

Abstract
Helicobacter pylori is a Gram-negative bacterium that specifically colonizes the gastric ecological niche. During the infectious process, which results in diseases ranging from chronic gastritis to gastric cancer, the host response is characterized by the activation of the innate immunity of gastric epithelial cells and macrophages. These cells thus produce effector molecules such as reactive oxygen species (ROS) to counteract the infection. The generation of ROS in response to H. pylori involves two canonical pathways: 1) the NADPH-dependent reduction of molecular oxygen to generate O2•−, which can dismute to generate ROS; and 2) the back-conversion of the polyamine spermine into spermidine through the enzyme spermine oxidase, leading to H2O2 production. Although these products have the potential to affect the survival of bacteria, H. pylori has acquired numerous strategies to counteract their deleterious effects. Nonetheless, ROS-mediated oxidative DNA damage and mutations may participate in the adaptation of H. pylori to its ecological niche. Lastly, ROS have been shown to play a major role in the development of the inflammation and carcinogenesis. It is the purpose of this review to summarize the literature about the production of ROS during H. pylori infection and their role in this infectious gastric disease.
Keywords: Helicobacter pylori, Reactive oxygen species, Polyamines, NADPH oxidase, Gastric cancer
Graphical Abstract
1. Infections with Helicobacter pylori
1.1. Epidemiologic aspects
Helicobacter pylori is a Gram-negative microaerophilic bacterium that colonizes the human stomach and it is estimated that half of world’s human population is infected. Although H. pylori potentially confers protection against diseases, notably in childhood [1], long-term infection has been associated with the development of chronic active gastritis. Moreover, approximately 10% of H. pylori-infected patients develop peptic ulcer disease, less than 0.1% develop mucosa-associated lymphoid tissue lymphoma, and 1 to 3% develop gastric adenocarcinoma [2, 3], the third leading cause of death by cancer worldwide corresponding to 10% of total cancer-related mortality. Therefore, H. pylori is considered as the most common etiologic agent of infection-related cancers and has been classified as a class I carcinogen.
Eradication of H. pylori by antibiotic-based therapy has been proposed to decrease the incidence of gastric malignant transformation [4]. However, the extraordinary genomic plasticity of H. pylori has led to considerable antibiotic resistance and the efficacy of the treatments has declined in recent years [5]. Thus, treatment failure is common and high rates of infection recurrence have been reported, principally in developing countries [6]. Lastly, the impact of H. pylori eradication on development of gastric cancer appears to be of little value once the development of precancerous lesions is observed [7].
This information highlights that H. pylori infection is a serious concern for human health in a world with increasing movement of populations and that non-antibiotic treatment therapy should be considered to limit the risk of development of gastric cancer. This could be achieved by a better understanding of the crosstalk of H. pylori and the host.
1.2. Determinism of pathogenicity
H. pylori strains express three major virulence factors. First, the persistence of the bacterium is principally due to the activity of the bacterial urease that neutralizes gastric acidity by generating ammonium from urea. Second, the cytotoxin-associated gene A (CagA) is a bacterial factor that belongs to the cag pathogenicity island (cagPAI) and is directly injected into human epithelial cells through a type 4 secretion system (T4SS). CagA is then sequentially phosphorylated on tyrosine residues of Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs by the host kinases SRC proto-oncogene (SRC) and ABL proto-oncogene 1 (ABL1) [8]. Four distinct EPIYA motifs (A-D) have been identified and most H. pylori strains exhibit EPIYA motif repeats on the C-terminal regions of CagA. The Western isolates exhibit the EPIYA-A/B/(1–3)C motifs, whereas the EPIYA-D motif replaces the C motif in East Asian strains [9]; in Amerindian strains, the last motif is a chimera of the D and C motifs [10]. It has been demonstrated that SRC phosphorylates EPIYA-C and EPIYA-D, whereas ABL phosphorylates all four motifs [8]. Epidemiological studies have emphasized that gastric cancer in East Asian countries is most often associated with the EPIYA-ABD motif [11] and more severe disease in Western countries is associated with multiple EPIYA-C motifs [12]. When CagA is phosphorylated in gastric epithelial cells, it activates the tyrosine-protein phosphatase non-receptor type 11 (also known as SHP2) [13], which then activates extracellular signal-regulated kinase 1/2 (ERK1/2)-dependent cytoskeletal rearrangements, increased motility, loss of cell polarity, resistance to apoptosis, and chromosomal instability, which have been linked to malignancy [14–16]. The phosphorylation of CagA has been also shown to stimulate the activation of the transcription factor nuclear factor-kappa B (NF-κB) [17–19]. It should be also underlined that CagA also possesses a CRPIA (conserved repeat responsible for phosphorylation-independent activity) motif, which is responsible for the phosphorylation-independent signaling of CagA that induces the phosphoinositide 3-kinase (PI3K) signaling pathway [18]. Consistent with these data, large epidemiologic studies have associated the strains harboring the gene cagA with higher rates of gastric cancer [20]. Third, the vacuolating cytotoxin A (VacA) contributes to H. pylori pathogenesis by regulating inflammatory process [21] and by reducing cell death by autophagy, thus favoring gastric colonization and oxidative damage [22]. Although the contribution of VacA to precancerous gastric intestinal metaplasia has not been directly demonstrated using animal models, epidemiological studies have emphasized that the signal region s1 and the middle region m1 of the vacA gene belong to strains that are associated with increased risk for the development of peptic ulcers and/or gastric cancer, compared to s2 or m2 strains [23].
However, the sole expression of virulence factors is not sufficient to explain H. pylori pathogenesis, mainly because only 1–3% of H. pylori-infected patients develop gastric cancer. Several studies have shown that an environmental component, including iron deficiency [24] or high-salt diets [25], may modulate H. pylori-induced inflammation and related carcinogenesis. In addition, the clinical outcome of H. pylori infection-induced gastric carcinogenesis is determined by the progression along the histologic cascade from non-atrophic gastritis to adenocarcinoma [26]. In this context, the level of gastritis and the regulation of proteins with potential effects on cell transformation are fundamental features that determine H. pylori-related diseases. Among the effectors that can affect cellular function or disrupt signaling, reactive oxygen species (ROS) have been unequivocally recognized as the yin and yang of the innate immune response towards pathogens. Identification of the source of ROS in H. pylori-infected mucosa, determination of the mechanism by which H. pylori survives the oxidative challenge imposed by the host, and comprehension of the deleterious effect of endogenous ROS are critical topics for a better understanding of H. pylori pathogenesis.
2. Polyamine-dependent ROS synthesis during H. pylori infection
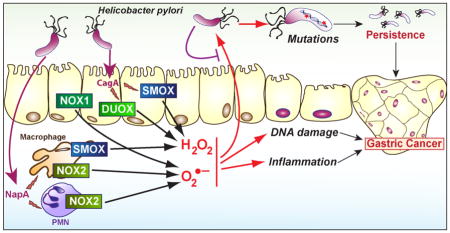
Polyamines are critical homeostatic regulators also involved in the modulation of pathogenesis of numerous diseases, including in the gastrointestinal tract. The inducible synthesis of the biogenic polyamines during H. pylori infection necessitates three steps (Fig. 1): First, the amino acid L-arginine is transported into the cells through the activity of the L-arginine transporter solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 (SLC7A2). Second, L-arginine is catabolized by the enzyme arginase into L-ornithine. Third, L-ornithine is converted by ornithine decarboxylase (ODC) into the first polyamine putrescine, which is then catabolized to spermidine and spermine by the action of spermidine synthase and spermine synthase, respectively. Importantly, the back-conversion of spermine to spermidine by spermine oxidase (SMOX) generates H2O2 (Fig. 1), which plays a critical role in H. pylori pathogenesis.
Fig. 1.

H. pylori-infected macrophages produces H2O2 from L-arginine. H. pylori induces SLC7A2 expression allowing the uptake of L-arginine in macrophages. The induction of ARG2, ODC, which requires a MYC-dependent pathway, and SMOX leads to the release of H2O2.
2.1. L-arginine bioavailability
The expression of the gene Slc7a2 and the transporter SLC7A2 is induced in the murine macrophage cell line RAW 264.7 and in murine peritoneal macrophages infected with H. pylori in vitro and in lamina propria mononuclear cells of mice and humans with H. pylori gastritis [27]; this increase in SLC7A2 protein expression enhances L-arginine uptake in the cells [27]. It has been shown that Slc7a2-deficient macrophages produce less polyamines than wild-type (WT) cells when stimulated with M-CSF [28]. Although this has never been reported in the context of H. pylori infection, it provides a strong rationale for the contention that polyamine synthesis depends on SLC7A2 activity.
Interestingly, it has been reported that spermine generated endogenously during H. pylori infection or added exogenously inhibits L-arginine uptake by macrophages [27]. Blocking Odc mRNA expression or ODC activity favors L-arginine entry in macrophages, but does not regulates Slc7a2 mRNA or SLC7A2 protein levels [27], suggesting that spermine is an inhibitor of SLC7A2 activity and may be considered as a negative feedback regulator of polyamine synthesis in immune cells.
Our lab has reported that H. pylori-infected Slc7a2−/− mice exhibit more colonization and less gastritis than WT animals. These events were accompanied by a defect in recruitment of macrophages in the infected gastric tissues and by an altered activation of dendritic cells and Th1 cells [29]. These data demonstrate that SLC7A2 plays a major function in the activation of the innate immune response during H. pylori infection; but the involvement of polyamines and/or SMOX-derived H2O2 in this mechanism remains unknown. Work is ongoing in our laboratory to address the contributions of myeloid versus epithelial SLC7A2 in mouse models of H. pylori infection. In that context, the potential role of modulation of L-arginine availability in the generation of ROS derived from polyamines that are downstream products of L-arginine metabolism is a theme that will be investigated.
2.2. Arginase activity
Two isoforms of arginase exist in mammals: the cytosolic arginase 1 (ARG1), and arginase 2 (ARG2) that is sublocalized to the mitochondria. ARG1, but not ARG2, is a marker of alternative (M2) macrophages and ARG2 is not induced by IFN-γ, the classical inducer of M1 phenotype, but can be express in M1 macrophages. Thus, it has been shown that cultured murine macrophages infected with H. pylori and gastric macrophages of infected mice display an M1/Mreg phenotype [30], associated with the expression of Arg2 mRNA expression and ARG2 protein synthesis through an NF-κB-dependent pathway [31–33]. However, the ARG1 isoform is not directly induced by H. pylori [31]. Arginase activity in infected RAW 264.7 macrophages has been principally detected in the mitochondrial fraction and not in the cytoplasm [32], further demonstrating that the ARG2 isoform is induced in response to H. pylori. In addition, it has been described using flow cytometry and immunostaining that ARG2, but not ARG1, localized to gastric macrophages isolated from C57BL/6 mice infected with H. pylori [33].
In mice infected with H. pylori, the genetic deletion of the gene Arg2 results in a decrease of putrescine synthesis, demonstrating that ARG2 regulates polyamine production during the infection [34]. Two studies have reported that H. pylori-infected Arg2−/− mice show more gastritis, characterized by enhanced M1 macrophage and Th1/Th17 responses, and less colonization [33, 34] than WT animals, suggesting that macrophage ARG2 decreases the inflammatory process during the infection. Therefore, the induction of ARG2 by H. pylori could represent a strategy by which this pathogen dampens and escapes the host immune response. Importantly, ARG2 is also a natural competitor for inducible nitric oxide synthase (NOS2), the enzyme that generates nitric oxide (NO) from L-arginine. NO is a free radical produced by H. pylori-infected macrophages that exhibits anti-microbial properties and can regulate inflammation and carcinogenesis [35]. In H. pylori-infected macrophages, blocking ARG2 activity results in increased NO production [32], highlighting that the induction of the arginase metabolic pathway by the bacterium is one way of controlling NO production. However, we have recently reported that Arg2−/− mice and Arg2−/−; Nos2−/− mice have similar levels of colonization and gastritis when infected with H. pylori [34], indicating that the role of ARG2 in controlling immune response is NOS2-independent. However, more evidence is needed to ascertain that the effect of ARG2 on H. pylori pathogenesis occurs through a polyamine-dependent pathway, in particular in human populations.
2.3. Induction of ODC
Studies have reported an increase of ODC mRNA expression in the gastric tissues of H. pylori-infected patients [27, 36], independently of the cagA status of the bacteria [36]. In one study, increased ODC activity was mainly observed in the areas of gastric atrophy in H. pylori-infected patients [37]. The concentration of total polyamines, but mainly spermidine, is enhanced by more than two times in the stomach of H. pylori-infected patients compared to H. pylori-negative subjects [38]. In contrast, Elitsur et al. reported no significant change in ODC activity in the mucosa of children infected with H. pylori compared to uninfected [39]; the low level of colonization and the absence of gastric atrophy, in which ODC was observed in adults [37], could be responsible for the lack of ODC induction. Interestingly, Chaturvedi et al. further demonstrated that ODC mRNA expression is increased patients with H. pylori gastritis, but not in those with H. pylori-negative gastritis [27]; in the same way, Patchett et al. reported that the presence of H. pylori, and not the severity of gastric inflammation is associated with increased ODC activity in humans [40]. These observations indicate that the presence of the bacteria is required for ODC induction, and that this is not a global response to inflammation. Supporting this, ODC mRNA expression [36], ODC activity in the corpus and antrum of the stomach [41, 42], and polyamine concentrations [38, 43] are decreased after successful eradication of H. pylori. In the same way, a concomitant decrease of H. pylori colonization, gastric ODC activity, and gastric polyamine concentration has been observed in patients treated with the probiotic Lactobacillus brevis strain CD2 [38].
Although it has been described that ODC can be induced in vitro in the gastric epithelial cell lines AGS and BGC-823 and in the immortalized epithelial cell line GES-1 through a phospho-CagA-dependent mechanism and a signal involving ERK1/2 and V-Myc avian myelocytomatosis viral oncogene homolog (MYC) [44], in vivo and in vitro evidence suggest that ODC induction is predominant in myeloid cells in response to H. pylori. Hence, ODC has been immunolocalized in gastric macrophages in both humans and mice infected with H. pylori [27]. Moreover, Odc mRNA expression and ODC activity are induced in the murine macrophage cell line RAW 264.7 stimulated with H. pylori SS1 [31]. This induction first requires the activation of the transcription factor activator protein 1 (AP-1), which corresponds to a phospho-FBJ murine osteosarcoma viral oncogene homolog (FOS)/Jun proto-oncogene (JUN) complex, through an ERK1/2-dependent pathway; then AP-1 initiates the transcription of the gene encoding the proto-oncogene MYC [45, 46]. Finally, this transcription factor binds the Odc promoter region and stimulates the expression of the gene [45]. Notably, chemical inhibition of ERK1/2 phosphorylation led to a decrease of ODC protein expression in gastric macrophages from H. pylori-infected mice [45], supporting the likelihood that this mechanism is not specific to macrophage cell lines. However, the bacterial factors involved in the AP-1/MYC signaling pathway in macrophages remain unknown.
2.4. Generation of ROS by SMOX
The homeostasis of polyamine content within a non-toxic range is a considerable challenge for cells, notably during a pathophysiological process in which the genes encoding enzymes involved in polyamine metabolism and catabolism are induced. In this regard, the back conversion of spermine to spermidine and putrescine is a critical step in the regulation of polyamine levels that can prevent the cytotoxic effects of spermine accumulation. Polyamine metabolism can involve several biochemical pathways that may have direct significance in H. pylori pathogenesis, ROS generation, and risk for carcinogenesis. First; the spermidine/spermine N(1)-acetyltransferase 1 (SAT1, also known as SSAT) catalyzes the acetylation of spermine into N(1)-acetylspermine and spermidine into N(1)-acetylspermidine; then, the oxidation of N(1)-acetylspermine and N(1)-acetylspermidine through the enzyme polyamine oxidase (PAOX) leads to the formation of spermidine and putrescine, respectively. However, this pathway is poorly induced in macrophages stimulated in vitro with H. pylori and in mice infected for 4 months with the strain SS1 [47]. In contrast, the flavoenzyme SMOX, which directly catalyzes the oxidation of spermine to spermidine and yields the generation of 3-aminopropanal and H2O2 [48], is overexpressed in macrophages [47] and in gastric epithelial cells [49] infected in vitro with H. pylori as well as in the gastric epithelium and monocytes of infected humans, mice, and gerbils [50].
Two cagA-deficient strains of H. pylori and a cagE mutant each induced less Smox mRNA expression in conditionally immortalized murine gastric epithelial cells and less SMOX mRNA in human epithelial cells than the WT strains [50], suggesting that CagA and the T4SS are involved in SMOX expression. These in vitro observations were confirmed by the analysis of gastric tissues from patients with H. pylori infection: individuals infected with cagA-positive H. pylori showed a significant increase of SMOX mRNA expression, and SMOX protein level in the gastric epithelium compared to patients harboring cagA-negative strains [50]. Similar data were obtained in mice infected with cagA-positive H. pylori versus a cagE mutant that cannot translocate CagA, and in gerbils infected with cagA-positive versus cagA-negative strains [50, 51].
3. NADPH oxidases
The mammalian NADPH oxidases (NOX) are enzymes that transport electrons across the plasma membrane. The electron acceptor is oxygen and therefore the product of the reaction is O2•−. Seven homologs have been identified, referred as five NOX enzymes (NOX1 to NOX5) and two dual oxidase (DUOX) enzymes. Expression of NOX1, NOX2, and DUOX2 have been reported during H. pylori infection (Fig. 2).
Fig. 2.

Regulation of NOX by H. pylori. NOX1 is induced in gastric epithelial cells through a PI3K/RAC1 signaling pathway; in these cells, the production of O2•− stimulate the expression of APEX that blocks RAC1. In myeloid cells, the expression of NOX2 requires a NapA/PI3K signals. DUOX2 has been shown to be induced in the gastric tissue of infected animals, but the subcellular localization of this enzyme remains unknown. PHD, peroxidase homology domain.
3.1. NOX1
The human and mouse NOX1 gene is located on the X chromosome and is mainly expressed in the human colon [52]. Although NOX1 expression and function have been evidenced in the gastric mucosa of the guinea pig [53], it has been first suggested that this isoform is not expressed in the human stomach [54]. However, Tominaga et al. have reported that the gene encoding NOX1 is induced in tissues from H. pylori-infected patients with gastric adenocarcinoma [55]. These authors also showed by immunofluorescence and confocal microscopy that the proteins NOX1 and the partners NOX organizer 1, NOX activator 1, and p22phox are sublocalized in the Golgi apparatus in gastric cancer cells; and that their expression levels are increased in gastric cancers compared to the surrounding tissue, and compared to areas of chronic atrophic gastritis or adenomas in patients without carcinoma [55]. This suggests that NOX1 could represent a marker of neoplastic transformation. In addition, non-transformed antral-derived primary epithelial cells and the gastric epithelial cell line AGS infected with H. pylori 26695 express NOX1 through a signal transduction process involving Ras-related C3 botulinum toxin substrate 1 (RAC1), which is also a protein partner for NOX1, and produce ROS rapidly and transiently [56]. Similarly, NOX1 induction and ROS production has been observed in primary guinea pig gastric mucosal cells infected with H. pylori lipopolysaccharide (LPS) [57] through a PI3K/RAC1-dependent pathway [58]. Therefore, RAC1 seems to play an important role in regulating NOX1 expression and activity in H. pylori-infected cells. It is exciting to note that the H. pylori protein CagL, which is a part of the T4SS, activates the host enzyme SRC [59], and that SRC is also a strong activator of RAC1.
Interestingly, the endonuclease apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1, also known as APE1), a multifunctional enzyme that plays a central role in the cellular response to oxidative stress, including DNA repair and redox regulation of transcriptional factors, is induced by endogenous ROS in H. pylori-infected AGS cells [56]. APEX1 inhibits RAC1 activation and consequently NOX1-dependent ROS production, and blocking APEX1 leads to sustained NOX1 activation and increased O2− generation [56]. This could represent a mechanism by which cells limit NOX1-dependent DNA damage and/or a strategy developed by the bacterium to promote its own survival by inhibiting ROS production. In addition, as an another striking example of host-pathogen interactions, it has been established that H. pylori superoxide dismutase and catalase decompose NOX1-mediated O2•− and H2O2, respectively, thus protecting gastric epithelial cells from ROS-induced cytotoxicity [60]. The authors proposed that the bacterium may use this mechanism to limit the elimination of the transformed cells, thus favoring tumor progression [60].
3.2. NOX2
The enzyme NOX2, also known as gp91phox or phagocyte NOX, is principally expressed in neutrophils and macrophages. H. pylori, H. pylori lysate, or H. pylori LPS can each induce a rapid oxidative burst in human primary blood-derived polymorphonuclear neutrophils (PMN) [61–63]. However, it has been reported that H. pylori does not efficiently induce the recruitment of the domains p47phox and p67phox to the phagosome, which normally leads to the release of large amounts of O2•− extracellularly [63]. This unique ability to prevent NOX2 assembly at the phagosome allows H. pylori to evade the oxidative killing and may result in neutrophil-derived oxidative damage to surrounding cells, mainly from the epithelium.
The gene napA of H. pylori [64] encodes the 17 kDa virulence factor called neutrophil-activating protein A (NapA) that forms hexagonal rings [65]. It has been shown that NapA is a chemoattractant for leukocytes and induces NOX2 in human neutrophils through a SRC/PI3K signaling pathway [66]. However, no alteration in gastric colonization has been observed in animals infected for 3 weeks with the napA mutant compared to the parental strain [67]. Chronic infection of mice or the use of animal models of H. pylori-mediated gastric dysplasia, e.g., transgenic FVB/N insulin-gastrin (INS-GAS) mice, or gerbils, would be useful to determine the role of NapA and the associated induction of NOX2 in carcinogenesis.
Similarly, the stimulation of blood-derived monocytes or differentiated THP-1 cells with H. pylori or its LPS yields a rapid and sustained production of O2•− [68]. More particularly, it has been reported that H. pylori cecropin-like peptide Hp(2–20) is a chemoattractant for monocytes through the N-formyl peptide receptors 1 and 2, and that monocytes stimulated with Hp(2–20) release ROS, which inhibit the function of antineoplastic lymphocytes [69].
3.3. DUOX2
DUOX1 and DUOX2 are NOX isoforms that generate H2O2 in a Ca2+- and NADPH-dependent manner [70]. DUOX(1–2) must interact with the activator proteins DUOXA(1–2) that organize DUOX for surface expression and activity. The mechanism by which DUOX isoforms produce H2O2 and not O2•− remains unknown, but it has been reported that the non mature DUOX2 releases O2•− whereas the glycosylated form of DUOX2 generates H2O2 [71].
A microarray analysis performed on the stomach of rhesus macaques (2–3 years-old) has shown that DUOX2 is one of the 119 genes induced in H. pylori-infected animals [72]. Moreover, the expression of DUOX2 is significantly decreased in animals infected with a cagPAI mutant strain although the level of colonization by the WT and the mutant is similar [72]. The authors also found that other factors with antimicrobial properties, e.g., defensins and the protein DMBT1 (for ‘deleted in malignant brain tumors 1’), have been induced in the infected monkeys and therefore proposed that the antimicrobial response activated by the cagPAI may affect the resident gastric microbiota to favor H. pylori colonization [72]. In this context, the study of DUOX2 deserves further investigation, even if it has been reported that H. pylori-infected patients, mainly those carrying a cagA/vacA-positive strains, show less DUOX2 levels than uninfected individuals [73].
4. Resistance of H. pylori to oxidative stress
The goal of the production of ROS by the host cells is to limit the development of pathogens. However, bacteria, including H. pylori, have elaborated strategies to counteract the deleterious effect of ROS by different mechanisms (Fig. 3). Superoxide dismutase (SOD) catalyzes the dismutation of O2•− to H2O2, which is subsequently dismutated into H2O through two electron reactions by catalase. H. pylori also express enzymes with reducing activity on peroxides.
Fig. 3.

Biochemical pathways used by H. pylori to counteract ROS. The enzyme KatA is transported to the periplasm by the action of KapA and the Tat system. O2•− is converted into H2O by SodB and KatA. Hydroperoxides, peroxynitrites, and H2O2 are reduced by the Prx/TrxA system. NapA protects H. pylori from DNA oxidation by an unknown mechanism. NpH and MutS are involved in oxidative DNA repair.
4.1. SOD
H. pylori expresses a single SOD encoded by the gene sodB [74, 75], which contrast to Escherichia coli that possesses three SODs [76]. H. pylori SOD consist of two identical subunits of 24 kDa and is an iron SOD [75]. H. pylori sodB mutants do not exhibit SOD activity and are highly susceptible to O2 and O2•− [77, 78] and, more surprisingly, to H2O2 [77]. Consequently, accumulation of ROS in an sodB-deficient strain led to an increase of mutagenesis frequency compared to WT strain [77]. Moreover, it has been reported that the enzyme SodB is essential for H. pylori to colonize mice [77], demonstrating that O2•− and/or H2O2 are effectively released in vivo and target the bacteria in the gastric mucosa. It has been also shown that the H. pylori strains that are associated with gastric cancer exhibit more SOD activity than those carried by patients with gastritis or gastric and duodenal ulcers, further evidencing that SodB has a major function in the virulence of H. pylori [79].
4.2. Catalase
The gene katA of H. pylori encodes a catalase that does not require NADPH binding for its activity [80]. Catalase possesses a high isoelectric point and resists a very high concentration of H2O2 [80, 81], but is inhibited by organic hydroperoxides [82]. The KatA-associated protein (KapA), which is encoded by the kapA gene located downstream of katA, is involved in the resistance of H. pylori to H2O2 [81]. Through a mechanism commonly described as “hitchhiking”, KatA binds to KapA and the heterologous protein complex is translocated to the periplasm by the twin-arginine translocation system [83, 84].
H. pylori katA− strains are viable in vitro [85], demonstrating that H2O2 endogenously generated by the bacterium itself is not a significant problem for H. pylori. However, catalase is essential for resistance to ROS because high concentrations of H2O2 kill katA- and kapA-deficient strains [81, 83]. In addition, strains lacking catalase activity and exposed to sublethal doses of ROS show more DNA oxidative damage than the parental H. pylori [78]. Accordingly, katA− strains cannot survive in neutrophils or macrophages after phagocytosis [86] and, as with the sodB mutant, the katA and kapA mutants fail to successfully colonize the stomach of mice when compared to the matched WT H. pylori [83]. This demonstrates that H. pylori catalase activity is implicated in the persistence of H. pylori within its ecological niche.
4.3. Peroxiredoxins
Bacterial peroxiredoxin (Prx) enzymes reduce H2O2, organic hydroperoxides, and peroxinitrite that is formed by the reaction of O2•− and NO. H. pylori contains three Prx forms: the thiol-specific peroxidase (Tpx), the alkyl hydroperoxide reductase (AhpC), and the bacterioferritin comigratory protein (BCP).
A two-dimensional gel analysis has evidenced that Tpx is an abundant protein in H. pylori [87]. The increased sensitivity of the tpx mutant to O2•−, H2O2, and organic hydroperoxide [88] confirms an in vivo role for Tpx as an important antioxidant protein in H. pylori. Hence, the H. pylori SS1 Δtpx strain was recovered from the stomach of only 5% of the inoculated mice at 3-weeks post-infection, whereas the WT strain colonized 78% of animals in the same experiment [89].
AhpC is the most abundant antioxidant protein in H. pylori. Oligomers of AhpC exhibit a peroxide-reductase activity, whereas high-molecular-weight complexes are molecular chaperones for prevention of protein misfolding under oxidative stress [90]. A mutant of H. pylori lacking the gene ahpC was more sensitive to oxidative stress conditions induced by H2O2 and also contained significantly higher amounts of 8-OHdG associated with its DNA, compared to WT bacteria [78, 82]. It has also been shown that AhpC is involved in the resistance to O2•− [91], but the molecular mechanism has not been demonstrated and it may correspond to the resistance to H2O2 generated by the activity of SOD. Interestingly, the ahpC mutant contains three times more hydroperoxides than WT [82]; since hydroperoxides inhibit KatA, the ΔahpC strain exhibits a significant decrease of catalase activity [82], demonstrating a cooperation between the different systems of resistance to ROS to further improve the survival of the bacterium against hostile oxidative conditions. Finally, it has been described that the ahpC mutant does not colonize mice [89].
Comtois et al. reported that the role of BCP in resistance to oxidative stress is minimal [88]. This could be attributed to the fact that BCP is less abundant than AhpC in H. pylori [91]. Wang et al. further demonstrated that BCP preferentially reduces linoleic acid hydroperoxide, and not H2O2 [91]. The deletion of bcp in H. pylori is associated with a modest reduction of gastric colonization compared to WT [91]. The specificity of BCP for organic hydroperoxide substrates probably justifies why the bcp deletion has a less dramatic effect on gastric colonization than the mutation of tpx or ahpC genes.
H. pylori lacks the glutathione system, which is essential for cellular thiol:disulfide balance and survival under oxidative stress in many Gram-negative bacteria [92]. Consequently, this bacterium uses the thioredoxin (Trx):Trx reductase (TrxR) system as an electron donor [91]. Two Trx exist in H. pylori: TrxA (also called Trx1) and TrxC (Trx2). While TrxA is more involved in preventing nitrogen-dependent damage than TrxC [88], both reductases have been shown to play a major function in the resistance against oxygen stress from O2•− and H2O2 [88, 93]. However, if TrxA forms a reductase system for AhpC [94] and BCP [91] and is a chaperone for H. pylori arginase RocF [95], the mechanism by which TrxC protects bacteria remains unknown. Nonetheless, the deletion of trxA yields a diminution of stomach colonization by H. pylori [88, 93], while the trxC mutant does not colonize the animals [93], demonstrating that both Trx enzymes, but mainly TrxC, are essential for H. pylori colonization and persistence. In regard to this data, the study of TrxC deserves further investigation.
4.4. NapA
As described above, NapA is an essential bacterial factor that induces ROS production by PMNs. In addition to this inducing effect, NapA also displays a role in the bacterium itself. Thus, the napA mutant is more sensitive to oxygen [96] and oxidative stress [67, 96] and therefore exhibits more bacterial DNA damage than the WT strain [67]. It has been proposed that NapA could bind and protect bacterial DNA from oxidative damages [97]. Remarkably, NapA is mainly induced when the main proteins involved in ROS detoxification, e.g., SodB, AhpC, Tpx, are absent in H. pylori [98], demonstrating an elaborated transcriptional plasticity of the genes encoding antioxidant effectors.
In a co-infection model, the napA− strain exhibited reduced ability to survive in the mouse stomach when competing with the WT parental strain [67]. This suggests that NapA is also involved in the combat of H. pylori against oxidative stress by an unidentified mechanism.
4.5. NADPH quinone reductase
The NADPH quinone reductase MdaB is a flavoprotein that catalyzes two-electron transfer from NADPH to ubiquinones and menaquinones. MdaB is involved in the resistance of H. pylori against O2, H2O2, and the O2•− donor paraquat [98, 99], but the molecular mechanism remains unidentified. It could be proposed that MdaB may act as an O2•− scavenger as its human homolog NAD(P)H:quinone oxidoreductase 1 [100]. Moreover, the mdaB mutant colonized mice less efficiently than the WT [99].
5. Genomic plasticity in response to ROS
The oxidative environment that surrounds H. pylori can result in DNA alterations in the pathogen. Exposure of H. pylori to methyl viologen, an O2•− donor, significantly increases point mutations, intergenomic recombination, and rearrangements between direct DNA repeats [101]. These mutations are transient and limited to the duration of stress, and do not lead to a global hypermutator phenotype. Intriguingly, in silico analyses have demonstrated that DNA repeats are especially concentrated in the H. pylori cag pathogenicity island [101]. Intriguingly, a study performed in rhesus macaques has shown that a mutation burst is observed in the H. pylori genome during the acute infection phase that is over 10 times faster than the mutation rate during chronic infection [102], demonstrating that the innate immune response may play a critical role in H. pylori genomic changes.
Nevertheless, compared to other pathogenic bacteria, the arsenal of H. pylori to counteract DNA oxidative damages is limited: it lacks translesion DNA synthesis polymerases, the DNA methyl-directed mismatch repair system (MutSHL), the SOS response, and the stress-induced sigma factors RpoS and RpoH [103, 104]. O’Rourke et al. have demonstrated that an H. pylori mutant lacking the endonuclease III Nph, is more sensitive to H2O2, exhibits more mutation frequencies when exposed to H2O2 or to activated macrophages [105]. NpH is a DNA glycosylase that removes oxidized nucleobases and repairs oxidized pyrimidines [106], which are recognized to block DNA replication and transcription and are thus more cytotoxic. This highlights that the ROS produced by the host are effectively able to provoke lethal DNA adducts in the H. pylori genome and that the presence of NpH can favor H. pylori survival. A mutS homolog has also been identified in the H. pylori genome. H. pylori mutS− is more sensitive to H2O2 and paraquat and shows increased mutation (mainly G:C to T:A) rates under oxidative stress conditions [107], demonstrating that MutS is involved in DNA repair in response to ROS. The authors have also demonstrated that MutS has high specific affinity to dsDNA containing oxidized purines (such as 8-oxoguanosine), which are mutated bases that are mostly mutagenic [107].
Of importance, a 1–2-log order decreased colonization of the mutS mutant compared to the parental strain has been demonstrated at day 90 post-infection in mice [107]. In contrast, the nph-deficient strain completely fails to colonize mice after 60 days [105]. These data emphasize that DNA repair in H. pylori seems to have higher specificity for cytotoxic base lesions than mutagenic base lesions, thus contributing to the high genetic variability that is observed among H. pylori strains [102].
To conclude, ROS-induced high mutation rate, associated with a reduced proofreading capacity of its DNA polymerase I [108], increases the chance of H. pylori acquiring mutations. This can then confer an adaptive advantage in the gastric environment and in response to the selective pressure imposed by host immunity.
6. Endogenous effect of ROS on the host
6.1. Cell signaling and inflammation
Inflammation is an important hallmark of H. pylori infection that represents a universal response to the infection, and production of ROS has been strongly implicated in the risk for neoplastic progression of H. pylori-induced disease. There are numerous potential targets of ROS within the cell, and their reaction with thiol residue-containing proteins, such as phosphatase or thioredoxin, can therefore regulate cell signaling [109].
Hence, the stimulation of AGS cells by H. pylori results in a rapid phosphorylation of V-Akt murine thymoma viral oncogene homolog 1 (AKT1) independently of CagA, leading to the induction of autophagy through p53 downregulation [110]. In this study, the authors also demonstrate that the treatment of the cells with the antioxidant N-acetylcysteine inhibits AKT1 activation [110], demonstrating the role of ROS in this molecular process. In the same way, Ngo et al. demonstrated that the oxidative burst induced by H. pylori in human gastric epithelial cells is responsible for the PI3K/AKT1-dependent expression of the transcription factor Snail family zinc finger 1 (SNAI1, also known as SNAIL) [111], which is essential for epithelial-mesenchymal transition and therefore in gastric cancer progression and metastasis. Additionally, it has been shown that the transcription factor signal transducer and activator of transcription 3 (STAT3), which plays a major function in inflammation and angiogenesis, is also induced in gastric epithelial cells via upregulated autocrine IL-6 signaling, partially mediated by endogenous ROS [112]. Moreover, α-lipoic acid, a naturally occurring thiol compound that exhibits antioxidant properties, inhibits NOX-derived ROS production in AGS cells and concomitantly dampens NF-κB and AP-1 activation and expression of the oncogenes β-catenin (CTNNB1) and MYC [113]. Lastly, it has been described that the production of vascular endothelial growth factor by H. pylori-infected gastric epithelial cells is dependent on the stabilization of the transcription factor hypoxia-inducible factor 1-alpha (HIF-1α) through ROS synthesis [114]. These data indicate that ROS signaling in gastric epithelial cells may contribute to cell transformation and gastric cancer development.
ROS production may also regulate the signaling events in myeloid cells. Thus, H. pylori induces IL-1β secretion through the inflammasome in murine bone marrow-derived macrophages and human peripheral blood mononuclear cells; this is inhibited by the use of the anti-oxidant molecule ebselen, suggesting that H. pylori-induced ROS are involved in the activation of the immune response of myeloid cells [115]. This effect was observed in the early events post-infection [115], suggesting that the oxidative burst, rather than the effect of SMOX is involved. These results are also consistent with in vitro data showing that the partial scavenging of ROS by N-acetylcysteine led to a decrease of H. pylori-induced inflammasome formation and production of IL-1β and IL-18 by the human monocytic cell line THP-1 [116].
Tsugawa et al have shown that VacA signals in human gastric epithelial cells through the receptor LDL receptor related protein 1 (LRP1) to decrease glutathione (GSH) levels and therefore increase ROS accumulation, leading to CagA degradation by autophagy [110]. Of note, CagA accumulates in CD44-positive gastric cancer stem-like cells because they retain a high level of GSH and resist ROS [110]. This study further highlights the important role of ROS in H. pylori carcinogenesis, not only by inducing ROS producing enzyme, but by altering GSH content.
Chronic granulomatous disease (CGD) mice are animals with a targeted disruption of the NOX2/gp91phox subunit. At 12 and 30-weeks post-infection with H. pylori, CGD mice showed no differences in colonization, but more glandular atrophy, proliferation of gastric epithelial cells, and neutrophil infiltration [117]. But overall, there were no significant changes in gastritis score between WT and CGD mice [117]. In contrast, a second study reported that gp91phox−/− mice exhibit more inflammation and increased mononuculear cell infiltration in the mucosa during infection with H. pylori [118].
6.2. Apoptosis and DNA damage
Apoptosis and formation of DNA damage are critical for H. pylori-induced carcinogenesis. Thus, the implication of ROS in the regulation of these both events has been extensively studied.
An increase of 8-hydroxy-2-deoxyguanosine (8-OHdG), a reliable biomarker of DNA oxidative damage, has been observed in cagA-positive H. pylori-infected patients compared to those uninfected or infected with cagA-negative strains [50, 119–121]; further, a correlation between the production of ROS and the level of 8-OHdG [121] and between 8-OHdG and the severity of the gastritis [120] has been evidenced. In addition, increased 8-OHdG staining has been associated with more advanced histopathologic lesions of atrophic gastritis and intestinal metaplasia in subjects from a high cancer risk region of Colombia [51, 122].
The gastric epithelial cell lines AGS or Kato III infected with H. pylori undergo apoptosis through a mechanism involving mitochondrial depolarization caused by H2O2 [49, 123–125], and this can be blocked by catalase or by antioxidants like vitamin E or N-acetylcyteine [49, 124, 125]. Further, Hayakawa et al. have also described that the rapid oxidative burst of H. pylori-infected gastric epithelial cells in vitro activates the apoptosis signal-regulating kinase 1 (ASK1) that ultimately triggers c-Jun N-terminal kinase (JNK) phosphorylation and cell apoptosis [125]. Remarkably. Ding et al. have reported that generation of ROS by both the bacteria themselves and the infected gastric epithelial cells mediate apoptosis [123].
It has been shown that SMOX is associated with apoptosis in macrophages [47] and in gastric epithelial cells [49], because the use of the SMOX inhibitor MDL 72527 or gene silencing by Smox siRNA in the mouse cells and SMOX siRNA in human cells reduces H2O2 synthesis, mitochondrial membrane depolarization, cytochrome c release, and caspase-3 activation. Moreover, SMOX expression in isolated epithelial cells from the gastric tissues of H. pylori-infected gerbils correlates with 8-oxoguanosine formation, as detected by flow cytometry [50]. Of note, there is a subpopulation of SMOX-expressing cells with oxidative DNA damage that were resistant to apoptosis [50], which may contribute to the development of gastric cancer. Further, the treatment with either the ODC inhibitor α-difluoromethylornithine or MDL 72527, reduces the abundance of the subpopulation of 8-oxoguanosine+ cells that were resistant to apoptosis, and were associated with gastric dysplasia and carcinoma in H. pylori-infected gerbils [51]. Furthermore, reduced levels of SMOX, DNA damage, and DNA damagehigh, apoptosislow cells have been observed in H. pylori-stimulated gastric epithelial cells derived from mice lacking the gene encoding the epidermal growth factor receptor (EGFR) [126]. Similar results were obtained in cells isolated from infected mice with disruption of EGFR signaling (Egfrwa5 mice) when compared to WT mice [126]. Together, these data suggest that EGFR is involved in H. pylori-mediated SMOX expression. Additionally, the kinase Erb-B2 receptor tyrosine kinase 2 (ERBB2) was shown to be critical for the cellular events associated with EGFR signaling in gastric epithelial cells; specifically, heterodimerization of pEGFR and ERBB2 was shown to lead to phosphorylation of ERBB2 and apoptosis resistance [126]. These data strongly support the contention that SMOX-derived ROS are key metabolites that link H. pylori infection to gastric cancer. Further evidence should arise from studies in Smox-deficient mice, which are ongoing in our laboratory.
6.3. Regulation of CagA signaling
CagA is phosphorylated in gastric epithelial cells by SRC [8]. pCagA then activates SHP2 by binding to SH2 domains, causing conformation change and activation without phosphorylation of C-terminal tyrosine residues [13]. SHP2 activation leads to signaling through ERK1/2 to stimulate cell proliferation, differentiation, and survival [14–16].
It has been shown that ROS induce SRC activation [127] and that CagA-independent generation of ROS contributes to H. pylori-induced SHP2 activation [128]. Specifically, the treatment of the human gastric epithelial cell line, MKN45, and the human monocytic cell line, U937, by the anti-oxidant N-acetylcyteine decreases H. pylori-induced SHP2 phosphorylation [128]. In contrast, it has been proposed that ROS, and mainly H2O2, reversibly inactivates protein tyrosine phosphatases, such as SHP2, by causing the oxidation of cysteine residues and formation of disulfide bonds [129, 130].
The contribution of ROS in the regulation of CagA phosphorylation and signaling in H. pylori-infected gastric epithelial cells deserves further investigation. The purported paradoxical effects of ROS could represent another example of the bacterium manipulating host cells to affect signaling events that benefit the bacterium.
7. Conclusions
The parasite-like lifestyle of H. pylori has been shaped by hundreds of thousands of years of coevolution with the human host. Thus, since the host expresses multiple ROS-producing enzymes that are redundant in gastric somatic and immune cells, H. pylori has elaborated strategies to counteract oxidative damage and increase its own survival. However, it is interesting to note that this bacterium possesses a limited arsenal to repair DNA damage; the bacterium does not tend to respond to ROS-induced mutagenic mutations, thus contributing to the high genetic variability that is observed among H. pylori strains. Therefore, we can raise the question of whether H. pylori favors its own genetic variation and adaptability to its niche by inducing ROS production by the host.
The current knowledge about the resistance of H. pylori to the effectors of the innate immune response has evidenced one intriguing feature: the bacterium fights the nitrosative stress and also inhibits NO production by the host [35]; however, it appears to only inhibit ROS-dependent oxidative damage without attenuating the expression/activity of SMOX or NOX. This could be explained by the fact that the most reactive nitrogen species result from the reaction of NO and ROS (e.g. peroxinitrite or dinitrogen trioxide), and that H. pylori may conserve energy by inhibiting just NO production, and not both NO and ROS. Therefore, it is conceivable that ROS are beneficial for the bacterium by favoring genomic variation required for long-term persistence and are deleterious for the host since their production is not inhibited by H. pylori.
As experimental systems have continued to evolve, in addition to standard animal models (mouse, gerbils, and monkeys) and in vitro models, we now have access to various three and two dimensional gastric organoid systems from rodents and humans in which to recapitulate the microenvironment of the bacterium and the host, which will be very powerful. We are hopeful that these models and increased studies of human and bacterial interactions will counteract some of the inherent limitations of animal models, to better ascertain the role of polyamines and ROS in H. pylori infection.
Recent analysis of the phylogeographic origins of both the human hosts and the infecting H. pylori strains have highlighted that the risk of developing neoplastic lesions is mainly associated with a lack of coevolution between the host and the bacterium [131]. In this context, a future challenge will be to include human ancestry and H. pylori phylogeographic origin in the analysis of the expression and the role of ROS-producing systems.
Highlights.
ROS are locally produced in the gastric mucosa during H. pylori infection
Spermine oxidase and NADPH oxidases are the main sources of ROS in the stomach
H. pylori has multiple strategies to resist oxidative damage
ROS-induced mutagenic changes in H. pylori leads to genetic variation and adaptation
Oxidative stress contributes to H. pylori-induced inflammation and carcinogenesis
Acknowledgments
This work was supported by NIH grants R01DK053620, R01AT004821, R01CA190612, P01CA028842, and P01CA116087 (to K.T.W.), a Department of Veterans Affairs Merit Review grant I01BX001453 (to K.T.W.), the Thomas F. Frist Sr. Endowment (K.T.W.), the Vanderbilt Center for Mucosal Inflammation and Cancer (K.T.W. and A.P.G.) and the Vanderbilt Digestive Disease Research Center, funded by P30DK058404 (K.T.W.).
Abbreviations
- 8-OHdG
8-hydroxy-2-deoxyguanosine
- ABL1
ABL proto-oncogene 1
- AhpC
alkyl hydroperoxide reductase
- AKT1
V-Akt murine thymoma viral oncogene homolog 1
- APEX1
endonuclease apurinic/apyrimidinic endodeoxyribonuclease 1
- ARG1
arginase 1
- ARG2
arginase 2
- ASK1
apoptosis signal-regulating kinase
- BCP
bacterioferritin comigratory protein
- CagA
cytotoxin-associated gene A
- cagPAI
cag pathogenicity island
- CGD
chronic granulomatous disease
- DUOX
dual oxidase
- EGFR
epidermal growth factor receptor
- EPIYA
Glu-Pro-Ile-Tyr-Ala
- ERBB2
Erb-B2 receptor tyrosine kinase 2
- ERK1/2
extracellular signal-regulated kinase 1/2
- FOS
FBJ murine osteosarcoma viral oncogene homolog
- GSH
glutathione
- HIF-1α
hypoxia-inducible factor 1-alpha
- JNK
c-Jun N-terminal kinase
- JUN
Jun proto-oncogene
- LPS
lipopolysaccharide
- LRP1
receptor LDL receptor related protein 1
- MYC
V-Myc avian myelocytomatosis viral oncogene homolog
- NapA
neutrophil-activating protein A
- NF-κB
nuclear factor-kappa B
- NO
nitric oxide
- NOS2
inducible nitric oxide synthase 2
- NOX
NADPH oxidase
- ODC
ornithine decarboxylase
- PAOX
polyamine oxidase
- PI3K
phosphoinositide 3-kinase
- PMN
polymorphonuclear neutrophil
- Prx
peroxiredoxin
- RAC1
Ras-related C3 botulinum toxin substrate 1
- ROS
reactive oxygen species
- SAT1
spermidine/spermine N(1)-acetyltransferase 1
- SLC7A2
solute carrier family 7 (cationic amino acid transporter, y+ system), member 2
- SMOX
spermine oxidase
- SNAI1
snail family zinc finger 1
- SOD
superoxide dismutase
- SRC
SRC proto-oncogene
- STAT3
signal transducer and activator of transcription 3
- T4SS
type 4 secretion system
- Tpx
thiol-specific peroxidase
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- VacA
vacuolating cytotoxin A
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blaser MJ, Chen Y, Reibman J. Does Helicobacter pylori protect against asthma and allergy? Gut. 2008;57:561–7. doi: 10.1136/gut.2007.133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–9. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 3.Vohlonen I, Pukkala E, Malila N, Harkonen M, Hakama M, Koistinen V, Sipponen P. Risk of gastric cancer in Helicobacter pylori infection in a 15-year follow-up. Scand J Gastroenterol. 2016;51:1159–64. doi: 10.1080/00365521.2016.1183225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malfertheiner P, Megraud F, O’Morain CA, Atherton J, Axon AT, Bazzoli F, Gensini GF, Gisbert JP, Graham DY, Rokkas T, El-Omar EM, Kuipers EJ G. European Helicobacter Study. Management of Helicobacter pylori infection--the Maastricht IV/ Florence Consensus Report. Gut. 2012;61:646–64. doi: 10.1136/gutjnl-2012-302084. [DOI] [PubMed] [Google Scholar]
- 5.Marcus EA, Sachs G, Scott DR. Eradication of Helicobacter pylori infection. Curr Gastroenterol Rep. 2016;18:33. doi: 10.1007/s11894-016-0509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan DR, Torres J, Sexton R, Herrero R, Salazar-Martinez E, Greenberg ER, Bravo LE, Dominguez RL, Ferreccio C, Lazcano-Ponce EC, Meza-Montenegro MM, Pena EM, Pena R, Correa P, Martinez ME, Chey WD, Valdivieso M, Anderson GL, Goodman GE, Crowley JJ, Baker LH. Risk of recurrent Helicobacter pylori infection 1 year after initial eradication therapy in 7 Latin American communities. JAMA. 2013;309:578–86. doi: 10.1001/jama.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ford AC, Forman D, Hunt RH, Yuan Y, Moayyedi P. Helicobacter pylori eradication therapy to prevent gastric cancer in healthy asymptomatic infected individuals: systematic review and meta-analysis of randomised controlled trials. BMJ. 2014;348:g3174. doi: 10.1136/bmj.g3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553–66. doi: 10.1172/JCI61143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naito M, Yamazaki T, Tsutsumi R, Higashi H, Onoe K, Yamazaki S, Azuma T, Hatakeyama M. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology. 2006;130:1181–90. doi: 10.1053/j.gastro.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 10.Duncan SS, Valk PL, Shaffer CL, Bordenstein SR, Cover TL. J-Western forms of Helicobacter pylori cagA constitute a distinct phylogenetic group with a widespread geographic distribution. J Bacteriol. 2012;194:1593–604. doi: 10.1128/JB.06340-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones KR, Joo YM, Jang S, Yoo YJ, Lee HS, Chung IS, Olsen CH, Whitmire JM, Merrell DS, Cha JH. Polymorphism in the CagA EPIYA motif impacts development of gastric cancer. J Clin Microbiol. 2009;47:959–68. doi: 10.1128/JCM.02330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaoka Y, El-Zimaity HM, Gutierrez O, Figura N, Kim JG, Kodama T, Kashima K, Graham DY. Relationship between the cagA 3′ repeat region of Helicobacter pylori, gastric histology, and susceptibility to low pH. Gastroenterology. 1999;117:342–9. doi: 10.1053/gast.1999.0029900342. [DOI] [PubMed] [Google Scholar]
- 13.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–6. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 14.Backert S, Moese S, Selbach M, Brinkmann V, Meyer TF. Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol Microbiol. 2001;42:631–44. doi: 10.1046/j.1365-2958.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 15.Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, Higuchi M, Takahashi A, Kurashima Y, Teishikata Y, Tanaka S, Azuma T, Hatakeyama M. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205–16. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 16.Umeda M, Murata-Kamiya N, Saito Y, Ohba Y, Takahashi M, Hatakeyama M. Helicobacter pylori CagA causes mitotic impairment and induces chromosomal instability. J Biol Chem. 2009;284:22166–72. doi: 10.1074/jbc.M109.035766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segal ED, Lange C, Covacci A, Tompkins LS, Falkow S. Induction of host signal transduction pathways by Helicobacter pylori. Proc Natl Acad Sci USA. 1997;94:7595–9. doi: 10.1073/pnas.94.14.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, Nagai S, Koyasu S, Gilman RH, Kersulyte D, Berg DE, Sasakawa C. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5:23–34. doi: 10.1016/j.chom.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Gobert AP, Verriere T, de Sablet T, Peek RM, Jr, Chaturvedi R, Wilson KT. Haem oxygenase-1 inhibits phosphorylation of the Helicobacter pylori oncoprotein CagA in gastric epithelial cells. Cell Microbiol. 2013;15:145–56. doi: 10.1111/cmi.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–5. [PubMed] [Google Scholar]
- 21.Supajatura V, Ushio H, Wada A, Yahiro K, Okumura K, Ogawa H, Hirayama T, Ra C. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J Immunol. 2002;168:2603–7. doi: 10.4049/jimmunol.168.6.2603. [DOI] [PubMed] [Google Scholar]
- 22.Raju D, Hussey S, Ang M, Terebiznik MR, Sibony M, Galindo-Mata E, Gupta V, Blanke SR, Delgado A, Romero-Gallo J, Ramjeet MS, Mascarenhas H, Peek RM, Correa P, Streutker C, Hold G, Kunstmann E, Yoshimori T, Silverberg MS, Girardin SE, Philpott DJ, El Omar E, Jones NL. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology. 2012;142:1160–71. doi: 10.1053/j.gastro.2012.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atherton JC, Peek RM, Jr, Tham KT, Cover TL, Blaser MJ. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology. 1997;112:92–9. doi: 10.1016/s0016-5085(97)70223-3. [DOI] [PubMed] [Google Scholar]
- 24.Noto JM, Gaddy JA, Lee JY, Piazuelo MB, Friedman DB, Colvin DC, Romero-Gallo J, Suarez G, Loh J, Slaughter JC, Tan S, Morgan DR, Wilson KT, Bravo LE, Correa P, Cover TL, Amieva MR, Peek RM., Jr Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J Clin Invest. 2013;123:479–92. doi: 10.1172/JCI64373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fox JG, Rogers AB, Ihrig M, Taylor NS, Whary MT, Dockray G, Varro A, Wang TC. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res. 2003;63:942–50. [PubMed] [Google Scholar]
- 26.Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48:3554–60. [PubMed] [Google Scholar]
- 27.Chaturvedi R, Asim M, Hoge S, Lewis ND, Singh K, Barry DP, de Sablet T, Piazuelo MB, Sarvaria AR, Cheng Y, Closs EI, Casero RA, Jr, Gobert AP, Wilson KT. Polyamines impair immunity to Helicobacter pylori by inhibiting L-Arginine uptake required for nitric oxide production. Gastroenterology. 2010;139:1686–98. doi: 10.1053/j.gastro.2010.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeramian A, Martin L, Serrat N, Arpa L, Soler C, Bertran J, McLeod C, Palacin M, Modolell M, Lloberas J, Celada A. Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J Immunol. 2006;176:5918–24. doi: 10.4049/jimmunol.176.10.5918. [DOI] [PubMed] [Google Scholar]
- 29.Barry DP, Asim M, Scull BP, Piazuelo MB, de Sablet T, Lewis ND, Coburn LA, Singh K, Ellies LG, Gobert AP, Chaturvedi R, Wilson KT. Cationic amino acid transporter 2 enhances innate immunity during Helicobacter pylori infection. Plos One. 2011;6:e29046. doi: 10.1371/journal.pone.0029046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gobert AP, Verriere T, Asim M, Barry DP, Piazuelo MB, de Sablet T, Delgado AG, Bravo LE, Correa P, Peek RM, Jr, Chaturvedi R, Wilson KT. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to Helicobacter pylori. J Immunol. 2014;193:3013–22. doi: 10.4049/jimmunol.1401075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gobert AP, Cheng Y, Wang JY, Boucher JL, Iyer RK, Cederbaum SD, Casero RA, Jr, Newton JC, Wilson KT. Helicobacter pylori induces macrophage apoptosis by activation of arginase II. J Immunol. 2002;168:4692–700. doi: 10.4049/jimmunol.168.9.4692. [DOI] [PubMed] [Google Scholar]
- 32.Lewis ND, Asim M, Barry DP, Singh K, de Sablet T, Boucher JL, Gobert AP, Chaturvedi R, Wilson KT. Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J Immunol. 2010;184:2572–82. doi: 10.4049/jimmunol.0902436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, Gobert AP, Chaturvedi R, Wilson KT. Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. J Immunol. 2011;186:3632–41. doi: 10.4049/jimmunol.1003431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardbower DM, Asim M, Murray-Stewart T, Casero RA, Jr, Verriere T, Lewis ND, Chaturvedi R, Piazuelo MB, Wilson KT. Arginase 2 deletion leads to enhanced M1 macrophage activation and upregulated polyamine metabolism in response to Helicobacter pylori infection. Amino Acids. 2016 doi: 10.1007/s00726-016-2231-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gobert AP, Wilson KT. The immune battle against Helicobacter pylori infection: NO offense. Trends Microbiol. 2016;24:366–76. doi: 10.1016/j.tim.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konturek PC, Rembiasz K, Konturek SJ, Stachura J, Bielanski W, Galuschka K, Karcz D, Hahn EG. Gene expression of ornithine decarboxylase, cyclooxygenase-2, and gastrin in atrophic gastric mucosa infected with Helicobacter pylori before and after eradication therapy. Dig Dis Sci. 2003;48:36–46. doi: 10.1023/a:1021774029089. [DOI] [PubMed] [Google Scholar]
- 37.Patchett SE, Alstead EM, Butruk L, Przytulski K, Farthing MJ. Ornithine decarboxylase as a marker for premalignancy in the stomach. Gut. 1995;37:13–6. doi: 10.1136/gut.37.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linsalata M, Russo F, Notarnicola M, Berloco P, Di Leo A. Polyamine profile in human gastric mucosa infected by Helicobacter pylori. Ital J Gastroenterol Hepatol. 1998;30:484–9. [PubMed] [Google Scholar]
- 39.Elitsur Y, Majumdar AP, Tureaud J, Dosescu J, Neace C, Velusamy L, Moshier JA. Tryosine kinase and ornithine decarboxylase activation in children with Helicobacter pylori gastritis. Life Sci. 1999;65:1373–80. doi: 10.1016/s0024-3205(99)00376-8. [DOI] [PubMed] [Google Scholar]
- 40.Patchett SE, Katelaris PH, Zhang ZW, Alstead EM, Domizio P, Farthing MJ. Ornithine decarboxylase activity is a marker of premalignancy in longstanding Helicobacter pylori infection. Gut. 1996;39:807–10. doi: 10.1136/gut.39.6.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alam K, Arlow FL, Ma CK, Schubert TT. Decrease in ornithine decarboxylase activity after eradication of Helicobacter pylori. Am J Gastroenterol. 1994;89:888–93. [PubMed] [Google Scholar]
- 42.Hirasawa R, Tatsuta M, Iishi H, Yano H, Baba M, Uedo N, Sakai N. Increase in apoptosis and decrease in ornithine decarboxylase activity of the gastric mucosa in patients with atrophic gastritis and gastric ulcer after successful eradication of Helicobacter pylori. Am J Gastroenterol. 1999;94:2398–402. doi: 10.1111/j.1572-0241.1999.01350.x. [DOI] [PubMed] [Google Scholar]
- 43.Messa C, Di Leo A, Greco B, Caradonna L, Amati L, Linsalata M, Giorgio I, Jirillo E. Successful eradicating treatment of Helicobacter pylori in patients with chronic gastritis: gastric levels of cytokines, epidermal growth factor and polyamines before and after therapy. Immunopharmacol Immunotoxicol. 1996;18:1–13. doi: 10.3109/08923979609007106. [DOI] [PubMed] [Google Scholar]
- 44.Xu X, Liu Z, Fang M, Yu H, Liang X, Li X, Liu X, Chen C, Jia J. Helicobacter pylori CagA induces ornithine decarboxylase upregulation via Src/MEK/ERK/c-Myc pathway: implication for progression of gastric diseases. Exp Biol Med (Maywood) 2012;237:435–41. doi: 10.1258/ebm.2011.011199. [DOI] [PubMed] [Google Scholar]
- 45.Cheng Y, Chaturvedi R, Asim M, Bussiere FI, Scholz A, Xu H, Casero RA, Jr, Wilson KT. Helicobacter pylori-induced macrophage apoptosis requires activation of ornithine decarboxylase by c-Myc. J Biol Chem. 2005;280:22492–6. doi: 10.1074/jbc.C500122200. [DOI] [PubMed] [Google Scholar]
- 46.Asim M, Chaturvedi R, Hoge S, Lewis ND, Singh K, Barry DP, Algood HS, de Sablet T, Gobert AP, Wilson KT. Helicobacter pylori induces ERK-dependent formation of a phospho-c-Fos.c-Jun activator protein-1 complex that causes apoptosis in macrophages. J Biol Chem. 2010;285:20343–20357. doi: 10.1074/jbc.M110.116988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaturvedi R, Cheng Y, Asim M, Bussiere FI, Xu H, Gobert AP, Hacker A, Casero RA, Jr, Wilson KT. Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J Biol Chem. 2004;279:40161–73. doi: 10.1074/jbc.M401370200. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Devereux W, Woster PM, Stewart TM, Hacker A, Casero RA., Jr Cloning and characterization of a human polyamine oxidase that is inducible by polyamine analogue exposure. Cancer Res. 2001;61:5370–3. [PubMed] [Google Scholar]
- 49.Xu H, Chaturvedi R, Cheng Y, Bussiere FI, Asim M, Yao MD, Potosky D, Meltzer SJ, Rhee JG, Kim SS, Moss SF, Hacker A, Wang Y, Casero RA, Jr, Wilson KT. Spermine oxidation induced by Helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Res. 2004;64:8521–5. doi: 10.1158/0008-5472.CAN-04-3511. [DOI] [PubMed] [Google Scholar]
- 50.Chaturvedi R, Asim M, Romero-Gallo J, Barry DP, Hoge S, de Sablet T, Delgado AG, Wroblewski LE, Piazuelo MB, Yan F, Israel DA, Casero RA, Jr, Correa P, Gobert AP, Polk DB, Peek RM, Jr, Wilson KT. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology. 2011;141:1696–708. doi: 10.1053/j.gastro.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaturvedi R, de Sablet T, Asim M, Piazuelo MB, Barry DP, Verriere TG, Sierra JC, Hardbower DM, Delgado AG, Schneider BG, Israel DA, Romero-Gallo J, Nagy TA, Morgan DR, Murray-Stewart T, Bravo LE, Peek RM, Jr, Fox JG, Woster PM, Casero RA, Jr, Correa P, Wilson KT. Increased Helicobacter pylori-associated gastric cancer risk in the Andean region of Colombia is mediated by spermine oxidase. Oncogene. 2015;34:3429–40. doi: 10.1038/onc.2014.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 53.Teshima S, Kutsumi H, Kawahara T, Kishi K, Rokutan K. Regulation of growth and apoptosis of cultured guinea pig gastric mucosal cells by mitogenic oxidase 1. Am J Physiol Gastrointest Liver Physiol. 2000;279:G1169–76. doi: 10.1152/ajpgi.2000.279.6.G1169. [DOI] [PubMed] [Google Scholar]
- 54.Salles N, Szanto I, Herrmann F, Armenian B, Stumm M, Stauffer E, Michel JP, Krause KH. Expression of mRNA for ROS-generating NADPH oxidases in the aging stomach. Exp Gerontol. 2005;40:353–7. doi: 10.1016/j.exger.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 55.Tominaga K, Kawahara T, Sano T, Toida K, Kuwano Y, Sasaki H, Kawai T, Teshima-Kondo S, Rokutan K. Evidence for cancer-associated expression of NADPH oxidase 1 (Nox1)-based oxidase system in the human stomach. Free Radic Biol Med. 2007;43:1627–38. doi: 10.1016/j.freeradbiomed.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 56.den Hartog G, Chattopadhyay R, Ablack A, Hall EH, Butcher LD, Bhattacharyya A, Eckmann L, Harris PR, Das S, Ernst PB, Crowe SE. Regulation of Rac1 and reactive oxygen species production in response to infection of gastrointestinal epithelia. PLoS Pathog. 2016;12:e1005382. doi: 10.1371/journal.ppat.1005382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshida LS, Nishida S, Shimoyama T, Kawahara T, Kondo-Teshima S, Rokutan K, Kobayashi T, Tsunawaki S. Superoxide generation by Nox1 in guinea pig gastric mucosal cells involves a component with p67(phox)-ability. Biol Pharm Bull. 2004;27:147–55. doi: 10.1248/bpb.27.147. [DOI] [PubMed] [Google Scholar]
- 58.Kawahara T, Kohjima M, Kuwano Y, Mino H, Teshima-Kondo S, Takeya R, Tsunawaki S, Wada A, Sumimoto H, Rokutan K. Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol Cell Physiol. 2005;288:C450–7. doi: 10.1152/ajpcell.00319.2004. [DOI] [PubMed] [Google Scholar]
- 59.Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–6. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- 60.Bauer G, Bereswill S, Aichele P, Glocker E. Helicobacter pylori protects oncogenically transformed cells from reactive oxygen species-mediated intercellular induction of apoptosis. Carcinogenesis. 2014;35:1582–91. doi: 10.1093/carcin/bgu074. [DOI] [PubMed] [Google Scholar]
- 61.Nielsen H, Andersen LP. Activation of human phagocyte oxidative metabolism by Helicobacter pylori. Gastroenterology. 1992;103:1747–53. doi: 10.1016/0016-5085(92)91430-c. [DOI] [PubMed] [Google Scholar]
- 62.Nielsen H, Birkholz S, Andersen LP, Moran AP. Neutrophil activation by Helicobacter pylori lipopolysaccharides. J Infect Dis. 1994;170:135–9. doi: 10.1093/infdis/170.1.135. [DOI] [PubMed] [Google Scholar]
- 63.Allen LA, Beecher BR, Lynch JT, Rohner OV, Wittine LM. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J Immunol. 2005;174:3658–67. doi: 10.4049/jimmunol.174.6.3658. [DOI] [PubMed] [Google Scholar]
- 64.Evans DJ, Jr, Evans DG, Takemura T, Nakano H, Lampert HC, Graham DY, Granger DN, Kvietys PR. Characterization of a Helicobacter pylori neutrophil-activating protein. Infect Immun. 1995;63:2213–20. doi: 10.1128/iai.63.6.2213-2220.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tonello F, Dundon WG, Satin B, Molinari M, Tognon G, Grandi G, Del Giudice G, Rappuoli R, Montecucco C. The Helicobacter pylori neutrophil-activating protein is an iron-binding protein with dodecameric structure. Mol Microbiol. 1999;34:238–46. doi: 10.1046/j.1365-2958.1999.01584.x. [DOI] [PubMed] [Google Scholar]
- 66.Satin B, Del Giudice G, Della Bianca V, Dusi S, Laudanna C, Tonello F, Kelleher D, Rappuoli R, Montecucco C, Rossi F. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a protective antigen and a major virulence factor. J Exp Med. 2000;191:1467–76. doi: 10.1084/jem.191.9.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang G, Hong Y, Olczak A, Maier SE, Maier RJ. Dual Roles of Helicobacter pylori NapA in inducing and combating oxidative stress. Infect Immun. 2006;74:6839–46. doi: 10.1128/IAI.00991-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mai UE, Perez-Perez GI, Wahl LM, Wahl SM, Blaser MJ, Smith PD. Soluble surface proteins from Helicobacter pylori activate monocytes/macrophages by lipopolysaccharide-independent mechanism. J Clin Invest. 1991;87:894–900. doi: 10.1172/JCI115095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Betten A, Bylund J, Christophe T, Boulay F, Romero A, Hellstrand K, Dahlgren C. A proinflammatory peptide from Helicobacter pylori activates monocytes to induce lymphocyte dysfunction and apoptosis. J Clin Invest. 2001;108:1221–8. doi: 10.1172/JCI13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bjorkman U, Ekholm R. Generation of H2O2 in isolated porcine thyroid follicles. Endocrinology. 1984;115:392–8. doi: 10.1210/endo-115-1-392. [DOI] [PubMed] [Google Scholar]
- 71.Ameziane-El-Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D, Gnidehou S, Ohayon R, Noel-Hudson MS, Francon J, Lalaoui K, Virion A, Dupuy C. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J Biol Chem. 2005;280:30046–54. doi: 10.1074/jbc.M500516200. [DOI] [PubMed] [Google Scholar]
- 72.Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a cag pathogenicity island-dependent manner. Gastroenterology. 2008;134:1049–57. doi: 10.1053/j.gastro.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H, Zhou Y, Zheng Y, Guo H, Gao L, Chen P, Feng D, Wu L, Yang M, Qi Y, Guo H, Chang Y, Chu FF, Gao Q. The gastric mucosa from patients infected with CagA+ or VacA+ Helicobacter pylori has a lower level of dual oxidase-2 expression than uninfected or infected with CagA-/VacA- H. pylori. Dig Dis Sci. 2016;61:2328–37. doi: 10.1007/s10620-016-4144-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spiegelhalder C, Gerstenecker B, Kersten A, Schiltz E, Kist M. Purification of Helicobacter pylori superoxide dismutase and cloning and sequencing of the gene. Infect Immun. 1993;61:5315–25. doi: 10.1128/iai.61.12.5315-5325.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pesci EC, Pickett CL. Genetic organization and enzymatic activity of a superoxide dismutase from the microaerophilic human pathogen, Helicobacter pylori. Gene. 1994;143:111–6. doi: 10.1016/0378-1119(94)90614-9. [DOI] [PubMed] [Google Scholar]
- 76.Benov LT, Fridovich I. Escherichia coli expresses a copper- and zinc-containing superoxide dismutase. J Biol Chem. 1994;269:25310–4. [PubMed] [Google Scholar]
- 77.Seyler RW, Jr, Olson JW, Maier RJ. Superoxide dismutase-deficient mutants of Helicobacter pylori are hypersensitive to oxidative stress and defective in host colonization. Infect Immun. 2001;69:4034–40. doi: 10.1128/IAI.69.6.4034-4040.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang G, Conover RC, Olczak AA, Alamuri P, Johnson MK, Maier RJ. Oxidative stress defense mechanisms to counter iron-promoted DNA damage in Helicobacter pylori. Free Radic Res. 2005;39:1183–91. doi: 10.1080/10715760500194018. [DOI] [PubMed] [Google Scholar]
- 79.Morishita K, Takeuchi H, Morimoto N, Shimamura T, Kadota Y, Tsuda M, Taniguchi T, Ukeda H, Yamamoto T, Sugiura T. Superoxide dismutase activity of Helicobacter pylori per se from 158 clinical isolates and the characteristics. Microbiol Immunol. 2012;56:262–72. doi: 10.1111/j.1348-0421.2012.00433.x. [DOI] [PubMed] [Google Scholar]
- 80.Hazell SL, Evans DJ, Jr, Graham DY. Helicobacter pylori catalase. J Gen Microbiol. 1991;137:57–61. doi: 10.1099/00221287-137-1-57. [DOI] [PubMed] [Google Scholar]
- 81.Harris AG, Hinds FE, Beckhouse AG, Kolesnikow T, Hazell SL. Resistance to hydrogen peroxide in Helicobacter pylori: role of catalase (KatA) and Fur, and functional analysis of a novel gene product designated ‘KatA-associated protein’, KapA (HP0874) Microbiology. 2002;148:3813–25. doi: 10.1099/00221287-148-12-3813. [DOI] [PubMed] [Google Scholar]
- 82.Wang G, Conover RC, Benoit S, Olczak AA, Olson JW, Johnson MK, Maier RJ. Role of a bacterial organic hydroperoxide detoxification system in preventing catalase inactivation. J Biol Chem. 2004;279:51908–14. doi: 10.1074/jbc.M408450200. [DOI] [PubMed] [Google Scholar]
- 83.Harris AG, Hazell SL. Localisation of Helicobacter pylori catalase in both the periplasm and cytoplasm, and its dependence on the twin-arginine target protein, KapA, for activity. FEMS Microbiol Lett. 2003;229:283–9. doi: 10.1016/S0378-1097(03)00850-4. [DOI] [PubMed] [Google Scholar]
- 84.Benoit SL, Maier RJ. Twin-arginine translocation system in Helicobacter pylori: TatC, but not TatB, is essential for viability. MBio. 2014;5:e01016–13. doi: 10.1128/mBio.01016-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Odenbreit S, Wieland B, Haas R. Cloning and genetic characterization of Helicobacter pylori catalase and construction of a catalase-deficient mutant strain. J Bacteriol. 1996;178:6960–7. doi: 10.1128/jb.178.23.6960-6967.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ramarao N, Gray-Owen SD, Meyer TF. Helicobacter pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity. Mol Microbiol. 2000;38:103–13. doi: 10.1046/j.1365-2958.2000.02114.x. [DOI] [PubMed] [Google Scholar]
- 87.Jungblut PR, Bumann D, Haas G, Zimny-Arndt U, Holland P, Lamer S, Siejak F, Aebischer A, Meyer TF. Comparative proteome analysis of Helicobacter pylori. Mol Microbiol. 2000;36:710–25. doi: 10.1046/j.1365-2958.2000.01896.x. [DOI] [PubMed] [Google Scholar]
- 88.Comtois SL, Gidley MD, Kelly DJ. Role of the thioredoxin system and the thiol-peroxidases Tpx and Bcp in mediating resistance to oxidative and nitrosative stress in Helicobacter pylori. Microbiol. 2003;149:121–9. doi: 10.1099/mic.0.25896-0. [DOI] [PubMed] [Google Scholar]
- 89.Olczak AA, Seyler RW, Jr, Olson JW, Maier RJ. Association of Helicobacter pylori antioxidant activities with host colonization proficiency. Infect Immun. 2003;71:580–3. doi: 10.1128/IAI.71.1.580-583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chuang MH, Wu MS, Lo WL, Lin JT, Wong CH, Chiou SH. The antioxidant protein alkylhydroperoxide reductase of Helicobacter pylori switches from a peroxide reductase to a molecular chaperone function. Proc Natl Acad Sci USA. 2006;103:2552–7. doi: 10.1073/pnas.0510770103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang G, Olczak AA, Walton JP, Maier RJ. Contribution of the Helicobacter pylori thiol peroxidase bacterioferritin comigratory protein to oxidative stress resistance and host colonization. Infect Immun. 2005;73:378–84. doi: 10.1128/IAI.73.1.378-384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 93.Kuhns LG, Wang G, Maier RJ. Comparative roles of the two Helicobacter pylori thioredoxins in preventing macromolecule damage. Infect Immun. 2015;83:2935–43. doi: 10.1128/IAI.00232-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baker LM, Raudonikiene A, Hoffman PS, Poole LB. Essential thioredoxin-dependent peroxiredoxin system from Helicobacter pylori: genetic and kinetic characterization. J Bacteriol. 2001;183:1961–73. doi: 10.1128/JB.183.6.1961-1973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McGee DJ, Kumar S, Viator RJ, Bolland JR, Ruiz J, Spadafora D, Testerman TL, Kelly DJ, Pannell LK, Windle HJ. Helicobacter pylori thioredoxin is an arginase chaperone and guardian against oxidative and nitrosative stresses. J Biol Chem. 2006;281:3290–6. doi: 10.1074/jbc.M506139200. [DOI] [PubMed] [Google Scholar]
- 96.Olczak AA, Olson JW, Maier RJ. Oxidative-stress resistance mutants of Helicobacter pylori. J Bacteriol. 2002;184:3186–93. doi: 10.1128/JB.184.12.3186-3193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cooksley C, Jenks PJ, Green A, Cockayne A, Logan RP, Hardie KR. NapA protects Helicobacter pylori from oxidative stress damage, and its production is influenced by the ferric uptake regulator. J Med Microbiol. 2003;52:461–9. doi: 10.1099/jmm.0.05070-0. [DOI] [PubMed] [Google Scholar]
- 98.Olczak AA, Wang G, Maier RJ. Up-expression of NapA and other oxidative stress proteins is a compensatory response to loss of major Helicobacter pylori stress resistance factors. Free Radic Res. 2005;39:1173–82. doi: 10.1080/10715760500306729. [DOI] [PubMed] [Google Scholar]
- 99.Wang G, Maier RJ. An NADPH quinone reductase of Helicobacter pylori plays an important role in oxidative stress resistance and host colonization. Infect Immun. 2004;72:1391–6. doi: 10.1128/IAI.72.3.1391-1396.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Siegel D, Gustafson DL, Dehn DL, Han JY, Boonchoong P, Berliner LJ, Ross D. NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol Pharmacol. 2004;65:1238–47. doi: 10.1124/mol.65.5.1238. [DOI] [PubMed] [Google Scholar]
- 101.Kang JM, Iovine NM, Blaser MJ. A paradigm for direct stress-induced mutation in prokaryotes. Faseb J. 2006;20:2476–85. doi: 10.1096/fj.06-6209com. [DOI] [PubMed] [Google Scholar]
- 102.Linz B, Windsor HM, McGraw JJ, Hansen LM, Gajewski JP, Tomsho LP, Hake CM, Solnick JV, Schuster SC, Marshall BJ. A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nat Commun. 2014;5:4165. doi: 10.1038/ncomms5165. [DOI] [PubMed] [Google Scholar]
- 103.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–47. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 104.Alm RA, Trust TJ. Analysis of the genetic diversity of Helicobacter pylori: the tale of two genomes. J Mol Med (Berl) 1999;77:834–46. doi: 10.1007/s001099900067. [DOI] [PubMed] [Google Scholar]
- 105.O’Rourke EJ, Chevalier C, Pinto AV, Thiberge JM, Ielpi L, Labigne A, Radicella JP. Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. Proc Natl Acad Sci USA. 2003;100:2789–94. doi: 10.1073/pnas.0337641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Laval J, Jurado J, Saparbaev M, Sidorkina O. Antimutagenic role of base-excision repair enzymes upon free radical-induced DNA damage. Mutat Res. 1998;402:93–102. doi: 10.1016/s0027-5107(97)00286-8. [DOI] [PubMed] [Google Scholar]
- 107.Wang G, Alamuri P, Humayun MZ, Taylor DE, Maier RJ. The Helicobacter pylori MutS protein confers protection from oxidative DNA damage. Mol Microbiol. 2005;58:166–76. doi: 10.1111/j.1365-2958.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- 108.Garcia-Ortiz MV, Marsin S, Arana ME, Gasparutto D, Guerois R, Kunkel TA, Radicella JP. Unexpected role for Helicobacter pylori DNA polymerase I as a source of genetic variability. PLoS Genet. 2011;7:e1002152. doi: 10.1371/journal.pgen.1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tsugawa H, Suzuki H, Saya H, Hatakeyama M, Hirayama T, Hirata K, Nagano O, Matsuzaki J, Hibi T. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe. 2012;12:764–77. doi: 10.1016/j.chom.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 111.Ngo HK, Lee HG, Piao JY, Zhong X, Lee HN, Han HJ, Kim W, Kim DH, Cha YN, Na HK, Surh YJ. Helicobacter pylori induces Snail expression through ROS-mediated activation of Erk and inactivation of GSK-3beta in human gastric cancer cells. Mol Carcinog. 2016 doi: 10.1002/mc.22464. [DOI] [PubMed] [Google Scholar]
- 112.Piao JY, Lee HG, Kim SJ, Kim DH, Han HJ, Ngo HK, Park SA, Woo JH, Lee JS, Na HK, Cha YN, Surh YJ. Helicobacter pylori activates IL-6-STAT3 signaling in human gastric cancer cells: Potential roles for reactive oxygen species. Helicobacter. 2016 doi: 10.1111/hel.12298. [DOI] [PubMed] [Google Scholar]
- 113.Byun E, Lim JW, Kim JM, Kim H. alpha-Lipoic acid inhibits Helicobacter pylori-induced oncogene expression and hyperproliferation by suppressing the activation of NADPH oxidase in gastric epithelial cells. Mediators Inflamm. 2014;2014:380830. doi: 10.1155/2014/380830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kang MJ, Song EJ, Kim BY, Kim DJ, Park JH. Helicobacter pylori induces vascular endothelial growth factor production in gastric epithelial cells through hypoxia-inducible factor-1alpha-dependent pathway. Helicobacter. 2014;19:476–83. doi: 10.1111/hel.12169. [DOI] [PubMed] [Google Scholar]
- 115.Semper RP, Mejias-Luque R, Gross C, Anderl F, Muller A, Vieth M, Busch DH, Prazeres da Costa C, Ruland J, Gross O, Gerhard M. Helicobacter pylori-induced IL-1beta secretion in innate immune cells is regulated by the NLRP3 inflammasome and requires the cag pathogenicity island. J Immunol. 2014;193:3566–76. doi: 10.4049/jimmunol.1400362. [DOI] [PubMed] [Google Scholar]
- 116.Li X, Liu S, Luo J, Liu A, Tang S, Liu S, Yu M, Zhang Y. Helicobacter pylori induces IL-1beta and IL-18 production in human monocytic cell line through activation of NLRP3 inflammasome via ROS signaling pathway. Pathog Dis. 2015;73 doi: 10.1093/femspd/ftu024. [DOI] [PubMed] [Google Scholar]
- 117.Keenan JI, Peterson RA, 2nd, Hampton MB. NADPH oxidase involvement in the pathology of Helicobacter pylori infection. Free Radic Biol Med. 2005;38:1188–96. doi: 10.1016/j.freeradbiomed.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 118.Blanchard TG, Yu F, Hsieh CL, Redline RW. Severe inflammation and reduced bacteria load in murine Helicobacter infection caused by lack of phagocyte oxidase activity. J Infect Dis. 2003;187:1609–15. doi: 10.1086/374780. [DOI] [PubMed] [Google Scholar]
- 119.Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, Park CK, Kasai H, Rhee KH. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996;56:1279–82. [PubMed] [Google Scholar]
- 120.Farinati F, Cardin R, Degan P, Rugge M, Mario FD, Bonvicini P, Naccarato R. Oxidative DNA damage accumulation in gastric carcinogenesis. Gut. 1998;42:351–6. doi: 10.1136/gut.42.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Papa A, Danese S, Sgambato A, Ardito R, Zannoni G, Rinelli A, Vecchio FM, Gentiloni-Silveri N, Cittadini A, Gasbarrini G, Gasbarrini A. Role of Helicobacter pylori CagA+ infection in determining oxidative DNA damage in gastric mucosa. Scand J Gastroenterol. 2002;37:409–13. doi: 10.1080/003655202317316033. [DOI] [PubMed] [Google Scholar]
- 122.de Sablet T, Piazuelo MB, Shaffer CL, Schneider BG, Asim M, Chaturvedi R, Bravo LE, Sicinschi LA, Delgado AG, Mera RM, Israel DA, Romero-Gallo J, Peek RM, Jr, Cover TL, Correa P, Wilson KT. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut. 2011;60:1189–95. doi: 10.1136/gut.2010.234468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ding SZ, Minohara Y, Fan XJ, Wang J, Reyes VE, Patel J, Dirden-Kramer B, Boldogh I, Ernst PB, Crowe SE. Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect Immun. 2007;75:4030–9. doi: 10.1128/IAI.00172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Calvino-Fernandez M, Benito-Martinez S, Parra-Cid T. Oxidative stress by Helicobacter pylori causes apoptosis through mitochondrial pathway in gastric epithelial cells. Apoptosis. 2008;13:1267–80. doi: 10.1007/s10495-008-0255-0. [DOI] [PubMed] [Google Scholar]
- 125.Hayakawa Y, Hirata Y, Kinoshita H, Sakitani K, Nakagawa H, Nakata W, Takahashi R, Sakamoto K, Maeda S, Koike K. Differential roles of ASK1 and TAK1 in Helicobacter pylori-induced cellular responses. Infect Immun. 2013;81:4551–60. doi: 10.1128/IAI.00914-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chaturvedi R, Asim M, Piazuelo MB, Yan F, Barry DP, Sierra JC, Delgado AG, Hill S, Casero RA, Jr, Bravo LE, Dominguez RL, Correa P, Polk DB, Washington MK, Rose KL, Schey KL, Morgan DR, Peek RM, Jr, Wilson KT. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology. 2014;146:1739–51. doi: 10.1053/j.gastro.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mehdi MZ, Pandey NR, Pandey SK, Srivastava AK. H2O2-induced phosphorylation of ERK1/2 and PKB requires tyrosine kinase activity of insulin receptor and c-Src. Antioxid Redox Signal. 2005;7:1014–20. doi: 10.1089/ars.2005.7.1014. [DOI] [PubMed] [Google Scholar]
- 128.Wang YC, Chen CL, Sheu BS, Yang YJ, Tseng PC, Hsieh CY, Lin CF. Helicobacter pylori infection activates Src homology-2 domain-containing phosphatase 2 to suppress IFN-gamma signaling. J Immunol. 2014;193:4149–58. doi: 10.4049/jimmunol.1400594. [DOI] [PubMed] [Google Scholar]
- 129.Caselli A, Marzocchini R, Camici G, Manao G, Moneti G, Pieraccini G, Ramponi G. The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J Biol Chem. 1998;273:32554–60. doi: 10.1074/jbc.273.49.32554. [DOI] [PubMed] [Google Scholar]
- 130.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–99. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 131.Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, Sobota RS, Sicinschi LA, Shaffer CL, Romero-Gallo J, de Sablet T, Harder RH, Bravo LE, Peek RM, Jr, Wilson KT, Cover TL, Williams SM, Correa P. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci USA. 2014;111:1455–60. doi: 10.1073/pnas.1318093111. [DOI] [PMC free article] [PubMed] [Google Scholar]