

Abstract
Changes in the gastrointestinal microbial community are frequently associated with chronic diseases such as Inflammatory Bowel Diseases. However, understanding the relationship of any individual taxon within the community to host physiology is made complex due to the diversity and individuality of the gut microbiota. Defined microbial communities such as the Altered Schaedler Flora (ASF) help alleviate the challenges of a diverse microbiota by allowing one to interrogate the relationship between individual bacterial species and host responses. An important aspect of studying these relationships with defined microbial communities is the ability to measure the population abundance and dynamics of each member. Herein, we describe the development of an improved ASF species-specific and sensitive real-time quantitative polymerase chain reaction (qPCR) for use with SYBR Green chemistry to accurately assess individual ASF member abundance. This approach targets hypervariable regions V1 through V3 of the 16S rRNA gene of each ASF taxon to enhance assay specificity. We demonstrate the reproducibility, sensitivity and application of this new method by quantifying each ASF bacterium in two inbred mouse lines. We also used it to assess changes in ASF member abundance before and after acute antibiotic perturbation of the community as well as in mice fed two different diets. Additionally, we describe a nested PCR assay for the detection of lowly abundant ASF members. Altogether, this improved qPCR method will facilitate gnotobiotic research involving the ASF community by allowing for reproducible quantification of its members under various physiological conditions.
Keywords: Altered Schaedler Flora, antibiotics, diet, mice, gut microbiota, qPCR
1. Introduction
The mammalian gastrointestinal (GI) microbiota is comprised of a diverse and complex assortment of symbiotic bacterial species that provides numerous health benefits to the host, including nutrient digestion, vitamin synthesis, colonization resistance against pathogens and maturation of the mucosal immune system (Buffie et al., 2013; Brestoff et al., 2013; Dethlefsen et al., 2007; Relman, D.A., 2012). Changes in the composition and structure of this microbial community are associated with various chronic diseases such as metabolic syndrome, colorectal cancer and inflammatory bowel diseases (Parekh et al., 2015; Sartor et al., 2012, Sears et al., 2014). To date, studies describing disease associated “dysbiosis” have been largely observational, and the exact contributions of individual gut microbiota members to any of those pathologies are only beginning to be identified. Indeed, pinpointing specific microbial contributions to any given disease is, in part, confounded by the sheer complexity of the intestinal microbiota as well as its highly individualized nature among hosts (Walter et al., 2011).
Alternatively, a defined gut microbiota provides an experimental model to determine the exact functional role of individual community members that circumvents the challenges often faced when utilizing more complex microbial ecosystems. The Altered Schaedler Flora (ASF) microbiota is one example of a defined community that offers the opportunity to study and dissect host-and-microbe relationships at a mechanistic level (Wymore et al., 2015). Moreover, it permits the addition of other specific organisms such as pathogens (Brugiroux et al., 2016, Endt et al., 2010) or pathobionts (Atherly et al., 2016, Ge et al., 2006, Rooks et al., 2017), thereby allowing for the measurement of individual member contributions to host functions such as immune system maturation (i.e., development of regulatory CD4+ T cells) and the onset of intestinal inflammation (Geuking et al, 2011, Liu et al., 2011; Wymore et al., 2015). Although the ASF community is limited in its metabolic capabilities and phylogenetic representation as compared to a conventional gut microbiota (Brugiroux et al., 2016; Dewhirst et al., 1999), the ASF community is resilient, and its members are long-term colonizers (Stehr et al., 2009) despite varying in abundance across different anatomical sites and host genotypes (Alexander et al., 2006; Sarma-Rupavtarm et al. 2004). Such characteristics make the ASF microbiota a useful gnotobiotic model. However, to realize the full potential of defined microbial communities, reliable quantification of the individual members is necessary in order to characterize population dynamics under a variety of conditions.
Here, we present sensitive and ASF species-specific primers to be used in a real-time quantitative polymerase chain reaction (qPCR) assay with SYBR Green chemistry to assess individual ASF species abundance. Our newly-developed primers target hypervariable regions V1 through V3 of the 16S rRNA gene of each ASF taxon to ensure specificity. Additionally, we describe qPCR primers designed to limit primer dimerization and generate small amplicons, which should facilitate their use with a variety of commercially-available SYBR Green chemistries. We further describe a nested PCR assay for detecting and verifying the presence of lowly abundant ASF members. Finally, we demonstrate the reproducibility and application of this assay by quantifying each ASF taxon under normal physiological conditions in two inbred mouse lines, before and after an acute antibiotic perturbation, and in mice fed two different diets.
2. Material and Methods
2.1. Altered Schaedler Flora community
Members of the ASF microbial community include: ASF 356, Clostridium sp.; ASF 360, Lactobacillus intestinalis; ASF 361, Lactobacillus murinus; ASF 457, Mucispirillum schaedleri; ASF 492, Eubacterium plexicaudatum; ASF 500, Pseudoflavonifractor sp.; ASF 502, Clostridium sp.; and ASF 519, Parabacteroides goldsteinii (Dewhirst et al., 1999; Orcutt et al., 1987; Wymore et al., 2015). Pure cultures of ASF strains and founder C3H/HeN gnotobiotic mice harboring all eight ASF members were a kind gift from Dr. Michael Wannemuehler, Iowa State University.
2.2. Bacterial growth and genomic DNA isolation
To isolate genomic DNA for 16S rRNA amplification by PCR, all ASF bacteria were cultured under the same conditions. Specifically, each individual ASF member was grown under anaerobic conditions at 37°C by first streaking the isolate onto an agar plate and subsequently passaging into a sterile glass culture tube containing 5 mL broth. Both the agar medium and broth were made using a modified version of Schaedler's recipe (Schaedler et al., 1965). Escherichia coli (cloning vector) was grown aerobically in Brucella agar (BD BBL™ Brucella Agar, Difco™ Becton Dickinson, Sparks, MD) for 24 hours at 37°C prior to broth inoculation (i.e., Brucella broth, BD BBL™ Brucella Agar, Difco™ Becton Dickinson) under the same conditions. Genomic DNA isolation from each individual ASF taxon and E. coli was achieved by using the QIAamp DNA Blood and Tissue Kit as per manufacturer instructions (Qiagen®, Germantown, MD) with a minor modifications as previously described (Oh et al., 2010).
2.3. CFU quantification of lactobacilli species
To count colony forming units (CFU) and construct standard curves using extracted genomic DNA material, L. intestinalis-ASF360 and L. murinus-ASF 361 were grown on Tryptic Soy Agar (TSA, BD Tryptic Soy Agar, Difco™ Becton Dickinson) under aerobic and stationary conditions at 37°C. One mL of each pure culture was inoculated into 9 mL of Tryptic Soy Broth (BD Tryptic Soy Broth, Bacto™ Becton Dickinson) for 24 hours under aerobic and stationary conditions at 37°C. One hundred μL of each subsequent ten-fold dilution were then plated onto TSA (BD Tryptic Soy Agar, Difco™ Becton Dickinson) to determine the total number of viable bacteria in the initial inoculum (in CFU/mL). Colonies were only counted when 20 to 250 colonies were present, and all plating was done in triplicate. Genomic DNA was also isolated from the same series of diluted cultures as described in section 2.2. This genomic DNA was then used in the qPCR assay to construct a standard curve and determine the CFU/mL limit of detection for each Lactobacillus.
2.4. Mice
Germ-free and ASF-bearing C3H/HeN and C57BL/6 mice were bred and reared in flexible film isolators and maintained under gnotobiotic conditions at the University of Nebraska-Lincoln. Germ-free status of the breeding colony was routinely checked by analyzing fresh feces via bacterial 16S rRNA gene-specific PCR (30 cycles, universal bacteria primers 8F and 1391R) (Turner et al., 1999) in combination with aerobic and anaerobic culture of feces in Brain Heart Infusion, Wilkins-Chalgren, and Yeast Mold broths and on TSA plates (all media from Difco™ Becton Dickinson) at 37°C for 7 days. For both C3H/HeN and C57BL/6 mice, the ASF community was assembled by colonizing germ-free breeding pairs with cecal contents from ASF bearing donors. Prior to inoculation of the breeding pairs, the ASF status of the donors was confirmed by using the gel-based PCR assay described by Sarma-Rupavtarm et al. (2004). To prepare the inoculum, cecal contents were harvested from donors after euthanasia and homogenized in sterile phosphate-buffered saline (PBS 1X without calcium and magnesium, pH 7, Corning Cellgro, Manassas, VA) at a ratio of one cecum per 1 mL of PBS 1X with vigorous vortexing. All inocula were prepared under sterile aerobic conditions in a biosafety cabinet immediately prior to transplant. The final homogenate solution was passed through a 100 μM sterile cell strainer (Fisherbrand™, Fisher Scientific Inc., Fair Lawn, NJ) into a 50-mL sterile polypropylene conical tube (Corning™ Falcon™, Fisher Scientific Inc.); 200 μL of homogenate was used to inoculate each germ-free mouse. Cecal contents from one donor were sufficient to inoculate five germ-free recipients. The inoculation procedure was performed daily for three consecutive days. On each day, cecal contents from ASF-bearing donors were collected and prepared as described above. The absence of contaminating bacteria in the inoculum was confirmed by aerobic culture of fresh feces on TSA at 37°C for 7 days. Because the lactobacilli present in the ASF community will grow under these conditions, a morphological examination and gram staining were also performed to verify the absence of contaminating bacteria. The presence of all ASF species in the recipient breeding mice was confirmed via both a gel-based PCR assay described by Sarma-Rupavtarm et al. (2004) and via our newly-developed assay. All mice used in the studies reported herein were offspring from ASF-bearing parents and were colonized with the ASF via vertical transmission.
To test the qPCR assay under normal physiological conditions, we raised and maintained twelve-week-old ASF colonized male and female C3H/HeN (n = 11) and C57BL/6 (n = 15) mice in flexible film isolators under gnotobiotic conditions for the duration of the study. These mice were fed an autoclaved chow diet (LabDiet 5K67, Purina Foods, St. Louis, MO; Table S1a). To quantify ASF member abundance following an antibiotic perturbation, eight-week old ASF colonized male and female C57BL/6 mice (n = 7) were administered either streptomycin or neomycin sulfate (10 mg/mL for each antibiotic, Sigma-Aldrich Co. LLC., St. Louis, MO) via their drinking water for two consecutive days. All antibiotic solutions were made fresh daily, and cages were cleaned daily to prevent residual colonization due to coprophagic behavior. Fresh fecal samples were collected into individual sterile tubes (Fisherbrand™ Snap-Cap™ Flat-Top 2 mL microcentrifuge tubes, Fisher Scientific Inc., Fair Lawn, NJ) and homogenized by adding 1 mL PBS 1X (Corning Cellgro, Manassas, VA) and vortexing vigorously. To compare ASF member abundance between two different diets, nine-week old ASF colonized C57BL/6 male mice were fed either a low fat diet (LFD, D12450K; 10% kcal from fat and no sucrose; n = 10) or a Western diet (WD, 60% kcal from fat and 17% kcal from sucrose; n = 11) for five weeks. Experimental diets were prepared by Research Diets (New Brunswick, NJ) and sterilized by γ-irradiation (min 50 kGy, Neutron Products, Dickerson, MD) as described in Bindels et al., 2017). Irradiation efficacy was assessed by testing 50 spore strips of Bacillus pumillus (NAMSA, Northwood, OH) placed between bags of diet before irradiation and then incubated after irradiation in Soybean-Casein Digest broth at 33°C for 7 days. Mice were fed an autoclaved chow diet (LabDiet 5K67, Purina Foods) during the period of time between weaning and introduction of the experimental diets. Diet compositions are described in Supplemental Material (Table S1b). Mice used in the antibiotic and diet studies were raised and maintained inside flexible film isolators under gnotobiotic conditions until introduction of either the antibiotics or the experimental diets. For the duration of each experiment, mice were housed in autoclaved cages on a positive pressure, individually ventilated caging system (Allentown Inc., Allentown, NJ), maintained on autoclaved bedding, provided autoclaved water and fed either an autoclaved or sterilized diet as described above. Cages were only opened in a biosafety cabinet. At the end of each study, mice were euthanized via carbon dioxide asphyxiation. During necropsy, cecal contents from individual mice were aseptically harvested into sterile 2 mL microcentrifuge tubes (Fisherbrand™ Snap-Cap™, Fisher Scientific Inc.). All samples were stored at −80oC until DNA isolation was performed. The Institutional Animal Care and Use Committee at the University of Nebraska-Lincoln approved all procedures involving animals (Protocols 817 and 1215).
2.5. Cecal and fecal DNA isolation
Cecal and fecal DNA isolations were performed as previously described (Martinez et al., 2009). Briefly, samples were washed three times with 1 mL of ice-cold PBS 1X (Corning Cellgro) and centrifuged at 8,000 × g for 5 min. Pelleted samples were resuspended in 750 μL lysis buffer (200 mM NaCl, 100 mM Tris [pH 8.0], 20 mM EDTA, 20 mg/mL lysozyme) and transferred to a sterile microcentrifuge tube (Fisherbrand™ Snap-Cap™ Fisher Scientific Inc.) containing 300 mg of 0.1 mm sterile zirconium beads (BioSpec Products, Bartlesville, OK). Following incubation at 37°C for 20 min, 85 μL of 10% sodium dodecyl sulfate solution and 40 μL proteinase K (15 mg/mL, Qiagen®, Germantown, MD) were added, and the samples incubated for 15 min at 60°C. Thereafter, 500 μL of phenol-chloroform-isoamyl alcohol (25:24:1, Acros Organics, Fisher Scientific Inc., Fair Lawn, NJ) were added, and the samples homogenized in a MiniBeadbeater-8 (BioSpec Products) for 2 min at maximum speed; samples were then cooled on ice immediately before layer separation via centrifugation at 10,000 × g for 5 min. The top layer was extracted twice; each time it was transferred to a new sterile microcentrifuge tube (Fisherbrand™ Snap-Cap™ Fisher Scientific Inc.) prior to one extraction with phenol-chloroform-isoamyl alcohol (25:24:1) and then two extractions with chloroform-isoamyl alcohol (24:1, Acros Organics Fisher Scientific Inc.). DNA was recovered by standard ethanol precipitation. Finally, DNA pellets were dried for approximately 20 min at room temperature and subsequently resuspended in 100 μL of DNase, RNase and Protease free Tris-EDTA 1X buffer (Tris 10 mM, EDTA 1 mM [pH 8.0], Fisher Scientific Inc.). Prior to use of the DNA samples in the qPCR, dilutions to the final concentration needed were achieved by using 0.1 μM filter sterilized, nuclease free, deionized and distilled molecular grade water (Fisher Scientific Inc.).
DNA concentration was quantified using fluorescent molecule labeling (Mansfield et al., 1995) (Quant-iT™ PicoGreen® dsDNA Broad Range Reagent, Thermo Fisher Scientific Inc., Middletown, VA) and a fluorometric reader (QUBIT® 2.0 Fluorometric Quantification-Invitrogen™, Thermo Fisher Scientific Inc., Middletown, VA) (Halaihel et al., 2009) as per manufacturer instructions.
2.6. Conventional 16S rRNA gene PCR
All PCR reactions targeting the 16S rRNA gene were performed using 8F and 1391R universal primers (Turner et al., 1999). Reaction conditions included: (1) an initial denaturation step of 5 min at 94°C; (2) 25 cycles of 1 min at 94°C, 45 s at 55°C and 2 min at 72°C; and (3) 1 cycle of 10 min at 72°C using a Techne TC-5000 PCR Thermal Cycler (GMI, St. Paul, MN). Each reaction contained 40 μL PCR mix and 10 μL DNA template (100 ng). The PCR mix was comprised of the following reagents and contained the following final concentrations in the well: Taq DNA polymerase (1.5 U; New England Biolabs® Inc., Ipswich, MA), 10X Buffer (1X; ThermoPol® Buffer New England Biolabs® Inc.), dNTPs (0.8 mM; Thermo Fisher Scientific Inc.), bovine serum albumin (0.1 mg/mL; New England Biolabs® Inc.), and 8F and 1391R primers (800 nM each; IDT DNA Technologies, Coralville, IA).
2.7. Design of specific primers
In our hands, the primers described in Sarma-Rupavtarm et al., (2004) produced amplicons of the expected size (data not shown). However, we also observed some primer dimerization and low-levels of cross-reactivity. To that end, we manually designed new oligonucleotide primer sequences for each ASF species after using the Clustal W algorithm for multiple sequence alignment from the DNA Lasergene 11 Software package (DNASTAR Inc., Madison, WI) to align all 16S rRNA ASF gene sequences. All sequences used for the alignment were taken from the GeneBank-NCBI database (http://www.ncbi.nlm.nih.gov/genbank/) and can be identified using the following accession numbers: Clostridium sp. (ASF 356; AF157052.1), L. intestinalis (ASF 360; AF157050.1), L. murinus (ASF 361; AF157049.1), M. schaedleri (ASF 457; AF157055.1), E. plexicaudatum (ASF 492; AF157054.1), Pseudoflavonifractor sp. (ASF 500; AF157051.1), Clostridium sp. (ASF 502; AF157053.1), P. goldsteinii (ASF 519; AF157056.1). All primer sequences were chosen by targeting hypervariable regions V1 through V3 in the 16S rRNA gene. Initial in silico validation of primer sequence specificity was performed by assessing their sequence identity to other available bacterial 16S rRNA gene sequences using three different databases: (1) GeneBank BLASTn (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (Altschul et al., 1990), (2) probeCheck (http://131.130.66.200/cgi-bin/probecheck/probecheck.pl) (Loy et al., 2008), and (3) Silva high quality ribosomal RNA databases (http://www.arb-silva.de/search/testprime/) (Cole et al., 2014). Only oligonucleotides that consistently demonstrated specificity in all three databases were used for subsequent in vitro testing for final selection. The in silico specificity threshold was the absence of 100% sequence match with other known but unrelated taxa. Other parameters considered for primer design included length, dissociation temperature, and amplicon size considering that SYBR Green qPCR is generally more efficient when shorter PCR products (i.e., ranging from 50 to 250 bp) are produced (Thornton et al., 2011). All primers were synthesized by Integrated DNA Technologies (Coralville, IA), and complete primer details are provided in Table 1. Additional information regarding the in silico specificity of each primer set can be found in Tables S5-S12, which list the hits for forward and reverse primers for each ASF taxon, as per probeCheck analysis (Loy et al., 2008). For comparison purposes, the locations of our primer binding sites in the 16S rRNA gene of each ASF taxon are presented in parallel to the ones previously published by Sarma-Rupavtarm et al. (2004) after aligning multiple gene sequences using the Clustal W algorithm from BioEdit software (Hall, T.A., 1999) (Fig. S1).
Table 1.
Primer sequences and length and amplicon size (bp) for all ASF species.
| Bacterial species | Taxon ID | Primers | Amplicon size (bp) | |
|---|---|---|---|---|
|
| ||||
| Length (nt) | Sequence (5′ to 3′) | |||
| Clostridium sp. | 356 | 49-73F | AAAATAATTAGGAGCTTGCTTTTAA | 138 |
| Lactobacillus intestinalis | 360 | 72-89F 201-184R | GGTGATGACGCTGGGAAC AAGCAATAGCCATGCAGC | 130 |
| Lactobacillus murinus | 361 | 57-76F 202-182R | GAACGAAACTTCTTTATCACC TAGCATAGCCACCTTTTACA | 146 |
| Mucispirillum schaedleri | 457 | 54-74F 188-165R | TCTCTTCGGGGATGATTAAAC AACTTTTCCTATATAAACATGCAC | 135 |
| Eubacterium plexicaudatum | 492 | 63-82F 199-177R | AATTCCTTCGGGGAGGAAGC TAAAACCATGCGGTTTTAAAAAC | 137 |
| Pseudoflavonifractor sp. | 500 | 46-63F | ACGGAGGACCCCTGAAGG | 172 |
| Clostridium sp. | 502 | 44-65F | GAGCGAAGCACTTTTTTAGAAC | 177 |
| Parabacteroides goldsteinii | 519 | 49-69F 192-169R | GCAGCACGATGTAGCAATACA TTAACAAATATTTCCATGTGGAAC | 144 |
2.8. Construction of qPCR standards
Because culturing individual ASF members under anaerobic conditions can be a laborious process, we chose to use plasmids containing the 16S rRNA gene of each ASF member as qPCR standards for quantifying ASF bacterial abundances in cecal and fecal samples. Specifically, the 16S rRNA gene from each ASF taxa and Escherichia coli was amplified using the bacterial-specific 16S rRNA primers 8F and 1391R (Turner et al., 1999) and cloned into a pCR2.1 plasmid vector using the Topo-TA cloning kit (TOPO® TA Cloning® Invitrogen, Thermo Fisher Scientific Inc., Middletown, VA). All plasmids were transformed into the competent E. coli vector provided with the TOPO cloning kit. Plasmid DNA was isolated as described by the manufacturer (TOPO® TA Cloning® Invitrogen). To ensure that the inserted ASF 16S rRNA gene sequences were an exact match to the source DNA, a PCR amplification and Sanger sequencing-based approached was used. First, all ASF 16S rRNA sequences were amplified conventional PCR using universal primers (Turner et al., 1999). PCR products were then purified (QIAquick PCR Purification Kit, Qiagen®) and sequenced using the Sanger method to ensure insertion of the correct target gene (Sanger et al., 1977). Amplicon alignment of all ASF gene products was generated using the CLC sequence viewer software (CLC Bio Qiagen®). Sequences were blasted against the GeneBank-NCBI database (http://www.ncbi.nlm.nih.gov/genbank/) using BLASTn for confirmation of their specificity (Altschul et al., 1990). Circular plasmids containing the 16S rRNA gene of each bacterium were used as standards for all qPCR reactions. Concentrations of the plasmids were determined using the PicoGreen assay as described in section 2.5. The plasmid pCR2.1 that includes the 16S rRNA of E. coli was used as a negative control to assess any potential cross-reactivity with the vector organism.
Similarly to Alexander et al., 2006, purified genomic DNA from each ASF bacterium was also used to construct standard curves. Additionally, genomic DNA isolated from serially diluted cultures of L. intestinalis (ASF 360) and L. murinus (ASF 361) was also used to determine detection limits for both organisms based on CFU/mL, given that those two ASF members can be readily cultured (see section 2.3).
Both plasmid and genomic DNA standard curves were tested in two matrices: (1) in 0.1 μM filter sterilized, nuclease free, deionized and distilled molecular grade water (Fisher Scientific Inc.) and (2) in germ-free (GF) fecal samples from C3H/HeN and C57BL/6 mice (sample from each host genotype included at a 1:1 ratio) subjected to the DNA extraction procedure described in section 2.5. Either plasmid or genomic DNA was spiked into the GF fecal sample after it was subjected to the DNA extraction process to determine if the GF fecal DNA matrix would interfere with assay performance. Plasmid and genomic DNA concentrations used to construct all ASF standard curves are available in the Supplemental Material (Table S2). For the genomic DNA standard curves, total abundance calculations were based on each ASF genome size as described in the Supplemental Material (Table S3).
2.9. ASF qPCR conditions
All qPCR reactions were performed using a Mastercycler ep gradient realplex2 (Eppendorf, Hamburg-Eppendorf Hamburg, Germany) in a 25 μL reaction mixture volume (i.e., 24 μL of qPCR mix plus 1 μL of DNA template) using SYBR Green core reagents. The number of replicates included for each sample or standard is specified in the figure legends. Two SYBR Green reagents were compared for their analytical sensitivity and efficiency when used with DNA isolated from cecal contents: RealMasterMix SYBR ROX 2.5×, 5 PRIME, Gaithersburg, MD (Mix A) versus the Maxima SYBR Green qPCR Master Mix 2X, Thermo Fisher Scientific Inc., Middletown, VA (Mix B). The more efficient and sensitive SYBR Green chemistry (Mix A) was subsequently used to quantify ASF abundances when comparing between mouse strains and before and after antibiotic treatment. Mix B was used when comparing standard curves generated with either genomic or plasmid DNA and for quantifying ASF abundances in mice fed low fat and Western diets because Mix A was not commercially available at that time. The following conditions were used for all reactions performed with either SYBR Green Mix A or B: (1) an initial denaturation step of 10 min at 95°C; (2) 35 cycles of 20 s at 95°C, 30 s at either 60 or 62°C, and 45 s at 68°C; (3) one cycle of 20 s at 95°C; (4) 1 cycle of 30 s at 60 or 62°C; (5) 1 20 min interval to generate a melting curve; and (6) 1 cycle of 45 s at 95°C. An annealing temperature of 60°C was utilized for ASF members L. murinus (ASF 361), E. plexicaudatum (ASF 492) and Pseudoflavonifractor sp. (ASF 500) while an annealing temperature of 62°C was used for Clostridium sp. (ASF 356), L. intestinalis (ASF 360), M. schaedleri (ASF 457), Clostridium sp. (ASF 502), and P. goldsteinii (ASF 519). Annealing temperatures were optimally defined based on the results of a gradient PCR reaction. Of note, the ASF qPCR primers described herein were not designed to be used in a multiplex reaction that includes all eight primer sets for the simultaneous amplification of multiple targets. All ASF primers, both forward and reverse, were used at a final concentration of 300 nM. To ascertain specificity, melting curves were obtained and analyzed after performing the qPCR assay to identify the melting peaks consistent with true products based on the positive controls (i.e., plasmid DNA cloned individually with the 16s rRNA gene from each ASF member). Additionally, for all qPCR assays, genomic DNA extracted from cecal or fecal samples from germ-free C3H/HeN or germ-free C57BL/6 mice was included as a negative control. Reactions consisting of SYBR Green master mix, primers, molecular grade water but no DNA were also included as negative controls. The fluorescence level for determining the cycle threshold (Ct value) was set to the Mastercycler software default for all reactions (i.e., fluorescence cut-off of at least 110). When L. intestinalis (ASF 360) was undetectable in fecal or cecal samples via our qPCR assay, its presence was confirmed with a nested PCR assay (see section 2.12). When the presence of L. intestinalis (ASF 360) was confirmed by the nested approach, a Ct value of 36 was assigned to these samples to estimate its abundance.
Ten ng of DNA isolated from either cecal or fecal samples was utilized to determine ASF member abundance; this amount was selected after comparing DNA amounts ranging from 5 to 50 ng per reaction, whereby the use of 10 ng optimized detection without saturating the reaction and compromising the analytical sensitivity of the assay. Of note, we observed that 20-30 ng of DNA template could also be used in each reaction without inhibition. The total eluted DNA and weight of the initial raw material were taken into account when calculating the number of bacteria in cecal and fecal samples. Ultimately, total bacterial numbers were reported in logarithmic scale (i.e., base 10) per gram of raw wet material (i.e., cecal or fecal samples). Formulas and example calculations can be found in the Supplemental Material.
2.10. Determination of rRNA operon copy number
To facilitate calculation of an estimated total number of bacteria in each sample, we attempted to determine the number of 16S rRNA gene operons for all ASF species by BLAST analysis of all known 16S rRNA gene sequences of each ASF species against their respective contigs using available genomic assemblies (Wannemuehler et al., 2014). However, we observed that full-length coverage of the 16S rRNA genes and adjacent genomic segments was lacking in the draft contigs, probably due to the common occurrence of repetitive sequences in the flanking regions of these genes and/or the presence of broken junctions in the sequences. Therefore, accurate predictions of 16S rRNA copy number for any given species were not possible from the existing draft genomic assemblies. To address this limitation, we selected four different copy numbers (1, 5, 10, 20) based on the range of predicted numbers across all ASF as previously reported by Sarma-Rupavtarm et al., (2004). Using estimated linear equations from the standard curves, we then assessed whether using a value of either 1, 5, 10 or 20 for the 16S rRNA gene copy number influenced the actual log10 prediction for bacterial abundance (expressed as log10 of total gene copies) in the test samples. This approach allowed us to quantitatively establish a value that would not under- or over-estimate the bacterial load for each ASF member and could therefore be used for the final quantification of all taxa given the limitations of accurately determining exactly how many 16S rRNA gene copies are present in each ASF genome.
2.11. Sensitivity and specificity testing
Standard curves for individual ASF species were performed in triplicate and generated by serially diluting the plasmid standard solutions in 0.1 μm filter sterilized, nuclease free, deionized and distilled molecular grade water from either 3 to 15 ng DNA (∼ 109 copies) on the high end to either 0.3 or 0.5 femtograms DNA on the low end (∼ 10 copies). Conversion to total bacterial numbers provided an overall range from 1 to 10 × 108 total 16S rRNA gene copies (which, based on our approach, is equivalent to the total number of bacterial cells; see Supplemental Materials for additional explanation) considering five 16S rRNA genes per species/cell as per analysis described in section 2.10. Those calculations were then used to establish the limit of detection for each ASF member (see Supplemental Material for example calculations). To test the specificity of all primers, two approaches were employed: (1) a crosscheck of every single primer and plasmid combination individually (i.e., Clostridium sp., ASF 356 primers with 356 plasmids as well as 356 primers with each of the other seven ASF plasmids tested in individual reactions); and (2) confirmation that the specificity of each primer set would be maintained in a mock community assembled by combining all plasmids in the reaction with or without its corresponding target. For all cross-checks, 1 ng of plasmid DNA was used for either individual member or mock community testing. For the latter, the positive control community contained all eight ASF taxa plasmids, whereas the negative control contained all plasmids except that corresponding to the primer set being tested. All amplicons generated in the specificity check were then purified (QIAquick PCR Purification Kit, Qiagen®, Germantown, MD) and sequenced using the Sanger method (Sanger et al., 1977). Thereafter, an alignment of all products was generated using the CLC sequence viewer software (CLC Bio Qiagen®) and blasted against the GeneBank-NCBI database (http://www.ncbi.nlm.nih.gov/genbank/) using BLASTn for specificity confirmation (Altschul et al., 1990). In addition, all ASF primer sets were tested against the plasmid vector cloned with the E. coli 16S rRNA gene since this bacterium was used for producing the qPCR plasmid standards.
2.12. Nested PCR for detection of ASF members 356 and 360
Given the overall lower abundance of Clostridium sp. (ASF 356) and L intestinalis (ASF 360) in our C3H/HeN mouse line, a nested PCR approach was used to determine the presence or absence of these taxa in fecal/cecal samples when necessary. After DNA isolation, a universal 16S rRNA PCR was performed as described previously (Turner at al., 1999). A 2% agarose gel (Agarose, Molecular Biology Grade, IBI Scientific, IA) was then used to visualize the PCR products. All amplicons generated in the reaction were purified (QIAquick PCR Purification Kit, Qiagen®, Germantown, MD); 20 ng of each PCR product was then used in the species-specific qPCR reaction as described in section 2.9.
2.13. Statistics
Standard curves were generated and analyzed using GraphPad Prism 7.0a, whereby a linear regression model was run to determine slope, intercept, and statistics such as the R-squared and Pearson correlation coefficient. All statistical analyses to assess changes in log10 total number of 16S rRNA gene copies (i.e., predicted total bacterial abundances) of any ASF taxa were also performed using GraphPad Prism 7.0a (2016, GraphPad Software, Inc., La Jolla, CA). When comparing the abundance of each ASF taxon between mouse strains and between low fat and Western diet fed mice, a non-parametric unpaired Mann-Whitney test was performed using a two-tailed distribution for p-value calculations (p < 0.05). For similar comparisons in the antibiotic perturbation study or between the two SYBR Green Master Mixes (A versus B), a non-parametric Wilcoxon matched-pairs signed rank test was used in combination with a one- or two-tailed distribution, respectively, for p-value determination (p < 0.05). The one-tailed distribution was specifically used when analyzing data from the antibiotic study because all ASF taxa were expected to decrease in abundance after such a perturbation. Statistical comparison of each ASF taxon abundance across the four different values used for the 16S rRNA gene copy number test was performed using a non-parametric Friedman test accounting for matched values across groups (i.e., data points across rows and between treatments) in combination with a post-hoc Dunn's Test (p < 0.05). All final graphs were generated using GraphPad Prism 7.0a.
To visualize the ASF community under normal physiological (i.e., comparing host genotypes) or perturbed conditions (i.e., antibiotic use or different diets) and when comparing the two SYBR Green Mater Mixes, a Principal Component Analysis (PCA) was performed using R software, version 3.3.1 (R Core Team 2016, R Foundation for Statistical Computing, Vienna, Austria). Specifically, the prcomp() function from the stats package was used for the PC calculations, and the autoplot() function from the ggplot2 package was used for generating the final plot. PC analysis was performed after scaling the bacterial abundances to avoid the biased impact of heterogeneous variances across groups in the PC loadings. Scree plot analysis (i.e., visualization of eigenvalues associated with each PC) was used to determine the optimal number of PCs to be depicted on each graph. The ellipse generated to group data points was created based on a Multivariate T distribution. The box-and-whiskers and dot plots comparing ASF abundances between the two SYBR Green Master Mixes were generated using the ggplot2 package from R software version 3.3.1. Functions ggplot(), geom_boxplot(), geom_point() and geom_line were used to produce the final graph.
3. Results
3.1. ASF primer specificity
Species-specificity of all ASF primers was confirmed by sequencing the qPCR products generated during all individual and mock community-based cross-check reactions (data not shown). Specifically, no other taxon was observed during the BLAST search except the targeted one (100% identity and e-value of nearly 0 across all ASF targets). Additionally, melting curve evaluations provided a distinct pattern and peak for each ASF taxon, consistent with the specificity results obtained by sequencing products from individual and mock community reactions. We observed no cross-reactivity of any primers with E. coli 16S rRNA gene. Moreover, no detectable Ct values were generated using any of the ASF primers in qPCR reactions consisting of only molecular grade water (i.e., no DNA template) or in reactions containing DNA isolated from the feces or cecal contents of germ-free mice, indicating an absence of non-specific background signals from both sample matrices.
3.2. ASF qPCR assay performance
The plasmid DNA standard curves used to estimate ASF abundances exhibited strong linearity and good efficiencies across all ASF taxa, with R2 values of >0.9 using Mix A or Mix B and efficiencies ranging from 0.78-0.99 (Mix A) and 0.53-0.97 (Mix B) (Table S4, Figs. 1 and S2). Although the linearity of Mix A and Mix B was very similar, Mix A provided lower detection limits for six of the eight ASF taxa (varying from 10 to 2,700 total bacterial cells for Mix A versus 8 to 130,000 for Mix B). The enhanced sensitivity for Mix A was likely a reflection of the improved reaction efficiencies as compared to Mix B (Table S4).
Fig. 1.
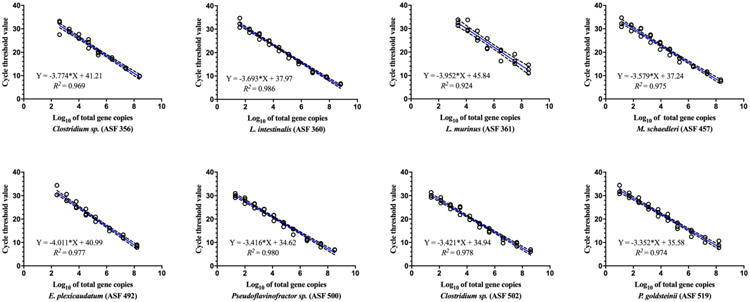
Linear regression model for SYBR Green Master Mix A depicting the standard curve parameters for ASF quantification. Standard curves were prepared using serial dilutions of purified vector-free plasmids containing the specific16S rRNA gene sequence for each ASF taxon and used to determine the limit of detection and efficiency of each reaction. The predicted linear model line (blue) and estimated equation are shown for each ASF taxon based on the log10 of total 16S rRNA gene copies, along with dotted lines representing the 95% confidence interval bands. Triplicates of each plasmid concentration were used to determine precision in quantification (open black circles). R-squared values are also shown for each curve. The final number for the log10 of total 16S rRNA gene copies for each ASF bacterium was calculated considering five copies of this gene per bacterial genome.
To verify the performance of Mix A and B using biological samples, we quantified the abundance of all ASF members using cecal contents from C57BL/6 mice harboring the ASF microbiota. All eight ASF members were detectable using both mixes with the exception of ASF 360, which was undetectable in some samples after 35 cycles. However, the presence of ASF 360 could be confirmed with a nested PCR approach. For those nested PCR-positive samples, a Ct value of 36 was assigned to estimate abundance. Principal Coordinate Analysis (PCA) revealed that ASF communities clustered according master mix and that significantly higher abundances for five of the eight ASF members were obtained with Mix A compared to Mix B (Figs. S3 and S4). Together, these data indicate that qPCR master mix chemistry can influence quantification results, and is a factor that should be considered when comparing ASF abundance values across studies.
For all assays, total bacterial numbers were calculated using five 16S rRNA copies per bacterium since there were no significant differences in cecal abundance of any ASF members when comparing values generated using 5 copies versus 1 or 5 copies versus 10 (Fig. S5). However, the predicted total bacterial abundances based on the log10 total 16S rRNA gene copies obtained when using 5 versus 20 gene copies was significantly less for all ASF members (∼ 0.5 to 1 log10 total bacteria per gram of cecal content; Fig. S5). In the end, we found the use of five copies per taxa for linear standard curve equations to be a reasonable and accurate estimate that could be used to quantify the abundance of all eight ASF members. This approach is also consistent with that proposed by (Sarma-Rupavtarm et al., 2004) who reported that five of the eight ASF taxa have 16S rRNA gene copy numbers ranging from four to six.
Standard curves were also generated using genomic DNA from cultures of individual ASF members. Genomic DNA curves exhibited both strong linearity and low limits of detection for all taxa (data expressed as log10 of total genome copies) when serially diluted in either molecular grade water (Fig. S6) or DNA extracted from germ-free mouse feces (Fig. S7). Because both lactobacilli (ASF 360 and 361) are readily cultivable in comparison to other ASF members, standard curves based on CFU/mL were also generated for these two species and confirmed the high sensitivity and linearity of those two qPCR primer sets (Fig. S6). Due to extended backorder of SYBR Green Mix A, all plasmid (Figs. S2 and S8) and genomic DNA standard curves (Figs. S6 and S7) were generated using Mix B. Together, these data demonstrate that our qPCR primers are highly sensitive and can be used to generate both plasmid and genomic DNA standard curves with strong linearity in a variety of sample matrices using two different SYBR Green formulations.
3.3. Quantification of ASF members in mouse cecal and fecal samples
To test the robustness and reproducibility of the qPCR assays in quantifying individual ASF members in the GI tract of mice, cecal samples were collected from two distinct inbred mouse lines, C3H/HeN and C57BL/6 mice. Comparative analysis of the abundance of each ASF member between age-matched C3H/HeN and C57BL/6 mice housed in separate gnotobiotic isolators revealed a significantly higher total number of all bacterial taxa for the C57BL/6 genotype with the exception of E. plexicaudatum (ASF 492) and Clostridium sp. (ASF 502) (Fig. 2). Noticeably, the variability in bacterial abundance was less in C57BL/6 mice, but the overall pattern of abundance of each taxon remained similar to those detected in the C3H/HeN line (Fig. 2). PCA revealed that ASF communities cultured by host genotype and confirmed that bacterial abundances were more widely dispersed in samples from C3H/HeN mice versus those from than C57BL/6 mice (Fig. S10). Together, these data demonstrate that although differences in ASF member abundances can be detected between the two mouse lines, the lowly, intermediate, or highly abundant taxa remain as such regardless of the host's genetic background.
Fig. 2.
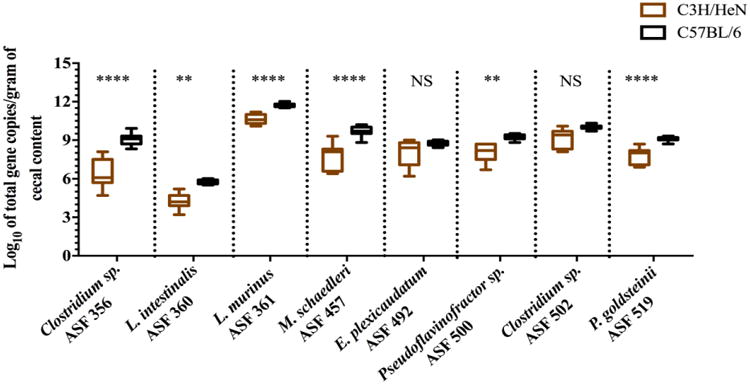
Box-and-whisker plots showing changes in ASF bacterial abundances (estimated using 16S rRNA copy numbers) in cecal contents of gnotobiotic C3H/HeN mice (n = 11) in comparison to age-matched, 12 week-old gnotobiotic C57BL/6 mice (n = 15). The y-axis indicates the ASF taxon log10 of total 16S rRNA gene copies per gram of cecal content, and the x-axis depicts each ASF member taxonomy and identification number. Whiskers depict the entire range of values (min to max); the horizontal bar in the middle of the box represents the median value. Asterisks refer to the degree of significance for the difference in bacterial abundance as determined by a non-parametric unpaired Mann-Whitney test using a two-tailed distribution for p-value calculations whereby comparing ranks across groups (NS = not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, and **** p ≤ 0.0001). All qPCRs were run in duplicate using SYBR Green Master Mix A. Estimated ASF abundances were achieved using a plasmid DNA standard curve.
To further assess the applicability of the newly-developed qPCR assays, an acute antibiotic perturbation of the ASF microbiota in C57BL/6 mice via either streptomycin or neomycin administration was performed. By using aminoglycoside antibiotics with the potential for broad-spectrum activity, we expected to decrease the abundance of multiple (but perhaps not all) taxa concomitantly and generate an altered community composition suitable for determining if our assay could still robustly quantify the ASF bacteria. PCA revealed that ASF communities clustered by antibiotic treatment status, indicating that administration of either streptomycin (Fig. 3 – Panel a) or neomycin (Fig. S11 – Panel a) induced global changes to the structure of the ASF community. Subsequent qPCR analyses demonstrated that both antibiotic treatments significantly decreased total ASF abundance across all taxa. In particular, Clostridium sp. (ASF 356), E. plexicaudatum (ASF 492), Pseudoflavonifractor sp. (ASF 500), Clostridium sp. (ASF 502), and P. goldsteinii (ASF 519) experienced marked reductions of 2 to 4 log10, with L. intestinalis (ASF 360) becoming undetectable in all samples following streptomycin treatment (Fig. 3 – Panel b). Neomycin treatment reduced the abundance of L. murinus (ASF 361) and P. goldsteinii (ASF 519) by 2 log10 (Fig. S11 – Panel b). Of note, a nested-PCR approach revealed that L. intestinalis (ASF 360) was still detectable, albeit in lower abundances, post-neomycin treatment.
Fig. 3.
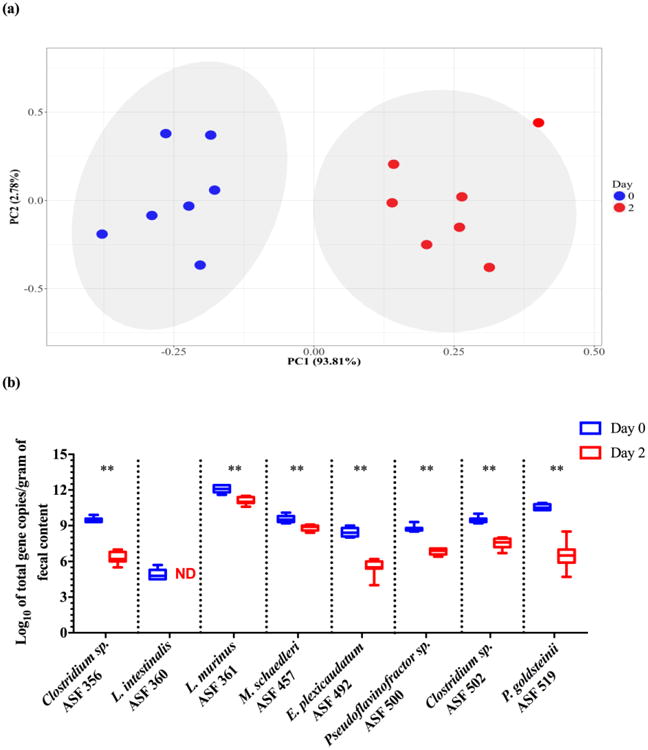
Changes in the ASF community following antibiotic perturbation are detectable with the newly-developed primers. Data represent the log10 of total 16S rRNA gene copies per gram of cecal contents between day 0 and 2 for streptomycin-treated mice. PCA plot depicting how the ASF community clusters following antibiotic perturbation. The x- and y-axes indicate the principal components 1 (PC1) and 2 (PC2), including the percent of variance explained by each PC. Each dot in the PCA plots represent an individual animal with its respective ASF community composition. The gray shaded areas in the plot show the dispersion of the data points within groups and were calculated based on a multivariate T distribution. For PC analysis, the log10 of total 16S rRNA gene copies for ASF 360 was excluded from the dataset because it was not undetected in any of the samples (Panel a). Box-and-whisker plots showing a significant decrease in the abundance of each individual ASF member in the feces of 8 week-old gnotobiotic C57BL/6 mice (n = 7 per treatment) following treatment with streptomycin for two consecutive days (10 mg/mL of drinking water) (Panel b). Abundances were estimated using 16S rRNA copy numbers. The y-axis indicates the ASF taxon log10 of total 16S rRNA gene copies per gram of feces, and the x-axis depicts each ASF member taxonomy and identification number. Whiskers depict the entire range of values (min to max); the horizontal bar in the middle of the box represents the median value. Asterisks refer to the degree of significance for the difference in bacterial 16S rRNA gene copies as determined by the non-parametric Wilcoxon matched-pairs signed rank test using a one-tailed distribution for p-value calculations (* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, and **** p ≤ 0.0001). All qPCRs were run in duplicate using the SYBR Green Master Mix A. Estimated ASF abundances were achieved using a plasmid DNA standard curve. ASF 360 was not detected (i.e., ND) at day 2 in streptomycin-treated mice.
An additional test to assess the performance of these qPCR assays included a comparison of the ASF communities in male C57BL/6 ASF-bearing mice fed either a low fat or a Western diet for five weeks. PCA revealed that ASF communities clustered by diet (Fig. 4 – Panel a), indicating that the two diets induced global changes to the ASF community structure. Subsequent qPCR analyses demonstrated that feeding a Western diet significantly increased the abundance of seven of the eight ASF members in comparison to feeding a low fat diet (Fig. 4 – Panel b). Of note, L. intestinalis (ASF 360) was undetectable in low fat diet fed mice, and L. murinus (ASF 361) decreased in abundance after Western diet feeding. Together, results from the antibiotic and dietary perturbation studies demonstrate the utility of our newly-developed qPCR assays to detect changes in ASF member abundances following both acute and chronic interventions that impact the gut microbiota.
Fig. 4.
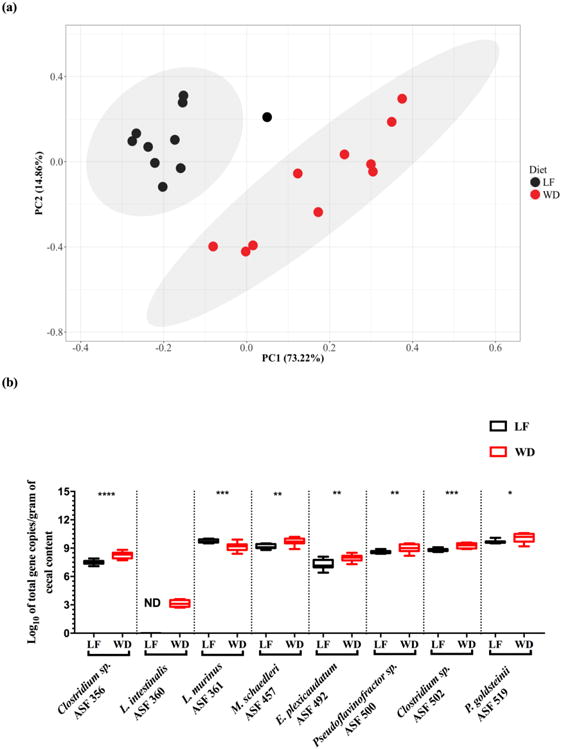
Newly-developed qPCR primers detect differences in ASF member abundances after feeding low fat and Western diets. Data represent the log10 of total 16S rRNA gene copies per gram of cecal contents in male C57BL/6 ASF-bearing mice fed either a low fat or a Western diet for five weeks. PCA plot depicting how the ASF community clusters by diet. The x- and y-axes indicate the principal components 1 (PC1) and 2 (PC2), including the percent of variance explained by each PC. Each dot in the PCA plot represents an individual animal with its respective ASF community composition. The gray shaded areas in the plot show the dispersion of the data points within groups and were calculated based on a multivariate T distribution. For PC analysis, the log10 of total 16S rRNA gene copies for ASF 360 was excluded from the dataset because it was not detected in any of the low fat diet fed mice (Panel a). Box-and-whisker plots showing significant alterations in the abundance of each individual ASF member in the feces of gnotobiotic C57BL/6 male mice fed either a low fat (LF; black boxes; n = 10) or Western (WD; red boxes; n = 11) diet for five weeks (Panel b). Abundances were estimated using 16S rRNA copy numbers. The y-axis indicates the ASF taxon log10 of total 16S rRNA gene copies per gram of feces, and the x-axis depicts each ASF member taxonomy and identification number. Whiskers depict the entire range of values (min to max); the horizontal bar in the middle of the box represents the median value. Asterisks refer to the degree of significance for the difference in bacterial 16S rRNA gene copies as determined by the non-parametric Wilcoxon matched-pairs signed rank test using a one-tailed distribution for p-value calculations (* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, and **** p ≤ 0.0001). All qPCRs were run in duplicate using SYBR Green Master Mix B. Estimated ASF abundances were achieved using a plasmid DNA standard curve. ASF 360 was not detected (i.e., ND) in LF fed mice.
4. Discussion
The original Schaedler flora was developed in 1965 to provide colonization resistance to newly derived germ-free rodents (Dewhirst et al., 1999; Orcutt et al., 1987; Schaedler et al., 1965; Trexler et al., 1999). Later, this community was modified to consist of eight species autochthonous to the murine GI tract that represent the Firmicutes, Bacteroidetes, and Deferribacteres phyla (Dewhirst et al., 1999; Orcutt et al., 1987) and was thus renamed as the Altered Schaedler Flora. Since then, the ASF microbiota has been used extensively to standardize the microbial communities of axenic animals across mouse vendors (Smith et al., 2007; Wymore et al., 2015). Upon colonization, each species likely occupies different niches of the mouse GI tract and contributes to immune maturation, colonization resistance and host physiology (Orcutt et al., 1987). In addition, this model GI microbial community offers a unique opportunity to assess the specific contribution of individual bacterial species during states of homeostasis and disease (Atherly et al., 2016; Brugiroux et al., 2016; Endt et al., 2010; Ge et al., 2006; Rooks et al., 2017). However, a robust molecular quantification method for this community is needed, especially considering that cultivating some ASF members can be cumbersome. Here, we present newly-designed qPCR primers for use with SYBR Green chemistry that are specific to each ASF member and limit primer dimerization.
For any quantitative assay aiming to determine the abundance of a single microbial species, reaction specificity is of utmost importance. Here, we have shown that cross-reactivity did not occur with any ASF primer set. Although our final assay was efficient and sensitive, differences in the performance of the two SYBR Green master mixes evaluated were observed with respect to detection limits and individual bacterial abundances. However, for samples where all members of the ASF community are present in relatively high abundance, differences among mixes are likely to be negligible. We anticipate that this qPCR assay should be reproducible with other SYBR Green master mixes, in part, because the amplicon size generated by all the ASF primers falls within the range recommended for SYBR Green assays (Thornton et al., 2011). However, as reaction efficiencies can vary, choice of commercial kit should be further validated in-house prior to use. Moreover, comparisons of ASF member abundances across studies should be done cautiously and consider whether different qPCR chemistries were used.
We also assessed the applicability of our qPCR assay under normal physiological conditions by evaluating ASF abundances in cecal samples obtained from healthy, gnotobiotic mice of two different genetic backgrounds colonized with the ASF since birth. Although most of the ASF taxa were present at significantly higher levels in the C57BL/6 mice compared to the C3H/HeN mice, the overall patterns of abundance remained similar regardless of the host's genetic background. Although host genotype had a significant impact on individual ASF member densities, confounding factors such as the source of the initial ASF inoculum, cage effects, and sex differences may have also influenced abundances (Alexander et al., 2006; Ge et al., 2006); such factors were not accounted for in this study.
Even though mouse genotype and environmental factors (i.e., husbandry practices, diet, etc.) often differ across studies, we and others (Alexander et al., 2006; Sarma-Rupavtarm et al., 2004; Stehr et al., 2009) have consistently found L. intestinalis (ASF 360) to be the least abundant member of the ASF community. The variable and often transient detection of this Lactobacillus species in feces and cecal contents may, in part, be explained not only by its low abundance in the ASF community, but also by its major niche in the rodent forestomach (Walter, 2008). Also consistent with other studies of the ASF community, we found Clostridium sp. (ASF 356), E. plexicaudatum (ASF 492), Pseudoflavonifractor sp. (ASF 500) and Clostridium sp. (ASF 502) to be among the most abundant members in the cecum—an expected finding as these four members are extremely oxygen sensitive (EOS) fusiform organisms who likely favor the c (Alexander et al., 2006; Sarma-Rupavtarm et al., 2004; Stehr et al., 2009).
Of interest, we and Alexander et al., 2006, found P. goldsteinii (ASF 519) to be lowly abundant in inbred C57BL/6 mice, while Sarma-Rupavtarm et al., 2004, and Ge et al., 2006, observed this taxon to be present at intermediate levels in C.B.-17 SCID and Swiss Webster mice, respectively. Stehr et al., 2009, found P. goldsteinii (ASF 519) to be one of the most abundant community members in outbred CD-1 mice. More recently, other laboratories have reported P. goldsteinii (ASF 519) as the most relatively abundant ASF community member in BALB/cByJ (Rooks et al., 2017) and C3H/HeN mice (Atherly et al., 2016) following colonization with either E. coli LF82 or Helicobacter bilis, respectively. The most abundant taxa in our healthy C3H/HeN and C57BL/6 mice fed normal chow was L. murinus (ASF 361), yet others have found its abundance in cecal contents to vary from low to intermediate levels in a variety of mouse lines (Alexander et al., 2006; Sarma-Rupavtarm et al., 2004; Stehr et al., 2009). At the moment, we cannot pinpoint a specific environmental factor that explains the high abundance of L. murinus (ASF 361) in our mice, but perhaps diet and other environmental factors may play a role.
Assessing bacterial abundance under normal physiological conditions is important, but the ability to quantify specific species following a perturbation of the GI ecosystem is also of great interest (Croswell et al., 2009; Dethlefsen et al., 2008; Ubeda et al., 2012). To that end, we chose to acutely perturb the ASF community by administering a two-day course of either streptomycin or neomycin, two aminoglycosides (a bactericidal protein synthesis inhibitor) with the potential to broadly impact both Gram positive and Gram negative ASF members (Schantz et al., 2004). Global changes in ASF community structure following treatment with either antibiotic were observed. Although each ASF taxon responded to the stress of both streptomycin and neomycin by decreasing in abundance, we did observe differences in the extent to which some ASF member densities were altered by the different antibiotic treatments.
We also performed a chronic dietary perturbation of the ASF community using low fat and Western diets to further test our qPCR method for quantifying ASF member abundance. Diet is a deterministic factor known to influence the composition of complex microbial ecosystems (Martinez et al., 2010). Similarly, our results show that diet can also significantly alter individual ASF densities and community structure. Of note, L. intestinalis (ASF 360) was undetectable in mice fed a low fat diet, an observation consistent with previous reports (Dubos and Schaedler, 1962; Dubos et al., 1965). Together, these findings indicate that our qPCR assay permits the detection of the majority of the ASF members when the gut microbial community is subject to perturbation, including antibiotic treatments and a dietary intervention.
5. Conclusions
In conclusion, we have developed an improved, species-specific and sensitive qPCR assay to be used with SYBR Green chemistry for the detection and quantification of all ASF organisms in gnotobiotic mice under varying physiological conditions. In parallel, we have also provided a nested PCR assay that can be used to verify the presence of two potentially lowly abundant ASF species, 356 and 360. Future work may focus on improving the throughput of our ASF qPCR assays, which is currently limited by the need for two different annealing temperatures and the inability to use multiple primer sets together in a multiplex assay. Altogether, we anticipate this improved qPCR assay will permit reproducible quantification of the ASF across laboratories, thereby facilitating research efforts employing this defined microbiota to answer a variety of experimental questions.
Supplementary Material
Highlights.
A species-specific, quantitative PCR assay for the Altered Schaedler Flora is described.
This SYBR green-based assay can be used with both fecal and cecal samples.
Primers target the hypervariable regions of the 16S rDNA of each ASF taxon to enhance assay specificity.
The presence of lowly abundant ASF taxa can be determined via a nested PCR assay.
This qPCR assay provided robust results under normal physiological conditions in two inbred mouse lines as well as both before and after perturbation of the community.
Acknowledgments
All authors are grateful for the technical expertise and skillful animal husbandry provided by Brandon White and all the staff at the UNL Gnotobiotic Mouse Facility. We also thank Dr. Michael Wannemuehler (Iowa State University) for providing the ASF strains and founder ASF-bearing C3H/HeN mice. This work was supported by a Career Development Award from the Crohn's and Colitis Foundation of America (grant # 3578), the National Institute of General Medical Sciences of the National Institutes of Health (1P20GM104320), and start-up funding from the University of Nebraska-Lincoln to ART. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander AD, Orcutt RP, Henry JC, Junior JB, Bissahoyo AC, Threadgill DW. Quantitative PCR assays for mouse enteric flora reveal strain-dependent differences in composition that are influenced by the microenvironment. Mamm Genome. 2006;17:1093–1104. doi: 10.1007/s00335-006-0063-1. [DOI] [PubMed] [Google Scholar]
- Atherly T, Mosher C, Wang C, Hostetter J, Proctor A, Brand MW, Phillips GJ, Wannemuehler M, Jergens AE. Helicobacter bilis infection alters mucosal bacteria and modulates colitis development in defined microbiota mice. Inflamm Bowel Dis. 2016;22:2571–2581. doi: 10.1097/MIB.0000000000000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bindels L, Segura Munoz R, Gomes-Neto JC, Mutemberezi V, Martinez I, Salazar N, Cody E, Quintero-Villegas M, de los Kittana H, Reyes-Gavilán CG, Schmaltz R, Muccioli GG, Walter J, Ramer-Tait AE. Resistant starch can improve insulin sensitivity independently of the gut microbiota. Microbiome. 2017 doi: 10.1186/s40168-017-0230-5. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. 2013;14:676–684. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugiroux S, Beutler M, Pfann C, Garzetti D, Ruscheweyh HJ, Ring D, Diehl M, Herp S, Lotscher Y, Hussain S, Bunk B, Pukall R, Huson DH, Munch PC, McHardy AC, McCoy KD, Macpherson AJ, Loy A, Clavel T, Berry D, Stecher B. Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat Microbiol. 2016;2:16215. doi: 10.1038/nmicrobiol.2016.215. [DOI] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croswell A, Amir E, Teggatz P, Barman M, Salzman NH. Prolonged impact of antibiotics on intestinal microbial ecology and susceptibility to enteric salmonella infection. Infect Immun. 2009;77:2741–2753. doi: 10.1128/IAI.00006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:2383–2400. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst F, Chien CC, Paster B, Ericson R, Orcutt R, Schauer D, Fox J. Phylogeny of the defined murine microbiota: altered Schaedler flora. Appl Environ Microbiol. 1999;65:3287–3292. doi: 10.1128/aem.65.8.3287-3292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubos RJ, Schaedler RW. The effect of diet on the fecal bacterial flora of mice and on their resistance to infection. J Exp Med. 1962;115:1161–1172. doi: 10.1084/jem.115.6.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubos R, Schaedler RW, Costello R, Hoet P. Indigenous, normal, and autochthonous flora of the gastrointestinal tract. J Exp Med. 1965;122:67–76. doi: 10.1084/jem.122.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endt K, Stecher B, Chaffron S, Slack E, Tchitchek N, Benecke A, Van Maele L, Sirard JC, Mueller AJ, Heikenwalder M, Macpherson AJ, Strugnell R, von Mering C, Hardt WD. The microbiota mediates pathogen clearance from the gut lumen after non-typhoidal Salmonella diarrhea. PLoS Pathogens. 2010;6:e1001097. doi: 10.1371/journal.ppat.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Z, Feng Y, Taylor NS, Ohtani M, Polz MF, Schauer DB, Fox JG. Colonization dynamics of altered Schaedler flora is influenced by gender, aging, and Helicobacter hepaticus infection in the intestines of Swiss Webster mice. Appl Environ Microbiol. 2006;72:5100–5103. doi: 10.1128/AEM.01934-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuking MB, Cahenzli J, Lawson MAE, Ng DCK, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- GraphPad Prism. La Jolla California USA; 2016. version 7.0a for Mac OS X, GraphPad Software. www.graphpad.com. [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Halaihel N, Vendrell D, Ruiz-Zaruela I, de Blas I, Alonso JL, Girones O, Perez T, Muzquiz JL. A new real time PCR-based assay for diagnosing Renibacterium salmoninarum in rainbow trout (Oncorhynchus mykiss) and comparison with other techniques. J Microbiol Methods. 2009;76:75–80. doi: 10.1016/j.mimet.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Liu Z, Ramer-Tait AE, Henderson AL, Demirkale CY, Nettleton D, Wang C, Hostetter JM, Jergens AE, Wannemuehler MJ. Helicobacter bilis colonization enhances susceptibility to typhlocolitis following an inflammatory trigger. Dig Dis Sci. 2011;56:2838–2848. doi: 10.1007/s10620-011-1701-3. [DOI] [PubMed] [Google Scholar]
- Loy A, Arnold R, Tischler P, Rattei T, Wagner M, Horn M. probeCheck – a central resource for evaluating oligonucleotide probe coverage and specificity. Environ Microbiol. 2008;10:2894–2898. doi: 10.1111/j.1462-2920.2008.01706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield ES, Worley JM, McKenzie SE, Surrey S, Rappaport E, Fortina P. Nucleic acid detection using non-radioactive labeling methods. Mol Cell Probes. 1995;9:145–156. doi: 10.1006/mcpr.1995.0023. [DOI] [PubMed] [Google Scholar]
- Martinez I, Kim J, Duffy PR, Schlegel VL, Walter J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS One. 2010;5:e15046. doi: 10.1371/journal.pone.0015046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez I, Wallace G, Zhang C, Legge R, Benson AK, Carr TP, Moriyama EN, Walter J. Diet-induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl Environ Microbiol. 2009;79:516–524. doi: 10.1128/AEM.00380-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh PL, Benson AK, Peterson DA, Patil PB, Moriyama EN, Roos S, Walter J. Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. ISME J. 2010;4:377–387. doi: 10.1038/ismej.2009.123. [DOI] [PubMed] [Google Scholar]
- Orcutt RP, Gianni FJ, Judge RJ. Development of an “Altered Schaedler Flora” for NCI gnotobiotic rodents. Microecol Ther. 1987;17:59. [Google Scholar]
- Parekh PJ, Balart LA, Johnson DA. The influence of the gut microbiome on obesity, metabolic syndrome and gastrointestinal disease. Clin Transl Gastroenterol. 2015;6:e91. doi: 10.1038/ctg.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R Foundation for Statistical Computing; Vienna, Austria: 2016. R: A language and environment for statistical computing. URL https://www.R-project.org/ [Google Scholar]
- Relman DA. The human microbiome: ecosystem resilience and health. Nutr Rev. 2012;70:S2–S9. doi: 10.1111/j.1753-4887.2012.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooks MG, Veiga P, Reeves AZ, Lavoie S, Yasuda K, Asano Y, Yoshihara K, Michaud M, Wardwell-Scott L, Gallini CA, Glickman JN, Sudo N, Huttenhower C, Lesser CF, Garrett WS. QseC inhibition as an antivirulence approach for colitis-associated bacteria. Proc Natl Acad Sci USA. 2017;114:142–147. doi: 10.1073/pnas.1612836114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Nat Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma-Rupavtarm RB, Ge Z, Schauer DB, Fox JG, Polz MF. Spatial distribution and stability of the eight microbial species of the Altered Schaedler Flora in the mouse gastrointestinal tract. Appl Environ Microbiol. 2004;70:2791–2800. doi: 10.1128/AEM.70.5.2791-2800.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor RB, Mazmanian SK. Intestinal microbes in inflammatory bowel diseases. Am J Gastroenterol Suppl. 2012;1:15–21. [Google Scholar]
- Schaedler RW, Dubs R, Costello R. Association of germfree mice with bacteria isolated from normal mice. J Exp Med. 1965;1:77–82. doi: 10.1084/jem.122.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schantz JT, Ng KW. A manual for primary human cell culture. World Scientific. 2004:89. [Google Scholar]
- Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15:317–328. doi: 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Stehr M, Greweling MC, Tischer S, Singh M, Blocker H, Monner DA, Muller W. Charles River altered Schaedler flora (CRASF®) remained stable for four years in a mouse colony housed in individually ventilated cages. Laboratory Anim. 2009;43:362–370. doi: 10.1258/la.2009.0080075. [DOI] [PubMed] [Google Scholar]
- Thornton B, Basu C. Real-time PCR (qPCR) primer design using free online software. Biochem Mol Biol Educ. 2011;39:145–154. doi: 10.1002/bmb.20461. [DOI] [PubMed] [Google Scholar]
- Trexler PC, Orcutt RP. Development of gnotobiotics and contamination control in laboratory animal science. In: Macpherson CW, Mattingly SF, editors. 50 Years of Laboratory Animal Science. Memphis: American Association for Laboratory Animal Science; 1999. pp. 121–128. [Google Scholar]
- Turner S, Pryer KM, Miao VPW, Palmer JD. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small submit rRNA sequence analysis. J Eukaryot Microbiol. 1999;46:327–338. doi: 10.1111/j.1550-7408.1999.tb04612.x. [DOI] [PubMed] [Google Scholar]
- Ubeda C, Pamer EG. Antibiotics, microbiota, and immune defense. Trends Immunol. 2012;33:459–466. doi: 10.1016/j.it.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J. Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl Environ Microbiol. 2008;74:4985–4996. doi: 10.1128/AEM.00753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Ley R. The human gut microbiome: ecology and recent evolutionary changes. Annu Rev Microbiol. 2011;65:411–429. doi: 10.1146/annurev-micro-090110-102830. [DOI] [PubMed] [Google Scholar]
- Wannemuehler MJ, Overstreet AM, Ward DV, Phillips GJ. Draft genome sequences of the Altered Schaedler Flora, a defined bacterial community from gnotobiotic mice. Genome Announc. 2014;2:1–2. doi: 10.1128/genomeA.00287-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymore BM, Wannemuehler MJ, Phillips GJ, Proctor A, Overstreet AM, Jergens AE, Orcutt RP, Fox JG. The Altered Schaedler Flora: continued applications of a defined murine microbial community. ILAR J. 2015;56:169–78. doi: 10.1093/ilar/ilv012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.