

Abstract
Cholesterol plays an important role in inducing pancreatic β-cell dysfunction, leading to an impaired insulin secretory response to glucose. This study aimed to determine the protective effects of sulforaphane, a natural isothiocyanate Nrf2-inducer, against cholesterol-induced pancreatic β-cells dysfunction, through molecular and cellular mechanisms involving mitochondrial bioenergetics. Sulforaphane prevented cholesterol-induced alterations in the coupling efficiency of mitochondrial respiration, improving ATP turnover and spare capacity, and averted the impairment of the electron flow at complexes I, II, and IV. Sulforaphane also attenuated the cholesterol-induced activation of the NFκB pathway, normalizing the expression of pro- and anti-inflammatory cytokines. In addition, it also inhibited the decrease in sirtuin 1 expression and greatly increased Pgc-1α expression in Min6 cells. Sulforaphane increased the expression of antioxidant enzymes downstream of the Nrf2 pathway and prevented lipid peroxidation induced by cholesterol. The antioxidant and anti-inflammatory properties of sulforaphane and its ability to protect and improve mitochondrial bioenergetic function contribute to its protective action against cholesterol-induced pancreatic β-cell dysfunction. Our data provide a scientifically tested foundation upon which sulforaphane can be developed as nutraceutical to preserve β-cell function and eventually control hyperglycemia.
1. Introduction
Sulforaphane (SFN) is a natural isothiocyanate derived from a glucosinolate found in cruciferous vegetables, especially broccoli, and is a potent activator of several NF-E2-related factor-2- (Nrf2-) regulated phase 2 enzymes [1]. Treatment of pancreatic β-cell lines, RINm5F, and INS-1(832/13) with SFN increases Nrf2 nuclear translocation and expression of phase 2 enzyme genes such as heme oxygenase-1 (Hmox-1) and glutamate-cysteine ligase catalytic subunit (Gclc) [2, 3]. Activation of Nrf2 by SFN prevented β-cell damage induced by cytokines downstream of the NFκB signaling pathway and restored insulin secreting responses to glucose [2]. Furthermore, pretreatment with SFN blocked the development of type 1 diabetes in streptozotocin-treated animals [2, 4]. It has also been shown that the activation of Nrf2 protects mitochondria from dysfunction and promotes mitochondrial biogenesis in neurodegenerative diseases [5]. In fact, recently we have shown that SFN is anticonvulsant and improves mitochondrial function in mouse hippocampal formations [6]. However, the effect of SFN on the mitochondrial function related to pancreatic β-cell function has not been studied yet.
Cholesterol is an essential structural component of cell membranes and serves as a precursor for the biosynthesis of steroid hormones and bile acids. High levels of cholesterol, however, contribute to pancreatic β-cell dysfunction [7, 8], leading to an impaired insulin secretory response to glucose, which is a hallmark of the transition from prediabetic to diabetic state [9]. Mice with specific inactivation of ABCA1 (ATP-binding cassette transporter subfamily A member 1), a transporter that mediates reverse cholesterol efflux in β-cells [10], and mice with LXR-β (liver X receptor beta) deficiency [11], a nuclear hormone receptor that increases ABCA1 expression in response to cholesterol, show impaired glucose tolerance and insulin secretion. Moreover, a direct link has been found between elevated cholesterol and reduced insulin secretion in islets isolated from C57BL/6J mice, in INS-1 rat pancreatic β-cells [12], and in Min6 cells [7, 8], whereby insulin secretion can be restored through cholesterol depletion [12]. Elevated cholesterol levels in pancreatic islets are also associated with β-cell dysfunction and reduced glucose-stimulated insulin secretion (GSIS) in LDL receptor deficient mice [13]. Given that pancreatic β-cells are particularly susceptible to oxidative stress due to their low antioxidant defenses [14], it has been suggested that cholesterol may induce β-cell dysfunction by increasing oxidative stress and causing mitochondrial damage [7, 8, 15, 16]. Although cholesterol has been shown to increase TNF-α (tumor necrosis factor alpha), interleukin-6 (IL-6), and macrophage colony-stimulating factor (M-CSF) in macrophages [17], it remains to be established to which extent inflammation contributes to β-cells dysfunction induced by cholesterol.
The present study aimed to determine the mechanism underlying the protective effect of SFN on impaired GSIS in a pancreatic β-cell line exposed to high concentration of cholesterol. This study also addressed the protective effects of SFN on mitochondrial bioenergetic dysfunction, inflammation, and oxidative stress induced by high levels of cholesterol and the molecular pathways involved. The effects of SFN were compared to those induced by 3,4-dihydroxyphenylacetic acid (ES), a microbiota-derived metabolite of quercetin that induces Nrf2 activation and that has been shown to protect against pancreatic β-cells dysfunction induced by high cholesterol [7].
2. Materials and Methods
2.1. Materials
3,4-dihydroxyphenylacetic acid (ES), water-soluble cholesterol, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), glucose, L-glutamine, pyruvate, sucrose, mannitol, fatty acid-free BSA, oligomycin, antimycin A, rotenone, N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD), ascorbic acid (AA), succinate, and malate were purchased from Sigma (MO, USA). Sulforaphane (SFN) was from Sapphire Bioscience Pty Ltd., (Sydney, NSW, Australia). Cytokine/Chemokine Magnetic Bead Panel MCYTOMAG-70K was from EMD Millipore Corporation (MI, USA). The TBARS Kit Assay, NFκB (p65) Transcription Factor Assay, and Nuclear Extraction Kit were obtained from Cayman Chemicals (MI, USA). The Rat Insulin ELISA Kit was from ALPCO (MA, USA). RNeasy Mini Kit was from Qiagen (NRW, Germany). RQ1 RNase-free DNase Kit was from Promega (WI, USA). Tetro cDNA Synthesis Kit was from Bioline (NSW, Australia). SYBR Green PCR Master Mix was from Applied Biosystems (CA, USA). The M-MLV Reverse Transcriptase, Oligo(dT)15 Primer, and Recombinant RNasin Ribonuclease Inhibitor were from Promega (WI, USA). The Pierce BCA Protein Assay Kit was from Thermo Scientific (IL, USA). All cell culture reagents (DMEM, FBS, penicillin, and streptomycin) were from GIBCO BRL (NY, USA).
2.2. Cell Culture
Min6 cells (p38-p51) (kindly provided by Dr. Francisco Pérez, University of Chile) were cultured in DMEM (25 mM glucose) supplemented with 10% heat-inactivated FBS, 100 IU/ml penicillin, 100 µg/ml streptomycin in a humidified atmosphere of 95% air, and 5% CO2. All the experiments were conducted in nonsupplemented DMEM medium. Protein content was determined by using the Pierce BCA Protein Assay Kit. The absorbance and fluorescence were measured using a Multi-Mode Microplate Reader (Synergy HT, BioTek, VT, USA). The “water-soluble cholesterol” containing 47 mg of cholesterol/g solid according to Certificate of Analysis (molar ratio, 1 : 6 cholesterol/methyl-β-cyclodextrin) was used to deliver cholesterol to the cells, as previously described [7, 8, 15, 18, 19]. Considering that methyl-β-cyclodextrin induces cholesterol depletion from cell membranes at very high concentration (~2% or 5 mM) [18, 20], a 10-time lower concentration of this compound was used here. Similar to previous studies we incubated Min6 cells for 6 h with 320 µM of cholesterol [7, 8, 15].
2.3. Mitochondrial Coupling Assay
Using the extracellular flux XFe96 analyzer (Seahorse Bioscience, MA, USA), the degree of coupling between the electron transport chain (ETC), the oxidative phosphorylation machinery, and ATP production was evaluated. Min6 cells plated at 1 × 105 cells/well on XFe96 well plates were treated for 6 h with cholesterol with/without SFN or ES, and oxygen consumption rate (OCR) was measured. After washing, the cells were incubated for 1 h at 37°C in XF Assay Modified Medium containing 25 mM glucose, 2 mM L-glutamine, and 2 mM pyruvate, pH 7.4. The same study was conducted in the absence of glucose, with XF Assay Modified Medium containing 2 mM L-glutamine and 2 mM pyruvate, pH 7.4. State 3 (basal respiration), state 4o (induced with 2 μM oligomycin), and maximal respiration (state 3u, stimulated with 1.5 μM FCCP, an uncoupling agent) were sequentially measured. Maximal respiration, spare capacity (state 3u minus state 3), ATP turnover (state 3 minus state 4o), and coupling efficiency (ATP turnover/state 3) were calculated as previously described [8]. All values were normalized to protein content. Rotenone and antimycin A (1 μM each) were added to block complex I and III, respectively, in order to determine the OCR unrelated to mitochondrial oxygen consumption. Only the mitochondrial dependent OCRs were considered for the calculation of mitochondrial function parameters which are expressed as nmol of oxygen consumed/min/mg protein.
2.4. Mitochondrial Electron Flow
The sequential electron flow through the complexes of the ETC was studied in mitochondria isolated from Min6 cells treated with cholesterol in the absence or in the presence of SFN or ES for 6 h, by using the extracellular flux XFe96 analyzer. This assay allows the study of the contribution and function of complexes I, II, and IV in the ETC in terms of OCR. Mitochondria were isolated as previously described [6, 7, 21]. Briefly, cells were harvested, washed in a Ca2+/Mg2+-free PBS, and centrifuged (10 min; 1,000g; 4°C). The pellet was resuspended and homogenized in MSHE solution (70 mM sucrose, 210 mM mannitol and 5 mM HEPES, 1 mM EGTA, and 0.5% (w/v) fatty acid-free BSA, pH 7.2). The homogenate was centrifuged at 1,000g for 10 min at 4°C and the resulting supernatant at 12,000g for 10 min. The pellet was washed and centrifuged at 10,000g for 10 min and resuspended in the same buffer. This preparation was used immediately for electron flow assay.
Freshly isolated mitochondria (4 µg) were plated in each well of XFe96 well plate and centrifuged at 2,000g for 20 min at 4°C in MAS buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, and 0.2% (w/v) fatty acid-free BSA, pH 7.2) in the presence of 10 mM pyruvate, 2 mM malate, and 4 µM FCCP (state 3u). Mitochondria were activated by the addition of 6 volumes of MAS at 37°C. Subsequently 2 µM rotenone (an inhibitor of complex I), 10 mM succinate (a substrate for complex II), 4 µM antimycin A (an inhibitor of complex III), and 1 mM AA/100 µM TMPD (AA/TMPD act as electron donors to cytochrome C in complex IV) were sequentially added. Rotenone inhibited the oxidation of pyruvate/malate mediated via complex I and, thus, the contribution of complex I to respiration was calculated as state 3u minus OCR after rotenone injection. Injection of succinate allows the mitochondria to respire via complex II; thus complex II-driven respiration was calculated as the increase in OCR after succinate injection. Electron flow is inhibited at complex III by antimycin A; then after the addition of AA/TMPD the activity of complex IV was calculated [22]. Values from electron flow assay are expressed as nmol of oxygen consumed/min/mg protein.
2.5. qPCR Measurements
Gene expression related to mitochondrial function and downstream of Nrf2 activation was studied. The mRNA levels of Sirt1, peroxisome proliferator-activated receptor gamma coactivator-1-alpha (Pgc-1α), Hmox-1, Gclc (the limiting enzyme in glutathione synthesis), and Cu/Zn superoxide dismutase (Sod1) were evaluated using qPCR as previously described [8, 23] in Min6 cells treated for 20 h with cholesterol in the presence or in absence of SFN or ES. The relative fold expression of each gene is expressed relative to the cycle thresholds of two housekeeping genes, Hmbs and Tbp in Min6 cells. Primer sequences are provided in Table 2.
Table 2.
List of primers used in this study for RT-PCR.
| Gene | Forward | Reverse |
|---|---|---|
| Sirt1 | GTTGATTGTGAAGCTGTTCGTG | TGGCTCTATGAAACTGTTCTGG |
| Pgc-1α | CACCAAACCCACAGAAAACAG | GGGTCAGAGGAAGAGATAAAGTTG |
| Hmox-1 | GTTCAAACAGCTCTATCGTGC | TCTTTGTGTTCCTCTGTCAGC |
| Gclc | CTCCAGTTCCTGCACATCTAC | AGAACATCGCCTCCATTCAG |
| Sod1 | AAGACTGGAAATGCTGGGAG | GGTTTGAGGGTAGCAGATGAG |
| Tbp | TTCTCGAAAGAATTGCGCTGT | GCCTTGTGAGTCATTTCAGTGA |
| Hmbs | AAGGGCTTTTCTGAGGCACC | AGTTGCCCATCTTTCATCACTG |
Hmbs: hydroxymethylbilane synthase; Hmox-1: heme oxygenase-1; Gclc: glutamate-cysteine ligase catalytic subunit; Pgc-1α: peroxisome proliferator-activated receptor gamma coactivator-1-alpha; Sirt1: sirtuin 1; Sod1: Cu/Zn superoxide dismutase; and Tbp: TATA box binding protein.
2.6. Lipid Peroxidation
Oxidative stress in Min6 cells was evaluated by measuring lipid peroxidation using a TBARS Assay Kit according to manufacturer's instructions. Cells were plated in 75 cm2 flasks at a density of 2 × 107 cells 24 h prior to incubation with cholesterol in the presence or absence of SFN or ES for 6 h.
2.7. Inflammatory Status Evaluation
Pro- and anti-inflammatory cytokines were measured in cell homogenates from Min6 treated for 20 h with cholesterol in the presence or absence of SFN or ES by using the MCYTOMAG-70K Cytokine/Chemokine Magnetic Bead Panel. We also evaluated the NFκB activation in nuclear protein extracts from Min6 cells treated with cholesterol and/or SFN or ES for 6 h. Nuclear extracts were obtained using a Nuclear Extraction Kit (Cayman). An NFκB (p65) Transcription Factor Assay Kit (Cayman) was used to evaluate NFκB p50 and p65 DNA binding activities to the response element by ELISA. The values were normalized to protein content.
2.8. Insulin Secretion Assay
Insulin secretion assay was performed as we previously described [7, 8]. Briefly, Min6 cells (p38–41) were incubated with cholesterol in the presence or absence of SFN or ES for 6 h, in serum-free and glucose-free DMEM supplemented with 2 mM L-glutamine and 25 mM HEPES, pH 7.4. Cells were then washed twice with PBS and placed immediately in serum-free DMEM with low glucose (5 mM) or high glucose (25 mM) for 1 h and medium was collected for the GSIS assay, according to Suzuki et al. [24]. Insulin levels in the medium were measured using a Rat Ultrasensitive Insulin ELISA Kit according to the manufacturer's instructions. Basal insulin secretion was measured in the absence of glucose in Min6 cells exposed to the different treatments. All values were normalized to protein content.
2.9. Statistical Analysis
Data were analyzed by two-way ANOVA (specified in results and each figure legend), followed by Bonferroni's Multiple Comparison Test using GraphPad Prism 6 statistical software (La Jolla, CA, USA). Unless indicated otherwise, the experiments were performed three times (three independent culture preparations) and in triplicate or quadruplicate. Values with different superscript letters (A, B, C, D, E, and F) indicate significant differences (p < 0.05) between groups. Values are expressed as mean ± SEM.
3. Results
3.1. Sulforaphane Prevents Cholesterol-Induced Mitochondrial Bioenergetic Dysfunction
Results of the coupling assay measuring OCRs in Min6 cells with the XFe96 analyzer are shown in Figure 1(a) for SFN and in Figure 1(b) for ES. Mitochondrial function parameters were quantified in Figures 1(c)–1(h). Cholesterol treatment in Min6 cells decreased basal respiration by 30% (Figure 1(c)), maximal respiration by 38% (Figure 1(d)), ATP turnover by 68% (Figure 1(e)), coupling efficiency by 55% (Figure 1(f)), and spare capacity by 53% (Figure 1(g)), but it did not alter proton leak (Figure 1(h)) compared to cells which were not treated with cholesterol (all two-way ANOVAs, post-tests p < 0.05, Figures 1(c)–1(g)). SFN and ES prevented the mitochondrial OCR impairments induced by cholesterol and improved the mitochondrial functions in both cholesterol treated cells and control cells in a concentration-dependent manner. SFN at both 2 µM and 10 µM, in the absence or presence of cholesterol, also improved the basal respiration by 33% and 67% (Figure 1(c)) and the maximal respiration by 28% and 56% (Figure 1(d)), respectively. SFN at both 2 and 10 µM increased ATP turnover and coupling efficiency by 210% and 130%, respectively, in comparison with untreated cells (Figures 1(e) and 1(f)). SFN 10 µM also enhanced the spare capacity by 45% (Figure 1(g)). In the presence or absence of cholesterol, ES at 2 µM and 10 µM expanded basal respiration by 35% and 90%, ATP turnover by 163% and 380% and coupling efficiency by 93% and 150%, respectively in comparison with untreated cells (Figures 1(c), 1(e), and 1(f)). ES at 10 µM, with or without cholesterol, improved the maximal respiration by 50% (Figure 1(d)) and both SFN and ES decreased the proton leak by around 30% with respect to the control cells (Figure 1(h)). No significant differences were found in the mitochondrial function parameters between the treatments when this coupling assay was conducted in the absence of glucose (data not shown).
Figure 1.

SFN improves mitochondrial bioenergetics in Min6 cells exposed to cholesterol. Min6 cells were incubated with 320 µM of cholesterol and/or 2 or 10 µM of SFN or ES. Mitochondrial functions with SFN (a) or ES (b) treatment were measured in the form of OCR and expressed as nmol of oxygen consumed/min/mg of protein using the XFe96 analyzer in cells treated for 6 h with vehicle (black solid circle) or with 320 µM cholesterol (chol) (red solid diamond), 2 µM SFN (green solid triangle), 10 µM SFN (blue solid square), 2 µM SFN + 320 µM chol (green vacant triangle), 10 µM SFN + 320 µM chol (blue vacant square), 2 µM ES (violet solid triangle), 10 µM ES (orange solid triangle), 2 µM ES + 320 µM chol (violet vacant triangle), and 10 µM ES + 320 µM chol (orange vacant triangle). (c) Basal respiration and (d) maximal respiration stimulated with 1.5 μM FCCP (State 3u) are shown. (e) ATP turnover was calculated as basal respiration minus respiration induced with 2 μM oligomycin (State 4o). (f) Coupling efficiency was calculated as the ratio of basal respiration minus State 4o relative to basal respiration. (g) Spare capacity was calculated as maximal respiration minus basal respiration. (h) Proton leak was calculated as State 4o minus nonmitochondrial OCR induced with 1 μM antimycin/rotenone. Control cells were not treated with cholesterol. All values are expressed as mean ± SEM, from three independent culture preparations, each treatment performed in quadruplicate. All two-ways ANOVAs and Bonferroni posttest. Values with different superscript letters indicate significant differences (p < 0.05) between groups. Chol: cholesterol; SFN: sulforaphane; ES: 3,4-dihydroxyphenylacetic acid; and OCR: oxygen consumption rate.
The electron flow assay measured as OCR in isolated mitochondria from Min6 is shown in Figure 2(a). Cholesterol pretreatment decreased the respiration driven by complex I, complex II, and complex IV by 51% (Figure 2(b)), 42% (Figure 2(c)), and 57% (Figure 2(d), all two-way ANOVAs, post-tests p < 0.05, Figures 2(b)–2(d)). Both SFN and ES at 10 µM, in the presence or absence of cholesterol, improved respiration due to the increased activity of complex I by 66% and 62% (Figure 2(b)), complex II by 62% and 59% (Figure 2(c)), and complex IV by 79% and 67% (Figure 2(d)), respectively.
Figure 2.

SFN increases mitochondrial electron flow in Min6 cells exposed to cholesterol. Min6 cells were incubated with 320 µM of cholesterol and/or 10 µM of SFN or ES and mitochondria were isolated. (a) Electron flow was measured in the form of OCR and expressed as nmol of oxygen consumed/min/mg of protein using the XFe96 analyzer in mitochondria from control cells (black solid circle) or cells after 6 h treatment with: 320 µM cholesterol (chol) (red solid diamond), 10 µM SFN (blue solid square), 10 µM SFN + 320 µM chol (blue vacant square), 10 µM ES (orange solid triangle), and 10 µM ES + 320 µM chol (orange vacant triangle). (b) Respiration driven by complex I was calculated as from the decrease in OCR after the addition of 2 μM rotenone. (c) Complex II- and (d) IV-driven respiration was calculated as the increase in OCR after the addition of 10 mM succinate or 1 mM AA/10 μM TMPD, respectively. Control cells were not treated with cholesterol. Values are expressed as mean ± SEM, from three independent culture preparations, each treatment performed in quadruplicate. All two-way ANOVAs and Bonferroni posttests. Values with different superscript letters indicate significant differences (p < 0.05) between groups. AA: ascorbate; Chol: cholesterol; SFN: sulforaphane; ES: 3,4-dihydroxyphenylacetic acid; and OCR: oxygen consumption rate.
3.2. Sulforaphane and Cholesterol Modulate Gene Expression Related to Mitochondrial Function
Sirtuin 1 and PCG-1α are key regulators of mitochondrial function [25]. Cholesterol treatment decreased Sirt1 gene expression by 15%, while SFN or ES, in the presence or absence of cholesterol, increased its expression by around 40% compared to vehicle treated Min6 cells (two-way ANOVA, posttest p < 0.05, Figure 3(a)). Cholesterol increased Pgc-1α gene expression by 46%, while SFN or ES at 10 µM, in the presence or absence of cholesterol, increased the expression by around 140% (two-way ANOVA, posttest p < 0.05, Figure 3(b)).
Figure 3.

SFN induces the expression of genes regulating mitochondrial function. Gene expression of (a) Sirt1 and (b) Pgc-1α in Min6 cells treated with 320 µM of cholesterol and/or 10 µM of SFN or ES for 20 h was evaluated. Values are expressed as relative fold expression of two housekeeping genes (Table 2). Values are expressed as mean ± SEM, from three independent culture preparations, each treatment performed in quadruplicate. All two-ways ANOVAs and Bonferroni post-tests. Values with different superscript letters indicate significant differences (p < 0.05) between groups. Chol: cholesterol; SFN: sulforaphane; ES: 3,4-dihydroxyphenylacetic acid; Sirt1: sirtuin 1; and Pgc-1α: peroxisome proliferator-activated receptor gamma coactivator-1-alpha.
3.3. Sulforaphane Prevents Cholesterol-Induced Lipid Peroxidation and Induces the Expression of Genes Related to Antioxidant Defences
The exposure to cholesterol doubled lipid peroxidation in Min6 cells. This was completely averted in the presence of 10 µM SFN or ES (two-way ANOVA, posttest p < 0.05, Figure 4(a)). SFN or ES at 10 µM, in the absence of cholesterol, had no effect on lipid peroxidation. Cholesterol increased the gene expression of Hmox-1, Gclc, and Sod1 by 135%, 35%, and 44%, respectively (all two-way ANOVAs, posttests p < 0.05, Figures 4(b)–4(d)). SFN or ES 10 µM, with or without cholesterol, increased the expression of Hmox-1 by 4.4-fold and 5.5-fold, respectively (Figure 4(b)), the expression of Gclc by around 2.7-fold, and of Sod1 by around 2.2-fold (Figures 4(c) and 4(d)).
Figure 4.

SFN prevents lipid peroxidation in Min6 cells exposed to cholesterol and induces the expression of genes related to antioxidant enzymes. (a) Oxidative stress was determined by measuring lipid oxidation using TBARS Assay Kit in Min6 cells incubated for 6 h with 320 µM of cholesterol and/or 10 µM of SFN or ES. Values are expressed as nmol of MDA/mg of protein. Gene expression of (b) Hmox-1, (c) Gclc, and (d) Sod1 in Min6 cells treated for 20 h with of 320 µM cholesterol and/or 10 µM of SFN or ES was evaluated. Values are expressed as relative fold expression of two housekeeping genes (Table 2). Values are expressed as mean ± SEM, from three independent culture preparations, each treatment performed in quadruplicate. All two-ways ANOVAs and Bonferroni post-tests. Values with different superscript letters indicate significant differences (p < 0.05) between groups. Chol: cholesterol; SFN: sulforaphane; ES; 3,4-dihydroxyphenylacetic acid; MDA: malondialdehyde; Hmox-1: heme oxygenase-1; Gclc; glutamate-cysteine ligase catalytic subunit; and Sod1: Cu/Zn superoxide dismutase.
3.4. Sulforaphane Prevents Cholesterol-Induced Inflammation
Cholesterol increased NFκB translocation to the nucleus by 39%, being this effect prevented in the presence of 10 µM SFN or ES (two-way ANOVA, posttest p < 0.05, Figure 5). In addition, cholesterol increased the levels of proinflammatory cytokines including IL-1β, TNFα, and IFNγ by 14%, 21%, and 17%, respectively, while it reduced the levels of anti-inflammatory cytokines such as IL-4 by 20% and IL-10 by 12% (All two-way ANOVAs, posttests p < 0.05, Table 1). In the presence of 10 µM SFN or ES the cholesterol-induced alterations of cytokines levels were completely prevented (Table 1).
Figure 5.
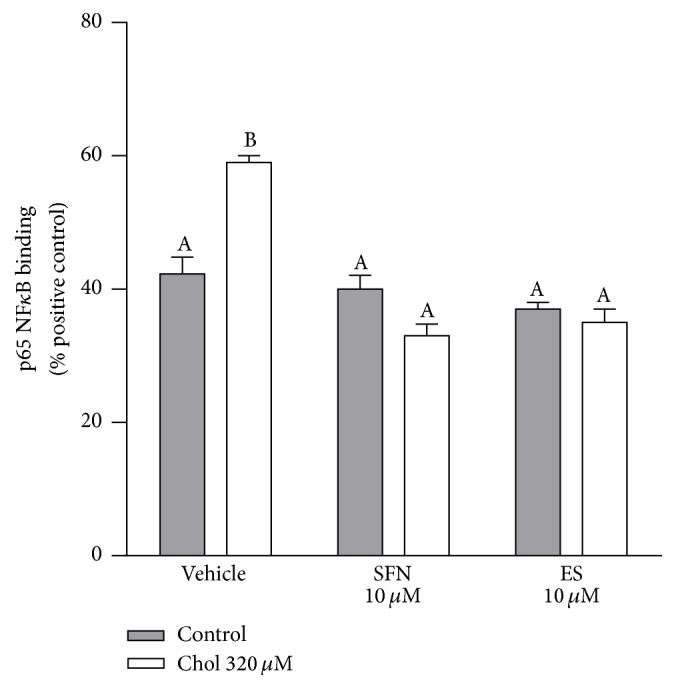
SFN inhibits the increase in nuclear NFκB translocation in Min6 cells exposed to cholesterol. NFκB (p65) binding to its cognate sequence was quantified in nuclear extracts from cells incubated for 6 h with 320 µM of cholesterol and/or 10 µM of SFN or ES. Values are expressed as percentage relative to positive control (HeLa cell lysate containing TNFα-activated NFκB (p65), 100%). Values are expressed as mean ± SEM, from three independent culture preparations, each treatment performed in triplicate. All two-ways ANOVAs and Bonferroni post-tests. Values with different superscript letters indicate significant differences (p < 0.05) between groups. Chol: cholesterol; SFN: sulforaphane; and ES: 3,4-dihydroxyphenylacetic acid.
Table 1.
Sulforaphane and ES prevent the changes in cytokines levels induced by cholesterol in Min6 cells.
| pg cytokines/mg protein | |||||
|---|---|---|---|---|---|
| IL-1β | TNFα | IFNγ | IL-4 | IL-10 | |
| Control | 74.15 ± 0.43A | 15.03 ± 0.40A | 31.22 ± 0.57A | 14.33 ± 0.59A | 85.20 ± 1.49A |
| Chol 320 µM | 82.02 ± 0.55B | 18.23 ± 1.85B | 36.66 ± 1.24B | 11.42 ± 1.10B | 75.13 ± 1.49B |
| SFN 10 µM | 69.90 ± 0.87A | 12.85 ± 0.25C | 27.51 ± 0.26A | 18.40 ± 0.58C | 89.00 ± 1.31A |
| SFN 10 µM + chol 320 µM | 65.25 ± 1.30C | 12.57 ± 0.35C | 29.23 ± 1.20A | 20.00 ± 0.22C | 85.32 ± 1.11A |
| ES 10 µM | 71.78 ± 1.16A | 11.86 ± 0.44C | 28.56 ± 0.34A | 16.39 ± 0.68D | 90.60 ± 1.64A |
| ES 10 µM + chol 320 µM | 71.28 ± 1.92A | 12.56 ± 0.38C | 28.50 ± 1.66A | 17.89 ± 0.39D | 86.25 ± 1.34A |
Proinflammatory and anti-inflammatory cytokines in Min6 cells treated for 20 h with 320 µM cholesterol and/or 10 µM SFN or ES. Values are expressed as mean ± SEM, from three independent culture preparations, each treatment performed in quadruplicate. All two-way ANOVA and Bonferroni posttests. Values with different superscript letters indicate significant differences (p < 0.05) between groups.
3.5. Sulforaphane Protects against Cholesterol Impaired Glucose-Stimulated Insulin Secretion
Basal secretion of insulin (without glucose) was similar for all the treatments (4.97 ± 0.05 ng/mg protein, mean ± SEM, Figure 6(a)). Insulin secretion in control cells increased by 2.6-fold in response to low glucose compared to the secretion in the absence of glucose (Figures 6(a) and 6(b)). The insulin secretion in response to low glucose was not different between any of the treatments (13.2 ± 0.6 ng/mg protein, mean ± SEM, Figure 6(b)). Insulin secretion of control cells increased by 30-fold in response to high glucose, compared to the secretion in the absence of glucose (Figures 6(a) and 6(c)), and by 11-fold compared to the secretion stimulated by low glucose (Figures 6(b) and 6(c)). With high glucose, cholesterol caused a 40% decrease in insulin secretion compared to the same condition in control cells (two-way ANOVA, posttest p < 0.05, Figure 6(c)). This effect of cholesterol was completely prevented by 10 µM SFN or ES (Figure 6(c)). SFN or ES alone had no effect on the insulin secretion induced by high glucose (Figure 6(c)).
Figure 6.

SFN protects against impaired glucose-stimulated insulin secretion in Min6 cells exposed to cholesterol. Insulin levels in the media of Min6 cells were determined in the (a) absence of glucose, (b) in response to 1 h exposure to low glucose (5 mM), or (c) high glucose (25 mM) after 6 h preincubation with 320 µM of cholesterol and/or 10 µM of SFN or ES, from three independent culture preparations, each treatment performed in triplicate. Values are expressed as mean ± SEM. N = 3. All two-ways ANOVAs and Bonferroni post-tests. Values with different superscript letters indicate significant differences (p < 0.05) between groups. Chol: cholesterol; SFN: sulforaphane; and ES: 3,4-dihydroxyphenylacetic acid.
4. Discussion
Pancreatic β-cell dysfunction, leading to an impaired insulin secretory response to glucose, plays a pivotal role in the transition from prediabetic state to the clinical type 2 diabetes mellitus (T2DM) [9, 26, 27]. In this study we found that SFN, to the same extent as ES, prevented GSIS impairment in a pancreatic β-cell line exposed to cholesterol. We demonstrated that SFN prevented cholesterol-induced mitochondrial bioenergetic dysfunctions, oxidative stress, and inflammation. The novel mechanisms of bioenergetics regulation by SFN described in the present study suggest that SFN is a potent protective agent of pancreatic β-cell function.
4.1. Sulforaphane Protects against Mitochondrial Dysfunctions Induced by Cholesterol
Our results demonstrate that cholesterol induces mitochondrial dysfunction by interfering with the ETC (Figures 2(a)–2(d)). We have recently shown that cholesterol reduces the activity of complex I [7], and in this study, we also showed for the first time that cholesterol impairs the respiration driven by complexes I, II, and IV during a state of major energy requirement induced by an uncoupler (further discussed below). A slowed electron flow reduces oxygen consumption and oxidative phosphorylation in mitochondria [28] as shown by the decrease in basal and maximal respiration, spare capacity, ATP turnover, and coupling efficiency observed in Min6 cells treated with cholesterol (Figures 1(c)–1(e)). Sulforaphane not only prevented this mitochondrial dysfunction, but also improved basal and maximal respiration as well as spare capacity and ATP turnover in Min6 cells. The protective effects of SFN on mitochondrial respiration may rely on its ability to improve complexes I-, II-, and IV-driven respiration. Although SFN has been previously shown to display hepatoprotective effects by preserving mitochondrial function, specifically the activities of mitochondrial complexes [29], here we have demonstrated for the first time enhanced respiration driven through complexes I, II, and IV by SFN in the pancreatic β-cell line and during increased energy demand. The latter is highly relevant in insulinoma, specifically under hyperglycemia condition, as β-cells constantly undergo an increased energy demand when insulin needs to be released. In addition, the spare capacity appears to be an important diagnostic measure of cell bioenergetics that experience high fluctuation in ATP demand [30]. SFN improves bioenergetics of the cells during high energy demands, with an increased spare capacity and an efficient electron flow resulting in higher ATP turnover. An elevation in the ATP/ADP ratio ensures continued exocytosis of insulin [31]. This is one important mechanism for the protection of SFN against the impairment on GSIS induced by cholesterol (Figure 6(c)).
4.2. Sulforaphane Improves the Expression of Genes Related to Mitochondrial Function
Our results also show that the improvement in mitochondrial function induced by SFN was associated with a rise in Sirt1 (Figure 3(a)) and Pgc-1α expression (Figure 3(b)) in both vehicle and cholesterol treated cells. Sulforaphane was shown to prevent the decreased expression of Sirt1 and Pgc-1α in an animal model of T2DM, as a protective mechanism against diabetic cardiomyopathy [32] and muscle atrophy [33]. However, this is the first study evaluating the modulation of the expression of these genes by SFN as another mechanism for the protection of pancreatic β-cells. In β-cells, Sirt1 regulates the expression of specific mitochondria-related genes that control metabolic coupling and a decrease in Sirt1 expression impairs glucose sensing and insulin secretion [34]. Sirtuin 1 activates PGC-1α by deacetylation, a key regulator of mitochondrial biogenesis and function [35]. Sulforaphane also promoted Pgc-1α expression (Figure 3(b)), probably through the increased expression of Sirt1 as evidenced by the improved mitochondrial function observed in the presence of SFN (Figures 1(a)–1(h) and Figures 2(a)–2(d)). In contrast, the increase in Pgc-1α expression induced by cholesterol (Figure 3(b)) may reflect a compensatory mechanism due to decreased levels of Sirt1 and thus lower activation of PGC-1α, which is reflected by the decreased mitochondrial activity (Figures 1(a)–1(h) and Figures 2(a)–2(d)). Alternatively, it might be an effort to directly restore the diminished energetic levels of the cell.
4.3. Sulforaphane Protects against the Oxidative Stress Induced by Cholesterol
Interestingly, we found that cholesterol, and to a higher extent SFN, induced the expression of antioxidant genes including Hmox-1, Gclc, and Sod1 and downstream Nrf2 activation (Figures 4(b)–4(d)). However cholesterol increased lipid peroxidation while SFN protected against this deleterious effect (Figure 4(a)). We have previously shown that cholesterol increases ROS levels and reduces SOD and glutathione peroxidase activities in Min6 cells, despite of increasing the translocation of Nrf2 to the nucleus [7]. This suggests that cholesterol promotes the inactivation of the antioxidant defences downstream Nrf2 activation by promoting the oxidation of the catalytic moieties of the antioxidant enzymes [36–38]. This is supported by the fact that, in the presence of an antioxidant such as ES, the cholesterol-induced oxidative stress as well as the inactivation of the antioxidant enzymes was totally prevented [7].
SFN, however, restored the redox homeostasis within the cell by increasing the expression of the antioxidant enzymes downstream Nrf2 activation (Figures 4(b)–4(d)) and, unlike cholesterol, preserved their activities by blocking oxidative stress, an event which is consistent with the protective effect of SFN on lipid peroxidation induced by cholesterol (Figure 4(a)). Interestingly, cobalt protoporphyrin, a known Hmox-1 inducer, protected INS-1 β-cells from high glucose-induced oxidative stress and apoptosis; however, it failed in restoring the GSIS in these cells [39]. The latter indicates that the mechanisms underlying the protective effect of SFN against the pancreatic β-cells dysfunction induced by cholesterol are beyond Nrf2 pathway induction and oxidative stress inhibition. The protective effects on mitochondrial bioenergetics provide a plausible mechanism supporting the antioxidant effects of SFN.
4.4. Sulforaphane Protects against Inflammation Induced by Cholesterol
SFN prevented the cholesterol-induced nuclear translocation of NFκB (Figure 5) and the increase in proinflammatory cytokine and the decrease in anti-inflammatory cytokine levels (Table 1). Therefore, SFN may protect against mitochondrial and pancreatic β-cells dysfunction by preventing inflammation. A combination of inflammatory cytokines (IL-1β, IFNγ, and TNF-α) decreases GSIS and promotes oxidative stress and mitochondrial dysfunction in INS-1 and RINm5F β-cells [40, 41]. SFN may protect β-cells via Hmox-1 (Figure 4(b)), since a cell-permeable heme oxygenase-1 (PEP-Hmox-1) protected INS-1 β-cells against apoptosis, oxidative stress, and inflammation induced by a cytokine mixture (IL-1β, IFNγ, and TNF-α) [42]. The antioxidant properties of SFN might also contribute to the prevention of cholesterol-induced activation of the NFκB pathway observed in this study, since ROS are associated with NFκB activation through the increase in IκB degradation [43]. SFN may also protect β-cells via Sirt1 (Figure 3(a)), since Sirt1 inhibits the transcriptional activity of NFκB, by deacetylating the RelA/p65 subunit [44] and Sirt1 overexpression downregulates NFκB activity in mice [45] and improves GSIS in β-cells [46].
4.5. Sulforaphane Protects Pancreatic Beta Cells against Cholesterol to the Same Extent as 3,4-Dihydroxyphenylacetic Acid
3,4-dihydroxyphenylacetic acid (ES) is the major microbial metabolite of quercetin and its glycosylated derivatives [47–54]. The flavonol quercetin is one of the most abundant polyphenol present in fruit and vegetables and in the Western diet [55–57]. It has been shown that ES is absorbable [58–60] and exerts strong antioxidant properties directly by scavenging free radicals [61] or indirectly by inducing the Nrf2 pathway [7, 62]. ES has been shown to protect against pancreatic β-cells dysfunction induced by cholesterol through its antioxidant and mitochondrial protective properties [7]. Our results show that ES and SFN exhibit similar effects, improving mitochondrial respiration during basal and high energetic demands by improving the electron flow through the ETC (Figures 1(a)–1(h) and Figures 2(a)–2(d)). Their anti-inflammatory activities (Figure 5 and Table 1) also appear to contribute to the protection against pancreatic β-cells dysfunction induced by cholesterol.
4.6. In Vivo Approach of the Health Effects of Sulforaphane and 3,4-Dihydroxyphenylacetic Acid in Terms of Their Effective Concentrations
Studies in both humans and rodents [63–65] have demonstrated that the intake of cruciferous vegetable containing active myrosinase results in a higher production of isothiocyanates than that of cruciferous lacking active myrosinase. For example, a plasma SFN concentration of 4 nM was detected in human volunteers after the intake of 200 g of blanched broccoli for 4 weeks [66] while a substantially higher plasma concentration of 2.3 µM of SFN was observed after consuming 40 g of fresh broccoli sprouts [67]. This can be explained by the inactivation of myrosinase during cooking. Increased SFN bioavailability can be attained after the intake of SFN-enriched broccoli sprout preparation (generated by quick steaming followed by myrosinase treatment) in mice [68]. Although the concentration of SFN used in our study cannot be reached after consuming broccoli in a normal diet, it could be attained through the intake of a diet rich in myrosinase-treated broccoli or SFN-based nutraceuticals.
It is unlikely that the effective concentration of ES studied in vitro here can be reached in vivo. However, additional experiments are needed to establish the plasma concentration that could be reached with a standard diet or a polyphenol rich diet, since the available information is mainly based on studies with limited sample sizes or carried out with subjects eating uncontrolled diets [58–60, 69, 70]. It seems that most of the ES absorbed is metabolized to 3-methoxy-4-hydroxyphenylacetic acid, since the concentration of this methyl derivative was found to be 24-fold higher than ES in the 24 h urine collections [60]. Thus, the intermediate metabolism is another important variable to consider since flavonoid bioavailability is intrinsically regulated by factors such as dietary intake, differences in host microbiota, polymorphism of intestinal transporters, metabolic pathways, and excretion. Moreover, the metabolic activity of the microbiota from diabetic subjects remains to be studied, specifically in terms of polyphenol metabolism.
In conclusion, our study demonstrates that SFN protects β-cells against cholesterol-induced impairments of their mitochondrial function by improving the electron flow in the ETC as well as the basal and maximal respiration, spare capacity, and ATP turnover. SFN promotes the expression of genes involved in antioxidant defense and averts cholesterol-induced lipid peroxidation and activation of the NFκB pathway, normalizing the expression of pro- and anti-inflammatory cytokines. The deleterious effects of cholesterol are associated with a decrease in Sirt1 expression while SFN increases it. These actions of SFN are similar to those of ES and are well suited to protect against cholesterol-induced pancreatic β-cell dysfunction, thereby preserving GSIS. Our results indicate that sulforaphane and ES are protective agents against cholesterol-induced alterations of pancreatic β-cell function. This study supports the consumption of glucosinolates and polyphenol-containing nutraceuticals, fruit, and vegetables to reduce the risk of diabetes.
Acknowledgments
The present study was supported by Fondecyt Initiation into Research Grant 11130232 to Catalina Carrasco-Pozo. The authors are grateful for support from IPRS and UQCent scholarships from the University of Queensland (Kah Ni Tan) and NHMRC Project Grant (1044007, Karin Borges).
Competing Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contributions
Catalina Carrasco-Pozo designed the experimental study. Catalina Carrasco-Pozo and Kah Ni Tan performed the experiments. Catalina Carrasco-Pozo, Karin Borges, and Martin Gotteland interpreted the results and drafted the manuscript. All authors commented and approved the submitted manuscript.
References
- 1.Jiménez-Osorio A. S., González-Reyes S., Pedraza-Chaverri J. Natural Nrf2 activators in diabetes. Clinica Chimica Acta. 2015;448:182–192. doi: 10.1016/j.cca.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Song M., Kim E., Moon W., et al. Sulforaphane protects against cytokine- and streptozotocin-induced β-cell damage by suppressing the NF-κB pathway. Toxicology and Applied Pharmacology. 2009;235(1):57–67. doi: 10.1016/j.taap.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Fu J., Zhang Q., Woods C. G., et al. Divergent effects of sulforaphane on basal and glucose-stimulated insulin secretion in β-cells: role of reactive oxygen species and induction of endogenous antioxidants. Pharmaceutical Research. 2013;30(9):2248–2259. doi: 10.1007/s11095-013-1013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Souza C. G., Da Motta L. L., De Assis A. M., et al. Sulforaphane ameliorates the insulin responsiveness and the lipid profile but does not alter the antioxidant response in diabetic rats. Food and Function. 2016;7(4):2060–2065. doi: 10.1039/c5fo01620g. [DOI] [PubMed] [Google Scholar]
- 5.Denzer I., Münch G., Friedland K. Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacological Research. 2016;103:80–94. doi: 10.1016/j.phrs.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 6.Carrasco-Pozo C., Tan K. N., Borges K. Sulforaphane is anticonvulsant and improves mitochondrial function. Journal of Neurochemistry. 2015;135(5):932–942. doi: 10.1111/jnc.13361. [DOI] [PubMed] [Google Scholar]
- 7.Carrasco-Pozo C., Gotteland M., Castillo R. L., Chen C. 3,4-dihydroxyphenylacetic acid, a microbiota-derived metabolite of quercetin, protects against pancreatic β-cells dysfunction induced by high cholesterol. Experimental Cell Research. 2015;334(2):270–282. doi: 10.1016/j.yexcr.2015.03.021. [DOI] [PubMed] [Google Scholar]
- 8.Carrasco-Pozo C., Tan K. N., Reyes-Farias M., et al. The deleterious effect of cholesterol and protection by quercetin on mitochondrial bioenergetics of pancreatic β-cells, glycemic control and inflammation: in vitro and in vivo studies. Redox Biology. 2016;9:229–243. doi: 10.1016/j.redox.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shulman G. I. Cellular mechanisms of insulin resistance. Journal of Clinical Investigation. 2000;106(2):171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunham L. R., Kruit J. K., Pape T. D., et al. β-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nature Medicine. 2007;13(3):340–347. doi: 10.1038/nm1546. [DOI] [PubMed] [Google Scholar]
- 11.Gerin I., Dolinsky V. W., Shackman J. G., et al. LXRβ is required for adipocyte growth, glucose homeostasis, and β cell function. Journal of Biological Chemistry. 2005;280(24):23024–23031. doi: 10.1074/jbc.m412564200. [DOI] [PubMed] [Google Scholar]
- 12.Hao M., Head W. S., Gunawardana S. C., Hasty A. H., Piston D. W. Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic β-cell dysfunction. Diabetes. 2007;56(9):2328–2338. doi: 10.2337/db07-0056. [DOI] [PubMed] [Google Scholar]
- 13.Bonfleur M. L., Vanzela E. C., Ribeiro R. A., et al. Primary hypercholesterolaemia impairs glucose homeostasis and insulin secretion in low-density lipoprotein receptor knockout mice independently of high-fat diet and obesity. Biochimica et Biophysica Acta—Molecular and Cell Biology of Lipids. 2010;1801(2):183–190. doi: 10.1016/j.bbalip.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Grankvist K., Marklund S. L., Taljedal I. B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochemical Journal. 1981;199(2):393–398. doi: 10.1042/bj1990393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y.-F., Wang L., Lee S., et al. Cholesterol induces mitochondrial dysfunction and apoptosis in mouse pancreatic beta-cell line MIN6 cells. Endocrine. 2010;37(1):76–82. doi: 10.1007/s12020-009-9275-y. [DOI] [PubMed] [Google Scholar]
- 16.Lu X., Liu J., Hou F., et al. Cholesterol induces pancreatic β cell apoptosis through oxidative stress pathway. Cell Stress and Chaperones. 2011;16(5):539–548. doi: 10.1007/s12192-011-0265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y., Tabas I. The inflammatory cytokine response of cholesterol-enriched macrophages is dampened by stimulated pinocytosis. Journal of Leukocyte Biology. 2007;81(2):483–491. doi: 10.1189/jlb.0806518. [DOI] [PubMed] [Google Scholar]
- 18.Lai C.-H., Chang Y.-C., Du S.-Y., et al. Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells. Infection and Immunity. 2008;76(7):3293–3303. doi: 10.1128/IAI.00365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khatibzadeh N., Spector A. A., Brownell W. E., Anvari B. Effects of plasma membrane cholesterol level and cytoskeleton f-actin on cell protrusion mechanics. PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0057147.e57147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziolkowski W., Szkatula M., Nurczyk A., et al. Methyl-beta-cyclodextrin induces mitochondrial cholesterol depletion and alters the mitochondrial structure and bioenergetics. FEBS Letters. 2010;584(22):4606–4610. doi: 10.1016/j.febslet.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 21.Tan K. N., Carrasco-Pozo C., McDonald T. S., Puchowicz M., Borges K. Tridecanoin is anticonvulsant, antioxidant, and improves mitochondrial function. Journal of Cerebral Blood Flow & Metabolism. 2016 doi: 10.1177/0271678x16659498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogers G. W., Brand M. D., Petrosyan S., et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE. 2011;6(7):p. 25. doi: 10.1371/journal.pone.0021746.e21746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hadera M. G., Smeland O. B., McDonald T. S., Tan K. N., Sonnewald U., Borges K. Triheptanoin partially restores levels of tricarboxylic acid cycle intermediates in the mouse pilocarpine model of epilepsy. Journal of Neurochemistry. 2014;129(1):107–119. doi: 10.1111/jnc.12610. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki K., Sato Y., Kai S., et al. Volatile anesthetics suppress glucose-stimulated insulin secretion in MIN6 cells by inhibiting glucose-induced activation of hypoxia-inducible factor 1. PeerJ. 2015 doi: 10.7717/peerj.1498.e1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cantó C., Auwerx J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current Opinion in Lipidology. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyssenko V., Almgren P., Anevski D., et al. Predictors of and longitudinal changes in insulin sensitivity and secretion preceding onset of type 2 diabetes. Diabetes. 2005;54(1):166–174. doi: 10.2337/diabetes.54.1.166. [DOI] [PubMed] [Google Scholar]
- 27.Tabák A. G., Herder C., Rathmann W., Brunner E. J., Kivimäki M. Prediabetes: a high-risk state for diabetes development. The Lancet. 2012;379(9833):2279–2290. doi: 10.1016/s0140-6736(12)60283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynafarje B. D., Ferreira J. Oxidative phosphorylation: kinetic and thermodynamic correlation between electron flow, proton translocation, oxygen consumption and ATP synthesis under close to in vivo concentrations of oxygen. International Journal of Medical Sciences. 2008;5(3):143–151. doi: 10.7150/ijms.5.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaona-Gaona L., Molina-Jijón E., Tapia E., et al. Protective effect of sulforaphane pretreatment against cisplatin-induced liver and mitochondrial oxidant damage in rats. Toxicology. 2011;286(1-3):20–27. doi: 10.1016/j.tox.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Brand M. D., Nicholls D. G. Assessing mitochondrial dysfunction in cells. The Biochemical Journal. 2011;435(2):297–312. doi: 10.1042/bj20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou J. C., Min L., Pessin J. E. Insulin granule biogenesis, trafficking and exocytosis. Vitamins and Hormones. 2009;80:473–506. doi: 10.1016/S0083-6729(08)00616-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z., Wang S., Zhou S., et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. Journal of Molecular and Cellular Cardiology. 2014;77:42–52. doi: 10.1016/j.yjmcc.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 33.Whitman S. A., Long M., Wondrak G. T., Zheng H., Zhang D. D. Nrf2 modulates contractile and metabolic properties of skeletal muscle in streptozotocin-induced diabetic atrophy. Experimental Cell Research. 2013;319(17):2673–2683. doi: 10.1016/j.yexcr.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luu L., Dai F. F., Prentice K. J., et al. The loss of Sirt1 in mouse pancreatic beta cells impairs insulin secretion by disrupting glucose sensing. Diabetologia. 2013;56(9):2010–2020. doi: 10.1007/s00125-013-2946-5. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Marcos P. J., Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. American Journal of Clinical Nutrition. 2011;93(4):884S–890S. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pigeolet E., Remacle J. Susceptibility of glutathione peroxidase to proteolysis after oxidative alteration by peroxides and hydroxyl radicals. Free Radical Biology & Medicine. 1991;11(2):191–195. doi: 10.1016/0891-5849(91)90171-x. [DOI] [PubMed] [Google Scholar]
- 37.Hodgson E. K., Fridovich I. The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: inactivation of the enzyme. Biochemistry. 1975;14(24):5294–5299. doi: 10.1021/bi00695a010. [DOI] [PubMed] [Google Scholar]
- 38.MacMillan-Crow L. A., Crow J. P., Thompson J. A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37(6):1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- 39.Lee E.-M., Lee Y.-E., Lee E., et al. Protective effect of heme oxygenase-1 on high glucose-induced pancreatic β-cell injury. Diabetes and Metabolism Journal. 2011;35(5):469–479. doi: 10.4093/dmj.2011.35.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai X., Ding Y., Zhang Z., Cai X., Li Y. Quercetin and quercitrin protect against cytokine-induced injuries in RINm5F β-cells via the mitochondrial pathway and NF-κB signaling. International Journal of Molecular Medicine. 2013;31(1):265–271. doi: 10.3892/ijmm.2012.1177. [DOI] [PubMed] [Google Scholar]
- 41.Lin C.-Y., Ni C.-C., Yin M.-C., Lii C.-K. Flavonoids protect pancreatic beta-cells from cytokines mediated apoptosis through the activation of PI3-kinase pathway. Cytokine. 2012;59(1):65–71. doi: 10.1016/j.cyto.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 42.Lee S. J., Kang H. K., Song D. K., et al. Transduction of PEP-1-heme oxygenase-1 into insulin-producing INS-1 cells protects them against cytokine-induced cell death. Biochemical and Biophysical Research Communications. 2015;461(3):549–554. doi: 10.1016/j.bbrc.2015.04.076. [DOI] [PubMed] [Google Scholar]
- 43.Li N., Karin M. Is NF-kappaB the sensor of oxidative stress? The FASEB Journal. 1999;13(10):1137–1143. [PubMed] [Google Scholar]
- 44.Yeung F., Hoberg J. E., Ramsey C. S., et al. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO Journal. 2004;23(12):2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfluger P. T., Herranz D., Velasco-Miguel S., Serrano M., Tschöp M. H. Sirt1 protects against high-fat diet-induced metabolic damage. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(28):9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moynihan K. A., Grimm A. A., Plueger M. M., et al. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metabolism. 2005;2(2):105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Aura A.-M., O'Leary K. A., Williamson G., et al. Quercetin derivatives are deconjugated and converted to hydroxyphenylacetic acids but not methylated by human fecal flora in vitro. Journal of Agricultural and Food Chemistry. 2002;50(6):1725–1730. doi: 10.1021/jf0108056. [DOI] [PubMed] [Google Scholar]
- 48.Benov L., Sztejnberg L., Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radical Biology and Medicine. 1998;25(7):826–831. doi: 10.1016/S0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- 49.Blaut M., Schoefer L., Braune A. Transformation of flavonoids by intestinal microorganisms. International Journal for Vitamin and Nutrition Research. 2003;73(2):79–87. doi: 10.1024/0300-9831.73.2.79. [DOI] [PubMed] [Google Scholar]
- 50.Braune A., Gütschow M., Engst W., Blaut M. Degradation of quercetin and luteolin by Eubacterium ramulus. Applied and Environmental Microbiology. 2001;67(12):5558–5567. doi: 10.1128/aem.67.12.5558-5567.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.LeBlanc L. M., Paré A. F., Jean-Francois J., Hébert M. J. G., Surette M. E., Touaibia M. Synthesis and antiradical/antioxidant activities of caffeic acid phenethyl ester and its related propionic, acetic, and benzoic acid analogues. Molecules. 2012;17(12):14637–14650. doi: 10.3390/molecules171214637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manach C., Williamson G., Morand C., Scalbert A., Rémésy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. The American Journal of Clinical Nutrition. 2005;81(1):230S–242S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 53.Schneider H., Schwiertz A., Collins M. D., Blaut M. Anaerobic transformation of quercetin-3-glucoside by bacteria from the human intestinal tract. Archives of Microbiology. 1999;171(2):81–91. doi: 10.1007/s002030050682. [DOI] [PubMed] [Google Scholar]
- 54.Schoefer L., Mohan R., Schwiertz A., Braune A., Blaut M. Anaerobic degradation of flavonoids by Clostridium orbiscindens. Applied and Environmental Microbiology. 2003;69(10):5849–5854. doi: 10.1128/aem.69.10.5849-5854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelly G. S. Quercetin. Alternative Medicine Review. 2011;16(2):172–194. [PubMed] [Google Scholar]
- 56.Ovaskainen M.-L., Törrönen R., Koponen J. M., et al. Dietary intake and major food sources of polyphenols in Finnish adults. Journal of Nutrition. 2008;138(3):562–566. doi: 10.1093/jn/138.3.562. [DOI] [PubMed] [Google Scholar]
- 57.Perez-Jimenez J., Fezeu L., Touvier M., et al. Dietary intake of 337 polyphenols in French adults. American Journal of Clinical Nutrition. 2011;93(6):1220–1228. doi: 10.3945/ajcn.110.007096. [DOI] [PubMed] [Google Scholar]
- 58.Gross M., Pfeiffer M., Martini M., Campbell D., Slavin J., Potter J. The quantitation of metabolites of quercetin flavonols in human urine. Cancer Epidemiology Biomarkers and Prevention. 1996;5(9):711–720. [PubMed] [Google Scholar]
- 59.Olthof M. R., Hollman P. C. H., Buijsman M. N. C. P., Van Amelsvoort J. M. M., Katan M. B. Chlorogenic acid, quercetin-3-rutinoside and black tea phenols are extensively metabolized in humans. The Journal of Nutrition. 2003;133(6):1806–1814. doi: 10.1093/jn/133.6.1806. [DOI] [PubMed] [Google Scholar]
- 60.Urpi-Sarda M., Monagas M., Khan N., et al. Targeted metabolic profiling of phenolics in urine and plasma after regular consumption of cocoa by liquid chromatography-tandem mass spectrometry. Journal of Chromatography A. 2009;1216(43):7258–7267. doi: 10.1016/j.chroma.2009.07.058. [DOI] [PubMed] [Google Scholar]
- 61.Rice-Evans C. A., Miller N. J., Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radical Biology and Medicine. 1996;20(7):933–956. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 62.Xue H., Xie W., Jiang Z., et al. 3,4-Dihydroxyphenylacetic acid, a microbiota-derived metabolite of quercetin, attenuates acetaminophen (APAP)-induced liver injury through activation of Nrf-2. Xenobiotica. 2016;46(10):931–939. doi: 10.3109/00498254.2016.1140847. [DOI] [PubMed] [Google Scholar]
- 63.Rouzaud G., Rabot S., Ratcliffe B., Duncan A. J. Influence of plant and bacterial myrosinase activity on the metabolic fate of glucosinolates in gnotobiotic rats. British Journal of Nutrition. 2003;90(2):395–404. doi: 10.1079/BJN2003900. [DOI] [PubMed] [Google Scholar]
- 64.Conaway C. C., Getahun S. M., Liebes L. L., et al. Disposition of glucosinolates and sulforaphane in humans after ingestion of steamed and fresh broccoli. Nutrition and Cancer. 2000;38(2):168–178. doi: 10.1207/S15327914NC382_5. [DOI] [PubMed] [Google Scholar]
- 65.Vermeulen M., Klöpping-Ketelaars I. W. A. A., Van Den Berg R., Vaes W. H. J. Bioavailability and kinetics of sulforaphane in humans after consumption of cooked versus raw broccoli. Journal of Agricultural and Food Chemistry. 2008;56(22):10505–10509. doi: 10.1021/jf801989e. [DOI] [PubMed] [Google Scholar]
- 66.Hauder J., Winkler S., Bub A., Rüfer C. E., Pignitter M., Somoza V. LC-MS/MS quantification of sulforaphane and indole-3-carbinol metabolites in human plasma and urine after dietary intake of selenium-fortified broccoli. Journal of Agricultural and Food Chemistry. 2011;59(15):8047–8057. doi: 10.1021/jf201501x. [DOI] [PubMed] [Google Scholar]
- 67.Clarke J. D., Hsu A., Riedl K., et al. Bioavailability and inter-conversion of sulforaphane and erucin in human subjects consuming broccoli sprouts or broccoli supplement in a cross-over study design. Pharmacological Research. 2011;64(5):456–463. doi: 10.1016/j.phrs.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y., Zhang T., Li X., Zou P., Schwartz S. J., Sun D. Kinetics of sulforaphane in mice after consumption of sulforaphane-enriched broccoli sprout preparation. Molecular Nutrition and Food Research. 2013;57(12):2128–2136. doi: 10.1002/mnfr.201300210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jenner A. M., Rafter J., Halliwell B. Human fecal water content of phenolics: the extent of colonic exposure to aromatic compounds. Free Radical Biology and Medicine. 2005;38(6):763–772. doi: 10.1016/j.freeradbiomed.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 70.Rios L. Y., Gonthier M.-P., Rémésy C., et al. Chocolate intake increases urinary excretion of polyphenol-derived phenolic acids in healthy human subjects. The American Journal of Clinical Nutrition. 2003;77(4):912–918. doi: 10.1093/ajcn/77.4.912. [DOI] [PubMed] [Google Scholar]