, Moore RE, Patterson GML. J Nat Prod. 1990;53:1533–1542. doi: 10.1021/np50072a021. [DOI] [PubMed] [Google Scholar]
- 9.Ishibashi M, Moore RE, Patterson GML, Xu C, Clardy J. J Org Chem. 1986;51:5300–5306. [Google Scholar]
- 10.Kitamura M, Schupp PJ, Nakano Y, Uemura D. Tetrahedron Lett. 2009;50:6606–6609. doi: 10.1016/j.tetlet.2009.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jansen R, Steinmetz H, Sasse F, Schubert WD, Hagelüken G, Albrecht SC, Müller R. Tetrahedron Lett. 2008;49:5796–5799. [Google Scholar]
- 12.Pattenden G, Ashweek NJ, Baker-Glenn CAG, Walker GM, Yee JGK. Angew Chem Int Ed. 2007;46:4356–4363. doi: 10.1002/anie.200700459. [DOI] [PubMed] [Google Scholar]
- 13.Dalisay DS, Rogers EW, Edison AS, Molinski TF. J Nat Prod. 2009;72:732–738. doi: 10.1021/np8007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsunaga S, Liu P, Celatka CA, Panek JS, Fusetani N. J Am Chem Soc. 1999;121:5605–5606. [Google Scholar]
- 15.Pawlik JR, Kernan MR, Molinski TF, Harper MK, Faulkner DJ. J Exp Mar Biol Ecol. 1988;119:99–109. [Google Scholar]
- 16.Pettit GR, Herald CL, Doubek DL, Herald DL. J Am Chem Soc. 1982;104:6846–6848. [Google Scholar]
- 17.Davidson SK, Allen SW, Lim GE, Anderson CM, Haygood MG. Appl Environ Microbiol. 2001;67:4531–4537. doi: 10.1128/AEM.67.10.4531-4537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharp KH, Davidson SK, Haygood MG. ISME J. 2007;1:693–702. doi: 10.1038/ismej.2007.78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.