

Abstract
Background
Severe hyperbilirubinemia is toxic during central nervous system development. Prolonged and uncontrolled high levels of unconjugated bilirubin lead to bilirubin-induced neurological damage and eventually death by kernicterus. Bilirubin neurotoxicity is characterized by a wide array of neurological deficits, including irreversible abnormalities in motor, sensitive and cognitive functions, due to bilirubin accumulation in the brain. Despite the abundant literature documenting the in vitro and in vivo toxic effects of bilirubin, it is unclear which molecular and cellular events actually characterize bilirubin-induced neurodegeneration in vivo.
Methods
We used a mouse model of neonatal hyperbilirubinemia to temporally and spatially define the response of the developing cerebellum to the bilirubin insult.
Results
We showed that the exposure of developing cerebellum to sustained bilirubin levels induces the activation of oxidative stress, ER stress and inflammatory markers at the early stages of the disease onset. In particular, we identified TNFα and NFKβ as key mediators of bilirubin-induced inflammatory response. Moreover, we reported that M1 type microglia is increasingly activated during disease progression.
Failure to counteract this overwhelming stress condition resulted in the induction of the apoptotic pathway and the generation of the glial scar. Finally, bilirubin induced the autophagy pathway in the stages preceding death of the animals.
Conclusions
This study demonstrates that inflammation is a key contributor to bilirubin damage that cooperates with ER stress in the onset of neurotoxicity. Pharmacological modulation of the inflammatory pathway may be a potential intervention target to ameliorate neonatal lethality in Ugt1 -/- mice.
Electronic supplementary material
The online version of this article (doi:10.1186/s12974-017-0838-1) contains supplementary material, which is available to authorized users.
Keywords: Ugt1a1, Crigler-Najjar syndrome, Astrocytes, Microglia, Oxidative stress, Autophagy, Apoptosis, ER stress
Background
Neonatal unconjugated hyperbilirubinemia is a common condition occurring in more than 60% of term newborns [1, 2] and almost all pre-term babies [3]. It is the result of the increased bilirubin production, mainly due to the high turnover of erythrocytes occurring after birth, and the delayed induction of the Ugt1a1 enzyme. It is usually considered a benign condition, but other concomitant causes may lead to uncontrolled acute hyperbilirubinemia [4, 5]. Prolonged high bilirubin levels are life threatening and often lead to permanent brain damage and death by kernicterus [6].
The incidence of kernicterus increases in underdeveloped and developing countries [5], and death by kernicterus is ranked as one of the three top causes of death among African newborns [7]. Clearly, the detailed identification of the neurological events and molecular targets triggering bilirubin neurotoxicity will help the understanding and management of this disease. To this aim, many in vitro and in vivo studies were carried out to increase the understanding of bilirubin neurotoxicity (for a review see [8]).
It has been shown that the free fraction of unbound bilirubin (Bf) causes neurotoxicity in vivo, and that the decrease of Bf by albumin supplementation increases neuro-protection and survival of the mutant mice and rats [9, 10].
Several studies using primary culture of neurons, glia cells or astrocytes, and neuronal-derived cell lines, showed that the sensitivity to bilirubin differs among cell types [11–14]. Moreover, bilirubin toxicity manifests in particular brain developmental stages, with less differentiated/immature neurons being more susceptible [15]. We showed in the Ugt1 -/- mice that neurotoxicity is cell specific, being the Purkinje cells (PCs) the most affected neuron in the cerebellum [16–18].
By differential proteomic analysis of affected cerebella, we showed that bilirubin affects antioxidant defences reducing the overall capacity of neurons to tackle toxicity [18]. This observation was in line with in vitro experiments using primary cultures of neurons [19] and immortalized hepatoma cell lines [20, 21].
In addition, toxic levels of bilirubin induce an inflammatory response through the release of pro-inflammatory cytokines and the activation of MAPK pathway in astrocytes and microglial cells primary cultures [22, 23], leading to cell death. Moreover, high bilirubin levels induce oxidative stress and activate the P38 MAPK pathway in vivo and in vitro [18, 24, 25]. Likewise, in a Ugt1a humanized mouse model, Yueh et al showed that glia activation and oxidative stress are hallmarks of bilirubin-induced toxicity, being Toll-like receptor 2 (TLR2) a key player of bilirubin-mediated inflammatory response [26].
However, a time course study of the molecular events leading to bilirubin neurotoxicity in vivo is still missing. To this aim, we took advantage of the mouse model of severe neonatal hyperbilirubinemia generated in our lab [16, 17]. Ugt1 -/- mice reproduce the major features of neonatal hyperbilirubinemia. In fact, consequent to the absence of Ugt1a1 bilirubin-glucuronidation activity in Ugt1 -/- mice [16], total plasma bilirubin levels rise immediately after birth leading to important cerebellar damage, cerebellar hypoplasia and early death by kernicterus, with 50% mortality at post-natal day 11 (P11) and no survivors after P14 [17].
Using temporary phototherapy (PT) treatment during the neonatal period, we identified a critical window of neuronal susceptibility to bilirubin toxicity, corresponding to post-natal day 8 [17]. We showed that the degree of bilirubin damage correlates with the developmental stage of the cerebellum, being the early phases the more susceptible.
Based on the previous findings, in this study, we made a step forward aiming to investigate the temporal progression of the molecular and cellular events leading to bilirubin-induced neurodegeneration in the Ugt1 -/- mouse model of neonatal hyperbilirubinemia.
Methods
Animals
Mice were housed and handled according to institutional guidelines, and experimental procedures approved by the ICGEB board, with full respect to the EU Directive 2010/63/EU for animal experimentation. Ugt1 -/- mice in the FVB/NJ background have been generated previously [17]. Homozygous mutant animals were obtained from heterozygous mating. Male and female pups were used for these studies. WT littermates were used as control. Animals used in this study were at least 99.8% FVB/NJ genetic background, obtained after more than ten backcrosses with wild-type FVB/NJ mice. Mice were kept in a temperature-controlled environment with 12/12-h light/dark cycle. They received a standard chow diet and water ad libitum.
Plasma bilirubin and UCB content measurement
Blood samples were collected at different time points in mutant and WT littermates by decapitation in EDTA-collecting tubes, at the moment of sacrificing the animals, as previously described [17]. Total bilirubin (TB) determination in plasma was performed using Direct and Total Bilirubin Reagent kit (BQ Kits, San Diego, CA) adapting the method to use minimal volumes (10 μl of plasma), as previously described [17].
Tissues for bilirubin content determination were collected and analysed as previously described [27].
Preparation of total RNA from the mouse cerebellum and real-time PCR analysis
Total RNA from mouse cerebellum was prepared using EuroGOLD Trifast (Euroclone, Milano, Italy). One microgram of total RNA was reverse-transcribed, as previously described [18]. Total cDNA (1 μL) was used to perform qPCR using the specific primers listed in Additional file 1. qPCR was performed using the iQ SYBR Green Supermix (Bio-Rad) and a C1000 Thermal Cycler CFX96 Real Time System (Bio-Rad). Expression of the gene of interest was normalized to the Gapdh housekeeping gene. Data were analysed using the ΔΔCt method. All primers are listed in Additional file 1.
Preparation of total protein extracts and western blot analysis
Cerebella were dissected, homogenized in lysis solution buffer (150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS, 50 mM Tris HCl pH 8, 2× protease inhibitors) and analysed by western blot as described previously [16]. Primary antibodies used were as follows: anti-calbindin 1:2000 (Synaptic Systems, Goettingen, Germany), anti-NeuN 1:2000 (Millipore, Temecula, CA), anti-Glial Fibrillary acidic protein (GFAP) 1:1000 (Sigma, St. Louis, MO), anti-Iba1 1:1000 (Wako, Neuss, Germany), anti-cleaved caspase3 1:500 (Cell Signalling), anti-caspase3 1:800 (Santa Cruz Biotechnology), anti-CHOP 1:1000 (Santa Cruz, sc-56107), anti-HO1 1:1000 (Enzo Life Sciences, ADI-OSA-110) and anti-LC3 1:1000 (Sigma, L7543). Antibody anti-tubulin mAb E7 (Developmental Studies Hybridoma Bank, Iowa City, IA) or anti-Actin (Sigma, A2066) was used for loading control determination. Representative images are shown in the figures. Each timepoint was run in a separate gel.
Brain histology and immunofluorescence
Nissl staining and immunofluorescence analysis of cerebellum samples were performed as previously described [17, 18]. The study was performed in a double-blind fashion: the genotype of the animals was unknown to the operator, while a different investigator analysed the data. Measurements were averaged for each animal.
Briefly, after blocking, specimens were incubated with the primary antibody for 2 h at RT in blocking solution with anti-calbindin 1:400 (Synaptic Systems, Goettingen, Germany), anti-NeuN 1:400 (Millipore, Temecula, CA), anti-glial fibrillary acidic protein, GFAP 1:200 (Sigma, St. Louis, MO), anti-iba1 1:200 (Wako, Neuss, Germany), anti-TNFα 1:100 (sc-1350, Santa Cruz), anti-NFKβ 1:100 (sc-8008, Santa Cruz), anti-MRC1 1:100 (CD206 clone MR5D3, MCA2235GA, BioRad), anti-CD68 1:100 (clone FA-11, BioRad), anti-CHOP 1:100 (GADD153, sc-56107, Santa Cruz), anti-HO1 1:200 (Enzo Life Sciences, ADI-OSA-110), anti-Fas 1:100 (CD95, NB120-13550, Novus Biologicals) and anti-HO1 1:200 (ADI-OSA-110, Enzo Life Sciences). Antigen retrieval with sodium citrate pH6 was performed for CHOP and HO1 immuno-stainings prior to blocking solution step. After 3× 5-min washes with blocking solution, specimens were incubated with secondary antibody (Alexa Fluor 488 or 568; Invitrogen Carlsbad, CA) for 2 h at RT. Nuclei were visualized by addition of Hoechst (10 μg/ml, Invitrogen) for 5 min after secondary antibody solution. Nissl-stained sections were mounted in Eukitt (Fluka, St. Louis, MO, USA) while immunofluorescence (IF) slides were mounted in Mowiol 4-88 (Sigma-Aldrich). Images were acquired on a Nikon Eclipse E-800 epi-fluorescent microscope with a charge-coupled device camera (DMX 1200 F; Nikon Amstelveen, The Netherlands). Digital images were collected using ACT-1 (Nikon) software.
Analysis of the layer thickness was performed on Nissl-stained sections by measuring the layer depth (μm) as previously described [16]. PC density analyses were performed as previously described [16, 17].
Statistics
The Prism package (GraphPad Software, La Jolla, CA) was used to analyse the data. Results are expressed as mean ± S.D. Values of p < 0.05 were considered statistically significant. Depending on the experiment, Student’s t test or two-way ANOVA, with Bonferroni’s post hoc comparison tests, were used, as indicated in the legends to the figures and text. Correlation analyses were done using the Pearson coefficient to assess the linearity between two variables and calculate two-tailed p value (95% of confidence interval).
Results
To better dissect the mechanisms leading to bilirubin neurotoxicity, we analysed three sequential time points: P5 at the onset of the pathology; P8 as a more advanced pathological condition, in which lethality is partially reversible by phototherapy (PT) application in a low proportion of mice [17]; and finally P10, as the latest and most severe phase, just one day before reaching 50% mortality [17].
Vulnerability to bilirubin depends on the developmental stage and the neuronal cell type
We have previously reported that bilirubin induces apoptosis in the cerebellum of C57Bl/6 Ugt1 -/- mice at P4 [18], a strain carrying the same genetic mutation but showing a more severe phenotype [16]. Here, we used a less severe mouse strain (Ugt1-/-/FVB/NJ) to study the apoptotic pathway in the cerebellum, which is the most vulnerable region to bilirubin toxicity in rodent animal models (Fig. 1a) [17, 28, 29]. No increase in caspase 3 cleavage was observed at P5 (Fig. 1b), but apoptosis occurred at P8, as demonstrated by a twofold increase of activated caspase 3 (Fig. 1b). These results are in line with our previous data showing that, despite some of the mice were rescued from mortality by PT starting at P8, the accumulated damage was not reversible [17].
Fig. 1.

Cerebellar neurons are differentially vulnerable to bilirubin. a Appearance of a jaundiced Ugt1 -/- mouse (Ugt1 -/-, red arrowed) and a wild-type (WT) littermate at P8. b WB analysis of total cerebellum protein extract using anti-cleaved and anti-total caspase3 antibodies at P5 and P8 of WT and Ugt1 -/- mice. The bar graphs show the mean of cleaved/total caspase3 ratio of the bands. c WB analysis and quantification of total cerebellum protein extracts of WT and Ugt1 -/- mice using an anti-calbindin antibody at the indicated time points. d Representative fluorescent immunohistochemistry of cerebellar sections from WT and Ugt1 -/- mice at P8 and P10, using an anti-calbindin antibody (red) to highlight PCs. Hoechst (blue) was used to mark nuclei. Scale bar 50 μm. The quantification of PC number at P8 and P10 of WT and Ugt1 -/- mice is represented in the bar graph (cell/mm). e Left panel, Nissl staining of cerebellar layers at P10 of WT and Ugt1 -/- mice. Scale bar 100 μm. Right panel, layer depth quantification of P8 and P10 WT and Ugt1 -/- (μm). f WB analysis of total cerebellum protein extracts of WT and Ugt1 -/- mice at P10 using an anti-NeuN antibody. g Representative fluorescent immunohistochemistry of cerebellar sections from WT and Ugt1 -/- mice using anti-NeuN antibody (green) to stain differentiated granule cells. Hoechst dye was used to stain nuclei (blue). Scale bar 50 μm. For all the experiments, values represent mean ± SD. Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001. The number of WT and Ugt1 -/- was ≥3 in all the experiments and time points. For WB analysis, β-tubulin was used as loading control. EGL external germinal layer, IGL internal granular layer, ML molecular layer
Neurodegeneration is a well-known feature of bilirubin-induced neurological dysfunction (BIND). To have a deeper insight in the mechanisms leading to cerebellar hypoplasia, we first estimated by western blot (WB) analysis the levels of calbindin, a Purkinje cell (PC)-specific marker [30]. We observed that PCs were not affected at P5, while at P8 and P10, there was a significant reduction in calbindin levels in Ugt1 -/- cerebellar protein extracts (56 and 61% decrease, at P8 and P10, respectively; Fig. 1c). Similarly, IF analysis showed a reduction in PC number of 30 and 50% at P8 and P10, respectively, and the almost complete disappearance of their dendritic arborization (Fig. 1d).
Likewise, we assessed the degree of cerebellar damage in brain sections of Ugt1 -/- mice over the phases preceding death (Fig. 1e and Additional file 2A). The long-term exposure of cerebellar neurons to high levels of bilirubin resulted in an overall reduction in cerebellar cortex thickness of 20 and 32%, at P8 and P10, respectively (Fig. 1e). Layer-thickness quantification showed that the external germinal layer (EGL)- and molecular layer (ML)-thickness were significantly reduced at both P8 and P10, while the internal granular layer (IGL)-thickness was reduced only at P10 (Additional file 2A).
Next, we verified whether bilirubin affected mature cerebellar granule cells (GCs) by determining NeuN expression, a nuclear protein antigen whose expression in the cerebellum is restricted to differentiated granule neurons [31]. Western blot experiments showed that bilirubin had no evident effects on differentiated GCs in all time points analysed (Fig. 1f and Additional file 2B). IF analysis performed at P8 and P10 showed no obvious reduction in cell density of neurons located in the IGL (Fig. 1g and Additional file 2C, respectively), suggesting that differentiated GCs were not affected by chronic exposure to bilirubin.
Collectively, our data support the concept that vulnerability of different neuronal cell types in vivo is associated with their developmental stage.
Then, we determined the levels of plasma and cerebellar bilirubin at different time points, ranging from P2 to P10 (Fig. 2). Mutant animals showed a time-dependent increase in plasma bilirubin levels, associated to the worsening of the phenotype. Importantly, there was a clear correlation between the levels of UCB in the cerebellum and those found in plasma (p < 0.001; Fig. 2f). These results demonstrate the tight association between the accumulated levels of UCB in the cerebellum and the increase in neurodegeneration and apoptosis.
Fig. 2.
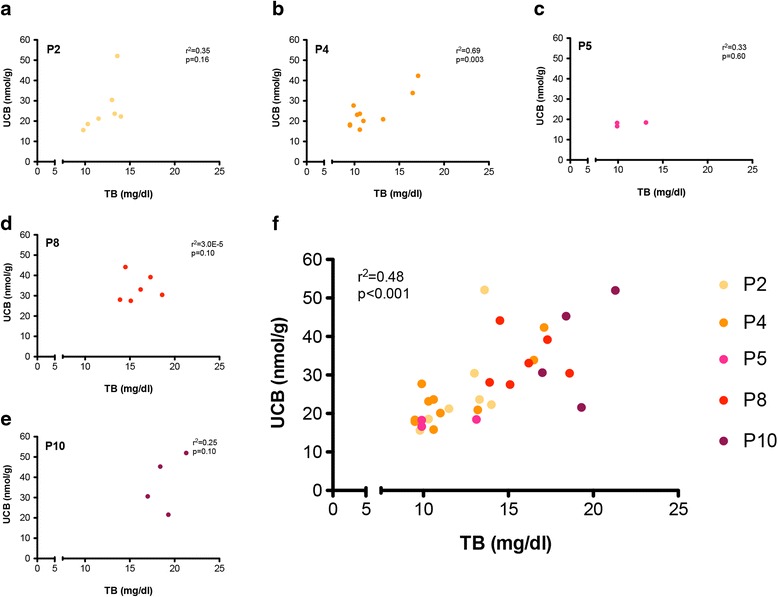
Correlation analysis between total plasma bilirubin and cerebellar bilirubin content at different post-natal ages. Plasma total bilirubin (mg/dL) and cerebellar UCB content (nmol/g) were plotted at each single time point: a P2, b P4, c P5, d P8 and e P10. f All time points in a single graph. Each dot represents a single animal. Correlation analyses were done using the Pearson coefficient to assess the linearity between two variables and calculate two-tailed p value (95% of confidence interval)
Bilirubin induces macro- and micro-gliosis in the cerebellum of Ugt1-/- mice
Since neurotoxicity is normally associated with neuroinflammation [32, 33], we investigated whether bilirubin-induced cell death was accompanied by an increase of inflammatory markers in the cerebellum of the Ugt1 -/- mice.
First, we analysed astrocyte response to bilirubin, by determining glial fibrillary acidic protein (GFAP) levels, an astrocyte-specific marker. No astrocytosis was detected in the early time point of bilirubin exposure (P5, Fig. 3a). Conversely, GFAP levels were two and threefold increased at P8 and P10, respectively, as determined by WB analysis (P8 and P10, Fig. 3a).
Fig. 3.
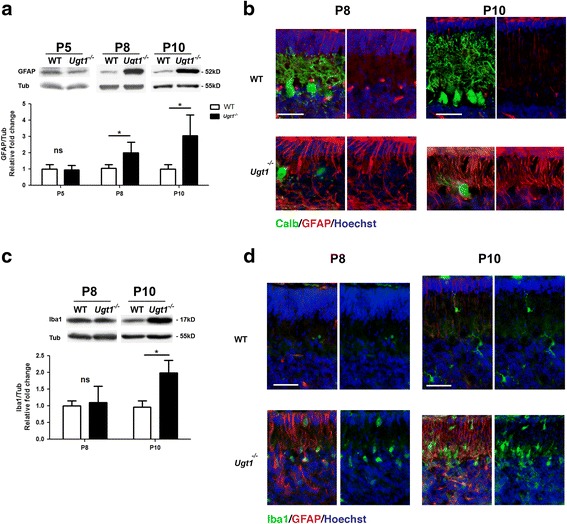
Bilirubin neurotoxicity activates astrocytes and microglia cells. a WB analysis of total cerebellum protein extract using an anti-GFAP antibody at indicated time points of WT and Ugt1 -/- mice. b Representative fluorescent immunohistochemistry of cerebellar sections from WT and Ugt1 -/- mice using anti-GFAP antibody (red) to detect astrocytes, co-stained with an anti-calbindin antibody (green) to highlight PCs. Scale bar 50 μm. c WB analysis of total cerebellum protein extracts using an anti-Iba1 antibody at P8 and P10 of WT and Ugt1 -/- mice. d Representative fluorescent immunohistochemistry of cerebellar sections from WT and Ugt1 -/- mice using an anti-Iba1 antibody (green) to detect microglia, co-stained with an anti-GFAP antibody (red) to highlight astrocytes. Scale bar 50 μm. For all the experiments, values represent the mean ± SD. Students t test, *p < 0.05. The number of WT and Ugt1 -/- was ≥3 in all the experiments and time points. For WB analysis, β-tubulin was used as loading control. For IF, Hoechst dye (blue) was used to mark nuclei
IF analysis performed at P8 and P10 revealed that the glial scar was selectively localized in those cerebellar fissures where PC loss was more prominent (Fig. 3b), such as fissures IV to VIII (Additional file 3A, section of whole cerebellum at P10). To note, the time-dependent increase in GFAP protein levels was also evident at mRNA level (Additional file 3B). These finding were in good correlation with the time-dependent loss of PCs (Fig. 1d).
Next, we determined the cerebellar microglia response to bilirubin by the analysis of ionized calcium-binding adapter molecule 1 (Iba1) as cell-specific marker. We observed that bilirubin triggered an increase in Iba1 mRNA levels starting from P8, and reaching up to threefold increase at P10, as determined by real-time RT-PCR (Additional file 4A). However, at protein levels, we observed a twofold increase in Iba1 only at P10 (Fig. 3c). Furthermore, IF staining of cerebellar sections showed that Iba1-positive cells were located in those zones more affected by bilirubin (Fig. 3d and Additional file 4B).
Interestingly, among all the inflammation markers tested (Fig. 4a and Additional file 5), only TNFα was differentially expressed in mutant animals compared to WT littermates, reaching up to ~10-fold (two-way ANOVA, p < 0.05) in very severe conditions, such as P10 (Fig. 4a). Immunofluorescence analysis of cerebellar sections at P10 showed the presence of TNFα- and NFKβ-positive cells in cerebellar samples from mutant mice, which co-stained with microglia-positive cells (Fig. 4b–d).
Fig. 4.
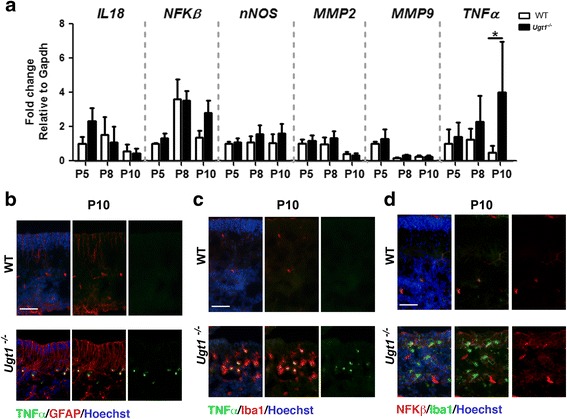
Inflammatory mediators of bilirubin neurotoxicity. a Relative mRNA expression of inflammatory markers. WT and Ugt1 -/- mice mRNA cerebellar expression levels of IL18, NFKβ, nNOS, MMP2, MMP9 and TNFα were analysed at the indicated time points by qRT-PCR. For each gene, data were normalized according to the values of the WT samples at P5. For all the experiments, values represent the mean ± SD. Two-way ANOVA test, *p < 0.05. The number of WT and Ugt1 -/- was ≥3 in all the experiments and time points. b Representative IF of cerebellar sections from WT and Ugt1 -/- mice at P10 using an anti-TNFα antibody (green), co-stained with an anti-GFAP antibody (red) to highlight astrocytes. c Representative IF of cerebellar sections at P10 using an anti-TNFα antibody (green), co-stained with an anti-Iba1 antibody (red) to highlight microglia. d Representative IF of cerebellar sections at P10 using an anti-NFKβ antibody (red), co-stained with an anti-Iba1 antibody (green) to highlight microglia. Scale bar 50 μm. For IF, Hoechst dye (blue) was used to mark nuclei
These data underline the complexity of the inflammatory response triggered by bilirubin in vivo and indicate that neuroinflammation is an important player contributing to each stage of the damage in our experimental model. Overall, these data show that the inflammatory response in vivo increases over time as bilirubin accumulates in the cerebellum and parallel the onset of neurodegeneration.
Time-dependent decrease of M2 is inversely proportional to M1 activation
To better characterize the microglia activation during prolonged bilirubin exposure, we performed real-time RT-PCR analysis of the molecular markers that discriminate the activation of microglia/macrophage states, such as those characterizing the pro-inflammatory M1 microglia (CD68 and CD86) and the anti-inflammatory M2 microglia (MRC1, Arg1 and MRC2).
We observed that the messenger RNA (mRNA) of the M1 pro-inflammatory microglia marker CD68 showed 2.0-, 2.5- and 4.0-fold increase at P5, P8 and P10, respectively, compared to WT samples at the same time points (Fig. 5a). Unexpectedly, when the mRNA expression of CD86, another component of M1 type microglia, was compared between Ugt1 -/- mice and WT littermates, no upregulation was observed.
Fig. 5.

Bilirubin induces activation of M1 and M2 microglia. a Time course of mRNA expression levels of M1 and M2 microglia markers. WT and Ugt1 -/- mice mRNA cerebellar expression levels of CD68, CD86, MCR1, Arg1 and MCR2 were analysed at the indicated time points by qRT-PCR. For each gene, data were normalized according to the values of the WT samples at P5. For all the experiments, values represent the mean ± SD. Two-way ANOVA test, *p < 0.05. The number of WT and Ugt1 -/- was ≥3 in all the experiments and time points. b Representative IF of cerebellar sections from WT and Ugt1 -/- mice at P5 using an anti-MRC1 antibody (red) to detect M2 microglia, co-stained with an anti-Iba1 antibody (green) to highlight microglia. c Representative IF of cerebellar sections at P10 using an anti-CD68 antibody (red) to detect M1 microglia, co-stained with an anti-Iba1 antibody (green). For IF, Hoechst dye (blue) was used to mark nuclei. Scale bar 50 μm
By contrast, real-time RT-PCR analysis revealed an inverted trend for the M2 type, which is considered to compose the neuroprotective counterpart of microglia. MRC1 mRNA in Ugt1 -/- mice was upregulated at P5 and showed decreased expression over time (the same pattern was observed for Arg1, but differences were not statistically significant), while MRC2 was significantly activated only at P8, and mRNA levels decreased at P10, both in WT and Ugt1-/- cerebella (Fig. 5a).
IF experiments reinforced the findings obtained by real-time PCR analysis showing MRC1-positive cells at P5 (Fig. 5b), and of CD68 at P10 (Fig. 5c). In particular, a portion of MRC1-positive cells co-stained with Iba1 at P5, confirming the activation of M2 microglia at the early stages of the disease. Likewise, we reported an important increase in CD68-positive cells at P10, providing evidence of pro-inflammatory M1 type of microglia activation at the later stages of the disease (Fig. 5c).
Since the transition from M2 to M1 phenotype is generally associated with inflammation-induced pathologies, taken together these data indicate that exposure to bilirubin triggers the activation of pro-inflammatory microglia with a parallel decrease in the neuroprotective one.
ER stress and oxidative stress activation in the early stages
It has been shown that bilirubin is involved in endoplasmic reticulum (ER) stress response activation in in vitro cell cultures [34–36]. Thus, we investigated whether ER stress is a key event in the onset of bilirubin toxicity in vivo.
Concomitantly to the activation of inflammation, we observed that the bilirubin insult also triggered the activation of different ER stress markers during the early phase of hyperbilirubinemia, as determined by real-time RT-PCR analysis (Fig. 6a and Additional file 6A). mRNA levels of ATF3, CD95/Fas and CHOP increased as aggravating stages of bilirubin toxicity moved forward. Since CHOP is a key effector in ER stress response, we performed IF analysis of cerebellar sections at P5 and at P10. IF analysis showed that CHOP signal was increased in mutant mice compared to WT littermates in all cerebellar layers at P5 (external germinal layer, Purkinje cell layer and internal granular layer, Fig. 6b).
Fig. 6.
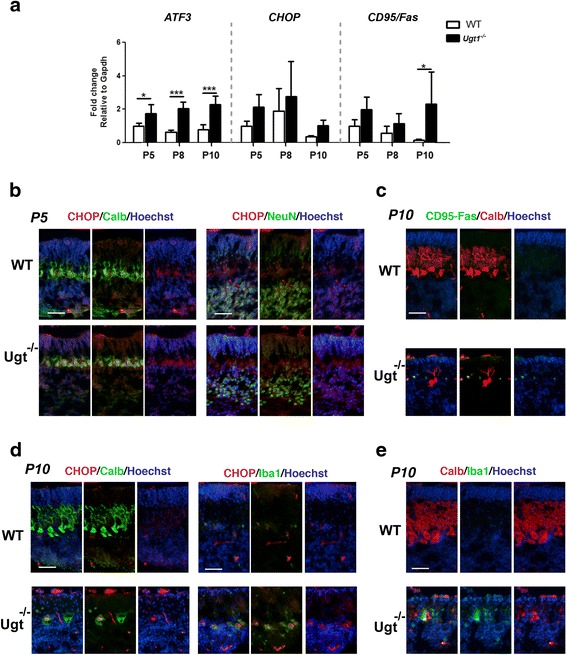
ER stress response to sustained bilirubin levels. a Relative mRNA expression analysis of different genes to assess the ER stress response over prolonged bilirubin exposure. WT and Ugt1 -/- mice mRNA expression levels of ATF3, CHOP and CD95/Fas were analysed at indicated time points by qRT-PCR. For each gene, data were normalized according to the values of the WT samples at P5. For all the experiments values represent the mean ± SD. Two-way ANOVA test, *p < 0.05. The number of WT and Ugt1 -/- was ≥3 in all the experiments and time points. b Representative IF of cerebellar sections from WT and Ugt1 -/- mice at P5 using an anti-CHOP antibody (red), co-stained with (left panel) an anti-calbindin antibody (green) to highlight Purkinje cells or (right panel) anti-NeuN to highlight granule cells. c Representative IF of cerebellar sections at P10 using an anti-CD95/Fas antibody (green), an anti-calbindin antibody (red) to highlight Purkinje cells. d Representative IF of cerebellar sections at P10 using an anti-CHOP antibody (red), co-stained with (left panel) an anti-calbindin antibody (green) or (right panel) anti-Iba1 to highlight microglia. e Representative IF of cerebellar sections at P10 using an anti-Calbindin antibody (red), co-stained with an anti-Iba1 antibody (green). For IF, Hoechst dye (blue) was used to mark nuclei. Scale bar 50 μm
As neurodegeneration moved forward, we observed that Fas/CD95 and CHOP signal concentrated in the PCL (Fig. 6c, d). In particular, at P10, CHOP was mainly localized in the nucleus of severely neurodegenerated PCs, but not in granule cell neurons (Fig. 6d and Additional file 6D). Moreover, we observed that CHOP-positive Purkinje cells were surrounded by Iba1-positive cells (Fig. 6d, e and Additional file 6E) indicating that microglia cells were actively engulfing degenerated neurons.
Finally, we focused our attention on oxidative stress, which is considered a major process triggered by bilirubin in vivo [18]. By real-time RT-PCR, different markers of oxidative stress were analysed to assess their activation status (Fig. 7a and Additional file 7A-B). We could not detect any variation in mRNA levels of the analysed cytochrome P450 family 1 subfamily A member 2 (Cyp1a1), and cytochrome P450 family 2 subfamily A member 5 (Cyp2a5) (Additional file 7B), nor in Nrf2, an important transcription factor involved in cellular response to oxidative stress (Fig. 7a). On the contrary, both the mRNA and protein expression levels of HO1 (heme oxygenase 1), a key enzyme in heme catabolism, were increased in RNA and protein extracts from Ugt1 -/- cerebella, in a time-dependent manner, starting from P8 and reaching more than fourfold increase at P10 at protein level (Fig. 7b). IF analysis showed an important increase of HO1-positive cells in the Purkinje cell area (P5 and P10, Additional file 7C and Fig. 7c, respectively). All HO1-positive cells were also Iba1 positive, but not Calbindin or NeuN positive, suggesting that some microglia cells experienced oxidative stress as a consequence of their phagocytic activity on degenerated PCs.
Fig. 7.

Bilirubin-induced oxidative stress response. a Relative mRNA expression of different genes to assess the oxidative stress response. Expression levels of Nrf2 and HO1 in WT and Ugt1 -/- mice were analysed at P5, P8 and P10 by qRT-PCR. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. Two-way ANOVA test, **p < 0.01. The number of WT and Ugt1 -/- was ≥3 in all the experiments and time points. b WB analysis and quantification of total cerebellum protein extracts of WT and Ugt1 -/- mice using an anti-HO1 antibody at the indicated time points. Actin was used as loading control. Student’s t test, *p < 0.05, **p < 0.01. c Representative IF of cerebellar sections from WT and Ugt1 -/- mice at P10 using an anti-HO1 antibody (red), co-stained with (left panel) an anti-calbindin antibody (green) to highlight Purkinje cells, (central panel) anti-NeuN antibody to highlight granule cells or (right panel) anti-Iba1 antibody to highlight microglia. For IF, Hoechst dye (blue) was used to mark nuclei. Scale bar 50 μm
Autophagy is a late event triggered by bilirubin
Because of the close connection between oxidative stress, neuro-apoptosis, inflammation and autophagy, we investigated whether the latter was activated in vivo in response to bilirubin.
To determine whether autophagy is triggered by hyperbilirubinemia, we performed a WB analysis and quantified the levels of the LC3-II form, as a marker of autophagy. We observed the increase the LC3-II form only in cerebellar total extracts of Ugt1 -/- animals at P10 (Fig. 8), corresponding to the most severe time point analysed.
Fig. 8.

Bilirubin triggers autophagy in the cerebellum of Ugt1 -/- mice. WB analysis of total cerebellum protein extracts using an anti-LC3 antibody at P5, P8 and P10 of WT and Ugt1 -/- mice. β-tubulin was used as a loading control. Lower panel: densitometric quantification of the bands; results are expressed as the mean of the LC3 II/I ratio. Student’s t test, ***p < 0.001. P5: WT n = 4, Ugt1 -/- n = 4; P8: WT n = 4, Ugt1 -/- n = 4; P10: WT n = 3; Ugt1 -/- n = 5
These data showed, for the first time, that bilirubin triggers autophagy in vivo in a mouse model of severe neonatal hyperbilirubinemia.
Discussion
Unveiling the mechanisms operating at the onset of a given disease is essential to develop potential pharmacological therapies against bilirubin-induced brain damage.
Our data indicate that long-term exposure of the developing cerebellum to high bilirubin levels induces the concomitant activation of different mechanism, ranging from inflammation to ER stress. In particular, we observe that the inflammatory response plus ER stress are activated in the early stages of the disease onset and, in turn, they affect survival of developing neurons. At last, bilirubin-induced neurotoxicity stimulates the autophagy pathway (Fig. 9a).
Fig. 9.

Molecular events leading to bilirubin neurodegeneration in a mouse model of neonatal hyperbilirubinemia. a The three sequential phases of the disease progression are indicated: P5, P8 and P10. Representative cartoon showing the morphology of cerebellar sections from WT (black line) and Ugt1 -/- (red line). Bilirubin affects cerebellar size of Ugt1 -/- mice in a time-dependent manner. Boxes indicate the main pathways and molecular players regulated by bilirubin. b Proposed interplay between the molecular events leading to cerebellar neurodegeneration. Exposure of the developing cerebellum to toxic bilirubin levels triggers the activation of neuroinflammation, ER stress, oxidative stress and autophagy pathways. Bold font indicates main mechanisms involved in the onset of the disease; arrows and molecular markers indicate the interconnection between different mechanisms. An image of a neurodegenerated Purkinje neuron (stained with Calb1, green) surrounded by glial scar (stained with GFAP, red) is represented
In the following sections, we will discuss in detail the interplay between the major players identified in the present work (Fig. 9b) and provide possible future directions.
Oxidative stress response
Although mildly elevated bilirubin concentrations are considered beneficial due to its antioxidant properties, high levels of bilirubin lead to considerable oxidative stress [37, 38]. This is particularly evident in the central nervous system when ROS production overwhelms antioxidant defences [39], resulting in neurodegeneration.
Typically, the redox-sensitive pathway is activated at the onset of brain injury as a protection to overcome oxidative stress. This was well demonstrated in tissue-culture studies showing that UCB-induced ROS excess impairs glutathione homeostasis [35, 40] and mitochondrial metabolism resulting in a cellular energetic crisis, swelling, cytochrome c release [41] and calcium perturbation [42]. All these events activate the mitochondrial apoptotic pathway [43] leading to increased oxidative stress and cellular redox status imbalance [44, 45]. Likewise, others and we showed that oxidative stress is a key feature of neurotoxicity animal models of systemic hyperbilirubinemia [18, 26, 46]. However, oxidative stress response to bilirubin did not elicit the induction of Cyps in our mouse model (Additional file 7B), differently to what observed in the Gunn rat [47], an animal model of the disease presenting a milder phenotype.
Interestingly, in our mouse model, the increase in ROS production resulted from the presence of activated microglia cells in the region where Purkinje cells degenerate. These cells, which are positive for HO-1, are involved in the removal of death cells and are associated with inflammation. We observed that HO-1 expression levels increment over time in the damaged tissue, with HO1-positive microglia cells found as early as P5. This observation is in agreement Silva and colleagues showing that an excess of activated microglia produces high levels of nitric oxide, which in turn mediates microglia-induced neuronal cell death [48].
ER stress response
The endoplasmic reticulum (ER) is very sensitive to perturbations of energy levels, intracellular calcium and redox state. Important modifications of its balance result in a condition named ER stress, characterized by an excess of unfolded proteins, and the induction of genes involved in cell survival and protein degradation, aiming to restore the normal state. However, when ER stress is sustained for long periods of time, cells are unable to overcome the accumulation of unfolded proteins and activate the apoptotic pathway.
We report that mRNA levels of key ER stress markers, such as ATF3 and Fas, were significantly induced in response to high bilirubin levels. However, no changes in transcriptional levels were observed in other genes related to unfolded protein response (UPR) and ER stress (GPR78, GRP94, P58 IPK , FKBP11 and DR5) at any time point analysed (Additional file 6A). At the early stages of the disease, CHOP signal was increased in cerebellar sections of mutant mice, with more intense signal in the Purkinje cell layer. At later stages, CHOP signal was localized in degenerating Purkinje cells, surrounded by microglia Iba1-positive cells, suggesting their involvement in removing cellular debris from death Purkinje cells. These results are in agreement with previous findings performed in tissue culture cell lines [20, 35, 36], showing that CHOP and ATF3 are key mediators in ER-stress bilirubin-induced response.
Interestingly, the increased expression of ATF3, Fas/CD95 and CHOP genes indicates that cells sense the overwhelming ER stress and activate the PERK-mediated pathway [49, 50]. Since bilirubin levels continuously increase over time [17], cells are not able to revert ER stress and activate the apoptotic pathway, a hypothesis supported by the increased levels of cleaved caspase 3 and TUNEL-positive cells observed in later stages of the disease [17].
Altogether, these observations provide evidence that ER stress is involved in the onset of bilirubin-induced neuro-apoptosis of Purkinje cells in vivo.
PC neurodegeneration and neuro-apoptosis
The neurodegenerative phenotype in the cerebellum of the mice used in this study is progressively enhanced after sustained high levels of bilirubin and correlates well with the reduced cerebellar development (Fig. 1e). Neurodegeneration is associated with reduced arborization of PC and progressive cell loss (Fig. 1d). In contrast, mature granule cells were not affected by bilirubin toxicity (Fig. 1f). These results are in line with previous observations performed by our lab in the Ugt1 -/- mouse model, in both C57Bl/6 and FVB/NJ genetic backgrounds [16–18]. In particular, we have previously demonstrated that PCs showed a neurodegenerative phenotype as determined by FluoroJade positivity, TUNEL assay and Sholl analysis [17, 18]. These data strengthen the concept that immature neurons are more sensitive to insults compared to more differentiated ones [51] and that bilirubin affects survival of neuronal precursors by impairing their neurogenesis, arborization and synaptic connectivity [17, 52].
Inflammation: pro-inflammatory mediators
In the nervous system, degenerating cells can stimulate inflammation. Although most neurodegenerative diseases involve inflammation, also ER stress is a critical mechanism that can regulate inflammation and is linked through several intersecting cellular pathways, particularly those mediated by ROS [50].
We showed that NFKβ, a well-known inflammatory mediator, responds to high bilirubin levels by the upregulation of its mRNA in a whole tissue extract and by its increase in microglia cells which migrate to the site of tissue injury, together with the increase of TNFα in microglia and astrocytes. To this respect, NFKβ plays a dual role because it exerts both neurotoxic and neuroprotective functions depending on the kinetics and the expression in the injured tissue. In fact, NFKβ regulates the defence response to stress stimuli (inflammation and oxidative stress) and cellular fate (apoptosis, proliferation and differentiation). However, prolonged inflammation leads to the activation of genes, mediated by NFKβ that boosts oxidative stress, inflammation and apoptosis [50]. This is in line with our results showing the exacerbation of neurotoxicity during the more severe conditions observed at P10 (increase in HO1, caspase 3, astrocytosis and gliosis). Similarly, NFKβ is activated in response to pathological condition like trauma, ischemia or different types of neurodegenerative disease [39].
Another key inflammatory factor is TNFα, activated in the more severe phases of the pathology in the Ugt1 -/- pups. This factor is released by activated microglia and astrocytes in the CNS and mediates the amplification of neuroinflammation events and the initiation of the apoptotic cascade. As in the cases of NFKβ, TNFα expression is increased in many neurodegenerative conditions [53].
Our findings support previous data by the laboratory of Brites and colleagues, showing that exposure of astrocytes and microglia primary cultures to bilirubin triggers the release of the pro-inflammatory cytokines TNFα and NFKβ [48, 54, 55]. However, in our model, increased transcriptional levels of TNFα and NFKβ occur after elevated and prolonged levels of tissue bilirubin, while in in vitro cultures these events occur within hours upon bilirubin induction. In addition, other well-known pro-inflammatory mediators are unchanged, such as IL1β, IL6 and INFγ [56]. The discrepancy between in vitro and in vivo data might be related to intensity of the insult, interplay between different cell types and developmental status of the brain, conditions not fully recapitulated by in vitro primary cultures.
Inflammation: cellular mediators
Activated microglia and astrocytes are the main effectors of the innate immune response in the CNS and are strongly activated in response to neurodegeneration, releasing neuroinflammatory factors. We showed that hyperbilirubinemia triggers the activation of both astrocytes and microglia in the cerebellum of Ugt1 -/- mice.
The activation of both astrocytes and microglia by bilirubin has been reported both in vitro [57] and in vivo. In particular, in vivo studies of Gunn rat cerebellum indicated that the increase in GFAP content is a common trait of hyperbilirubinemia [58]. Moreover, the involvement of glia and the role of TLR2 were shown in the humanized knockout mouse model of hyperbilirubinemia [26, 59], although the time course of the disease onset was not investigated. The block of inflammatory activation resulted in death of hyperbilirubinemic TLR2 KO mice, underlined the beneficial effects of inflammatory response in the context of bilirubin toxicity [26]. However, despite the important inflammatory response observed in our Ugt1 -/- animals, we could not detect TLR2 activation in response to bilirubin (Additional file 5D). In fact, because of the early neonatal lethality of our mouse model, we investigated the developmental stages within P5 and P10, while TLR2 activation was reported at later stages, namely P14 [26]. We speculate that the difference between the two models could be related to the extent and duration of the bilirubin insult, intrinsic differences between the two models, or a combination of both.
In addition, we investigated the interplay between inflammatory M1 and anti-inflammatory M2 microglia in the context of bilirubin toxicity. While M2 microglia was induced in at the early stages of the disease, decreasing over time as the severity of the phenotype increases, the trend of M1 microglia activation was the opposite, being progressively activated at later stages, as determined by Iba1 and CD68 M1 microglia markers. A similar trend of activation/deactivation of M1 and M2 microglia was also observed in other neurodegenerative pathologies such as spinal cord injury [60] and traumatic brain injury [61, 62].
Autophagy: the deathblow
Increasing evidences showed that autophagy is a crucial defence mechanism against neurodegenerative diseases and its deregulation is a key event that decides cell fate [63, 64].
Our results showed that autophagy is activated in response to bilirubin in the cerebellum of the Ugt1 -/- mice at P10 (Fig. 8). These observations are in line with experiments performed in neuroblastoma and endothelial cell lines exposed to high bilirubin levels [35, 65]. In both cases, the autophagy pathway was activated at very high levels of bilirubin (Bf = 100–140 nM) and at later stages (24–72 h) when cell death was evident.
Interestingly, autophagy is linked to ER stress through Fas/CD95, which is strongly induced in the cerebellum of Ugt1 -/- mice upon bilirubin exposure (Fig. 6). In fact, Fas/CD95 triggers cell death by the induction of caspase-mediated apoptosis but also stimulates autophagy through MAPK pathway in HeLa cells [66]. Moreover, CHOP is required to increase transcriptional levels of genes responsible for the correct formation and function of the autophagosome [67].
Taken together, these results indicate that autophagy is the latest pro-survival mechanism activated by cells to overcome bilirubin cellular stress prior to death.
Conclusions
The dissection of the molecular mechanisms operating at the onset of neonatal hyperbilirubinemia revealed that different pathways are simultaneously regulated in response to the bilirubin insult, rather than being activated in a sequential time frame. Inflammation, together with ER stress, appear to be the leading processes resulting in neurodegeneration. The more precise understanding of the molecular interactions is crucial to set up therapeutic strategies.
To this respect 4-phenylbutyrate (4-PBA), an ER stress inhibitor, has been shown to reduce ER stress and neurodegeneration in a mouse model of Parkinson’s disease and tissue culture cells treated with bilirubin [36, 68]. Moreover, minocycline, a tetracycline with anti-inflammatory properties has been used in many neurodegenerative diseases to reduce inflammation and ameliorate the outcome of the disease [69], and in the Gunn rat, it reduced cerebellar abnormalities [24]. Administration of minocycline to juvenile Gunn rats resulted in the prevention of acute brainstem auditory evoked potential abnormalities produced by sulfadimethoxine [70, 71]. The concept that several components participate in the response to bilirubin neurotoxicity is supported by the observation that the treatment with antioxidants of Gunn rats having sulfadimethoxine-induced encephalopathy was not sufficient to reduce BIND. On the contrary, minocycline, a molecule with multiple properties in addition to the antioxidant effect, was able to prevent BIND [46]. It remains to be tested if the use of these compounds will ameliorate neurodegeneration and increase survival of the lethal mouse model presented in this study.
Further studies are needed to demonstrated whether modulation of one or more of them are required to reduce neurodegeneration and death, before this knowledge could be translated to patients.
Additional files
Table listing the oligonucleotides used. (XLSX 13 kb)
A) Left panel, representative Nissl staining of cerebellar layers at P8 of WT and Ugt1 -/- mice. Right panel, layer depth quantification (μm) at P8 and P10 of WT and Ugt1 -/- mice. Scale bar 100 μm. B) WB analysis of total cerebellum protein extracts using an anti-NeuN antibody at P8. β-tubulin was used as a loading control. Values represent mean ± SD. C) Representative fluorescent immunohistochemistry of cerebellar sections from WT and Ugt1 -/- mice using anti-NeuN antibody (green) to stain differentiated granule cells at P8. Hoechst (blue) was used to mark nuclei. Scale bar: 50 μm. For all the experiments the values represent the mean ± SD. Student t test, ns not significant; *p < 0.05, **p < 0.01. WT n = 4, Ugt1 -/- n = 4. EGL, external germinal layer; IGL internal granular layer; ML, molecular layer. (TIF 6110 kb)
A) Representative fluorescent immunohistochemistry of WT and Ugt1 -/- cerebellum sections using anti-GFAP antibody (red) to highlight astrocytes, co-stained with an anti-calbindin antibody (green) to highlight PCs. Hoechst (blue) was used to mark nuclei. Scale bar: 500 μm. Boxed areas indicate fields shown in Fig. 3b. IV, VI, VIb, VII and IX indicate the cerebellar fissures. B) mRNA expression levels of GFAP at P5, P8 and P10 in total RNA preparations of WT and Ugt1 -/- cerebella. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. Two-way ANOVA, **p < 0.01, ***p < 0.001. Number of WT and Ugt1 -/- was ≥3 in all the experiments. (TIF 11619 kb)
A) mRNA expression levels of Iba1 at P5, P8 and P10 in total RNA preparations of WT and Ugt1 -/- cerebella. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. One-way ANOVA test, *p < 0.05. B) Representative fluorescent immunohistochemistry of WT and Ugt1 -/- cerebellum using an anti-Iba1 antibody (green) to highlight microglia, co-stained with an anti-GFAP antibody (red) to highlight astrocytes. Hoechst (blue) was used to mark nuclei. Scale bar: 500 μm. Boxed areas indicate fields shown in Fig. 3d. Number of WT and Ugt1 -/- was ≥3 in all the experiments. (TIF 10480 kb)
A) Example of expression levels under the detection limit of the technique. PCR product of IL1β mRNA expression. As positive control dendritic cells (DC) were treated with LPS for 24 hs. (lane 2). B) Example of detectable mRNA: TNFα expression. C) List of mRNAs that were not detected by qRT-PCR in cerebellar total RNA extracts. D) TLR2 and E) iNOS mRNA relative expression levels in WT and Ugt1 -/- animals at the indicated time points. For each gene, data were normalized according to the values of the WT samples at P5. Values represent mean ± SD. Two-way ANOVA, ns not significant. For representative agarose gels, lane 1: 1Kb ladder; lane 2: PCR product from dendritic cells (DCs) treated with LPS for 24 h; lane 3: CB, Ugt1 -/- cerebellar total RNA extract; lane 4, blank. (TIF 2216 kb)
A) Relative mRNA expression of ER stress response genes showing no changes at the different time points. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. Two-way ANOVA, ns not significant. Number of WT and Ugt1 -/- was ≥3 in all the experiments B) WB analysis and quantification of total cerebellum protein extracts of WT and Ugt1 -/- mice using an anti-CHOP antibody at the indicated time points. Actin was used as loading control. Student t test, ns not significant. C) Representative IF of cerebellar sections from Ugt1 -/- mice at P10 using an anti-CHOP antibody (red), co-stained with an anti-calbindin antibody (green) to highlight Purkinje cells; D) representative IF of cerebellar sections from WT and Ugt1 -/- mice at P10 using an anti-CHOP antibody (red), co-stained with an anti-NeuN antibody to highlight granule cells; E) representative IF of cerebellar sections from Ugt1 -/- mice at P10 using an anti-CHOP antibody (red), co-stained with an anti-Iba1 antibody to highlight microglia. For IF, Hoechst dye (blue) was used to mark nuclei. Scale bar: 50 μm. (TIF 24031 kb)
Relative mRNA expression of A) oxidative stress response genes and B) Cyp1a1 and Cyp2a5 showing no changes at the different time points. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. Two-way ANOVA, ns not significant. Number of WT and Ugt1 -/- was ≥3 in all the experiments. C) Representative IF of cerebellar sections from WT and Ugt1 -/- mice at P5 using an anti-HO1 antibody (red), co-stained with anti-Iba1 antibody to highlight microglia. For IF, Hoechst dye (blue) was used to mark nuclei. Scale bar: 50 μm. (TIF 10064 kb)
Acknowledgements
The authors thank Prof. E. Tongiorgi for the microscope facility resources and the bioExperimentation Facility for help with animal care.
Funding
This work was supported by Telethon (GGP10051), by Friuli-Venezia Giulia Regional Grant and by Beneficentia Stiftung to A. F. Muro (ICGEB), by AXA Research Fund to G. Bortolussi (ICGEB), and by the Czech Ministry of Health (RVO VFN64165) and Czech Ministry of Education (KONTAKT LH15097) to L. Vitek.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Authors’ contributions
S.V. and G.B. carried out the experiments. S.V., J.J. and L.V. determined bilirubin in tissues. G.B. and A.F.M conceived the experiments, analysed the data and wrote the paper. All authors revised the final manuscript and had final approval of the submitted and published versions.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
Mice were housed and handled according to institutional guidelines, and experimental procedures are approved by the ICGEB board, with full respect to the EU Directive 2010/63/EU for animal experimentation.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- 4-PBA
4-phenylbutyrate
- Arg1
Arginase 1
- ATF3
Activating transcription factor 3
- Bf
Free fraction of unbound bilirubin
- BIND
Bilirubin-induced neurological dysfunction
- CD68
Cluster of differentiation 68
- CD86
Cluster of differentiation 86
- CD95
Cluster of differentiation 95
- Cyp1a2
Cytochrome P450 family 1 subfamily A member 2
- Cyp2a5
Cytochrome P450 family 2 subfamily A member 5
- EGL
External germinal layer
- ER
Endoplasmic reticulum
- GCs
Granule cells
- GFAP
Glial fibrillary acidic protein
- HO1
Heme oxygenase 1
- Iba1
Ionized calcium-binding adapter molecule 1
- IF
Immunofluorescence
- IGL
Internal granular layer
- IL18
Interleukin 18
- IL1β
Interleukin 1β
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- ML
Molecular layer
- MMP2
Matrix-metallo-protease 2
- MMP9
Matrix-metallo-protease 9
- MRC1
Mannose receptor, C type 1
- MRC2
Mannose receptor, C type 2
- NFKβ
Nuclear factor kappa-light-chain-enhancer of activated B cells
- nNOS
Neuronal nitric oxide synthase
- Nrf2
Nuclear factor (erythroid-derived 2)-like 2
- P
Post-natal day
- PCR
Polymerase chain reaction
- PCs
Purkinje cells
- PT
Phototherapy
- TNFα
Tumor necrosis factor alpha
- Ugt1a1
UDP glucuronosyltransferase family 1 member A1
- WB
Western blot
Contributor Information
Simone Vodret, Email: simone.vodret@icgeb.org.
Giulia Bortolussi, Phone: +39-040-3757312, Email: giulia.bortolussi@icgeb.org.
Jana Jašprová, Email: janinnava@seznam.cz.
Libor Vitek, Email: vitek@cesnet.cz.
Andrés F. Muro, Phone: +39-040-3757369, Email: muro@icgeb.org
References
- 1.Johnson LH, Bhutani VK, Brown AK. System-based approach to management of neonatal jaundice and prevention of kernicterus. J Pediatr. 2002;140:396–403. doi: 10.1067/mpd.2002.123098. [DOI] [PubMed] [Google Scholar]
- 2.Pediatrics AAo Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114:297–316. doi: 10.1542/peds.114.1.297. [DOI] [PubMed] [Google Scholar]
- 3.Reichman NE, Teitler JO, Moullin S, Ostfeld BM, Hegyi T. Late-preterm birth and neonatal morbidities: population-level and within-family estimates. Ann Epidemiol. 2015;25:126–132. doi: 10.1016/j.annepidem.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Bhutani VK, Wong RJ. Bilirubin neurotoxicity in preterm infants: risk and prevention. J Clin Neonatol. 2013;2:61–69. doi: 10.4103/2249-4847.116402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greco C, Arnolda G, Boo NY, Iskander IF, Okolo AA, Rohsiswatmo R, Shapiro SM, Watchko J, Wennberg RP, Tiribelli C, Coda Zabetta CD. Neonatal jaundice in Low- and middle-income countries: lessons and future directions from the 2015 don ostrow trieste yellow retreat. Neonatology. 2016;110:172–180. doi: 10.1159/000445708. [DOI] [PubMed] [Google Scholar]
- 6.Watchko JF. Kernicterus and the molecular mechanisms of bilirubin-induced CNS injury in newborns. Neuromolecular Med. 2006;8:513–529. doi: 10.1385/NMM:8:4:513. [DOI] [PubMed] [Google Scholar]
- 7.Olusanya BO, Ogunlesi TA, Slusher TM. Why is kernicterus still a major cause of death and disability in low-income and middle-income countries? Arch Dis Child. 2014;99:1117–1121. doi: 10.1136/archdischild-2013-305506. [DOI] [PubMed] [Google Scholar]
- 8.Watchko JF, Tiribelli C. Bilirubin-induced neurologic damage—mechanisms and management approaches. N Engl J Med. 2013;369:2021–2030. doi: 10.1056/NEJMra1308124. [DOI] [PubMed] [Google Scholar]
- 9.Schreuder AB, Rice AC, Vanikova J, Vitek L, Shapiro SM, Verkade HJ. Albumin administration protects against bilirubin-induced auditory brainstem dysfunction in Gunn rat pups. Liver Int. 2013;33:1557–1565. doi: 10.1111/liv.12219. [DOI] [PubMed] [Google Scholar]
- 10.Vodret S, Bortolussi G, Schreuder AB, Jasprova J, Vitek L, Verkade HJ, Muro AF. Albumin administration prevents neurological damage and death in a mouse model of severe neonatal hyperbilirubinemia. Sci Rep. 2015;5:16203. doi: 10.1038/srep16203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falcao AS, Bellarosa C, Fernandes A, Brito MA, Silva RF, Tiribelli C, Brites D. Role of multidrug resistance-associated protein 1 expression in the in vitro susceptibility of rat nerve cell to unconjugated bilirubin. Neuroscience. 2007;144:878–888. doi: 10.1016/j.neuroscience.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 12.Falcao AS, Fernandes A, Brito MA, Silva RF, Brites D. Bilirubin-induced immunostimulant effects and toxicity vary with neural cell type and maturation state. Acta Neuropathol. 2006;112:95–105. doi: 10.1007/s00401-006-0078-4. [DOI] [PubMed] [Google Scholar]
- 13.Notter MF, Kendig JW. Differential sensitivity of neural cells to bilirubin toxicity. Exp Neurol. 1986;94:670–682. doi: 10.1016/0014-4886(86)90246-3. [DOI] [PubMed] [Google Scholar]
- 14.Silva RF, Rodrigues CM, Brites D. Rat cultured neuronal and glial cells respond differently to toxicity of unconjugated bilirubin. Pediatr Res. 2002;51:535–541. doi: 10.1203/00006450-200204000-00022. [DOI] [PubMed] [Google Scholar]
- 15.Vaz AR, Silva SL, Barateiro A, Falcao AS, Fernandes A, Brito MA, Brites D. Selective vulnerability of rat brain regions to unconjugated bilirubin. Mol Cell Neurosci. 2011;48:82–93. doi: 10.1016/j.mcn.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Bortolussi G, Zentilin L, Baj G, Giraudi P, Bellarosa C, Giacca M, Tiribelli C, Muro AF. Rescue of bilirubin-induced neonatal lethality in a mouse model of Crigler-Najjar syndrome type I by AAV9-mediated gene transfer. FASEB J. 2012;26:1052–1063. doi: 10.1096/fj.11-195461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bortolussi G, Baj G, Vodret S, Viviani G, Bittolo T, Muro AF. Age-dependent pattern of cerebellar susceptibility to bilirubin neurotoxicity in vivo in mice. Dis Model Mech. 2014;7:1057–1068. doi: 10.1242/dmm.016535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bortolussi G, Codarin E, Antoniali G, Vascotto C, Vodret S, Arena S, Cesaratto L, Scaloni A, Tell G, Muro AF. Impairment of enzymatic antioxidant defenses is associated with bilirubin-induced neuronal cell death in the cerebellum of Ugt1 KO mice. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brito MA, Brites D, Butterfield DA. A link between hyperbilirubinemia, oxidative stress and injury to neocortical synaptosomes. Brain Res. 2004;1026:33–43. doi: 10.1016/j.brainres.2004.07.063. [DOI] [PubMed] [Google Scholar]
- 20.Oakes GH, Bend JR. Global changes in gene regulation demonstrate that unconjugated bilirubin is able to upregulate and activate select components of the endoplasmic reticulum stress response pathway. J Biochem Mol Toxicol. 2010;24:73–88. doi: 10.1002/jbt.20313. [DOI] [PubMed] [Google Scholar]
- 21.Oakes GH, Bend JR. Early steps in bilirubin-mediated apoptosis in murine hepatoma (Hepa 1c1c7) cells are characterized by aryl hydrocarbon receptor-independent oxidative stress and activation of the mitochondrial pathway. J Biochem Mol Toxicol. 2005;19:244–255. doi: 10.1002/jbt.20086. [DOI] [PubMed] [Google Scholar]
- 22.Fernandes A, Falcao AS, Silva RF, Gordo AC, Gama MJ, Brito MA, Brites D. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J Neurochem. 2006;96:1667–1679. doi: 10.1111/j.1471-4159.2006.03680.x. [DOI] [PubMed] [Google Scholar]
- 23.Fernandes A, Falcao AS, Silva RF, Brito MA, Brites D. MAPKs are key players in mediating cytokine release and cell death induced by unconjugated bilirubin in cultured rat cortical astrocytes. Eur J Neurosci. 2007;25:1058–1068. doi: 10.1111/j.1460-9568.2007.05340.x. [DOI] [PubMed] [Google Scholar]
- 24.Lin S, Wei X, Bales KR, Paul AB, Ma Z, Yan G, Paul SM, Du Y. Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur J Neurosci. 2005;22:21–27. doi: 10.1111/j.1460-9568.2005.04182.x. [DOI] [PubMed] [Google Scholar]
- 25.Lin S, Yan C, Wei X, Paul SM, Du Y. p38 MAP kinase mediates bilirubin-induced neuronal death of cultured rat cerebellar granule neurons. Neurosci Lett. 2003;353:209–212. doi: 10.1016/j.neulet.2003.09.053. [DOI] [PubMed] [Google Scholar]
- 26.Yueh MF, Chen S, Nguyen N, Tukey RH. Developmental onset of bilirubin-induced neurotoxicity involves Toll-like receptor 2-dependent signaling in humanized UDP-glucuronosyltransferase1 mice. J Biol Chem. 2014;289:4699–4709. doi: 10.1074/jbc.M113.518613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bockor L, Bortolussi G, Vodret S, Iaconcig A, Jasprova J, Zelenka J, Vitek L, Tiribelli C, Muro AF. Modulation of bilirubin neurotoxicity by the Abcb1 transporter in the Ugt1-/- lethal mouse model of neonatal hyperbilirubinemia. Hum Mol Genet. 2016. [DOI] [PubMed]
- 28.Schutta HS, Johnson L. Bilirubin encephalopathy in the Gunn rat: a fine structure study of the cerebellar cortex. J Neuropathol Exp Neurol. 1967;26:377–396. doi: 10.1097/00005072-196707000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Fujiwara R, Nguyen N, Chen S, Tukey RH. Developmental hyperbilirubinemia and CNS toxicity in mice humanized with the UDP glucuronosyltransferase 1 (UGT1) locus. Proc Natl Acad Sci U S A. 2010;107:5024–5029. doi: 10.1073/pnas.0913290107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitney ER, Kemper TL, Rosene DL, Bauman ML, Blatt GJ. Calbindin-D28k is a more reliable marker of human Purkinje cells than standard Nissl stains: a stereological experiment. J Neurosci Methods. 2008;168:42–47. doi: 10.1016/j.jneumeth.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 31.Sarnat HB, Nochlin D, Born DE. Neuronal nuclear antigen (NeuN): a marker of neuronal maturation in early human fetal nervous system. Brain Dev. 1998;20:88–94. doi: 10.1016/S0387-7604(97)00111-3. [DOI] [PubMed] [Google Scholar]
- 32.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease—a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/S0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 34.Barateiro A, Vaz AR, Silva SL, Fernandes A, Brites D. ER stress, mitochondrial dysfunction and calpain/JNK activation are involved in oligodendrocyte precursor cell death by unconjugated bilirubin. Neuromolecular Med. 2012;14:285–302. doi: 10.1007/s12017-012-8187-9. [DOI] [PubMed] [Google Scholar]
- 35.Calligaris R, Bellarosa C, Foti R, Roncaglia P, Giraudi P, Krmac H, Tiribelli C, Gustincich S. A transcriptome analysis identifies molecular effectors of unconjugated bilirubin in human neuroblastoma SH-SY5Y cells. BMC Genomics. 2009;10:543. doi: 10.1186/1471-2164-10-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qaisiya M, Brischetto C, Jasprova J, Vitek L, Tiribelli C, Bellarosa C. Bilirubin-induced ER stress contributes to the inflammatory response and apoptosis in neuronal cells. Arch Toxicol. 2016. [DOI] [PubMed]
- 37.Brito MA, Lima S, Fernandes A, Falcao AS, Silva RF, Butterfield DA, Brites D. Bilirubin injury to neurons: contribution of oxidative stress and rescue by glycoursodeoxycholic acid. Neurotoxicology. 2008;29:259–269. doi: 10.1016/j.neuro.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 38.Brito MA, Rosa AI, Falcao AS, Fernandes A, Silva RF, Butterfield DA, Brites D. Unconjugated bilirubin differentially affects the redox status of neuronal and astroglial cells. Neurobiol Dis. 2008;29:30–40. doi: 10.1016/j.nbd.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 39.Fischer R, Maier O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid Med Cell Longev. 2015;2015:610813. doi: 10.1155/2015/610813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giraudi PJ, Bellarosa C, Coda-Zabetta CD, Peruzzo P, Tiribelli C. Functional induction of the cystine-glutamate exchanger system Xc(-) activity in SH-SY5Y cells by unconjugated bilirubin. PLoS One. 2011;6 doi: 10.1371/journal.pone.0029078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodrigues CM, Sola S, Silva R, Brites D. Bilirubin and amyloid-beta peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol Med. 2000;6:936–946. [PMC free article] [PubMed] [Google Scholar]
- 42.Stoeckius M, Erat A, Fujikawa T, Hiromura M, Koulova A, Otterbein L, Bianchi C, Tobiasch E, Dagon Y, Sellke FW, Usheva A. Essential roles of Raf/extracellular signal-regulated kinase/mitogen-activated protein kinase pathway, YY1, and Ca2+ influx in growth arrest of human vascular smooth muscle cells by bilirubin. J Biol Chem. 2012;287:15418–15426. doi: 10.1074/jbc.M111.266510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brites D. Bilirubin injury to neurons and glial cells: new players, novel targets, and newer insights. Semin Perinatol. 2011;35:114–120. doi: 10.1053/j.semperi.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Tell G, Gustincich S. Redox state, oxidative stress, and molecular mechanisms of protective and toxic effects of bilirubin on cells. Curr Pharm Des. 2009;15:2908–2914. doi: 10.2174/138161209789058174. [DOI] [PubMed] [Google Scholar]
- 45.Qaisiya M, Coda Zabetta CD, Bellarosa C, Tiribelli C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell Signal. 2014;26:512–520. doi: 10.1016/j.cellsig.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 46.Daood MJ, Hoyson M, Watchko JF. Lipid peroxidation is not the primary mechanism of bilirubin-induced neurologic dysfunction in jaundiced Gunn rat pups. Pediatr Res. 2012;72:455–459. doi: 10.1038/pr.2012.111. [DOI] [PubMed] [Google Scholar]
- 47.Gazzin S, Zelenka J, Zdrahalova L, Konickova R, Zabetta CC, Giraudi PJ, Berengeno AL, Raseni A, Robert MC, Vitek L, Tiribelli C. Bilirubin accumulation and Cyp mRNA expression in selected brain regions of jaundiced Gunn rat pups. Pediatr Res. 2012;71:653–660. doi: 10.1038/pr.2012.23. [DOI] [PubMed] [Google Scholar]
- 48.Silva SL, Vaz AR, Diogenes MJ, van Rooijen N, Sebastiao AM, Fernandes A, Silva RF, Brites D. Neuritic growth impairment and cell death by unconjugated bilirubin is mediated by NO and glutamate, modulated by microglia, and prevented by glycoursodeoxycholic acid and interleukin-10. Neuropharmacology. 2012;62:2398–2408. doi: 10.1016/j.neuropharm.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Kim KH, Jeong JY, Surh YJ, Kim KW. Expression of stress-response ATF3 is mediated by Nrf2 in astrocytes. Nucleic Acids Res. 2010;38:48–59. doi: 10.1093/nar/gkp865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaudhari N, Talwar P, Parimisetty A, Lefebvre d'Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213. doi: 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Falcao AS, Silva RF, Pancadas S, Fernandes A, Brito MA, Brites D. Apoptosis and impairment of neurite network by short exposure of immature rat cortical neurons to unconjugated bilirubin increase with cell differentiation and are additionally enhanced by an inflammatory stimulus. J Neurosci Res. 2007;85:1229–1239. doi: 10.1002/jnr.21227. [DOI] [PubMed] [Google Scholar]
- 52.Fernandes A, Falcao AS, Abranches E, Bekman E, Henrique D, Lanier LM, Brites D. Bilirubin as a determinant for altered neurogenesis, neuritogenesis, and synaptogenesis. Dev Neurobiol. 2009;69:568–582. doi: 10.1002/dneu.20727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olmos G, Llado J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014;2014:861231. doi: 10.1155/2014/861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernandes A, Silva RF, Falcao AS, Brito MA, Brites D. Cytokine production, glutamate release and cell death in rat cultured astrocytes treated with unconjugated bilirubin and LPS. J Neuroimmunol. 2004;153:64–75. doi: 10.1016/j.jneuroim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 55.Gordo AC, Falcao AS, Fernandes A, Brito MA, Silva RF, Brites D. Unconjugated bilirubin activates and damages microglia. J Neurosci Res. 2006;84:194–201. doi: 10.1002/jnr.20857. [DOI] [PubMed] [Google Scholar]
- 56.Fernandes A, Brites D. Contribution of inflammatory processes to nerve cell toxicity by bilirubin and efficacy of potential therapeutic agents. Curr Pharm Des. 2009;15:2915–2926. doi: 10.2174/138161209789058165. [DOI] [PubMed] [Google Scholar]
- 57.Brites D. The evolving landscape of neurotoxicity by unconjugated bilirubin: role of glial cells and inflammation. Front Pharmacol. 2012;3:88. doi: 10.3389/fphar.2012.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mikoshiba K, Kohsaka S, Takamatsu K, Tsukada Y. Cerebellar hypoplasia in the Gunn rat with hereditary hyperbilirubinemia: immunohistochemical and neurochemical studies. J Neurochem. 1980;35:1309–1318. doi: 10.1111/j.1471-4159.1980.tb09003.x. [DOI] [PubMed] [Google Scholar]
- 59.Barateiro A, Chen S, Yueh MF, Fernandes A, Domingues HS, Relvas J, Barbier O, Nguyen N, Tukey RH, Brites D. Reduced myelination and increased glia reactivity resulting from severe neonatal hyperbilirubinemia. Mol Pharmacol. 2016;89:84–93. doi: 10.1124/mol.115.098228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsieh CL, Kim CC, Ryba BE, Niemi EC, Bando JK, Locksley RM, Liu J, Nakamura MC, Seaman WE. Traumatic brain injury induces macrophage subsets in the brain. Eur J Immunol. 2013;43:2010–2022. doi: 10.1002/eji.201243084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turtzo LC, Lescher J, Janes L, Dean DD, Budde MD, Frank JA. Macrophagic and microglial responses after focal traumatic brain injury in the female rat. J Neuroinflammation. 2014;11:82. doi: 10.1186/1742-2094-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 64.Alirezaei M, Kemball CC, Whitton JL. Autophagy, inflammation and neurodegenerative disease. Eur J Neurosci. 2011;33:197–204. doi: 10.1111/j.1460-9568.2010.07500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palmela I, Sasaki H, Cardoso FL, Moutinho M, Kim KS, Brites D, Brito MA. Time-dependent dual effects of high levels of unconjugated bilirubin on the human blood-brain barrier lining. Front Cell Neurosci. 2012;6:22. doi: 10.3389/fncel.2012.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, Wu Y, Cheng Y, Zhao Z, Tashiro S, Onodera S, Ikejima T. Fas-mediated autophagy requires JNK activation in HeLa cells. Biochem Biophys Res Commun. 2008;377:1205–1210. doi: 10.1016/j.bbrc.2008.10.151. [DOI] [PubMed] [Google Scholar]
- 67.B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–7699. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Inden M, Kitamura Y, Takeuchi H, Yanagida T, Takata K, Kobayashi Y, Taniguchi T, Yoshimoto K, Kaneko M, Okuma Y, et al. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J Neurochem. 2007;101:1491–1504. doi: 10.1111/j.1471-4159.2006.04440.x. [DOI] [PubMed] [Google Scholar]
- 69.Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. 2009;196:168–179. doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 70.Geiger AS, Rice AC, Shapiro SM. Minocycline blocks acute bilirubin-induced neurological dysfunction in jaundiced Gunn rats. Neonatology. 2007;92:219–226. doi: 10.1159/000103740. [DOI] [PubMed] [Google Scholar]
- 71.Rice AC, Chiou VL, Zuckoff SB, Shapiro SM. Profile of minocycline neuroprotection in bilirubin-induced auditory system dysfunction. Brain Res. 2011;1368:290–298. doi: 10.1016/j.brainres.2010.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table listing the oligonucleotides used. (XLSX 13 kb)
A) Left panel, representative Nissl staining of cerebellar layers at P8 of WT and Ugt1 -/- mice. Right panel, layer depth quantification (μm) at P8 and P10 of WT and Ugt1 -/- mice. Scale bar 100 μm. B) WB analysis of total cerebellum protein extracts using an anti-NeuN antibody at P8. β-tubulin was used as a loading control. Values represent mean ± SD. C) Representative fluorescent immunohistochemistry of cerebellar sections from WT and Ugt1 -/- mice using anti-NeuN antibody (green) to stain differentiated granule cells at P8. Hoechst (blue) was used to mark nuclei. Scale bar: 50 μm. For all the experiments the values represent the mean ± SD. Student t test, ns not significant; *p < 0.05, **p < 0.01. WT n = 4, Ugt1 -/- n = 4. EGL, external germinal layer; IGL internal granular layer; ML, molecular layer. (TIF 6110 kb)
A) Representative fluorescent immunohistochemistry of WT and Ugt1 -/- cerebellum sections using anti-GFAP antibody (red) to highlight astrocytes, co-stained with an anti-calbindin antibody (green) to highlight PCs. Hoechst (blue) was used to mark nuclei. Scale bar: 500 μm. Boxed areas indicate fields shown in Fig. 3b. IV, VI, VIb, VII and IX indicate the cerebellar fissures. B) mRNA expression levels of GFAP at P5, P8 and P10 in total RNA preparations of WT and Ugt1 -/- cerebella. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. Two-way ANOVA, **p < 0.01, ***p < 0.001. Number of WT and Ugt1 -/- was ≥3 in all the experiments. (TIF 11619 kb)
A) mRNA expression levels of Iba1 at P5, P8 and P10 in total RNA preparations of WT and Ugt1 -/- cerebella. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. One-way ANOVA test, *p < 0.05. B) Representative fluorescent immunohistochemistry of WT and Ugt1 -/- cerebellum using an anti-Iba1 antibody (green) to highlight microglia, co-stained with an anti-GFAP antibody (red) to highlight astrocytes. Hoechst (blue) was used to mark nuclei. Scale bar: 500 μm. Boxed areas indicate fields shown in Fig. 3d. Number of WT and Ugt1 -/- was ≥3 in all the experiments. (TIF 10480 kb)
A) Example of expression levels under the detection limit of the technique. PCR product of IL1β mRNA expression. As positive control dendritic cells (DC) were treated with LPS for 24 hs. (lane 2). B) Example of detectable mRNA: TNFα expression. C) List of mRNAs that were not detected by qRT-PCR in cerebellar total RNA extracts. D) TLR2 and E) iNOS mRNA relative expression levels in WT and Ugt1 -/- animals at the indicated time points. For each gene, data were normalized according to the values of the WT samples at P5. Values represent mean ± SD. Two-way ANOVA, ns not significant. For representative agarose gels, lane 1: 1Kb ladder; lane 2: PCR product from dendritic cells (DCs) treated with LPS for 24 h; lane 3: CB, Ugt1 -/- cerebellar total RNA extract; lane 4, blank. (TIF 2216 kb)
A) Relative mRNA expression of ER stress response genes showing no changes at the different time points. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. Two-way ANOVA, ns not significant. Number of WT and Ugt1 -/- was ≥3 in all the experiments B) WB analysis and quantification of total cerebellum protein extracts of WT and Ugt1 -/- mice using an anti-CHOP antibody at the indicated time points. Actin was used as loading control. Student t test, ns not significant. C) Representative IF of cerebellar sections from Ugt1 -/- mice at P10 using an anti-CHOP antibody (red), co-stained with an anti-calbindin antibody (green) to highlight Purkinje cells; D) representative IF of cerebellar sections from WT and Ugt1 -/- mice at P10 using an anti-CHOP antibody (red), co-stained with an anti-NeuN antibody to highlight granule cells; E) representative IF of cerebellar sections from Ugt1 -/- mice at P10 using an anti-CHOP antibody (red), co-stained with an anti-Iba1 antibody to highlight microglia. For IF, Hoechst dye (blue) was used to mark nuclei. Scale bar: 50 μm. (TIF 24031 kb)
Relative mRNA expression of A) oxidative stress response genes and B) Cyp1a1 and Cyp2a5 showing no changes at the different time points. For each gene, data were normalized according to the values of the WT samples at P5. Values represent the mean ± S.D. Two-way ANOVA, ns not significant. Number of WT and Ugt1 -/- was ≥3 in all the experiments. C) Representative IF of cerebellar sections from WT and Ugt1 -/- mice at P5 using an anti-HO1 antibody (red), co-stained with anti-Iba1 antibody to highlight microglia. For IF, Hoechst dye (blue) was used to mark nuclei. Scale bar: 50 μm. (TIF 10064 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article.