

ABSTRACT
Artemisinin-based combination therapies are a key pillar in global malaria control and are recommended as a first-line Plasmodium falciparum treatment. They rely upon a rapid 4-log-unit reduction in parasitemia by artemisinin compounds with a short half-life and the killing of remaining parasites by a partner compound with a longer half-life. Current treatment guidelines stipulate giving three 24-h-interval doses or six 12-h-interval doses over a 3-day period. Due to the short half-life of artesunate and artemether, almost all of the resulting cytocidal activity is confined within a single 48-h asexual P. falciparum cycle. Here, we utilized a luciferase reporter, Plasmodium berghei ANKA, in a cytocidal model in which treatment was initiated at high parasitemia, allowing us to monitor a greater than 3-log-unit reduction in parasite density, as well as 30-day survival. In this study, we demonstrated that increasing the artesunate duration from spanning one asexual cycle to spanning three asexual cycles while keeping the total dose constant results in enhanced cytocidal activity. Single daily artesunate doses at 50 mg/kg of body weight over 7 days were the minimum necessary for curative monotherapy. In combination with a single sub-human-equivalent dose of the partner drug amodiaquine or piperaquine, the three-asexual-cycle artesunate duration was able to cure 75% and 100% of mice, respectively, whereas 0% and 33% cures were achieved with the single-asexual-cycle artesunate duration. In summary, cytocidal activity of the artemisinin compounds, such as artesunate, can be improved solely by altering the dosing duration.
KEYWORDS: antimalarial agents, artemisinin, pharmacodynamics
INTRODUCTION
Infection with the malaria parasite resulted in approximately 438,000 deaths in 2015, with 99% of the cases attributable to the most lethal species, Plasmodium falciparum (1). As there is no available vaccine, antimalarial drugs remain the primary defense against this deadly disease. Current guidelines from the World Health Organization recommend the use of artemisinin-based combination therapy (ACT) for cases of uncomplicated malaria caused by P. falciparum (2). Naturally occurring artemisinin is not used clinically; rather, it has been replaced by its semisynthetic derivatives artesunate and artemether or by the metabolite of the semisynthetic derivatives, dihydroartemisinin (DHA) (3). Dihydroartemisinin is activated by iron and or heme to cause radical damage to surrounding proteins, resulting in reduction of the total parasite burden more rapidly than other antimalarials (4). However, both dihydroartemisinin and the parent drugs have a short plasma half-life of less than a few hours, necessitating a partner drug with a longer plasma half-life to clear residual parasites (5, 6). Over the past decade, the adoption of ACT as a first-line treatment has been integral in a promising 30% decrease in malaria-associated mortality rates (1).
Malaria control efforts now face new challenges with the increasing spread of P. falciparum isolates with a delayed clearance phenotype throughout the greater Mekong subregion of Southeast Asia (7, 8). This phenotype is genetically associated with mutations in the P. falciparum Kelch-13 gene. Delayed clearance is defined as the presence of parasites in the blood 72 h following initiation of chemotherapy, which corresponds to a parasite clearance half-life of greater than 5 h (8). Importantly, there is no correlation between the parasite clearance half-life and an increased standard 72-hour constant drug 50% inhibitory concentration (IC50) of artesunate or artemether (9–11). However, a ring stage survival assay, which involves exposing early ring stage parasites to short, 6-h drug pulses of pharmacologically relevant concentrations of DHA, can discriminate between the tolerant and sensitive parasites (12). Early ring stages of P. falciparum are less metabolically active and therefore less susceptible to drugs than are the more metabolically active trophozoites and schizonts (13, 14). P. falciparum isolates with the Kelch-13 mutations maintain a 48-hour asexual erythrocytic cycle but have a prolonged ring stage and compensatory shorter trophozoite stage (15). Changes in antimalarial drug sensitivity in accordance with the stage of the parasite life cycle underscore the need to understand the effect of artemisinin compound treatment duration on the parasitological outcome (16). The consequences of decreasing efficacy of ACT are 2-fold: (i) individuals fail to clear the infection within a standard course of treatment, resulting in significant human and economic costs, and (ii) increased selective pressures are placed on the partner drug, thereby compromising the future efficacy of the quinolines or other partner compounds (17).
Considerations for antimicrobial treatment regimens include the amount of drug administered (dose), the frequency of dosing (schedule), and the length of treatment (duration). Current ACT regimens in Southeast Asia involve 3 doses of 4 mg/kg of body weight artesunate given at the time of the clinic visit (day 1) and the following 2 days (9). Although this treatment regimen is designed to target the parasite over 3 days and two life cycles, this is often not the case (18). Most patients are diagnosed between 2 and 5 p.m. (19). The second and third doses of artemisinin are typically consumed the next morning (16 h) and the subsequent morning (40 h). The artemisinin derivatives artesunate and artemether and their metabolite dihydroartemisinin have elimination half-lives (t1/2) of approximately 0.5, 2, and 0.5 h, respectively (20, 21). Due to the short plasma half-lives of artesunate, artemether, and dihydroartemisinin, patients clear almost all of the drug within a single 48-hour life cycle of P. falciparum, as the drug is found at subnanomolar concentrations 12 h after the last dose (22).
We hypothesize that spacing three identical artesunate doses over three separate asexual cycles, rather than confining them to a single asexual cycle, results in a greater log reduction in Plasmodium berghei parasites, making the difference between cure and no cure when combined with partner compounds. The P. berghei model has previously been utilized to characterize single-dose pharmacokinetics and pharmacodynamics (PK/PD) of dihydroartemisinin and piperaquine (23–25). Additionally, a 14-day duration of 100 mg/kg artesunate was found to be curative in C57BL/6 mice in a suppressive test, whereas a 14-day duration of 10 mg/kg or a 7-day duration of 100 mg/kg was not curative (26). Also, in a Plasmodium vinckei suppression test in C57BL/6 mice, 4 consecutive daily doses of artesunate were curative when given intraperitoneally at 30 mg/kg or orally at 80 mg/kg (27).
To investigate the optimization of antimalarial dosing regimens, we adapted a transgenic luciferase reporter, P. berghei, to study cytocidal drug activity in a murine model in which drug treatment is initiated at a high parasitemia (∼10%) and daily parasitemia and survival are monitored for at least 30 days. The transgenic parasites have previously been used to study parasite sequestration and parasite inhibition in vivo (28, 29). Our parasiticidal model more accurately mimics infections and treatment outcomes in the field and is superior to currently used in vitro drug suppression assays, which often investigate parasite inhibition at a low parasitemia compared to untreated controls. In the present study, we compared the efficacy of a single-life-cycle duration of artesunate treatment with that of a triple-life-cycle duration of artesunate treatment in our cytocidal P. berghei model. We also assessed the cytocidal activity of the two durations in combination with the 4-aminoquinolines piperaquine and amodiaquine.
RESULTS
Luciferase assay and analysis.
Based on the standard curve generated from the dilution series, we identified the limit of quantification of the luciferase assay at 52,000 photons/s, corresponding to 300 parasites per well or 600 parasites per μl (Fig. 1). A limit of detection at 1,000 photons per second corresponds to 0.1 parasites per ml using the equation derived in Fig. 1. We were able to compare blood films to luciferase robustly above 600 parasites per μl, which, with approximately 10 million mouse erythrocytes per μl, is 0.006% parasitemia. We graphed the radiant flux, transformed to parasites per µl, between the limit of detection and the limit of quantification.
FIG 1.
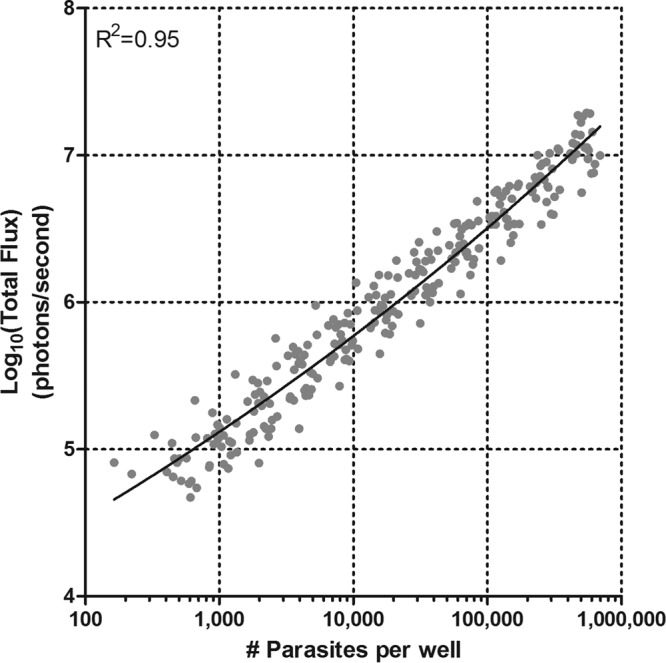
Relationship between parasite density and total flux. A dilution series was performed on blood samples from 24 mice ranging from 6 to 12% parasitemia. The data were fitted with a nonlinear regression of a log-log line (R2 = 0.95), giving the following equation: x = 10(log y − 0.55)/0.05, where y = log total flux. From this equation, the number of parasites in a given well can be quantitatively estimated within a range of 300 to 600,000 parasites/well.
Increased duration of artesunate treatment.
In order to determine the effect of an increased artesunate treatment duration on the parasitological outcome, we evaluated the cytocidal activities of two different regimens of artesunate. We varied the dosing interval of artesunate but held several variables constant: starting parasitemia (∼10%), total dose of artesunate, and route of administration. As the three 50-mg/kg doses of artesunate are noncurative by themselves, recrudescence can be used as a surrogate measure of total parasite killing. That is, a longer delay in parasite recrudescence indicates greater overall cytocidal activity. The mice that received the single-asexual-cycle duration of artesunate treatment (0, 8, and 20 h) all had a quantifiable recrudescent infection by day 4, while those that received the triple-asexual-cycle duration of artesunate treatment (0, 24, and 48 h) had a quantifiable recrudescent infection by day 6 or 7 (Fig. 2). An observed 4-log-unit kill was seen over 48 h (2-log-unit kill in the single 24-h life cycle) for both regimens, with similar rates of initial parasite reduction. There was a difference of 3 days in return to initial parasitemia. Two-way analysis of variance (ANOVA) was performed to determine any statistically significant differences in time to return of initial parasitemia, and in all four trials, the time to return of initial parasitemia was significantly less in the mice receiving all of the artesunate within a 20-h period.
FIG 2.

Effect of increased artesunate (AS) duration on P. berghei ANKA infection. (A to D) Three doses of 50 mg/kg of artesunate were administered at either 0, 8, and 20 h or 0, 24, and 48 h to female BALB/c mice. Time zero corresponds to a parasitemia of ∼10%, as determined by the percentage of infected erythrocytes in Giemsa-stained blood films. Four independent trials are represented as means ± standard errors of the mean (SEM) and were analyzed using two-way ANOVA. Each graph is annotated with the initial parasitemia (Pi) and the luciferase assay limit of quantitation (LQ). (E) Time of recrudescence data summarized for all four trials and analyzed using an unpaired t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Mean and standard error of the mean are shown.
Real-time PCR quantification of infection.
In order to demonstrate in another manner the enhanced cytocidal activity of the increased artesunate interval observed in the luciferase assay, we performed real-time PCR concurrently with the luciferase assay trial depicted in Fig. 2A. At the same time blood was collected for the luciferase assay, 50 μl of blood was collected for PCR. The PCR results confirmed what was observed in the luciferase assay, as recrudescence occurred 2 to 3 days earlier in the single-asexual-cycle duration group relative to the extended-duration group (Fig. 3).
FIG 3.
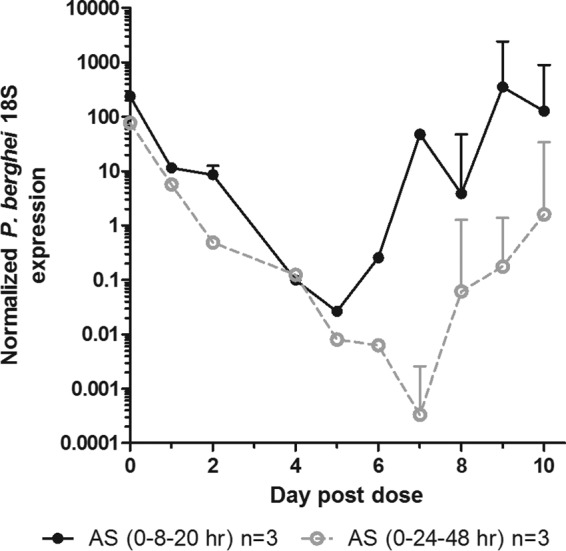
qPCR verification of the effect of increased artesunate duration on P. berghei ANKA infection. At the time of blood sampling for the luciferase assay, DNA was extracted and infection was quantified using TaqMan primers and probes for P. berghei ANKA 18S and normalized to M. musculus β-actin. The data are represented as the means and SEM for the normalized P. berghei 18S expression.
Increasing daily doses of 50 mg/kg artesunate.
Additionally, we wanted to identify in this model a curative duration of human equivalent artesunate when dosed daily. Administering artesunate for 4, 5, or 6 days resulted in 66%, 50%, and 33% recrudescence and return to initial parasitemia by days 13, 11 to 13, and 14, respectively (Table 1). Administering artesunate for 7, 9, 11, or 13 days all resulted in 0% recrudescent infections, returning to the level of initial parasitemia by 30 days. With approximately 109 initial parasites in the 2 ml of blood in a single mouse, we had slightly less than a 2-log-unit kill every 24 h in this model. A single mouse in the 7-day-duration group did experience transient recrudescent parasitemia after day 30 but later cleared the infection. Upon transfer of the infected blood to a naive mouse at day 45, the parasitemia multiplied at a log unit each 24 h, the normal rate, and was fully susceptible to treatment with artesunate (data not shown). We then further explored the impact of duration of drug treatment versus total dose of drug administered. We summed the total dose of drug received over 7 days (350 mg/kg × 0.02 kg = 7 mg) and administered this in a single amount. Although the same total amount of drug was given, recrudescence occurred in all 3 mice, and return to initial parasitemia occurred by day 10 or 11.
TABLE 1.
Identifying a curative, cytocidal dose of artesunate on P. berghei ANKA infectiona
| Duration of artesunate treatment (days) | Total dose of artesunate (mg/kg) | Proportion of mice with recrudescent parasitemia | Day of return to initial parasitemia |
|---|---|---|---|
| 3 | 150 | 5/5 | 9, 10, 11, 11, 6 |
| 4 | 200 | 2/3 | 13, 13 |
| 5 | 250 | 3/6 | 13, 13, 11 |
| 6 | 300 | 1/3 | 14 |
| 7 | 350 | 0/3 | NA |
| 1 | 350 | 3/3 | 10, 11, 10 |
| 9 | 450 | 0/3 | NA |
| 11 | 550 | 0/3 | NA |
| 13 | 650 | 0/3 | NA |
Artesunate (50 mg/kg) was administered at 24-hour intervals to female BALB/c mice for durations of 3, 4, 5, 6, 7, 9, 11, or 13 consecutive days; 350 mg/kg of artesunate was administered once to one group of mice. The starting parasitemia was ∼10%, as determined by the percentage of infected erythrocytes in Giemsa-stained blood films.
Increased duration of artesunate treatment in combination with 4-aminoquinolines.
To determine the effect of an increased artesunate treatment duration in combination therapy, we added the 4-aminoquinolines amodiaquine and piperaquine at each of the artesunate intervals. Single doses of amodiaquine at 6 and 12 mg/kg resulted in recrudescent parasitemia exceeding that of the initial infection by days 4 and 6, respectively (Fig. 4A and B). When combinations of 6 mg/kg or 12 mg/kg amodiaquine with both durations of artesunate were administered, enhanced cytocidal activity was observed with the extended duration of artesunate administration (Fig. 4A and B). This was evident from the delay in parasite recrudescence that occurred with the enhanced duration of artesunate treatment, as analyzed by two-way ANOVA. Enhanced cytocidal activity was also observed with amodiaquine (18 mg/kg) in combination with the extended duration of artesunate treatment (Fig. 4C). All of the mice that received either artesunate treatment duration in combination with amodiaquine (6, 12, or 18 mg/kg) eventually experienced recrudescent parasitemia exceeding pretreatment levels and were euthanized. However, when combination therapy included 24 mg/kg amodiaquine, 66% of the mice receiving the extended duration of artesunate treatment cleared the infection and survived to day 30. In contrast, 100% of the mice receiving the single-asexual-cycle duration of artesunate treatment with amodiaquine (24 mg/kg) experienced recrudescent parasitemia exceeding pretreatment levels by day 11 (Fig. 4D and 5A). When administered combination therapy, including 42 mg/kg amodiaquine, 75% of the mice receiving the extended duration of artesunate treatment cleared the infection and survived to day 30, while 100% of the mice receiving the single-asexual-cycle duration of artesunate and amodiaquine treatment (42 mg/kg) experienced recrudescent parasitemia exceeding pretreatment levels by day 11 (Fig. 4E and 5B). All of the mice that survived until day 30 were parasite negative by Giemsa-stained blood films on day 14, counting at least 1,000 erythrocytes for a limit of detection of 0.1% parasitemia.
FIG 4.

Effect of increased artesunate duration in combination with amodiaquine. In combination with a single dose of amodiaquine (Amq), three doses of 50 mg/kg of artesunate (AS) were administered at either 0, 8, and 20 h or 0, 24, and 48 h to female BALB/c mice. Time zero corresponds to a parasitemia of ∼10%, as determined by the percentage of infected erythrocytes in Giemsa-stained blood films. (A and B) Single doses of Amq at 6 mg/kg (A) or 12 mg/kg (B) were administered alone and in combination with AS. (C to E) Single doses of Amq at 18 mg/kg (C), 24 mg/kg (D), or 42 mg/kg (E) were administered in combination with AS. The data are represented as means ± SEM (or individual mice when group survival varied) and were analyzed by two-way ANOVA. (F) Time of recrudescence data summarized for all five trials and analyzed using an unpaired t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
FIG 5.

Kaplan-Meier curves without return to initial parasitemia for two artesunate treatment durations in combination with amodiaquine. Single doses of Amq (24 mg/kg [A]; 42 mg/kg [B]) were administered at time zero with three doses of 50 mg/kg of artesunate administered at either 0, 8, and 20 h or 0, 24, and 48 h to female BALB/c mice. Upon return to initial parasitemia (∼10%), the mice were euthanized. The mice were monitored until day 30 for recrudescent parasitemia.
In order to determine if this observation was reproducible with another ACT partner compound, we tested both durations of artesunate treatment in combination with piperaquine. Single doses of piperaquine at 9 and 18 mg/kg resulted in a return to initial parasitemia by days 4 and 6, respectively (Fig. 6A). As 9 mg/kg piperaquine alone did not result in substantial parasite killing, we chose 18 mg/kg to test in combination with both of the artesunate durations. In combination with the extended interval of artesunate treatment and 18 mg/kg piperaquine, 100% of the mice cured the infection and survived until day 30 (Fig. 6B and C). In contrast, only 33% of the mice receiving the single-asexual-cycle duration of artesunate treatment and 18 mg/kg piperaquine were able to cure the infection and survive until day 30. Of the mice that did not survive the infection, recrudescent parasitemia exceeded pretreatment levels by day 12.
FIG 6.

Effect of increased artesunate duration in combination with piperaquine. (A) Single doses of piperaquine (Pip) at either 9 mg/kg or 18 mg/kg were administered at time zero to female BALB/c mice. Time zero corresponds to a parasitemia of ∼10%, as determined by the percentage of infected erythrocytes in Giemsa-stained blood films. (B) A single dose of Pip (18 mg/kg) was administered at time zero, with three doses of 50 mg/kg artesunate (AS) administered at either 0, 8, and 20 h or 0, 24, and 48 hours to female BALB/c mice. The data are represented as means ± SEM or as individual mice. (C) Lack of return to initial parasitemia is represented on the Kaplan-Meier curve and was analyzed using the log-rank test. Upon return to initial parasitemia (∼10%), the mice were euthanized. The mice were monitored until day 30 for recrudescent parasitemia.
DISCUSSION
By the mid-1990s, chloroquine-resistant and sulfadoxine-pyrimethamine-resistant P. falciparum had emerged and spread on every malarial continent, requiring a new approach to antimalarial treatment regimens (30). Several clinical trials have compared the efficacies of different treatment durations for artemether-lumefantrine. A 3-day duration of 4 doses resulted in 28-day cure rates of 69% to 85% in Thailand and 71% in the Gambia (31–33). A comparison of a 3-day and a 5-day duration of 6 doses revealed 28-day cure rates of 93% and 97% in Bangkok, Thailand, and 99% and 100% in Mae La, Thailand (34). This led to the conclusion that no significant difference in efficacy existed between treatment regimens of 3 or 5 days, and thus, the 93% to 99% cure rate was determined to be an acceptable standard. In 2001, the World Health Organization convened a technical consultation on antimalarial combination therapy to address the dwindling of effective treatment options. Combining the artemisinin derivatives with a short half-life with a partner drug with a longer half-life was determined to allow a reduction in the duration of treatment. It was concluded that, if costs allowed, the following options were available for implementation: 3 days of artemether-lumefantrine, artesunate (3 days) plus amodiaquine, and artesunate (3 days) plus sulfadoxine-pyrimethamine, where high partner drug efficacy remained (35). For over a decade, the 3-day dosing regimen of ACTs was sufficiently effective. However, initial reports of decreased in vitro susceptibility to the artemisinin compounds arose in 2005, and reports from Southeast Asia continue to document the decreasing in vivo efficacy of dihydroartemisinin-piperaquine against P. falciparum infections (36–39). This emphasizes the need for both novel antimalarials and more effective implementation of current antimalarials.
Our study aimed to address the latter by focusing on the role that treatment duration has on the cytocidal activity of the artemisinin derivative artesunate in a mouse model of malaria with a 24-hour asexual life cycle. The 3-day dosing regimen of ACTs and the very short plasma half-life of artesunate result in approximately a single-life-cycle exposure of the parasite to artesunate. In terms of antimicrobial therapy, this single-life-cycle exposure of P. falciparum is an anomaly. With a 24-hour doubling time, Mycobacterium tuberculosis is treated with 6 months of combination therapy (40). With a 30-min doubling time, Streptococcus pneumoniae infections are treated with a recommended minimum 5-day course of therapy (41). We hypothesized that the same total dose of artesunate would achieve greater reduction in the P. berghei biomass when administered over 3 asexual life cycles versus a single asexual life cycle.
We adapted the luciferase reporter P. berghei for use in evaluating the cytocidal activities of artesunate, amodiaquine, and piperaquine over a 3-log-unit range of parasite density. In mice, dihydroartemisinin, the metabolite of artesunate, has a half-life of 25 min, while piperaquine has a significantly longer half-life of 18 days (42, 43). Amodiaquine has a half-life of 2.8 h in mice, but it is rapidly metabolized to desethylamodiaquine, which has a half-life in humans of 10 days (44, 45). By using this cytocidal murine model of malaria, we determined that extending the interval of 3 doses of artesunate from spanning one asexual cycle to spanning three asexual cycles resulted in a significantly greater reduction in parasite density from the same total amount of drug. This was evident from the 2- to 3-day delay in time to return to initial parasitemia in both the luciferase assay and quantitative PCR (qPCR). Comparing the strikingly different efficacies of the same total dose of artesunate administered at a single time point or spread out evenly across 7 days further highlights the importance of artesunate treatment duration. Our results support the findings of a previous study demonstrating that increased survival was achieved when the same total dose of artesunate was administered in 3 or 4 daily doses versus a single dose in a P. vinckei model (46). Furthermore, a previous study on pharmacodynamic modeling in P. berghei found that three doses of 30 mg/kg dihydroartemisinin would produce a 12-fold-lower nadir than a single dose of 100 mg/kg (25). However, to our knowledge, our study is the first to demonstrate the importance of the specific timing of 3 artesunate doses with respect to parasite life cycle progression.
In addition to investigating the effect of monotherapy treatment duration, we determined the effect of an increased artesunate duration in combination with subcurative and sub-human-equivalent doses of two widely used 4-aminoquinolines: amodiaquine and piperaquine. With two different doses of amodiaquine at 24 mg/kg and 42 mg/kg, we have demonstrated that increasing the duration of artesunate enables cures in 66% and 75% of mice, respectively. In contrast, neither 24 mg/kg nor 42 mg/kg of amodiaquine was sufficient to cure any mice when combined with a single-asexual-cycle exposure to artesunate. Furthermore, 18 mg/kg of piperaquine in combination with a single-asexual-cycle exposure to artesunate cured 33% of mice but was 100% curative in combination with the increased artesunate treatment duration. By improving noncurative dosing regimens with amodiaquine and piperaquine to curative dosing regimens through an alteration in treatment duration, we have demonstrated the biological significance of targeting the parasite with artesunate over multiple life cycles. Although we have analyzed the effect of treatment duration in a P. berghei model, the concept of a single- versus multiple-life-cycle hit of the artemisinin compounds may have strategic application to P. falciparum chemotherapy. Our results support those of a recent PK/PD modeling study of dihydroartemisinin-piperaquine that suggested that administering the same total dose over an increased duration would result in increased P. falciparum killing and a 3-fold reduction in treatment failure rates in humans (47).
Early studies on the efficacy of artemisinin monotherapy for human P. falciparum cases have shown, not surprisingly, that an increased duration of treatment and subsequent increase in the total dose are associated with increasing 28-day cure rates. Ittarat et al. reported a 69% cure rate when 600 mg artesunate was administered over 3 days (48). The same total dose of artesunate was reported by Bunnag et al. to result in a 92.5% cure rate with a 7-day duration and an 85% cure rate with a 5-day duration. However, it is worth noting that the 7-day duration involved placebo administered on days 3 and 4, with only 5 actual days of drug administration (49). In a similar study, Bunnag et al. investigated the schedule of artesunate monotherapy by comparing the same total dose for a 5-day duration with daily or twice daily dosing. They observed 28-day cure rates of 72% for daily dosing and 76% for split daily dosing. Although a slight increase in efficacy was observed, the authors concluded that duration, rather than dosing schedule, may be a more important factor for antimalarial efficacy (50). More recently, in western Cambodia, Bethell et al. compared the efficacies of a 7-day duration of artesunate monotherapy at 2, 4, or 6 mg/kg, for an approximate total dose of 700, 1,400, or 2,100 mg. At 28 days, the cure rates were 92%, 94%, and 84%, respectively, with the 6-mg/kg arm halted due to several individuals developing neutropenia (51). This study indicates that dose escalation of artesunate is insufficient to combat treatment failures, as 2,100 mg of artesunate is not well tolerated and doubling the total dose from 700 mg to 1,400 mg did not significantly improve the outcome. Although the duration of artemisinin compound administration was recognized as a key factor for successful treatment in monotherapy, it has been largely overlooked in recent discussions of improving combination therapy.
As was shown in the ring stage survival assays, artemisinin-resistant parasites exhibit stage-specific drug susceptibility (12). Similarly, other studies have demonstrated the stage-specific drug susceptibility of wild-type P. falciparum with respect to the artemisinins and quinolines (13, 14). Given that ACTs remain the current first-line therapy against P. falciparum infections, both drug sensitive and drug resistant, modifying the duration of artemisinin derivatives deserves consideration. It has previously been suggested that the artemisinin derivatives, despite their rapid cytocidal activity, must be present over three P. falciparum asexual life cycles in order to effect combination drug cure (18). If we are able to increase drug efficacy by extending treatment duration and achieving a greater reduction in the total parasite biomass, the opportunity remains to increase the efficacy of the ACTs and subsequently decrease selective pressures on the partner compounds. While there are several promising leads in the antimalarial pipeline, such as KAE609 and KAF156, these compounds have not yet entered phase 3 clinical trials (30). Thus, the lack of novel compounds on the immediate horizon requires a reevaluation of how to best implement ACTs, specifically the artemisinin derivatives. Triple ACTs involving an artemisinin component plus two partner compounds, as well as cycling the use of ACTs on a population level, have been suggested as possible solutions (52, 53). Artemether-lumefantrine plus amodiaquine and dihydroartemisinin-piperaquine plus mefloquine are under investigation and are registered at ClinicalTrials.gov as NCT02453308. Based on our data, we suggest doubling the dosing duration of current ACTs from 3 days to 6 days in order to expose multiple life cycles of the P. falciparum parasite to drugs.
By adapting a luciferase reporter, P. berghei, to study the cytocidal activity of antimalarials over at least a 30-day period, we have demonstrated the importance of artesunate treatment duration in combination therapy. Our data suggest that, keeping the total dose constant, greater parasite killing can be achieved by extending the duration of treatment with artemisinin derivatives. This work may have relevance for current dosing guidelines of WHO-recommended antimalarials and future efforts geared toward the development of novel antimalarial treatment regimens.
MATERIALS AND METHODS
Ethics statement.
All experimentation was carried out under a protocol approved by the Animal Care and Use Committee of the Johns Hopkins University (MO15H319).
Drug preparation and dosing.
Artesunate (Sigma-Aldrich) was dissolved in 5% NaHCO3. Amodiaquine dihydrochloride dehydrate (Sigma-Aldrich) was dissolved in nuclease-free water. Piperaquine tetraphosphate tetrahydrate (AvaChem Scientific) was dissolved in 75 mM HCl in nuclease-free water. Artesunate was administered via intraperitoneal injection in 200 μl; amodiaquine and piperaquine were administered via oral gavage in 200 μl. The human-equivalent doses of each drug in mice were calculated using a Km factor of 12, a ratio of body weight (kilograms) to surface area (square meters) (54). The quinolines were dosed on salt weight.
In vivo cytocidal model of murine malaria.
Female BALB/c mice (Jackson Laboratory) weighing 20 g ± 20% were used for all experimentation. BALB/c mice were chosen as the inbred mouse strain for our experiments because, unlike C57BL/6 mice, BALB/c mice are much less susceptible to early death on days 7 to 10 by experimental cerebral malaria with P. berghei ANKA infection. For malaria parasite blood-stage infections and drug responses, there are no sex differences in BALB/c mice. As male and female BALB/c mice display differences in tissue iron levels, female mice were used for consistency throughout all the experiments (55). Mice were infected with 500,000 erythrocytes infected with P. berghei ANKA green fluorescent protein (GFP)-luciferase from a donor mouse. For all experiments, P. berghei parasites were passaged through 3 or fewer mice. We obtained original stock from BEI Resources/American Type Culture Collection (ATCC) and transmitted the parasite through Anopheles stephensi mosquitoes to generate fresh stocks in 5 mice, for which blood was aliquoted for frozen stocks. A frozen stock was injected into a donor mouse, which was then passaged through no more than two mice before starting from a new fresh-frozen stock for a new set of experiments. For all experiments, parasitemia was monitored by Giemsa-stained thin blood film and luciferase analysis until approximately 10% parasitemia was reached. There were no significant differences in starting parasitemia between treatment groups. Mice were grouped by initial weight with no significant differences between treatment groups (unpaired t test). After drug treatment, the mice were followed up for 30 days with regular blood sampling for the luciferase assay. Mice were euthanized when recrudescent parasitemia exceeded the original parasitemia or when >20% weight loss occurred.
Luciferase assay and analysis.
A dilution series was performed using P. berghei-infected blood to establish a standard curve for the translation of luciferase signal (total flux) into number of parasites per well. Five microliters of blood was drawn from 24 mice with parasitemia ranging from approximately 6 to 12% and added to 45 μl of lysis buffer. Twofold dilutions in lysis buffer (20 mM Tris [pH 7.5], 5 mM EDTA, 0.008% [wt./vol] saponin, and 0.08% [vol/vol] Triton X-100) were performed 15 times. The impact of decreasing hematocrit was found to be insignificant (data not shown). Samples were then processed for the luciferase assay, with the number of parasites per well ranging from 300 to 600,000. A total of 0.5 μl of whole blood was analyzed per well in the luciferase assay. During the drug treatment, 5 μl of blood was collected from the tail of each mouse at regular intervals and deposited into 45 μl of lysis buffer in a 96-well plate (29). Samples were stored at −80°C until processed. A total of 5 μl of blood-lysis buffer (whole-blood equivalent, 0.5 μl) was transferred to a black, opaque 96-well plate, and 95 μl of luciferase buffer (20 mM Tricine, 100 μM EDTA, 1.07 mM K2CO3, 2.67 mM MgSO4, 17 mM dithiothreitol [DTT], 250 μM ATP, 250 μM d-luciferin) was added. Luciferase activity was measured in the Ivis Spectrum in vivo imaging system and analyzed using Living Image v. 4.4 software. The raw luciferase activity is reported as radiant flux in photons/second. Less than 1,000 photons/second was below the limit of detection. Total radiant flux was compared to the number of parasites per well using GraphPad Prism 5 software and the standard-curve equation derived in Fig. 1. Blood film parasitemia was compared to the luciferase assay when parasitemia was detectable by microscopy. Radiant flux above 52,000 photons/second, corresponding to 300 parasites per well or 600 parasites per μl, was the robust limit of quantification.
Increased duration of artesunate treatment.
Mice were administered 50 mg/kg of artesunate at 3 separate time points to best replicate the human ACT regimen, as well as to test an extended duration of the ACT regimen. Five days following infection, the first group of mice were given three artesunate doses within a single asexual life cycle of P. berghei, at time zero and 8 and 20 h. The second group of mice were given three artesunate doses within 3 asexual life cycles of P. berghei, at time zero and 24 and 48 h. We performed 4 independent trials with >12 mice in each group.
Real-time PCR quantification of infection.
The level of P. berghei infection was quantified during the in vivo experimentation on the increased duration of artesunate treatment, with 3 mice in each treatment group. At regular intervals, 50 μl of blood was collected from the tail of each mouse. It was deposited into 150 μl of 1-mg/ml heparin sodium salt in phosphate-buffered saline (PBS), and DNA extraction was performed with the QIAamp DNA blood minikit. The level of infection was measured using oligonucleotide primers for P. berghei ANKA 18S and Mus musculus β-actin (Table 2). Amplification was performed using Bio-Rad IQ Multiplex Powermix with the Bio-Rad CFX384 real-time PCR detection system. The standard curves for 18S and β-actin were generated using a cDNA plasmid and known concentrations of mouse genomic DNA, respectively. The qPCR was carried out concurrently with the luciferase assay (represented in Fig. 2A) and thin blood film analysis. Data analysis was carried out using GraphPad Prism 5.
TABLE 2.
Sequences of primers and probes used to quantify P. berghei infection
| Target | Primer/probe | Sequence (5′-3′) |
|---|---|---|
| P. berghei 18S rRNA | Forward | AAG CAT TAA ATA AAG CGA ATA CAT CCT TA |
| Reverse | GGA GAT TGG TTT TGA CGT TTA TGC G | |
| Probe | 6-FAM CAA TTG GTT TAC CTT TTG CTC TTT | |
| M. musculus β-actin | Forward | GTA TCC CGG GTA ACC CTT CT |
| Reverse | GCA GAA ACT GCA AAG ATC CA | |
| Probe | Cy5 TGG CCA GCT TCT CAG CCA CG |
Increasing daily doses of 50 mg/kg artesunate.
We administered 50 mg/kg of artesunate to groups of 3 mice for durations spanning 3 to 13 days (Table 1). We then monitored parasite recrudescence and the number of days until initial parasitemia was reached, at which point the mice were euthanized. A single dose of 350 mg/kg of artesunate was administered to a single group of 3 mice to identify any differences in cytocidal activity compared to the 7-day duration of 50 mg/kg.
Increased duration of artesunate in combination with 4-aminoquinolines.
First, we sought to identify a subcurative dose of amodiaquine and piperaquine that would result in cytocidal activity when administered alone but that would not be curative in combination with artesunate administered within a single asexual life cycle. For this, we administered single doses of each drug at 0 h and monitored parasitemia using the luciferase assay. Amodiaquine was administered at 6 and 12 mg/kg, and piperaquine was administered at 9 and 18 mg/kg, which are approximately 1/10 and 1/20 of the human-equivalent dose. Second, we combined a single dose of the quinoline compound with both durations of the three-dose artesunate regimens. The 4-aminoquinoline compounds have half-lives far exceeding those of artemisinin compounds, and thus, we administered only a single dose of both amodiaquine and piperaquine at time zero, coinciding with 10% parasitemia and the first dose of artesunate.
ACKNOWLEDGMENTS
We thank the Johns Hopkins Malaria Research Institute and the Bloomberg Family Foundation (D.J.S.).
REFERENCES
- 1.World Health Organization. 2014. World Malaria Report. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.World Health Organization. 2010. Guidelines for the treatment of malaria, 2nd ed World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.German PI, Aweeka FT. 2008. Clinical pharmacology of artemisinin-based combination therapies. Clin Pharmacokinet 47:91–102. doi: 10.2165/00003088-200847020-00002. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Zhou B. 2010. Biological actions of artemisinin: insights from medicinal chemistry studies. Molecules 15:1378–1397. doi: 10.3390/molecules15031378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balint GA. 2001. Artemisinin and its derivatives: an important new class of antimalarial agents. Pharmacol Ther 90:261–265. doi: 10.1016/S0163-7258(01)00140-1. [DOI] [PubMed] [Google Scholar]
- 6.Bloland PB, Ettling M, Meek S. 2000. Combination therapy for malaria in Africa: hype or hope? Bull World Health Organ 78:1378–1388. [PMC free article] [PubMed] [Google Scholar]
- 7.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han K-T, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amaratunga C, Sreng S, Suon S, Phelps ES, Stepniewska K, Lim P, Zhou C, Mao S, Anderson JM, Lindegardh N, Jiang H, Song J, Su X, White NJ, Dondorp AM, Anderson TJC, Fay MP, Mu J, Duong S, Fairhurst RM. 2012. Artemisinin-resistant Plasmodium falciparum in Pursat province, western Cambodia: a parasite clearance rate study. Lancet Infect Dis 12:851–858. doi: 10.1016/S1473-3099(12)70181-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phyo AP, Nkhoma S, Stepniewska K, Ashley EA, Nair S, McGready R, Ler Moo C, Al-Saai S, Dondorp AM, Lwin KM, Singhasivanon P, Day NPJ, White NJ, Anderson TJC, Nosten F. 2012. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet 379:1960–1966. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witkowski B, Khim N, Chim P, Kim S, Ke S, Kloeung N, Chy S, Duong S, Leang R, Ringwald P, Dondorp AM, Tripura R, Benoit-Vical F, Berry A, Gorgette O, Ariey F, Barale J-C, Mercereau-Puijalon O, Menard D. 2013. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob Agents Chemother 57:914–923. doi: 10.1128/AAC.01868-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, Kim S, Lim P, Mao S, Sopha C, Sam B, Anderson JM, Duong S, Chuor M, Taylor WRJ, Suon S, Mercereau-Puijalon O, Fairhurst RM, Menard D. 2013. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 13:1043–1049. doi: 10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ter Kuile F, White NJ, Holloway P, Pasvol G, Krishna S. 1993. Plasmodium falciparum: in vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Exp Parasitol 76:85–95. doi: 10.1006/expr.1993.1010. [DOI] [PubMed] [Google Scholar]
- 14.Klonis N, Xie SC, McCaw JM, Crespo-Ortiz MP, Zaloumis SG, Simpson JA, Tilley L. 2013. Altered temporal response of malaria parasites determines differential sensitivity to artemisinin. Proc Natl Acad Sci U S A 110:5157–5162. doi: 10.1073/pnas.1217452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hott A, Casandra D, Sparks KN, Morton LC, Castanares G-G, Rutter A, Kyle DE. 2015. Artemisinin-resistant Plasmodium falciparum parasites exhibit altered patterns of development in infected erythrocytes. Antimicrob Agents Chemother 59:3156–3167. doi: 10.1128/AAC.00197-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White NJ, Krishna S. 1989. Treatment of malaria: some considerations and limitations of the current methods of assessment. Trans R Soc Trop Med Hyg 83:767–777. doi: 10.1016/0035-9203(89)90322-2. [DOI] [PubMed] [Google Scholar]
- 17.Lubell Y, Dondorp A, Guérin PJ, Drake T, Meek S, Ashley E, Day NPJ, White NJ, White LJ. 2014. Artemisinin resistance–modelling the potential human and economic costs. Malar J 13:452. doi: 10.1186/1475-2875-13-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White NJ. 1997. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob Agents Chemother 41:1413–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Menard D, Fidock DA. 2015. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris CA, Duparc S, Borghini-Fuhrer I, Jung D, Shin C-S, Fleckenstein L. 2011. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar J 10:263. doi: 10.1186/1475-2875-10-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Djimdé A, Lefèvre G. 2009. Understanding the pharmacokinetics of Coartem. Malar J 8:S4. doi: 10.1186/1475-2875-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tarning J, Rijken MJ, McGready R, Phyo AP, Hanpithakpong W, Day NPJ, White NJ, Nosten F, Lindegardh N. 2012. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated malaria. Antimicrob Agents Chemother 56:1997–2007. doi: 10.1128/AAC.05756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel K, Batty KT, Moore BR, Gibbons PL, Kirkpatrick CM. 2014. Predicting the parasite killing effect of artemisinin combination therapy in a murine malaria model. J Antimicrob Chemother 69:2155–2163. doi: 10.1093/jac/dku120. [DOI] [PubMed] [Google Scholar]
- 24.Patel K, Batty KT, Moore BR, Gibbons PL, Bulitta JB, Kirkpatrick CM. 2013. Mechanism-based model of parasite growth and dihydroartemisinin pharmacodynamics in murine malaria. Antimicrob Agents Chemother 57:508–516. doi: 10.1128/AAC.01463-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibbons PL, Batty KT, Barrett PHR, Davis TME, Ilett KF. 2007. Development of a pharmacodynamic model of murine malaria and antimalarial treatment with dihydroartemisinin. Int J Parasitol 37:1569–1576. doi: 10.1016/j.ijpara.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Gumede B, Folb P, Ryffel B. 2003. Oral artesunate prevents Plasmodium berghei Anka infection in mice. Parasitol Int 52:53–59. doi: 10.1016/S1383-5769(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 27.Lombard MC, N′Da DD, Tran Van Ba C, Wein S, Norman J, Wiesner L, Vial H. 2013. Potent in vivo anti-malarial activity and representative snapshot pharmacokinetic evaluation of artemisinin-quinoline hybrids. Malar J 12:71. doi: 10.1186/1475-2875-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franke-Fayard B, Waters AP, Janse CJ. 2006. Real-time in vivo imaging of transgenic bioluminescent blood stages of rodent malaria parasites in mice. Nat Protoc 1:476–485. doi: 10.1038/nprot.2006.69. [DOI] [PubMed] [Google Scholar]
- 29.Franke-Fayard B, Djokovic D, Dooren MW, Ramesar J, Waters AP, Falade MO, Kranendonk M, Martinelli A, Cravo P, Janse CJ. 2008. Simple and sensitive antimalarial drug screening in vitro and in vivo using transgenic luciferase expressing Plasmodium berghei parasites. Int J Parasitol 38:1651–1662. doi: 10.1016/j.ijpara.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 30.Wells TNC, van Huijsduijnen RH, Van Voorhis WC. 2015. Malaria medicines: a glass half full? Nat Rev Drug Discov 14:424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 31.Looareesuwan S, Wilairatana P, Chokejindachai W, Chalermrut K, Wernsdorfer W, Gemperli B, Gathmann I, Royce C. 1999. A randomized, double-blind, comparative trial of a new oral combination of artemether and benflumetol (CGP 56697) with mefloquine in the treatment of acute Plasmodium falciparum malaria in Thailand. Am J Trop Med Hyg 60:238–243. [DOI] [PubMed] [Google Scholar]
- 32.van Vugt M, Brockman A, Gemperli B, Luxemburger C, Gathmann I, Royce C, Slight T, Looareesuwan S, White NJ, Nosten F. 1998. Randomized comparison of artemether-benflumetol and artesunate-mefloquine in treatment of multidrug-resistant falciparum malaria. Antimicrob Agents Chemother 42:135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Seidlein L, Jaffar S, Pinder M, Haywood M, Snounou G, Gemperli B, Gathmann I, Royce C, Greenwood B. 1997. Treatment of African children with uncomplicated falciparum malaria with a new antimalarial drug, CGP 56697. J Infect Dis 176:1113–1116. doi: 10.1086/516524. [DOI] [PubMed] [Google Scholar]
- 34.Vugt MV, Wilairatana P, Gemperli B, Gathmann I, Phaipun L, Brockman A, Luxemburger C, White NJ, Nosten F, Looareesuwan S. 1999. Efficacy of six doses of artemether-lumefantrine (benflumetol) in multidrug-resistant Plasmodium falciparum malaria. Am J Trop Med Hyg 60:936–942. [DOI] [PubMed] [Google Scholar]
- 35.World Health Organization. 2001. Antimalarial drug combination therapy. Report of a WHO Technical Consult. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 36.World Health Organization. 2005. Susceptibility of Plasmodium falciparum to antimalarial drugs: report on global monitoring 1996-2004, p 1–133. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 37.Leang R, Barrette A, Bouth DM, Menard D, Abdur R, Duong S, Ringwald P. 2013. Efficacy of dihydroartemisinin-piperaquine for treatment of uncomplicated Plasmodium falciparum and Plasmodium vivax in Cambodia, 2008 to 2010. Antimicrob Agents Chemother 57:818–826. doi: 10.1128/AAC.00686-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaorattanakawee S, Lon C, Jongsakul K, Gawee J, Sok S, Sundrakes S, Kong N, Thamnurak C, Chann S, Chattrakarn S, Praditpol C, Buathong N, Uthaimongkol N, Smith P, Sirisopana N, Huy R, Prom S, Fukuda MM, Bethell D, Walsh DS, Lanteri C, Saunders D. 2016. Ex vivo piperaquine resistance developed rapidly in Plasmodium falciparum isolates in northern Cambodia compared to Thailand. Malar J 15:519. doi: 10.1186/s12936-016-1569-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amaratunga C, Lim P, Suon S, Sreng S, Mao S, Sopha C, Sam B, Dek D, Try V, Amato R, Blessborn D, Song L, Tullo GS, Fay MP, Anderson JM, Tarning J, Fairhurst RM. 2016. Dihydroartemisinin–piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis 16:357–365. doi: 10.1016/S1473-3099(15)00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nahid P, Dorman SE, Alipanah N, Barry PM, Brozek JL, Cattamanchi A, Chaisson LH, Chaisson RE, Daley CL, Grzemska M, Higashi JM, Ho CS, Hopewell PC, Keshavjee SA, Lienhardt C, Menzies R, Merrifield C, Narita M, O'Brien R, Peloquin CA, Raftery A, Saukkonen J, Schaaf HS, Sotgiu G, Starke JR, Migliori GB, Vernon A. 2016. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America clinical practice guidelines: treatment of drug-susceptible tuberculosis. Clin Infect Dis 63:e147–e195. doi: 10.1093/cid/ciw376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell GD, Dean NC, Dowell SF, File TM, Musher DM, Niederman MS, Torres A, Whitney CG. 2007. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 44:S27–S72. doi: 10.1086/511159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batty KT, Gibbons PL, Davis TME, Ilett KF. 2008. Pharmacokinetics of dihydroartemisinin in a murine malaria model. Am J Trop Med Hyg 78:641–642. [PubMed] [Google Scholar]
- 43.Moore BR, Batty KT, Andrzejewski C, Jago JD, Page-Sharp M, Ilett KF. 2008. Pharmacokinetics and pharmacodynamics of piperaquine in a murine malaria model. Antimicrob Agents Chemother 52:306–311. doi: 10.1128/AAC.00878-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurawattimath V, Pocha K, Mariappan TT, Trivedi RK, Mandlekar S. 2012. A modified serial blood sampling technique and utility of dried-blood spot technique in estimation of blood concentration: application in mouse pharmacokinetics. Eur J Drug Metab Pharmacokinet 37:23–30. doi: 10.1007/s13318-011-0066-5. [DOI] [PubMed] [Google Scholar]
- 45.Orrell C, Little F, Smith P, Folb P, Taylor W, Olliaro P, Barnes KI. 2008. Pharmacokinetics and tolerability of artesunate and amodiaquine alone and in combination in healthy volunteers. Eur J Clin Pharmacol 64:683–690. doi: 10.1007/s00228-007-0452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LaCrue AN, Scheel M, Kennedy K, Kumar N, Kyle DE. 2011. Effects of artesunate on parasite recrudescence and dormancy in the rodent malaria model Plasmodium vinckei. PLoS One 6:e26689. doi: 10.1371/journal.pone.0026689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kay K, Hodel EM, Hastings IM. 2015. Altering antimalarial drug regimens may dramatically enhance and restore drug effectiveness. Antimicrob Agents Chemother 59:6419–6427. doi: 10.1128/AAC.00482-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ittarat W, Pickard AL, Rattanasinganchan P, Wilairatana P, Looareesuwan S, Emery K, Low J, Udomsangpetch R, Meshnick SR. 2003. Recrudescence in artesunate-treated patients with falciparum malaria is dependent on parasite burden not on parasite factors. Am J Trop Med Hyg 68:147–152. [PubMed] [Google Scholar]
- 49.Bunnag D, Viravan C, Looareesuwan S, Karbwang J, Harinasuta T. 1991. Double blind randomised clinical trial of two different regimens of oral artesunate in falciparum malaria. Southeast Asian J Trop Med Public Health 22:534–538. [PubMed] [Google Scholar]
- 50.Bunnag D, Viravan C, Looareesuwan S, Karbwang J, Harinasuta T. 1991. Double blind randomised clinical trial of oral artesunate at once or twice daily dose in falciparum malaria. Southeast Asian J Trop Med Public Health 22:539–543. [PubMed] [Google Scholar]
- 51.Bethell D, Se Y, Lon C, Tyner S, Saunders D, Sriwichai S, Darapiseth S, Teja-Isavadharm P, Khemawoot P, Schaecher K, Ruttvisutinunt W, Lin J, Kuntawungin W, Gosi P, Timmermans A, Smith B, Socheat D, Fukuda MM. 2011. Artesunate dose escalation for the treatment of uncomplicated malaria in a region of reported artemisinin resistance: a randomized clinical trial. PLoS One 6:e19283. doi: 10.1371/journal.pone.0019283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boni MF, Smith DL, Laxminarayan R. 2008. Benefits of using multiple first-line therapies against malaria. Proc Natl Acad Sci U S A 105:14216–14221. doi: 10.1073/pnas.0804628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dondorp AM, Yeung S, White L, Nguon C, Day NPJ, Socheat D, von Seidlein L. 2010. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol 8:272–280. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 54.Reagan-Shaw S, Nihal M, Ahmad N. 2008. Dose translation from animal to human studies revisited. FASEB J 22:659–661. [DOI] [PubMed] [Google Scholar]
- 55.Hahn P, Song Y, Ying G, He X, Beard J, Dunaief JL. 2009. Age-dependent and gender-specific changes in mouse tissue iron by strain. Exp Gerontol 44:594–600. doi: 10.1016/j.exger.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]