

ABSTRACT
Visceral leishmaniasis is a fatal parasitic neglected disease affecting 1.5 million people worldwide. Based on a drug repositioning approach, the aim of this work was to investigate the in vitro immunomodulatory potential of buparvaquone (BPQ) and to establish a safe regimen to evaluate the in vivo efficacy of BPQ entrapped by negatively charged nanoliposomes (BPQ-LP) in Leishmania infantum-infected hamsters. Small-angle X-ray scattering, dynamic light scattering, and the ζ-potential were applied in order to study the influence of BPQ on the liposome structure. Our data revealed that BPQ was located in the polar-apolar interface, snorkeling the polar region, and protected against aggregation inside the lipophilic region. The presence of BPQ also decreased the Z-average hydrodynamic diameter and increased the surface charge. Compared to intravenous and intramuscular administration, a subcutaneous route was a more effective route for BPQ-LP; at 0.4 mg/kg, BPQ-LP reduced infection in the spleen and liver by 98 and 96%, respectively. Treatment for 5 days resulted in limited efficacy, but 10 days of treatment resulted in an efficacy similar to that of a 15-day regimen. The nanoliposomal drug was highly effective, with a mean 50% effective dose of 0.25 mg/kg, reducing the parasite load in bone marrow by 80%, as detected using quantitative PCR analysis. In addition, flow cytometry studies showed that BPQ upregulated cytokines as tumor necrosis factor, monocyte chemoattractant protein 1, interleukin-10 (IL-10), and IL-6 in Leishmania-infected macrophages, eliminating the parasites via a nitric oxide-independent mechanism. This new formulation proved to be a safe and effective treatment for murine leishmaniasis that could be a useful candidate against visceral leishmaniasis.
KEYWORDS: Leishmania, drug delivery, liposomes, therapy
INTRODUCTION
The worldwide prevalence and severity of leishmaniasis has led the World Health Organization to consider it a priority infectious disease (1). Due to the complexity of host-parasite interaction, as well as contradictory results from vaccination models, the control of infection is mainly dependent on chemotherapeutic intervention (2). There are now a number of therapies for various forms of leishmaniasis; the preferences for first-line and second-line treatment vary based on the type of disease and are often guided by regional practice. Pentavalent antimonials, such as Pentostam and Glucantime, have been used to treat leishmaniasis for the last 70 years (3). However, these drugs have multiple adverse effects and decreasing effectiveness due to the development of parasite resistance, limiting their application (4). In Brazil, pentavalent antimony is still effective for treating most infections, despite its high toxicity. Until now, limited data about the resistance to these drugs have been available. Other drugs used for treatment include amphotericin B, miltefosine, and pentamidine, but all of these current treatments have limitations. Therefore, novel, safe, and low-cost treatments are still needed (5).
Finding new uses for approved drugs, also known as drug repositioning or repurposing, is an important approach for the introduction of novel treatments against neglected parasitic diseases and has the advantage of decreasing the costs and the time of the drug discovery and development process (6). Buparvaquone (BPQ), a hydroxynaphthoquinone, marketed as Butalex, is a safe drug currently used for the veterinary treatment of theileriosis in cattle. Croft et al. (7) demonstrated for the first time the in vitro activity of BPQ against Leishmania donovani; however, the drug showed limited in vivo efficacy in a BALB/c mouse model. Our previous study demonstrated a potent in vitro efficacy of BPQ against Leishmania infantum; though that was a preliminary study, we also showed that BPQ effectiveness could be improved using a liposomal formulation (8).
Liposomes have been widely used for the sustained and targeted delivery of drugs. Amphotericin B has been used in clinical therapy for leishmaniasis for many years in the form of a safe liposomal formulation named Ambisome (9). However, liposomal formulations must be adequately designed for delivery or for a specific disease, and this requires includes previous knowledge of the target cells, as well as of the physical-chemical properties, including (i) the size, (ii) the surface charge, (iii) the phase transition temperature of phospholipids, (iv) the amount of cholesterol, and (v) the host cell surface receptors (10). Previous studies have demonstrated that the addition of phosphatidylserine to liposome membranes results in preferential phagocytosis by macrophages via surface receptors designated “scavenger receptors” (11). The use of phosphatidylserine-treated liposomes has yielded promising results against visceral leishmaniasis, increasing the in vitro selectivity and therapeutic index of antimony (12) and furazolidone (13).
The drugs and compounds used to kill Leishmania can directly affect the parasites or indirectly affect the parasites via activated macrophages. Leishmania uses these cells as a host; however, the same cells, when activated, can also elicit an immunomodulatory action against them, through the production of nitric oxide (NO), reactive oxygen species, and cytokines such as gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) (14, 15). In the present study, BPQ was entrapped in negatively charged liposomes containing phosphatidylserine as a delivery approach to macrophages, and a detailed physicochemical analysis of liposomal buparvaquone (BPQ-LP) formulation was performed using small-angle X-ray scattering (SAXS), dynamic light scattering (DLS), and the ζ-potential. The BPQ-LP formulation was administered for different periods using different routes of administration and doses, and its efficacy was observed in the spleen, liver and also in the bone marrow, using real-time quantitative PCR (qPCR) for parasite detection. Finally, using a flow cytometry analysis, we evaluated the immunomodulatory potential of BPQ-LP in macrophages infected with Leishmania.
RESULTS
Physicochemical characterizations.
In order to characterize the influence of BPQ in PS-containing liposomes (BPQ-LP), DLS, ζ-potential, and SAXS experiments were evaluated. DLS measurements indicated that BPQ was able to alter the overall size of the liposomes, by decreasing the average diameter (Z-average) from 242 ± 3 nm to 145 ± 3 nm in the absence and presence of BPQ, respectively. Such change represents a decrease of ∼40% in the liposome hydrodynamic diameter. The liposome ζ-potential also changed due to the presence of BPQ. In the absence or presence of BPQ, the values were −61 ± 2 mV or −48 ± 1 mV, respectively, demonstrating an ∼20% increase in the ζ-potential value due to the presence of BPQ. In order to get more information on the BPQ and the liposome interaction, SAXS measurements were performed.
Figure 1A shows the SAXS curves of liposomes in the absence (○) and presence (□) of BPQ. The arrow in the inset of Fig. 1A indicates small changes in the membrane scattering curve due to the presence of BPQ. Figure 1B and C show the SAXS curves of liposomal phosphatidylserine (PS-LP) and BPQ-LP, respectively, along with the best model able to fit these SAXS curves. The fitting parameters were described in Table 1. In the absence of BPQ (i.e., the liposomal system), the SAXS curves were suggestive of unilamellar vesicles in solution, i.e., the absence of sharp peaks at large (or middle) q values. The fitting parameters indicated that the membrane thickness was equal to 57.8 ± 1.3 Å.
FIG 1.
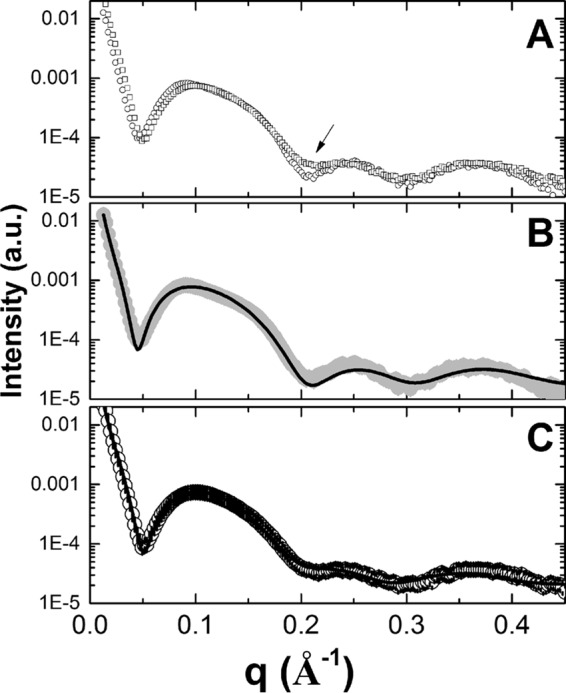
(A) SAXS curves of PS-LP (○) and BPQ-LP (□). (B) SAXS curve of LP (solid gray circles), along with the best-fit data using only one theoretical model. (C) SAXS curve of BPQ-LP (○), along with the best-fit data using the combination of two theoretical models. The arrow in panel A indicates small changes in the membrane scattering curve due to the presence of BPQ.
TABLE 1.
SAXS data determined using one or two theoretical models to describe the scattering intensity
| Parametera | Mean ± SD |
||
|---|---|---|---|
| Absence of BPQ (model 1) | Presence of BPQ |
||
| Model 1 | Model 2 | ||
| wt (%) | 100 | 90 ± 5 | 10 ± 5 |
| Rpol (Å) | 9.3 ± 0.6 | 10.0 ± 0.5 | 12.1 ± 0.5 |
| RCH2 (Å) | 17.2 ± 0.4 | 17.2 ± 0.3 | 11.1 ± 0.3 |
| RCH3 (Å) | 2.4 ± 0.9 | 2.7 ± 0.8 | 1.1 ± 0.8 |
| ρpol (e/Å3) | 0.41 ± 0.02 | 0.41 ± 0.01 | 0.41 ± 0.01 |
| ρCH2 (e/Å3) | 0.33 ± 0.01 | 0.33 ± 0.01 | 0.33 ± 0.01 |
| ρCH3 (e/Å3) | 0.22 ± 0.01 | 0.22 ± 0.01 | 0.22 ± 0.01 |
“wt (%)” is the weight (percent) of each model in the scattering curve, and Ri and ρi are the thickness and electron density, respectively, of the ith region. These are divided into polar region (pol; lipid head group) and CH2 and CH3 (lipid chains), respectively.
It was not possible to fit the experimental curve of BPQ-LP using only one theoretical model (data not shown). Thus, in order to obtain a reasonable fitting, we used two different theoretical models to fit this scattering curve. As observed in Table 1, model 1 is dominant in the scattering profile for the liposome in the presence of BPQ, being responsible for almost 90% of the scattering (Table 1). Moreover, model 1 has structural parameters similar to those of the one observed for the system in the absence of BPQ (Table 1). The second model used in the fitting (model 2), however, has different parameters compared to model 1, in particular for the thickness of each region (Table 1). In order to better understand the influence of this second model on the scattering curve for the system in the presence of BPQ, the electron density profile (EDP) curve was calculated.
Figure 2 shows the EDP for the PS-LP (dashed line) and BPQ-LP (solid line) systems; both curves were quite similar, except in the region indicated by the arrows. The small change observed in that specific region for the system in the presence of BPQ could indicate that the drug is located in that region. Taken together, the SAXS and ζ-potential results indicated that BPQ could be located at the apolar-polar interface, snorkeling the aqueous solution, and also be protected from aggregation inside the apolar region. One should bear in mind that BPQ is highly lipophilic (due to the presence of the heteroaromatic rings) and has a log P = 6.5 and a pKa of 5.2; thus, an interfacial location for the drug should be facilitated in order to accommodate both hydrophilic and hydrophobic regions of BPQ. A schematic representation of the bilayer, along with the most probable location of BPQ, is illustrated in Fig. 2.
FIG 2.
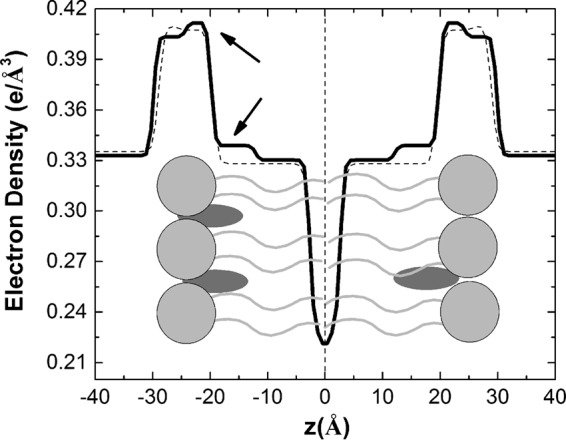
Electron density profiles for the membrane in the absence (dashed line) or presence (solid line) of BPQ. Inside the plot there is a schematic representation of the bilayer composed by the lipids (light gray) and the BPQ molecule (dark gray). Arrows indicate altered regions.
By using ultra-high-performance liquid chromatography (UPLC) for the quantification of BPQ in liposomes, we determined the encapsulation efficiency (%EE), which resulted in a mean value of 89.7% ± 3.6% (considering 10 batches). The drug/lipid ratio (D/L) was then calculated using the colorimetric assay of Stewart (49), which resulted in a D/L of 0.035 (mole ratio) and a final concentration of buparvaquone of 0.91 ± 0.25 mg/ml in the liposomes.
In vivo efficacy of nanoliposomal buparvaquone.
L. infantum-infected hamsters were treated with BPQ-LP using different regimens. Initially, three different routes of administration were chosen: subcutaneous (s.c.), intramuscular (i.m.), and intravenous (i.v.). qPCR analysis demonstrated that the s.c. route was the most effective, reducing the infection by 98% (P < 0.05) and 96% (P < 0.05) in the spleen and liver, respectively, at 0.4 mg/kg (Fig. 3) for 10 consecutive days. The i.v. route was also effective in the spleen, reducing infection by 88% (P < 0.05) (Fig. 3A), but showed limited efficacy in the liver, reducing the parasite load in the liver by 45% (Fig. 3B) compared to the untreated group. Administration i.m. yielded a lack of efficacy and resulted in similar parasite loads to untreated animals. The s.c. route was chosen for the remaining experiments.
FIG 3.

Evaluation of different routes of administration of BPQ-LP at 0.4 mg/kg in hamsters previously infected s.c., i.m., or i.v. with L. infantum amastigotes (5 hamsters/group). The parasite load was evaluated by RNA using RT-qPCR. *, P < 0.05 compared to the untreated group. Parasite load is expressed as normalized data in the spleen (A) and liver (B).
Infected animals were then assessed in three different periods of BPQ-LP administration via the s.c. route: 5, 10, and 15 consecutive days. Treatment for 5 days did not reduce the numbers of parasites in either the spleen or the liver (Fig. 4A and B). Conversely, treatment for 15 days was highly effective but yielded nonsignificant differences compared to the 10-day regimen (Fig. 4C to F). When BPQ-LP was administered for 15 days, it reduced infection by 94.58% (P < 0.05) in the spleen and 99.34% (P > 0.05) in the liver.
FIG 4.

Evaluation of treatment regimen for BPQ-LP at 0.4 mg/kg in hamsters previously infected with L. infantum (5 hamsters/group) as follows: 5 days (spleen [A] and liver [B]), 10 days (spleen [C] and liver [D]), and 15 days (spleen [E] and liver [F]) following administration by the s.c. route. The parasite load was evaluated by measuring RNA using qPCR. *, P < 0.05 compared to the untreated group. The parasite load is expressed as normalized data.
Based on previous data, the 50% effective dose (ED50) of BPQ-LP was determined using three different doses, administered for 10 consecutive days by the s.c. route. The treatment at 0.6 mg/kg/day was the most effective, reducing the parasite load by 97.54% in the spleen and 93.30% in the liver. At 0.3 mg/kg/day, BQP-LP reduced the parasite load by 43.57% in the spleen and 53.3% in the liver. At 0.1 mg/kg/day, BQP-LP reduced the parasite load by 5.28% in the spleen and 33.64% in the liver (P < 0.05) (Table 2). A t test analysis showed a significant difference (P < 0.05) for the BPQ-LP treatment in the spleen and liver, with P values of 0.0145 and 0.0362, respectively. According to the ED50 curves, BQP-LP yielded ED50 values of 0.31 mg/kg in the spleen (95% confidence interval [CI] = 0.19 to 0.50 mg/kg) and 0.19 mg/kg in the liver (95% CI = 0.04 to 0.83 mg/kg), with a mean ED50 of 0.25 mg/kg (Table 2).
TABLE 2.
ED50 values for liposomal BPQ in L. infantum-infected hamstersa
| Treatment group (mg/kg/day) | Mean % infection ± SD |
Mean ED50 (mg/kg) ± SD | |
|---|---|---|---|
| Spleen | Liver | ||
| 0.6 | 2.46 ± 1.45 | 6.70 ± 5.51 | |
| 0.3 | 56.43 ± 20.43 | 46.70 ± 36.28 | |
| 0.1 | 94.72 ± 31.59 | 66.36 ± 38.88 | |
| Control (untreated) | 100.00 ± 43.40 | 100.00 ± 40.15 | |
| ED50 | 0.31 | 0.19 | 0.25 ± 0.06 |
Treatment was performed using different doses administered s.c. for 10 consecutive days. The parasite load was evaluated using reverse transcription-quantitative PCR in the liver and spleen. ED50, 50% effective dose.
Efficacy of nanoliposomal buparvaquone in bone marrow.
The efficacy of BQP-LP was also verified in the bone marrow of L. infantum-infected hamsters after 10 consecutive days of s.c. treatment at 0.4 mg/kg. The qPCR analyses of bone marrow showed that BPQ-LP significantly reduced the parasite load in this site, reducing the levels by 80.23% (P < 0.05) compared to the untreated group (Fig. 5).
FIG 5.
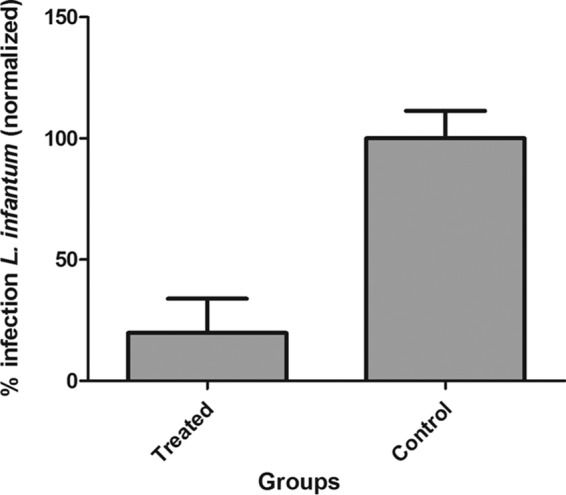
Evaluation of treatment regimen of BPQ-LP at 0.4 mg/kg in hamsters previously infected with L. infantum (5 hamsters/group) as follows: 15 days of administration by the s.c. route in the bone marrow. Data were analyzed by RT-qPCR in the bone marrow. *, P < 0.05 compared to the untreated group. The parasite load is expressed as normalized data.
In vitro immunomodulatory potential of buparvaquone.
L. infantum-infected and uninfected macrophages were treated with BPQ, and the production of cytokines was evaluated by flow cytometry. Buparvaquone upregulated the production of TNF, monocyte chemoattractant protein 1 (MCP-1), interkeukin-10 (IL-10), and IL-6 in L. infantum-infected macrophages compared to untreated L. infantum-infected macrophages (Fig. 6). No alteration could be observed in noninfected macrophages. Liposomal buparvaquone (BPQ-LP) was also incubated with uninfected macrophages and with Leishmania-infected macrophages, but it demonstrated an immunomodulatory effect similar to that of free BPQ, with no statistical differences.
FIG 6.

Effects of BPQ treatment on the production of proinflammatory cytokines (TNF, MCP-1, IL-6, and IL-10) in L. infantum-infected macrophages at 100 μM. The cytokines IL-12p70 and IFN-γ could not be detected. Mϕ, macrophages; MϕI, Leishmania-infected macrophages; Mϕ+BPQ, macrophages treated with buparvaquone; MϕI+BPQ, Leishmania-infected macrophages treated with buparvaquone; Mϕ+BPQ-LP, macrophages treated with liposomal buparvaquone; MϕI+BPQ-LP, Leishmania-infected macrophages treated with liposomal buparvaquone. The results are expressed in pg/ml, and cytokines were measured by CBA assay.
Quantification of NO.
Macrophages were incubated with BPQ or BPQ-LP, and the nitric oxide (NO) content evaluated using a Griess colorimetric assay. The NO levels of macrophages treated with BPQ or BPQ-LP were similar to those for untreated cells (control), demonstrating a lack of NO regulation (data not shown).
DISCUSSION
Phosphatidylserine-treated liposomes have proved an efficient tool for delivering drugs to Leishmania-infected macrophages (9, 12). The association of negatively charged phosphatidylserine with liposomes improves the selective uptake by macrophages via scavenger receptors, resulting in higher in vitro (12) and in vivo efficacies against Leishmania parasites (13). Two additional important characteristics of this formulation may also contribute to higher efficiency in macrophages: (i) saturated phospholipids at an elevated transition temperature, which yields a gel state at a physiological temperature (37°C), facilitate uptake by macrophages, and (ii) a reduced size of vesicles (<200 nm) confers greater stability for formulation and bone marrow targeting (13). In order to study the physicochemical parameters of this formulation with buparvaquone, our DLS, ζ-potential, and SAXS analyses indicated that, although BPQ was not able to alter the inner structure of the membrane, it altered the net charge on the liposome surface, as well as its hydrodynamic radius. Such a change in size represented a reduction of ∼40% in the liposome hydrodynamic radius value. Taken together, these results may indicate that BPQ is located near the membrane surface in such a position that could favor a decrease in the liposome superficial charge. SAXS is an important tool for studying nanometric systems, such as liposomes or vesicles. It provides structural information regarding membrane thickness and the electron density profile (EDP). Our analysis demonstrated that in the absence of BPQ, the SAXS curves corresponded to unilamellar vesicles, with an absence of sharp peaks at large (or middle) q values (16, 17). Our data also demonstrated that the membrane thickness value was slightly larger than other values reported in the literature, such as 43 Å for the POPC phospholipid (18) or the 34 ± 4 Å for the cationic lipid DODAB (17). Note that the liposomes used here were composed of different phospholipids (PC and PS) rather than representing a single-lipid system. In association with drug delivery systems, drug repositioning has been an important tool for neglected parasitic diseases. From the old antimony salts to the newest oral drug miltefosine, all drugs used to clinically treat visceral leishmaniasis were introduced by the repositioning approach (19–21). Buparvaquone (BPQ), a veterinary drug used to treat bovine theileriosis, demonstrated antileishmanial activity; Croft et al. demonstrated not only a potent in vitro activity but also a limited efficacy to reduce the parasite load in an L. donovani-BALB/c mouse model (7). These authors demonstrated the low efficacy of free buparvaquone when administered at 100 mg/kg by the s.c. route (i.e., a visceral leishmaniasis murine model), which ranged from 35 to 62%, with a reduced efficacy (43%) when administered by perorally. BPQ failure was also observed in Leishmania-infected dogs treated with a drug regimen of 5 mg/kg for 12 days (22). BPQ at 20 mg/kg also failed against L. infantum-infected hamsters when administered by the intraperitoneal (i.p.) route (8). Several BPQ derivatives have been proposed to overcome the elevated lipophilicity (log P = 5.3) problem. This lipophilic feature of BPQ makes the drug an interesting candidate for drug delivery systems as liposomes.
In the present study, BPQ was entrapped in a liposomal formulation containing a 20% (molar ratio) of negatively charged phosphatidylserine, and the safety and efficacy of this administration regimen was investigated using a hamster model. Initially, L. infantum-infected hamsters were treated using three different routes (s.c., i.m., and i.v.), and the parasite load was evaluated via quantitative real-time PCR. The s.c. administration of BPQ-LP for 10 consecutive days was the most effective route for reducing the parasite load in both organs; compared to i.v. route, the s.c. resulted in greater efficiency (11 and 213% greater in the spleen and liver, respectively). Our data demonstrated that BPQ-LP was also effective when administered via the i.v. route, but to a lesser extent compared to s.c. route. This could be the result of a differential bioavailability of the liposomal buparvaquone when administered via the s.c. route.
The inoculation sites could strongly influence the pharmacokinetic (PK) properties of drugs, resulting in higher or reduced efficiencies. Dhanikula et al. (23) demonstrated that the PK parameters of a liposomal formulation entrapping the anticancer drug paclitaxel was highly altered when administered via s.c. and i.v. routes (23). The bioavailability was improved when the drug was administered via the s.c. route, with the plasma half-life (t1/2) varying from 3.4 h (i.v.) to 18 h (s.c.), with the area under the first moment curve being ∼3-fold higher by the s.c. route. These PK parameters could directly contribute to a higher bioavailability of the drug, improving the efficacy in the affected organs. A successful treatment using drug-loaded liposomes depends also on the route of administration (e.g., s.c., perorally, and i.v.). Conventional liposomes, administered s.c., aim to target the lymphatic system for the distribution of therapeutic agents or vaccination (24). Liposomes administered i.v. face barriers such as the endothelial lining of the vasculature (24). Allen et al. (25), studying the administration of liposomes via s.c., i.v., and i.p. routes, demonstrated that conventional liposomes (administered s.c.) appeared to be intact in the circulation after moving down the lymph node chains that drain the site of administration. These authors also demonstrated that, although the liposome levels were significantly higher in the draining lymph nodes after s.c. administration, the levels associated with other tissues were proportionately reduced relative to the i.v. and i.p. routes of administration. Although the half-lives in blood presented significant differences, the liposomes demonstrated similar biodistribution and PK profiles among the three selected routes. A similar work using free and chloroquine-entrapped liposomes demonstrated that liposomes administered s.c. acted as a depot for a slow release of the drug (26). Oussoren et al. also demonstrated that the inclusion of phosphatidylserine improved localization at the lymph nodes (27), which are important sites of Leishmania infection (28). Conversely, our study using the i.m. route demonstrated the failure of BPQ-LP to control infection in the spleen and liver. Differences between i.m. and s.c. administrations can be ascribed to differences in the structure of the tissue at the injection sites and to local physiological processes. Muscle tissue has a rich supply of capillaries. If diffusion through the blood capillary walls is the major pathway and the rate-limiting step in drug transport, i.m. absorption will be faster than s.c. absorption once the drug is released from the depot (29). In our assays, although BPQ-LP was not quantified in blood or tissues, the superior efficacy of BPQ-LP administered s.c. might be ascribed to a depot effect after administration, allowing a sustained release of the possible intact nanoparticles. Further determination of PK parameters may help to explain such differences.
The establishment of adequate drug regimens is indispensable for a successful therapy, avoiding excessive administration of the drugs and a consequently higher risk of toxicity. Our studies confirmed that 5 days of treatment was insufficient to reduce the numbers of parasites in L. infantum-infected hamsters. This result could be ascribed to the possible slow release of the drug when administered s.c., demanding a prolonged administration to accumulate in the spleen and liver. However, the administration of BPQ-LP for 15 days resulted in an efficacy similar to that for 10 days of treatment, which resulted in a >90% reduction in the parasite load. Since shorter periods of therapy are advantageous and cost-effective, resulting in higher patient compliance, all experiments were further performed within 10 consecutive days. In addition, the target product profiles for visceral leishmaniasis published by the Drugs for Neglected Disease initiative recommend a drug candidate if it demonstrates an efficacy that is >90% within 10 days of treatment (30).
The natural course of visceral leishmaniasis establishes an intense infection in the liver, spleen, and bone marrow, with parasite persistence mainly in the last two sites by mechanisms that are poorly understood (31). Targeting Leishmania parasites inside bone marrow is crucial in order to avoid disease recrudescence. Bone marrow is a primary lymphoid tissue and a major hematopoietic organ. The blood vessels between the hematopoietic compartment and the circulation form a barrier, referred to as the marrow-blood barrier. The movement of particles through fenestrations in the endothelial wall is size dependent, ranging from 85 to 150 nm (32). In our studies, the liposomal formulation was able to deliver the buparvaquone to bone marrow macrophages, resulting in the elimination of 80% of parasites after 10 days of treatment. The efficiency of BPQ-LP at this site may be useful for controlling the recurrence of the disease, and future experiments could demonstrate whether this efficacy in bone marrow is related to the targetability of PS-nanoliposomes. The size and inclusion of phosphatidylserine in liposomes has been described to be essential when one considers the bone marrow site. Studies examining rat bone marrow delivery demonstrated that increasing phosphatidylserine from 0 to 10% in liposomes enhanced the bone marrow accumulation (33, 34). In addition, Sou et al. demonstrated that an optimized formulation to target the bone marrow, with vesicles of ∼250 nm, localized in the endosomes and lysosomes of resident macrophages (35).
In order to investigate the ED50 of BPQ-LP, a drug regimen consisting of 10 days of treatment was performed with three different doses. Considering the mean ED50 in the spleen and liver, BPQ-LP demonstrated an efficacy similar to that of liposomal amphotericin B in the murine model. Currently, the only liposomal formulation in clinical use for treating visceral leishmaniasis is Ambisome (amphotericin B). Studies conducted by Yardley and Croft in a visceral model with L. donovani-infected mice, using a 5-day treatment regimen administered via the i.v. route, demonstrated an ED50 of 0.7 mg/kg (36). Based in our data, it was also possible to observe that even at higher doses of BPQ (0.6 mg/kg), no increase in the efficacy could be observed. Our previous studies with L. infantum-infected hamsters treated with the standard drug Glucantime (pentavalent antimony) also demonstrated no sterile cure, even at elevated doses of 50 mg/kg, as detected by qPCR (13). Mendonça et al. suggested that the clinical cure of leishmaniasis is rarely associated with a sterile cure in humans, as well as the persistence of parasites and relapses (37).
Drugs can exert an antileishmanial effect by a lethal action against the parasites inside the host cells, or through an indirect modulatory effect on host cells. Muniz-Junqueira et al. (38) demonstrated that meglumine antimoniate, a drug currently in clinical use to treat leishmaniasis, had no influence on the NO production of monocytes but significantly increased the production of TNF by 230%. Our results demonstrated for the first time that buparvaquone (or BPQ-LP) has an immunomodulatory effect in macrophages, the host cells for Leishmania. Our findings showed that BPQ (or BPQ-LP) elicited the production of TNF, MCP-1, IL-10, and (mainly) IL-6. We also observed that, similarly to meglumine antimoniate, there was no increase in NO levels after buparvaquone (or BPQ-LP) incubation with macrophages, but there was an elevated production of TNF. This effect might be ascribed to the elevated levels of the IL-10, an anti-inflammatory cytokine that catalyzes arginase activity over inducible nitric oxide synthase (iNOS), preventing the production of NO (39). Although exacerbated upregulation of IL-6 and IL-10 has been correlated with a worse prognosis for visceral leishmaniasis (40), the increased production of CCL2 (MCP-1) can accelerate granuloma maturation in the liver and reduce the parasite load during experimental visceral leishmaniasis (41, 42). In addition, studies using TNF receptor-deficient mice demonstrated that an increase in endogenous TNF-α contributed to macrophage activation and the enhancement of host antimicrobial defenses (43–45). Therefore, considering this balance of cytokine production, we suggest that the antiparasitic effect of buparvaquone correlate with the upregulation of MCP-1 and TNF in macrophages.
Buparvaquone has been extensively studied for the treatment of visceral leishmaniasis. We reported here that buparvaquone has an immunomodulatory effect in host cells and, when entrapped in PS-liposomes, the drug is highly effective at low doses at eliminating L. infantum parasites in a hamster model. The phosphatidylserine-liposomes used here proved to be a useful tool for delivering BPQ to Leishmania-infected macrophages in the liver and spleen, as well as the bone marrow, an important site of parasite persistence. Future evaluation of PK parameters is needed in order to assess the bioavailability of BPQ-LP.
MATERIALS AND METHODS
Drugs and chemicals.
3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (Thiazol Blue; MTT), sodium dodecyl sulfate (SDS), M-199 medium, RPMI 1640 medium without phenol red (RPMI-PR-1640), and cholesterol were purchased from Sigma-Aldrich. Hydrogenated phospholipids were kindly provided by Lipoid GmbH (Ludwigshafen, Germany). 2-[(4-tert-Butylcyclohexyl)methyl]-3-hydroxy-1,4-naphthoquinone (buparvaquone) and other analytical reagents were purchased from Sigma (St. Louis, MO). Molecular biology reagents are purchased from Life Technologies, and the cytometric bead array (CBA) assay was purchased from Becton Dickinson (BD).
Experimental animals.
Golden hamsters (Mesocricetus auratus) and BALB/c mice were obtained from the Instituto Adolfo Lutz of São Paulo State (São Paulo, Brazil), kept in sterile boxes with absorbent material, and received food and water ad libitum. Golden hamsters were infected each month with amastigotes from the spleen to maintain the Leishmania strain. BALB/c mice were used to obtain peritoneal macrophages. Animal experiments were performed with the approval of the Ethics Committee of the Instituto Adolfo Lutz (project CEUA-IAL/Pasteur 05/2011) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publications 8023).
Parasites and macrophages.
L. infantum (MHOM/BR/1972 strain 46LD) was obtained from a Brazilian patient presenting visceral leishmaniasis. This strain was identified by Fundação Oswaldo Cruz from Rio de Janeiro (Fiocruz, Brazil), using multilocus enzyme electrophoresis methods. This strain was used as a standard strain referenced in GenBank. L. infantum amastigotes were maintained by serial passage of amastigotes isolated from the spleens of previously infected hamsters. The animals were infected with 108 amastigotes (300 μl) by the i.p. route. At 60 days postinfection, the animals were euthanized, and the spleens were analyzed by light microscopy using Giemsa-stained smears. The parasite load was expressed in Leishman-Donovan units (i.e., the number of amastigotes per 1,000 nucleated cells × organ weight [in g] × 2 × 105) (46). Briefly, the spleen was macerated in a tissue grinder tube containing 5 ml of phosphate-buffered saline (PBS), and the amastigotes were separated by differential centrifugation to obtain a suspension of parasites. Promastigotes were maintained in M-199 medium supplemented with 10% fetal calf serum (FCS) and 0.25% hemin at 24°C. Macrophages were collected from the peritoneal cavities of BALB/c mice by washing with RPMI 1640 medium supplemented with 10% FCS and maintained in a 5% CO2-humidified incubator at 37°C (13).
BPQ entrapment in liposomes.
Encapsulation of BPQ was performed as described by Reimão et al. (8). Briefly, for the liposome preparation, BPQ (6 mg) was dissolved in methanol and sonicated in a bath sonicator for 10 min at 25°C (solution A). Solution B consisted of saturated egg phosphatidylcholine (180 mg), saturated egg phosphatidylserine (54.93 mg), and cholesterol (13.05 mg) at a 7:2:1 molar ratio, dissolved in 8.4 ml of chloroform-methanol (1:1). The total amount of lipid was 234.93 mg. The mixture of solutions A and B was further sonicated for 10 min. The mixture was evaporated in a rotary evaporator at 55°C for 40 min in a vacuum and protected from light. A preheated (55°C) isotonic solution of 2.25% (vol/vol) glycerol (6 ml) was added to the lipid film using glass beads. The swelling process of the preformed liposomes was performed in a rotary evaporator at 55°C for 60 min without vacuum. The liposomes were sonicated in a bath sonicator under heating (55°C) for 30 min, followed by a freeze-thaw process using three consecutive cycles of liquid nitrogen. The untrapped BPQ was separated from the liposomes by centrifugation (4,000 × g for 15 min).
Quantification of BPQ into liposomes.
The concentration of the encapsulated BPQ was determined using by UPLC with a binary AT system (Prominence LC-20; Shimadzu Corp., Kyoto, Japan) and a UV photodiode detector array (SPDM20A) on a reversed-phase ACE C18 column (4.6 mm by 250 mm, 5-μm particle size). The wavelength was set at 251 nm. The flow rate was 1 ml/min using an isocratic method with acetonitrile-methanol (7:3 [vol/vol]) and 0.1% trifluoroacetic acid. The BPQ was diluted in methanol (1 mg/ml) to obtain standard solutions in a range of 1.56 to 100 μg/ml, and 20-μl samples were injected into the column. The results obtained were extrapolated from a standard curve using a linear regression curve. The encapsulation efficiency (%EE) was calculated using the following equation (47):
| (1) |
The drug/lipid (D/L) ratio (48) was determined from the final amount of the drug (detected by UPLC) and the final amount of lipid, which was determined by the colorimetric method of Stewart (49).
DLS.
Dynamic light scattering (DLS) measurements were applied to obtain the vesicles mean diameter in the absence or presence of BPQ through the apparent diffusion coefficient measurement. The DLS was performed using ZetaSizer ZS90 equipment (Malvern, United Kingdom) with the detector positioned at 90° and an He-Ne laser (λ = 632.8 nm) as a light source. All measurements were performed at 22°C. Samples were diluted 20-fold to ensure there was no multiple scattering inside the cuvette during the experiments. The results showed a unimodal distribution and represent the average of at least four experiments.
The apparent hydrodynamic diameter values, which are related to the diffusional dynamics of a vesicle, were obtained with the time correlation functions and analyzed by the cumulative analysis software provided by Malvern. The Dh determination was calculated by means of a Stokes-Einstein calculation, as in the following equation:
| (2) |
where kB is the Boltzmann constant, T is the absolute temperature, η is the viscosity of the solvent, and Dh and Dapp are the hydrodynamic diameter and the apparent diffusion coefficient, respectively.
ζ-Potential.
ζ-Potential measurements of PS-containing vesicles in the absence or presence of BPQ were evaluated in order to check the effect of the drug on the vesicle effective surface charge. The ζ-potential was performed using ZetaSizer ZS90 equipment (Malvern) with the detector positioned at 17° and an He-Ne laser (λ = 632.8 nm) as a light source and applying PALS (phase analysis light scattering) and LDV (laser Doppler velocity) to calculate the electrophoretic mobility and the ζ-potential. The ζ-potential is a measure of the system stability, since it is able to give information about the net change at the vesicle surface. Actually, the ζ-potential is defined as the electrical potential in the beginning of the so-called double layer, i.e., the slipping plane of the vesicle surface. In the present study, the ζ-potential was obtained using the electrophoretic mobility, based on the Helmholtz-Smoluchowski equation (50), which is used in water-based systems in the presence of an ionic strength (I) of >1 mM and a particle diameter (Dh) of >100 nm. The measurement for each sample was repeated at least three times. In order to avoid multiple scattering, the samples were diluted 20-fold in the appropriate buffer before the measurements.
SAXS.
SAXS measurements were performed in the SAXS1 beam line in the National Synchrotron Light Laboratory (Campinas, São Paulo, Brazil). The X-ray wavelength used was λ = 0.1488 nm, and the sample-to-detector distance was ∼1,000 mm, providing a q-range of 0.1 < q < 4 nm−1, where q is the scattering vector, which is defined as q = (4π/λ)sin(θ), i.e., 2θ the scattering angle. A bidimensional PILATUS 300k detector was used. Samples were set between two flat mica walls with a 1-mm spacer and a thermal bath was used for temperature control, set to 22 ± 0.5°C. Scattering data were normalized for the acquisition time (100 s) and checked for radiation damage by measuring several consecutive frames. No effect of radiation damage was observed in the present study. The final scattering curves were subtracted from the aqueous solution and corrected by the sample attenuation.
SAXS data analysis for liposome-like structures can be theoretically evaluated considering that the liposome has a larger size than the X-ray wavelength (λ). Under this assumption, the scattering curve can be described by an infinite plane (in the x and y directions) with a nonhomogeneous cross section, along the z-axis, representing the membrane thickness (16, 17). The scattering intensity can be calculated as follows:
| (3) |
where k is a normalization constant related to the experimental setup, np is the lipid concentration, q is the scattering vector, and Pt(q) is the bilayer cross-section form factor. In the present study, Pt(q) was calculated based on a three-step model (16, 17). In this case, each leaflet let of the bilayer is composed of three distinct regions: the polar head group, the paraffinic chains, and the innermost methyl group or the CH3 region. Moreover, each region has its own electron density, as well as its own thickness: ρpol is the electron density for the polar head group, whereas ρCH2 and ρCH3 are the electron densities of the paraffinic and the CH3 regions, respectively. The thicknesses are designated Rpol, RCH2, and RCH3 for the polar head group, the paraffinic chains, and the CH3 region, respectively. The theoretical scattering profiles were fitted against the experimental SAXS curves using the GENFIT software (51). This software also allows the combination (sum) of different theoretical models in order to better describe the studied system. According to data from the literature (16, 17), the fitting parameters were set to vary in a specific range, i.e., RCH3 was set to vary within the “0.15 nm < RCH3 < 0.35 nm” range, ρCH3 was set to vary within the “150 nm−3 < ρCH3 < 200 nm−3” range, and ρpar was set to vary within the “250 nm−3 < ρpar < 330 nm−3” range. The remaining fitting parameters were allowed to vary in a broader range in order to ensure a reasonable description of the systems. As shown below, in order to fit the SAXS curve for the system in the presence of BPQ, two distinct fitting models were used. Thus, SAXS curves are discussed in terms of changes in the electron density profile function, ρ(z) (with z being the direction perpendicular to the membrane), in order to propose the site location for BPQ in the membrane.
Analysis of different routes of administration of BPQ-LP with L. infantum-infected hamsters.
Young male Golden hamsters (110 g) were infected i.p. with L. infantum amastigotes (108/animal, 300 μl). At 40 days postinfection, the hamsters were treated with BPQ entrapped in PS-liposomes (BPQ-LP) for 10 consecutive days. The animal groups (n = 5/group) were divided by treatment administration route—s.c., i.v., or i.m.—at 0.4 mg/kg/day (200 μl). The control group (n = 5/group) received vehicle (200 μl, 2.25% glycerol solution without drug). The animals were euthanized at 51 days postinfection. A tissue sample of the spleen and liver (∼200 mg) was removed, weighed, and used for RNA extraction and cDNA synthesis for analysis of the parasitic load (8). Euthanasia was performed in a procedure room away from other rodent housing, using an initial administration of ketamine (50 mg/kg) and xylazine (5 mg/kg) by the i.p. route. The animals were euthanized in a CO2 chamber, which was gradually filled at a rate of 20% of the chamber volume per min.
Treatment analysis.
The BPQ-LP treatment was analyzed by real-time PCR as previously reported (52, 53). All amplification runs with cDNA contained two negative controls (pure water and a negative DNA sample collected from a healthy hamster) and one positive control (DNA sample extracted from L. infantum).
DNA extractions.
Promastigotes of L. infantum were used as positive controls in the log curve phase; 108 parasites were harvested, washed twice in PBS (pH 7.2), and centrifuged at 1,000 × g for 10 min. Different concentrations of promastigotes were used to obtain the points of the standard curve ranging from 108 to 102 forms. The parasite pellet was dissolved in a 10 mM Tris-HCl (pH 8.0) solution containing 10 mM EDTA, 0.5% SDS, 0.01% N-laurylsarcosine, and proteinase K at 100 μg/ml, followed by incubation at 56°C until complete cell lysis. DNA was extracted by using a PureLink genomic DNA kit (Invitrogen) according to the manufacturer's instructions.
RNA extraction and cDNA synthesis.
Hamster RNA samples were obtained from spleen, liver, and bone marrow samples. RNA molecules were extracted and purified by using a Spin Tissue RNA minikit (Invisorb) according to the manufacturer's instructions. After RNA extraction, the samples were immediately processed for determining the cDNA. DNA, and RNA purities in a NanoDrop ND100 (Thermo Scientific). Each real-time PCR was performed according to the manufacturer's instructions (Invitrogen) using a Verity 96-well thermal cycler (Thermo-Fisher Scientific, USA). The RNA samples (11 μl) were mixed with 1 mM deoxynucleoside triphosphate, 3 μg of random primers, 10 mM dithiothreitol, and 1 μl of Moloney murine leukemia virus reverse transcriptase in a buffer (250 mM Tris-HCl [pH 8.3], 375 mM KCl, 15 mM MgCl2) with a final volume of 20 μl. The cDNA samples were stored at −20°C.
The genetic material (DNA and cDNA) was first tested with marker 150-152 for the Leishmania genus as a screening test that amplifies a 120-bp fragment of the conserved-region kDNA minicircles. Identification of L. infantum was performed using the marker RV1-RV2, which amplifies a 145-pb fragment of a variable region of the kDNA minicircle (region LT1) (52).
Primers.
The following primers, probe, and quencher were designed using the OligoPrimer analysis software (v7): (i) forward primer (5′-CCGCGTGCCTGTCG-3′), 18 μM; (ii) reverse primer (5′-CCCACACAAGCGGGAACT-3′), 18 μM; (iii) TaqMan FAM-dye-labeled probe (5′-CCTCCTTGGACTTTGC 3), 5 μM; and reporter quencher NFQ. Amplification runs contained two negative controls and one positive control. The thermal profile was performed at 50°C for 2 min for optimal AmpliErase UNG activity and 95°C for 10 min. Then, 40 cycles of 95°C for 15 s and 60°C for 1 min were performed. This analysis was performed using the StepOne real-time PCR system (Thermo-Fisher Scientific) (52, 53).
Analysis of different administration regimens.
After selecting the most effective route of administration (s.c.) for BPQ-LP, the effectiveness of the treatment was evaluated for different for periods (5, 10, and 15 consecutive days) at 0.4 mg/kg/day (200 μl) by real-time PCR as described above.
Efficacy of BPQ-LP treatment in the bone marrow.
After 10 days of BPQ-LP treatment at 0.4 mg/kg/day (group 1, n = 5), the parasite load was evaluated in the bone marrow of the hamsters. The control group (n = 5) received vehicle (200 μl, 2.25% glycerol solution without drug). The material was collected from femurs and tibias, and RNA extraction was performed for the qPCR analysis (52, 53). Animals were euthanized as described above, and the animal surfaces were cleaned with 70% ethanol. Skin from the distal part of the mouse was removed; the muscles from the lower extremities were cut off using scissors, and the acetabulum was carefully dislocated from the hip joint. The remaining muscles from the femur and tibia were removed using a scalpel and scissors, and the femur was separated from the tibia at the knee joint. The bones were placed in a petri dish containing ice-cold RPMI 1640 supplemented with 10% fetal bovine serum and gentamicin (20 μg/ml). A 25-gauge needle and a 12-ml syringe filled with PBS and 2 mM EDTA was used to flush the bone marrow cells from the bone. The cells were removed by scraping the inner surfaces of the bones (54), and RNA extraction was performed for the qPCR analysis (52, 53).
Determination of the ED50 of BPQ-LP in L. infantum-infected hamsters.
Young male Golden hamsters previously infected were treated s.c. for 10 consecutive days as follows: group 1, BPQ-LP at 0.6 mg/kg/day; group 2, BPQ-LP at 0.3 mg/kg/day; group 3, BPQ-LP at 0.1 mg/kg/day; and group 4, untreated group (vehicle 2.25% glycerol solution, 200 μl). Each group consisted of five animals. The animals were euthanized at 51 days postinfection, and the parasitic loads were analyzed as described previously by real-time PCR.
Immunomodulatory studies in macrophages.
Peritoneal macrophages were seeded into 24-well plates at 105 cells/ml in RPMI medium supplemented with 10% calf serum and incubated overnight at 37°C in a 5% CO2 humidified incubator. After being washed with PBS, the cells were infected (overnight) with L. infantum amastigotes (10:1 amastigote/macrophage ratio). After 10 h, the cells were washed and treated with BPQ and BPQ-LP (10 μM) for 48 h. The culture supernatants were analyzed at 48 h posttreatment to determine the cytokine levels. The concentrations of the released mediators—IL-6, IL-10, MCP-1, and TNF—were determined by using an inflammatory CBA assay (mouse inflammation kit; BD Biosciences, San Jose, CA) in accordance with the manufacturer's protocols.
Quantification of NO.
Macrophages were collected as described above and treated with BPQ and BPQ-LP for 72 h at 10 μM. The nitric oxide (NO) content was analyzed by using a Griess assay (55) in the supernatants of macrophages. The results obtained were extrapolated from a standard curve prepared with NaNO2 in concentrations ranging from 1 to 400 μM.
Statistical analysis.
The data obtained were reported as the mean and standard deviation of duplicate samples from two or three independent assays. The data were analyzed using GraphPad Prism 5.0 software by a t test analysis for significance (P < 0.05).
ACKNOWLEDGMENTS
We thank CNPq for a scientific research award given to A.G.T. and L.R.S.B. This work was supported by GlaxoSmithKline and by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) grants 2015/23403-9, 2015/15822-1, 2013/07275-5, and 2013/50228-8.
We thank Paolo Mariani and Francesco Spinozzi, who provided GENFIT software, and Albane Kessler and Silvia Gonzalez-Del Valle (GlaxoSmithKline, Tres Cantos, Spain) for data evaluation. Finally, we thank Nahiara E. Zorgi for flow cytometry data analysis.
REFERENCES
- 1.World Health Organization. 2016. Leishmaniasis fact sheet. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs375/en/. [Google Scholar]
- 2.Kedzierski L, Zhu Y, Handman E. 2006. Leishmania vaccines: progress and problems. Parasitology 133:S87–S112. doi: 10.1017/S0031182006001831. [DOI] [PubMed] [Google Scholar]
- 3.Tiuman TS, Santos AO, Ueda-Nakamura T, Filho BP, Nakamura CV. 2011. Recent advances in leishmaniasis treatment. Int J Infect Dis 15:e525–e532. doi: 10.1016/j.ijid.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 4.McGwire BS, Satoskar AR. 2014. Leishmaniasis: clinical syndromes and treatment. QJM 107:7–14. doi: 10.1093/qjmed/hct116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres DC, Adaui V, Ribeiro-Alves M, Romero GA, Arévalo J, Cupolillo E, Dujardin JC. 2010. Targeted gene expression profiling in Leishmania braziliensis and Leishmania guyanensis parasites isolated from Brazilian patients with different antimonial treatment outcomes. Infect Genet Evol 10:727–733. doi: 10.1016/j.meegid.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Ekins S, Williams AJ, Krasowski MD, Freundlich JS. 2011. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov Today 16:298–310. doi: 10.1016/j.drudis.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 7.Croft SL, Hogg J, Gutteridge WE, Hudson AT, Randall AW. 1992. The activity of hydroxynaphthoquinones against Leishmania donovani. J Antimicrob Chemother 30:827–832. doi: 10.1093/jac/30.6.827. [DOI] [PubMed] [Google Scholar]
- 8.Reimão JQ, Colombo FA, Pereira-Chioccola VL, Tempone AG. 2012. Effectiveness of liposomal buparvaquone in an experimental hamster model of Leishmania infantum chagasi. Exp Parasitol 130:195–199. doi: 10.1016/j.exppara.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Burza S, Sinha PK, Mahajan R, Lima MA, Mitra G, Verma N, Balasegaram M, Das P. 2014. Five-year field results and long-term effectiveness of 20 mg/kg liposomal amphotericin B (Ambisome) for visceral leishmaniasis in Bihar, India. PLoS Negl Trop Dis 8:e2603. doi: 10.1371/journal.pntd.0002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang HI, Yeh MK. 2012. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine 7:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi A, Yamamoto N, Akamatsu N, Saido TC, Kaneda M, Umeda M, Tanoue K. 2000. PS-liposome and ox-LDL bind to different sites of the immunodominant domain (155-183) of CD36: a study with GS95, a new anti-CD36 monoclonal antibody. Thromb Res 97:317–326. doi: 10.1016/S0049-3848(99)00179-6. [DOI] [PubMed] [Google Scholar]
- 12.Tempone AG, Perez D, Rath S, Vilarinho AL, Mortara RA, de Andrade HF. 2004. Targeting Leishmania chagasi amastigotes through macrophage scavenger receptors: the use of drugs entrapped in liposomes containing phosphatidylserine. J Antimicrob Chemother 54:60–68. doi: 10.1093/jac/dkh281. [DOI] [PubMed] [Google Scholar]
- 13.Tempone AG, Mortara RA, de Andrade HF Jr, Reimão JQ. 2010. Therapeutic evaluation of free and liposome-loaded furazolidone in experimental visceral leishmaniasis. Int J Antimicrob Agents 36:159–163. doi: 10.1016/j.ijantimicag.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Binker MG, Zhao DY, Pang JY, Harrison RE. 2007. Cytoplasmic linker protein-170 enhances spreading and phagocytosis in activated macrophages by stabilizing microtubules. J Immunol 179:3780–3791. doi: 10.4049/jimmunol.179.6.3780. [DOI] [PubMed] [Google Scholar]
- 15.Kolodziej H, Kiderlen AF. 2005. Antileishmanial activity and immune modulatory effects of tannins and related compounds on Leishmania parasitized RAW 264.7 cells. Phytochemistry 66:2056–2071. doi: 10.1016/j.phytochem.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez RM, Riske KA, Amaral LQ, Itri R, Teresa Lamy M. 2008. Influence of salt on the structure of DMPG studied by SAXS and optical microscopy. Biochim Biophys Acta 1778:907–916. doi: 10.1016/j.bbamem.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Rozenfeld J, Duarte EL, Barbosa LRS, Lamy MT. 2015. Effect of oligonucleotide on the structure of cationic DODAB vesicles. Phys Chem Physics 17:7498–7506. doi: 10.1039/C4CP05652C. [DOI] [PubMed] [Google Scholar]
- 18.Domingues MM, Bianconi ML, Barbosa LRS, Santiago PS, Tabak M, Castanho MA, Itri R, Santos NC. 2013. rBPI21 interacts with negative membranes endothermically promoting the formation of rigid multilamellar structures. Biochim Biophys Acta 11:2419–2427. doi: 10.1016/j.bbamem.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Sanderson L, Yardley V, Croft SL. 2014. Activity of anti-cancer protein kinase inhibitors against Leishmania spp. J Antimicrob Chemother 69:1888–1891. doi: 10.1093/jac/dku069. [DOI] [PubMed] [Google Scholar]
- 20.Croft SL, Snowdon D, Yardley V. 1996. The activities of four anticancer alkyllysophospholipids against Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei. J Antimicrob Chemother 38:1041–1047. doi: 10.1093/jac/38.6.1041. [DOI] [PubMed] [Google Scholar]
- 21.Lamy L, Wonde T, Lamy H. 1966. Action of amphotericin B on Leishmania donovani during multiplication in mouse macrophages maintained in vitro. Bull Soc Pathol Exot Filiales 59:964–968. [PubMed] [Google Scholar]
- 22.Vexenat JA, Croft SL, Furtado-Campos JH, Miles MA. 1998. Failure of buparvaquone (Butalex) in the treatment of canine visceral leishmaniasis. Vet Parasitol 77:71–73. doi: 10.1016/S0304-4017(96)01150-8. [DOI] [PubMed] [Google Scholar]
- 23.Dhanikula AB, Singh DR, Panchagnula R. 2005. In vivo pharmacokinetic and tissue distribution studies in mice of alternative formulations for local and systemic delivery of Paclitaxel: gel, film, prodrug, liposomes, and micelles. Curr Drug Deliv 2:35–44. doi: 10.2174/1567201052772852. [DOI] [PubMed] [Google Scholar]
- 24.Monteiro N, Martins A, Reis RL, Neves NM. 2014. Liposomes in tissue engineering and regenerative medicine. J R Soc Interface 6:20140459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen TM, Hansen CB, Guo LS. 1993. Subcutaneous administration of liposomes: a comparison with the intravenous and intraperitoneal routes of injection. Biochim Biophys Acta 25:9–16. [DOI] [PubMed] [Google Scholar]
- 26.Titulaer HA, Eling WM, Crommelin DJ, Peeters PA, Zuidema J. 1990. The parenteral controlled release of liposome encapsulated chloroquine in mice. J Pharm Pharmacol 42:529–532. doi: 10.1111/j.2042-7158.1990.tb07052.x. [DOI] [PubMed] [Google Scholar]
- 27.Oussoren C, Zuidema J, Crommelin DJ, Storm G. 1997. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid composition, and lipid dose. Biochim Biophys Acta 1328:261–272. [DOI] [PubMed] [Google Scholar]
- 28.de Mello CX, Figueiredo FB, Mendes AA, Miranda LF, de Oliveira RV, Madeira MF. 2016. Thick smear is a good substitute for the thin smear in parasitological confirmation of canine visceral leishmaniasis. Am J Trop Med Hyg 99:99–103. doi: 10.4269/ajtmh.15-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Yang L, Cheng G, Wei HY, Zeng Q. 2011. Encapsulation, pharmacokinetics, and tissue distribution of interferon α-2b liposomes after intramuscular injection to rats. Arch Pharm Res 34:941–948. doi: 10.1007/s12272-011-0611-4. [DOI] [PubMed] [Google Scholar]
- 30.Katsuno K, Burrows JN, Duncan K, Hooft van Huijsduijnen R, Kaneko T, Kita K, Mowbray CE, Schmatz D, Warner P, Slingsby BT. 2015. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat Rev Drug Discov 14:751–758. doi: 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- 31.Kaye PM, Svensson M, Ato M, Maroof A, Polley R, Stager S, Zubairi S, Engwerda CR. 2004. The immunopathology of experimental visceral leishmaniasis. Immunol Rev 201:239–253. doi: 10.1111/j.0105-2896.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- 32.Tardi P, Wan CP, Mayer L. 2016. Passive and semi-active targeting of bone marrow and leukemia cells using anionic low cholesterol liposomes. J Drug Target 16:1–11. doi: 10.2174/1871527316666161205123123. [DOI] [PubMed] [Google Scholar]
- 33.Boerman OC, Oyen WJ, van Bloois L, Koenders EB, van der Meer JW, Corstens FH, Storm G. 1997. Optimization of technetium-99m-labeled PEG liposomes to image focal infection: effects of particle size and circulation time. J Nucl Med 38:489–493. [PubMed] [Google Scholar]
- 34.Schettini DA, Ribeiro RR, Demicheli C, Rocha OG, Melo MN, Michalick MS, Frézard F. 2006. Improved targeting of antimony to the bone marrow of dogs using liposomes of reduced size. Int J Pharm 315:140–147. doi: 10.1016/j.ijpharm.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 35.Sou K, Goins B, Takeoka S, Tsuchida E, Phillips WT. 2007. Selective uptake of surface-modified phospholipid vesicles by bone marrow macrophages in vivo. Biomaterials 28:2655–2666. doi: 10.1016/j.biomaterials.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 36.Yardley V, Croft SL. 2000. A comparison of the activities of three amphotericin B lipid formulations against experimental visceral and cutaneous leishmaniasis. Int J Antimicrob Agents 13:243–248. doi: 10.1016/S0924-8579(99)00133-8. [DOI] [PubMed] [Google Scholar]
- 37.Mendonça MG, de Brito ME, Rodrigues EH, Bandeira V, Jardim ML, Abath FG. 2004. Persistence of Leishmania parasites in scars after clinical cure of American cutaneous leishmaniasis: is there a sterile cure? J Infect Dis 189:1018–1023. doi: 10.1086/382135. [DOI] [PubMed] [Google Scholar]
- 38.Muniz-Junqueira MI, de Paula-Coelho VN. 2008. Meglumine antimonate directly increases phagocytosis, superoxide anion and TNF-alpha production, but only via TNF-alpha it indirectly increases nitric oxide production by phagocytes of healthy individuals, in vitro. Int Immuno Pharmacol 8:1633–1638. doi: 10.1016/j.intimp.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 39.Tuladhar R, Natarajan G, Satoskar AR. 2011. Role of costimulation in leishmaniasis. Int J Biol Sci 7:1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maia C, Campino L. 2012. Cytokine and phenotypic cell profiles of Leishmania infantum infection in the dog. J Trop Med 2012:541571. doi: 10.1155/2012/541571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cotterell SE, Engwerda CR, Kaye PM. 1999. Leishmania donovani infection initiates T-cell-independent chemokine responses, which are subsequently amplified in a T-cell-dependent manner. Eur J Immunol 29:203–224. doi:. [DOI] [PubMed] [Google Scholar]
- 42.Dey R, Majumder N, Bhattacharyya Majumdar S, Bhattacharjee S, Banerjee S, Roy S, Majumdar S. 2007. Induction of host protective Th1 immune response by chemokines in Leishmania donovani-infected BALB/c mice. Scand J Immunol 66:671–683. doi: 10.1111/j.1365-3083.2007.02025.x. [DOI] [PubMed] [Google Scholar]
- 43.Nashleanas M, Kanaly S, Scott P. 1998. Control of Leishmania major infection in mice lacking TNF receptors. J Immunol 160:5506–5513. [PubMed] [Google Scholar]
- 44.Rothe J, Lesslauer W, Lötscher H, Lang Y, Koebel P, Köntgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. 1993. Mice lacking the tumor necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 45.Yap GS, Scharton-Kersten T, Charest H, Sher A. 1998. Decreased resistance of TNF receptor p55- and p75-deficient mice to chronic toxoplasmosis despite normal activation of inducible nitric oxide synthase in vivo. J Immunol 160:1340–1345. [PubMed] [Google Scholar]
- 46.Bradley DJ, Kirkley J. 1977. Regulation of Leishmania populations within the host. I. The variable course of Leishmania donovani infections in mice. Clin Exp Immunol 30:119–129. [PMC free article] [PubMed] [Google Scholar]
- 47.Ong SG, Ming LC, Lee KS, Yuen KH. 2016. Influence of the encapsulation efficiency and size of liposome on the oral bioavailability of griseofulvin-loaded liposomes. Pharmaceutics 26:pii: E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnston MJ, Edwards K, Karlsson G, Cullis PR. 2008. Influence of drug-to-lipid ratio on drug release properties and liposome integrity in liposomal doxorubicin formulations. J Liposome Res 18:145–157. doi: 10.1080/08982100802129372. [DOI] [PubMed] [Google Scholar]
- 49.Stewart JCM. 1980. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem 104:10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 50.Park HM, Lee WM. 2008. Helmholtz-Smoluchowski velocity for viscoelastic electro-osmotic flows. J Colloid Interface Sci 317:631–636. doi: 10.1016/j.jcis.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 51.Spinozzi F, Ferrero C, Ortore MG, De Maria Antolinos A, Mariani P. 2014. GENFIT: software for the analysis of small-angle X-ray and neutron scattering data of macro-molecules in solution. J Appl Crystallogr 47:1132–1139. doi: 10.1107/S1600576714005147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colombo FA, Odorizzi RM, Laurenti MD, Galati EA, Canavez F, Pereira-Chioccola VL. 2011. Detection of Leishmania infantum RNA in fleas and ticks collected from naturally infected dogs. Parasitol Res 109:267–274. doi: 10.1007/s00436-010-2247-6. [DOI] [PubMed] [Google Scholar]
- 53.Reimão JQ, Colombo FA, Pereira-Chioccola VL, Tempone AG. 2011. In vitro and experimental therapeutic studies of the calcium channel blocker bepridil: detection of viable Leishmania chagasi by real-time PCR. Exp Parasitol 128:111–115. doi: 10.1016/j.exppara.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 54.Swamydas M, Lionakis MS. 2013. Isolation, purification, and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp 10:e50586. doi: 10.3791/50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lezama-Dávila CM, Isaac-Márquez AP, Barbi J, Cummings HE, Lu B, Satoskar AR. 2008. Role of phosphatidylinositol-3-kinase-gamma (PI3Kγ)-mediated pathway in 17β-estradiol-induced killing of Leishmania mexicana in macrophages from C57BL/6 mice. Immunol Cell Biol 86:539–543. doi: 10.1038/icb.2008.39. [DOI] [PubMed] [Google Scholar]