

Abstract
Due to their widespread incorporation into a range of biomedical and consumer products, the ingestion of silver nanoparticles (AgNPs) is of considerable concern to human health. However, the extent to which AgNPs will be modified within the gastric compartment of the gastrointestinal tract is still poorly understood. Studies have yet to fully evaluate the extent of physicochemical changes to AgNPs in the presence of biological macromolecules, such as pepsin, the most abundant protein in the stomach, or the influence of AgNPs on protein structure and activity. Herein, AgNPs of two different sizes and surface coatings (20 and 110 nm, citrate or polyvinylpyrrolidone) were added to simulated gastric fluid (SGF) with or without porcine pepsin at three pHs (2.0, 3.5, and 5.0), representing a range of values between preprandial (fasted) and postprandial (fed) conditions. Rapid increases in diameter were observed for all AgNPs, with a greater increase in diameter in the presence of pepsin, indicating that pepsin facilitated AgNPs aggregation. AgNPs interaction with pepsin only minimally reduced the protein’s proteolytic functioning capability, with the greatest inhibitory effect caused by smaller (20 nm) particles of both coatings. No changes in pepsin secondary structural elements were observed for the different AgNPs, even at high particle concentrations. This research highlights the size-dependent kinetics of nanoparticle aggregation or dissolution from interaction with biological elements such as proteins in the gastrointestinal tract. Further, these results demonstrate that, in addition to mass, knowing the chemical form and aggregation state of nanoparticles is critical when evaluating toxicological effects from nanoparticle exposure in the body.
Introduction
Engineered nanoparticles are used in a wide variety of consumer, industrial, pharmaceutical, and biomechanical products because of their unique physical properties.1–3 In particular, silver nanoparticles (AgNPs) have gathered interest as potential antimicrobial agents in consumer products (i.e. clothing and food packaging) owing to the ability of the silver ions (Ag+) released from the particle to penetrate the bacterial cell wall.4–6 As AgNPs are incorporated into more consumer products, ingestion of AgNPs, either accidentally or from the deliberate incorporation into food-related products, is likely to increase. However, the toxicity of AgNPs remains a controversial issue. While exposure of different cell lines to AgNPs to has been shown to induce varying degrees cytotoxicity,7–10 in vivo studies have shown inconsistent results.11–13 Nonetheless, only limited research has examined modification of AgNPs within the gastrointestinal (GI) tract.14–17 A limitation of most in vitro toxicological studies is the direct use of laboratory generated or commercially available nanoparticles, without consideration of the potential for AgNPs to be modified during passage through the stomach.18 Physicochemical characteristics, including size, surface charge, coating, and agglomeration state will affect how nanoparticles interact with one another.19 More importantly, these physical characteristics can affect the interaction with biological moieties, such as proteins, within the body. Therefore, characterizing the extent of dissolution or aggregation of ingested nanoparticles is essential for directing and prioritizing future toxicological studies of nanoparticle-related exposure.4
When a nanoparticle is introduced into biological media, a protein corona quickly forms around the nanoparticle, an interaction dependent upon the surface properties of both the biomolecules in the media and the nanoparticles.20, 21 The layers have been termed the “soft” corona, consisting of loosely bound proteins, and the “hard” corona, consisting of tightly bound proteins.22–25 The formation of a protein corona is a dynamic process; adsorption and desorption of proteins occurs as the nanoparticle is transported through the media.26, 27 The proteins with the highest binding affinity will preferentially bind and form the hard corona, which subsequently can be characterized via analytical methods.26, 27 It is the presence of these biological components within the hard corona that are of the greatest interest, as these can either directly or indirectly modify the bioreactivity of the particle.28 For example, the adsorption of proteins to the nanoparticle interface has been shown to modify protein function, by either inhibiting enzymatic activity or inducing conformational changes.29–31 This structural rearrangement has the potential to prevent binding to target receptors or may trigger an immune response in the body, thereby enhancing nanoparticle toxicity.32 Furthermore, the composition of the corona has been shown to modulate cellular interactions, including membrane affinity, uptake, and retention of nanoparticles.33–35 Ultimately, changes in nanoparticle reactivity can affect signalling pathways that may lead to altered cellular function.22, 27, 33 Therefore, understanding the extent of the nanoparticle-protein interaction and subsequent changes to the protein structure are essential for assessing biological alterations due to exposure.
The high salt content and acidity of the GI tract have great potential to drastically alter the physicochemical properties of AgNPs after ingestion, but limited research examining these processes in model systems has been conducted.14–17 In high salt synthetic biological fluids silver nanoparticles have been shown to undergo physical transformations resulting in aggregation or dissolution.36, 37 Previous studies have shown that exposure of AgNPs to simple simulated gastric fluid (SGF) can modify aggregation state, zeta potential, and morphology.38–41 These changes can occur within seconds to minutes after exposure, via the immediate release of small amounts of Ag+, followed by the precipitation of AgCl with excess Cl−, and subsequent aggregation of AgNPs through Van der Waal’s attraction.41 Thus, in this inorganic only system, AgNPs appear primarily in aggregated form, with low Ag+ concentration present. Nanoparticle transformations studied through changes in pH, salts and enzymes will provide a greater understanding and ability to interpret toxicity data from in vitro and in vivo studies.
While studies using simple gastric media formulations have provided insight into mechanistic processes regarding transformations of AgNPs,14–16 they lack biological macromolecules, specifically the protein component, which represent an important aspect of physiologically relevant conditions. Previous in vitro research has indicated that nanoparticles can be taken up into intestinal epithelial cells via macropinocytosis or endocytosis.42, 43 Yet, it remains unclear whether these nanoparticles reach the intestine in dissolved ionic (Ag+) or nanoparticle form.44 Dissolution studies of AgNPs in SGF with porcine pepsin have observed increases in the hydrodynamic diameter attributed partially to the formation of a protein corona, though the relative importance and characteristics of corona formation were not investigated.17, 45 Both studies suggested that the pepsin corona may protect AgNPs from the acidic environment of the stomach, which was proposed to aid the particles in reaching the intestine at their initial size. Given the complexity of nanoparticle modifications in the GI tract and the influence of a protein corona, understanding the interaction between pepsin and silver nanoparticles is essential for ultimately deciphering the cause of toxicity.
In this study, two sizes of AgNPs with two different coatings (20 nm and 110 nm citrate and polyvinylpyrrolidone (PVP)-coated) were exposed to SGF with and without proteins to determine the modification of AgNPs and properties of the protein corona. The SGF was prepared at three different pH levels (fasted pH 2, late fed pH 3.5, and early fed pH 5), which represent a range of values between the prandial and post-prandial conditions. SGF with pepsin (SGF-P), the primary proteolytic enzyme in the stomach, was observed to immediately influence the size distribution of the nanoparticles, indicating rapid protein corona formation and a greater increase in diameter over a 60 minutes than for SGF without pepsin. Furthermore, the influence of AgNPs on the protein itself was explored by assessing the effect on protein catalytic activity and conformational structure.
Experimental
Materials
Four AgNPs were evaluated in this study, including citrate-coated 20 and 110 nm (C20 and C110), and PVP-coated 20 and 110 nm (P20 and P110) particles each containing a 7 nm gold core (Nanocomposix, Inc.). The AgNPs were stored in stock solutions of 1 mg mL−1 at 4 °C in the dark. Initial characterization for the AgNPs was provided by the Nanoparticle Characterization Laboratory (NCL), with additional characterization reported in recent literature in which these particles were utilized.12, 14 This characterization included transmission electron microscopy (TEM), ultraviolet-visible spectroscopy (UV-Vis), dynamic light scattering and zeta potential (DLS and ZP), nanoparticle tracking analysis (NTA), and inductively coupled plasma mass spectrometry (ICP-MS). The initial sizes and zeta potentials of each type of the AgNPs in MilliQ water (18 MΩ) were confirmed using NTA and DLS/ZP prior to use in this work, at 31.8 nm for C20, 28.5 nm for P20, 113.1 nm for C110, and 106.9 nm for P110 (Table S1). Manufacturer concentrations were reported as 1 mg/mL and precise silver mass concentrations from NCL are given in Table S1.
Simple SGF was prepared using 34.2 mM NaCl (Sigma Aldrich) in 37% w/v HCl (Sigma Aldrich, Inc.) and MilliQ water (18 MΩ).46 The SGF pH was adjusted to 2.0, 3.5, and 5.0 using HCl, and was monitored with a pH meter (Mettler Toledo FE20). The SGF was then filtered through a 0.22 μm polyethersulfone filter membrane (Corning Life Sciences, Tewksbury, MA). Porcine pepsin (Sigma Aldrich, Inc.) was used to represent stomach protein, and was added in a concentration of 0.19 mg mL−1 to the SGF, referred to as SGF-P, approximately 45 minutes prior to experiments. This concentration of pepsin was chosen because the pepsin concentration in the human fasted state ranges from 0.11 to 0.22 mg mL−1.47
Dissolution Apparatus
The United States Pharmacopeia Apparatus 2 (USP 2) is a standard method used in pharmaceutical testing to evaluate the dissolution properties of orally administered solid dosage forms (Figure 1).46 Its simple design, consisting of a vessel and paddle, makes it easy to use and appropriate for quality control and research and development purposes.48 Literature values for the volume of liquid in the stomach have been recorded to range from 18 – 54 mL in the fasted condition,49 while averaging 250 mL in the fed condition.50 Due to the small volume within the stomach, a USP 2 mini vessel of 150 mL (72600573, Hanson Research), the smallest available, was used. To mimic movement and digestion within the stomach, a PTFE-coated mini paddle (72800460, Hanson Research) was inserted into the vessel and rotated using a motorized stirrer (EW-50006-01, Caframo Limited) at a constant 65 rotations per minute (RPM), a speed within the range of USP 2 recommendations (50 – 75 RPM).46 The vessel was placed in a glass aquarium containing water, an immersion heater (Cole Palmer), and Techne™ hollow plastic balls (Fischer Science), which were used to maintain a constant temperature of 37 °C, normal human body temperature. Prior to experiments, the SGF and SGF-P were allowed to warm and mix in the USP 2 apparatus for 45 minutes prior to the addition of AgNPs. Additionally, SGF-P at each pH was characterized in the NTA prior to each trial and over a 2 hour time period to ensure that the pepsin was not significantly changing the size distribution through aggregation without AgNPs (Figure S1). The concentration of AgNPs added to the vessel was determined based on the particle concentration (particles mL−1) range of the NTA and was between 108 and 109 particles mL−1 in a volume of 25 – 75 μL, of the stock solutions of AgNPs, depending on the size of the initial AgNPs.
Figure 1.

The USP 2 apparatus set-up including a 150 mL mini vessel and PTFE-coated mini paddle which are immersed in an aquarium holding water heated to 37 °C. The motorized stirrer rotates at a constant 65 RPM during experiments. a) shows the entire apparatus, while b) zooms in on the USP vessel. The Techne™ white plastic balls (Fischer Science) assist in maintaining an even 37 °C across the apparatus.
Nanoparticle Tracking Analysis
Nanoparticle tracking analysis (NTA) was used to assess change in the hydrodynamic diameter of AgNPs over the course of one hour after introduction to SGF or SGF-P within the USP 2. The NanoSight LM10 used in this study and the theory behind its operation has been described elsewhere in detail.51–55 Briefly, the LM10 was equipped with a 20× objective microscope and a 405 nm laser diode module. The laser is directed into a 350-μL sampling chamber and light scattering from nanoparticles is captured with a high sensitivity sCMOS camera at 30 frames per second. This set up is capable of detecting particles within the size range of 10 – 1000 nm. Particle displacement due to Brownian motion is converted using the Stokes equation to hydrodynamic diameter.56
At time points of 1, 10, 30, and 60 minutes after AgNPs were introduced, 1 mL aliquots were extracted from the vessel and injected into the LM10 sampling chamber using a syringe pump (NTA1014 NanoSight), which allowed for greater precision in determining the hydrodynamic diameters of AgNPs due to steady sample flow. All samples were measured within 5 minutes of extraction from the vessel. The measurement for time zero for all AgNPs was performed separately in MilliQ water due to the rapid modification of the nanoparticles in the SGF. To determine average size distributions, 10 videos of 30 seconds each were captured for a total of 300 seconds at each sampling time point. Videos were batch analyzed using the NTA 3.0 (Build 60) software and plotted to obtain the peak mode.
Transmission Electron Microscopy
A JEOL 3011 TEM was used to collect high resolution images of each of the AgNPs after reaction in SGF-P at the different pHs. After 10 minutes of AgNPs reacting in SGF-P, a 10 μL droplet was extracted and deposited onto a carbon -coated 200-mesh copper TEM grid (Ted Pella, Inc.) and allowed to coat the grid for 20 minutes. After which, excess liquid was wicked away using a Kim-wipe and the grid was quickly dip-rinsed in MilliQ to remove any remaining acidic media. Grids dried for 24 hours prior to collecting images. The JEOL 3011 was operated at 1.5 × 10−7 Pa and at an accelerating voltage of 100 kV. Images collected from the JEOL 3011 were processed using ImageJ version 1.48 (http://imagej.nih.gov/ij/) and projected area diameters were calculated from measured particle perimeter.
UV-Vis Spectroscopy
A UV-Vis spectrometer (Cary 50; Agilent Technologies) was used to assess the modification of the AgNPs by pepsin and formation of complexes between pepsin and AgNPs. For assessing modification of AgNPs by pepsin, a 100 μg mL−1 sample of stock solution was added to 10 mL of each SGF or SGF-P (containing 1 mg mL−1 of pepsin) and incubated at 37 °C in a water bath. After 10 minutes, samples were removed from the bath and a 2.5 mL aliquot was used for absorbance measurements. Spectra were collected in the range of 300 – 800 nm in a 1 cm path length quartz cuvette. To monitor the formation of pepsin-AgNP complexes, a 100 μg mL−1 sample of the stock solution of AgNPs was added to 10 mL of SGF-P (containing 0.19 mg mL−1 of pepsin) and incubated in a water bath for 10 minutes at 37 °C. After 10 min, 2.5 mL of sample was extracted and absorbance measurements were recorded from 200 – 360 nm in a 1 cm quartz cuvette. Initial background absorbance of SGF was used to blank the spectrometer for each experiment and all experiments were run in triplicate.
The Anson method was used to assess the change in pepsin catalytic activity in the stomach upon introduction of the AgNPs.57 Three different concentrations (10, 50, and 100 μg mL−1) of C20, P20, C110, P110, and AgNO3 were used. Each was mixed individually in a solution of HCl at pH 2 containing 0.5 mg mL−1 of pepsin. The mixtures were then incubated for 10 minutes in a water bath at 37 °C. Following the first incubation, a 1-mL aliquot of bovine hemoglobin (Sigma Aldrich, Inc.) was added to the mixture and allowed to incubate for 10 minutes, after which, the reaction was terminated by adding 2 mL of 5 % trichloroacetic acid. Upon termination, the mixture remained in the water bath for 5 minutes. After this time, a 2-mL aliquot of the mixture was extracted and centrifuged (Baxter Scientific Products, Biofuge A) at 5,000 RPM for 5 minutes. The supernatant was collected and placed in a 1 cm quartz cuvette and absorbance at 280 nm was measured using the UV-Vis spectrometer. Initial background absorbance of the pH 2 HCl solution was used as a blank and pepsin alone was used as a control. Experiments were repeated in triplicate and data are presented as percentage of control:
Circular Dichroism of Pepsin
Circular dichroism (CD; Jasco CD J-1500 spectrometer) was used to assess changes in pepsin secondary structure in the presence of C20, P20, C110, and P110. Concentrations of 100, 200, and 500 μg mL−1 of each of the AgNPs were added individually to a mixture consisting of 1 mg mL−1 pepsin and HCl at either pH 2 or 5. Upon addition of AgNPs, the media was incubated at 37 °C in a water bath for 60 min. After incubation, the mixtures were removed and centrifuged at 12,000 RPM for 20 min. Approximately 500 μL of the supernatant was removed from the mixture, and the pellet was resuspended in 10 mM HCl. The concentration of pepsin was confirmed using UV-Vis spectroscopy with the Beer-Lambert law using a molar extinction coefficient for pepsin of 51,300 cm−1 M−1.58, 59
CD spectra were recorded in a 1 cm quartz cuvette from 190 – 250 nm. The scanning speed was set at 50 nm min−1 for 3 accumulations. Data was converted into mean residual ellipticity (MRE; deg-cm2 dmol−1) and analyzed using CDPro software (http://lamar.colostate.edu/~sreeram/CDPro/main.html), which was available online to examine protein structure using CD. SDP48 was selected as the reference set because of the inclusion of both soluble and denatured proteins.60 The fractions of the structural elements outputs for the Continll program were summed together. For the statistical analysis, p-values were calculated for pepsin activity decrease at the 95% confidence interval assuming a normal distribution.
Results
Size Distributions of AgNPs in SGF using NTA
The size distributions collected using the NTA for C20, P20, C110, and P110 in SGF pH 2, respectively, is shown in Figure 2a–d. Figure 2e–h shows size distributions for C20, P20, C110, and P110 in SGF-P pH 2. In SGF pH 2, C20 and P20 more than double their diameter, growing by 55 nm and 47 nm after 1 minute (Table S1 and S2). In contrast, the C110 and P110 show a relatively small growth in diameter, by only 19 nm and 29 nm, respectively. The growth in diameter of the AgNPs in SGF over 60 minutes was observed to decrease with increasing pH. In SGF-P pH2, there was an immediate growth for each AgNP to even larger diameters when compared to the corresponding SGF. To more easily see the change in diameter of the AgNPs as a function of time, Figure 3 shows the peak mode diameter (log-normal fit) versus time for C20 (Figure 3a), P20 (Figure 3b), C110 (Figure 3c), and P110 (Figure 3d). The C20 and P20 underwent the greatest growth within the first minute in SGF-P pH 2, indicating aggregates of roughly ~100–200 particles. C20 and P20 then appeared to reach equilibrium, whereas in SGF pH 2 they had a slower (though still rapid) initial growth followed by gradual growth over the 60 minute period. Again, the observed growth for C110 and P110 was much less than that of the 20 nm AgNPs. For all four AgNPs, greater growth was observed in the presence of pepsin. The C20 was observed to grow more in diameter after incubation in SGF and SGF-P pH 2 when compared to P20, and both 20 nm particles are observed to grow by 55 – 67 % more than the 110 nm particles in either media. These results indicate that the increase in the diameter of the AgNPs was dependent on the initial particle diameter, not the surface coating.
Figure 2.
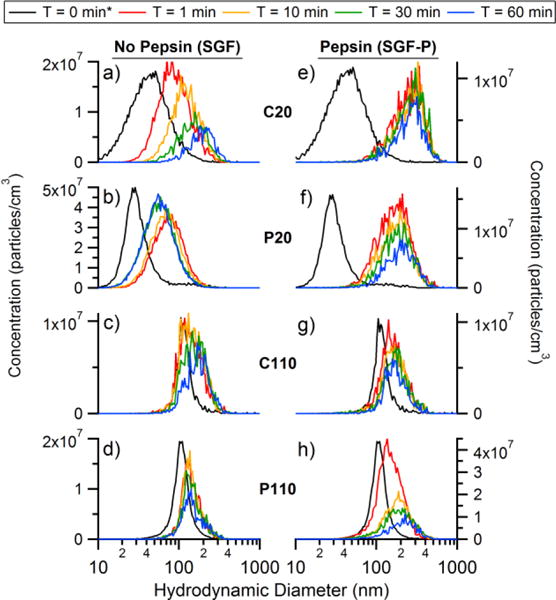
a) C20, b) P20, c) C110, and d) P110 NTA size distributions in pH 2 SGF and e) C20, f) P20, g) C110, h) P110 NTA in SGF-P. *Time = 0 min measurement was performed separately in MilliQ water due to the fast reactivity of the AgNPs at low pH.
Figure 3.
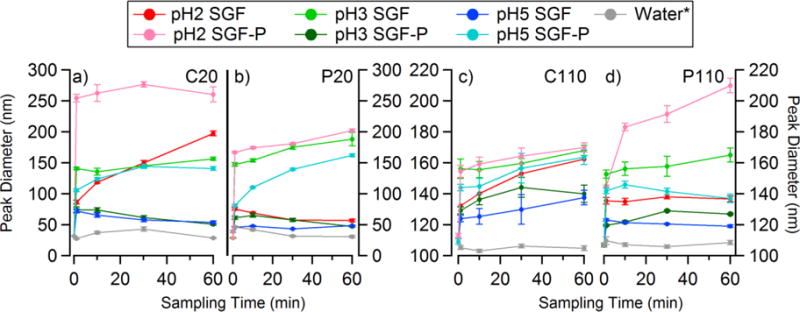
The change in the peak diameter for a) C20, b) P20, c) C110, and d) P110 in both SGF and SGF-P at pH 2, 3.5, and 5. *Time = 0 min measurement was performed separately in MilliQ water due to the fast reactivity of the AgNPs at low pH (same as Figure 2).
TEM images were collected for each type of the AgNPs in SGF-P at all pHs after 10 minutes of incubation (Figure S2). This time point was chosen because very little change in diameter was observed after this point. The TEM images show aggregation at the initial time point most likely due to drying prior to imaging and when under the vacuum of the SEM. The C20 and P20 showed large (> 10 particles) agglomerates and single isolated particles were rarely observed. The C110 and P110 were also aggregated, though less so than the C20 and P20, and individual particles were easier to locate on sample TEM grids. These images indicated that there were minimal changes in initial primary particle diameter, due to dissolution of the Ag metal to Ag+, within the larger aggregate (Figure 4). This was determined by measuring the projected area diameters for individual AgNPs with in the aggregate using Image-J. The primary particle diameter is then compared with initial NCL characterization. Only C110 could have a real difference in primary particle size, but has limited statistics due to experimental difficulties. Ultimately, these results indicate the AgNPs, and in particular the 20 nm AgNPs, retain their initial diameter when added to SGF-P and do not dissolve to form Ag+ in appreciable quantities.
Figure 4.
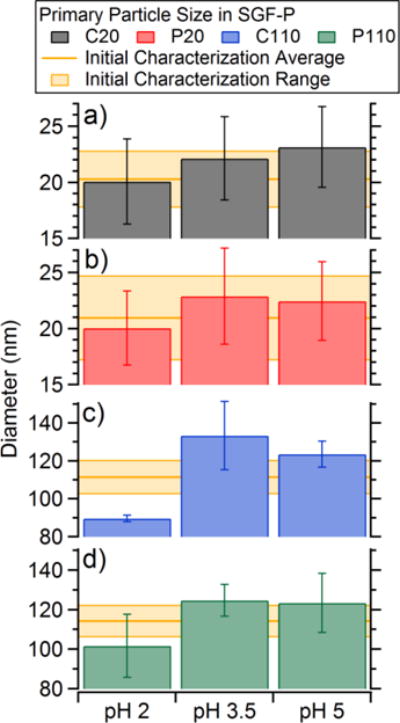
Average primary particle projected area diameters determined from TEM images of a) C20, b) P20, c) C110, and d) P110 after 10 minutes of incubation in SGF-P at pH 2, 3.5, and 5. The yellow bars represent the average diameter and standard deviation from the initial NCL characterization.
Physical changes to AgNPs characterized using UV-Vis
To determine the influence of pepsin on the physical properties of the AgNPs, each of the AgNPs were individually incubated for 10 minutes in SGF and SGF-P at all pHs, and their absorbance was measured using a UV-Vis spectrometer (Figure 5). Images were collected of each sample to visually show the colour and indicate the change in absorption. In water, C20 and P20 each had a large peak in absorbance present at 405 and 395 nm, respectively. This was due to their localized surface plasmon resonance (Figures 5a–b, respectively) and correlated with initial NCL characterization. In addition to this, P20 had a second peak around 600 nm. In SGF, both the C20 and P20 peaks were observed to decrease in intensity with decreasing pH. Additionally, in SGF pH 2 the C20 peak at 405 nm was no longer observed. For both C20 and P20 in SGF-P at all pHs, there was a drastic decrease in absorbance intensity and a red shift of the higher intensity (~400 nm) peak. There was also the emergence of a lower intensity peak around 605 nm, indicating both aggregation and increased particle diameter.61, 62 “Manna, 2001 #583” In water, C110 and P110 both showed two peaks in absorbance at 410 and 508 nm (Figure 5c), and 410 and 509 nm (Figure 5d), respectively. In the SGF at all pHs, there was a complete absence of the absorbance at 508 nm for C110. In the presence of pepsin, however, these peaks remained for C110, but each showed a slight broadening and decrease in intensity. In both cases with and without pepsin, the C110 absorbance did not appear to be greatly influenced by pH. In contrast, both P110 absorption peaks were observed to decrease in intensity with decreasing pH down to pH 3.5, irrespective of the presence of pepsin. In SGF and SGF-P pH 2, these two peaks were observed to broaden and have a large decrease in intensity when compared to other pHs. The presence of pepsin was observed to influence the spectral properties of each type of AgNPs, with the greatest effect on the smaller, C20 and P20 particles. This indicates that pepsin was interacting with each of the AgNPs over the range of pHs and forming pepsin-AgNP complexes.
Figure 5.
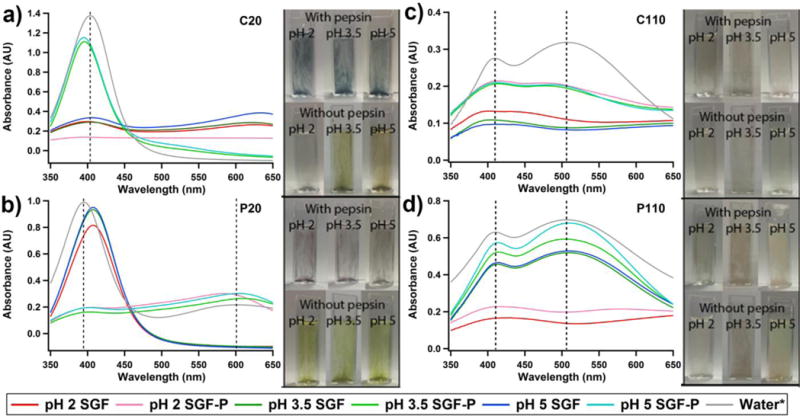
UV-Vis spectra of a) C20, b) P20, c) C110, and d) P110 after 10 minutes in SGF and SGF-P at pH 2, 3.5, and 5. The photographs show colour changes for the particles in media when either the pH is changed or pepsin is added.
In order to study changes in pepsin tertiary structure, UV-Vis spectra were collected from 200 – 360 nm, a range where pepsin absorbs, after a 10 minute incubation with each of the AgNPs at all pHs (Figure S3). The initial UV-Vis spectrum for pepsin in water showed a maximum absorption in the region from 275 – 280 nm, which is primarily due to the aromatic amino acids, tryptophan, and tyrosine, with some contribution from cysteine residues.63–65 Upon addition of the AgNPs with SGF-P, there was an observed decrease in absorbance for all four AgNPs, with very minor blue shifts in the absorbance peak. No clear trend was observed as a function of pH. These modifications in absorbance represent tertiary structure changes of pepsin via transformation of the microenvironment of the residues.66 Incubation with C20 showed the greatest decrease in absorbance.
Influence of AgNPs on Pepsin Secondary Structure
To assess changes in pepsin secondary structure upon the introduction of AgNPs, circular dichroism (CD) spectra were collected in the far-UV region from 190 – 250 nm (spectra in Supplemental Information, Figure S4). The calculated percentages of secondary structural elements are given in Table S3. Pepsin is a β-sheet-rich protein, which also has a significant amount of disordered structure.63, 67 After a 60 minute incubation in SGF-P pH 2 and pH 5 mixtures, the fractions of secondary structural elements did not change beyond ± 1%, indicating the AgNPs did not induce significant changes in secondary structure.
Influence of AgNPs on Pepsin Catalytic Activity
The potential for altering the biochemical activity of pepsin was assessed, since the AgNP-pepsin interaction was shown to influence pepsin’s tertiary structure. After a 10 minute incubation with each type of the AgNPs, moderate decreases in pepsin catalytic activity were observed (Figure 6). These decreases in activity were observed in a dose-dependent manner, with C20 and P20 exhibiting greater potential inhibition. Based on manufacturer specifications from Nanocomposix, the concentrations of nanoparticles (0.09, 0.46, and 0.93 μmol) corresponded to 2.3 × 1013, 4. 6 × 1013, and 1.15 × 1014 particles mL−1 for C20 and P20, and 1.0 × 1010, 3.0 × 1010, and 7.0 × 1010 particles mL−1 for C110 and P110. At the highest concentration (1:1), the C20 and P20 reduced pepsin activity by approximately 20%, whereas C110 and P110 reduced pepsin activity by 13% and 10%, respectively. At the highest concentration of AgNO3, there was minimal reduction of pepsin activity, approximately 5%. Interestingly, Ag+ ions have been shown to preferentially interact with sulfhydryl groups in the cysteine residues of proteins and can subsequently inhibit enzymatic activity.68, 69 However, the decrease in activity due to Ag+ ions is less than for all AgNPs, suggesting that interaction with the sulfhydryl groups does not have as large of an effect on activity as binding to the nanoparticle within the protein corona. Overall, the results indicates that the AgNPs have a greater inhibitory effect on pepsin catalytic activity than Ag+ ions.
Figure 6.
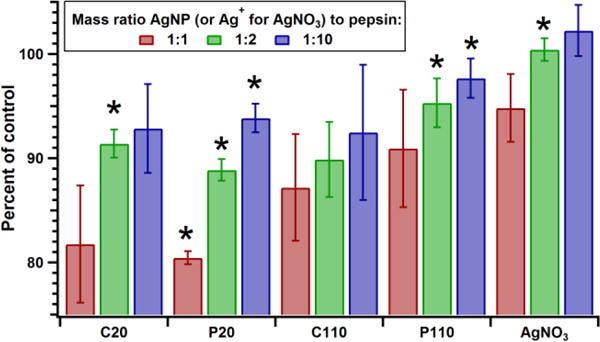
The change in pepsin catalytic activity after incubation for 10 minutes in an HCl/H2O solution of pH 2 with C20, P20, C110, P110, and AgNO3 of three concentrations (10, 50, and 100 μg mL−1). Activity is reported as percent of the pepsin-only control. *Indicates values with p < 0.05 compared to those for pepsin in SGF
Discussion
Size, surface coating, and GI media components can greatly influence physicochemical transformations of AgNPs within the GI tract. In the present study, changes in the size distribution of four different AgNPs in SGF and SGF-P at three pHs were evaluated over a 60 minute period using a USP 2 dissolution apparatus. This was the first use of the USP 2 approach to study nanoparticle modification (dissolution or aggregation). With each of the trials, the AgNPs underwent transformations dependent on the type of dissolution media and initial characteristics of the nanoparticle. Overall, the AgNPs were observed to reach larger diameters when added to SGF-P. The greatest increase in size occurred in the order of: fasted (pH 2), late fed (pH 3.5), and early fed (pH 5). Within fasted media, the 20 nm particles underwent the greatest increase in size; whereas, the 110 nm particles reached slightly less than double their initial size. The greatest rate of growth for the AgNPs was observed within the first minute of being added to either SGF or SGF-P, which is consistent with our previous study of SGF over shorter time periods, which showed that particle growth in SGF occurred rapidly on a 30 second time scale.14
AgNPs have been shown to dissolve when they are added to increasingly acidic solutions of HNO3.36 However, when NaCl is present, a significant increase in particle size due to aggregation has been observed.14, 38–40 AgNPs are highly susceptible to aggregation in the presence of chloride anions (Cl−), which can interact with free Ag+ or Ag+ on the nanoparticle surface to form insoluble AgCl.14, 70 Our previous work has proposed that this aggregation in SGF occurs via a multi-step process: first, Ag+ is generated from the acid stripping the coating and dissolving a small portion of the metal surface, then the Cl− (which is in excess in the stomach) precipitates with the Ag+ due to low solubility (1.92 × 10−4 g/100g H2O), which forms AgCl.14 The AgCl precipitate contributes to the aggregation process by decreasing surface charge and allowing Van der Waal’s forces to hold the nanoparticles together as aggregates after collisions in the media.14
Increasing the complexity of the system through the addition of pepsin to the SGF dramatically increased the growth rate and final diameter of the AgNPs by facilitating aggregation. Additionally, further NTA trials using fasted SGF and SGF-P indicated that C20 and P20 remained aggregated for up to 4 hours after incubation in the gastric system (Figures S4). Current studies of AgNPs in cell culture media have shown contradictory results regarding the aggregation of AgNPs in the presence of proteins.71–73 For example, Kittler et al.72 found that interactions of AgNPs with proteins present in cell culture media increased the degree of aggregation for AgNPs. In contrast, other studies have reported that proteins can stabilize individual pure AgNPs against aggregation while in cell culture media, primarily by acting as a coating agent that shields particle from modification.71, 73 Modifications of the isomeric conformation of proteins, such as bovine serum albumin, have been reported and may induce or prevent aggregation of nanoparticles.74
However, in gastric media of high ionic strength and low pH, aggregation has been observed to be enhanced. Both Walzack et al.17 and Bohmert et al.45 observed large agglomerates of AgNPs when digested in acidic gastric media with pepsin, which was attributed to bound proteins or aggregates. Present results indicate that the addition of pepsin increased aggregation of AgNPs in a pH-dependent manner. Changes in the gastric media acidity influenced the electrostatic and hydrophobic interactions between the protein and the AgNPs, such that aggregation was enhanced. UV-Vis data confirmed the influence of pepsin, in the form of a protein corona, on the SGF-induced physical changes of AgNPs. The shifts in peak absorbance of the AgNPs and changes in peak intensity indicated interactions between the pepsin and AgNPs, indicative of pepsin-AgNP binding. This was consistent with previous literature, which have shown red shifts in absorption spectra of AgNPs after incubation with proteins.68, 75
Adsorption of a protein onto a nanoparticle is a complex process, dependent on multiple factors, including nanoparticle size, material, surface charge, as well as the protein characteristics and the GI media used.28, 76, 77 Pepsin is the primary proteolytic enzyme in the stomach, responsible for the degradation of peptide bonds.78 It is secreted as a zymogen, and in its activated form, at acidic pH levels (2–3), it has a molecular weight of 34 kDa.79 Pepsin has been shown to form complexes with small molecules, indicated by red shifts in wavelength and changes absorbance intensity in UV-vis spectra.29, 63, 64 Reductions in absorption intensity indicate that the chromophores, tryptophan and tyrosine, are less exposed to the solvent, thus contributing differently to the adsorption coefficient.65 The present study suggests that the absorption of pepsin to the AgNPs reduced exposure of these chromophores in such a way that the formation of the pepsin-AgNP complex ultimately influenced the tertiary structure of the protein.
Previous reports have indicated there are conformational changes in secondary structure when proteins bind to or interact with nanoparticles.29, 68, 80 Banerjee et al.31 observed a minor change (≤ 8%) in secondary structural elements of the protein alpha A-Crystallin at a stoichiometric ratio of AgNPs to protein. At ratios of excess protein to AgNPs, these shifts in percentages were again observed to be minor (≤ 3%). Kakinen et al.68 observed modest, up to 15.4%, conformational changes in protein secondary structure after adsorption to AgNPs, attributed to the formation of multiple layers of protein coating, with the outer layers protecting inner one. In the present study, no significant change was observed in the percentages of structural elements even at the highest concentration of AgNPs (6.3:1 molar ratio of pepsin to AgNPs). Given the reductions in pepsin activity at lower molar ratios of pepsin to AgNPs, this suggests that when pepsin binds to the AgNPs within the corona, the secondary structure of the protein is preserved. The formation of the corona has been shown to modify the interaction with cells and translocation in gastric and duodenal fluids,81, 82 which emphasizes the importance of understanding protein corona properties under different conditions.83–85
The activity of a protein is highly dependent on proper conformational structure, as well as access of the substrate to the active site. The incubation of pepsin with AgNPs showed both a concentration and size-dependent relationship of pepsin activity inhibition. Interestingly, C20 and P20 were observed to reduce the pepsin activity to a greater extent than C110 and P110. Smaller nanoparticles (<50 nm) have been shown to preferentially bind a greater number of proteins than similar nanoparticles of larger (~100 nm) sizes, due to a greater surface area to mass ratio.24, 86 It is also possible that since C20 and P20 have a greater number of particles per given volume, the inhibitory effect may be in part due to a crowding out effect.
The affinity of Ag+ ions for thiol groups in proteins has been reported in several studies.87–89 Since Ag+ ions have previously been reported to inhibit enzyme activity via interaction with cysteine residues, pepsin was incubated with AgNO3.68, 69 Only minor reductions in activity were observed. Pepsin has two aspartic residues, Asp32 and Asp215, within the catalytic site, as well as 6 cysteine residues within the entire protein.90–92 The substrate binding cleft is located between two homologous lobes (N-terminal lobe and C-terminal lobe) and is protected by a β-hairpin loop.78 It is possible that these Ag-thiol interactions can happen without altering proteolytic function, due to their distance from the catalytic cleft. Since the nanoparticles exhibited greater inhibition than the Ag+ ions, our data suggests that the inhibition of activity was due to the physical adsorption onto the nanoparticle, rather than conformational changes in structure from Ag+ binding.
Conclusions
This study demonstrated that aggregation rates and terminal sizes of AgNPs of different sizes and coatings were enhanced in SGF containing protein versus SGF without protein. The presence of pepsin in the SGF was observed to lead to a significant enhancement in aggregation of AgNPs. This aggregation occurred rapidly, within the first minute of being added to SGF-P, and showed a pH-dependent relationship: fasted (pH 2) > late fed (pH 3.5) > early fed (pH 5). Changes in UV-Vis absorption for each of these AgNP in SGF-P confirmed these physical changes, and indicated the formation of a protein corona, which was confirmed with electron microscopy analysis. The binding of pepsin to the AgNPs was shown to induce minor shifts in protein tertiary structure, with no discernible changes in secondary structure. At high concentrations of AgNPs, proteolytic activity of pepsin was reduced. The higher standard deviations for the 1:1 samples run in triplicate explains the lack of statistical significance, but would be expected to be significant were it not for experimental challenges related to the higher AgNPs concentrations. Since changes in pepsin secondary structure were not observed, these results suggest that the inhibition of pepsin activity was a result of the physical adsorption of the protein onto the nanoparticle. The studies across different pHs, and thus prandial states, indicate increased particle growth with increasing acidity.
Future studies will expand the current artificial stomach model to contain multiple vessels representing the fluid composition of the small intestine, as well as increasing the complexity to more physiologically relevant conditions of the gastrointestinal tract (multiple enzymes, surfactants, etc.). This will enable us to explore both the transformation of AgNPs in the complex GI environment and the subsequent influence it has on the properties of the protein corona. Ultimately, understanding the nature of these interactions within the GI tract can provide insight for assessing risk to downstream biochemical and cellular processes that may be impacted from nanoparticle exposure.
Supplementary Material
Acknowledgments
This project was supported through National Institute of Health Grant U01ES020128. Silver nanoparticles used in this study where procured, characterized, and provided to investigators by NIEHS Center for Nanotechnology Health Implications Research (NCNHIR) Consortium. D.I.S was supported by the National Institute of Environmental Health Sciences under Award Number T32ES007062. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The University of Michigan Center for Materials Characterization (MC)2 is acknowledged for use of the JEOL 3011 High Resolution Electron Microscope. The Biophysics Department at the University of Michigan is acknowledged for use of the Jasco CD-Spectropolarimeter.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Chaudhry Q, Scotter M, Blackburn J, Ross B, Boxall A, Castle L, Aitken R, Watkins R. Food Addit Contam Part A. 2008;25:241–258. doi: 10.1080/02652030701744538. [DOI] [PubMed] [Google Scholar]
- 2.Mitrano DM, Rimmele E, Wichser A, Erni R, Height M, Nowack B. ACS Nano. 2014;8:7208–7219. doi: 10.1021/nn502228w. [DOI] [PubMed] [Google Scholar]
- 3.Varner KE, El-Badawy A, Feldhake D, Venkatapathy R. 2010 [Google Scholar]
- 4.Peters R, Kramer E, Oomen AG, Rivera ZE, Oegema G, Tromp PC, Fokkink R, Rietveld A, Marvin HJ, Weigel S, Peijnenburg AA, Bouwmeester H. ACS Nano. 2012;6:2441–2451. doi: 10.1021/nn204728k. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhry Q, Scotter M, Blackburn J, Ross B, Boxall A, Castle L, Aitken R, Watkins R. Food additives & contaminants Part A, Chemistry, analysis, control, exposure & risk assessment. 2008;25:241–258. doi: 10.1080/02652030701744538. [DOI] [PubMed] [Google Scholar]
- 6.Pal S, Tak YK, Song JM. Applied and environmental microbiology. 2007;73:1712–1720. doi: 10.1128/AEM.02218-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim TH, Kim M, Park HS, Shin US, Gong MS, Kim HW. Journal of biomedical materials research Part A. 2012;100:1033–1043. doi: 10.1002/jbm.a.34053. [DOI] [PubMed] [Google Scholar]
- 8.De Matteis V, Malvindi MA, Galeone A, Brunetti V, De Luca E, Kote S, Kshirsagar P, Sabella S, Bardi G, Pompa PP. Nanomedicine: nanotechnology, biology, and medicine. 2015;11:731–739. doi: 10.1016/j.nano.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Gliga AR, Skoglund S, Wallinder IO, Fadeel B, Karlsson HL. Part Fibre Toxicol. 2014;11:11. doi: 10.1186/1743-8977-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haase A, Rott S, Mantion A, Graf P, Plendl J, Thunemann AF, Meier WP, Taubert A, Luch A, Reiser G. Toxicological sciences: an official journal of the Society of Toxicology. 2012;126:457–468. doi: 10.1093/toxsci/kfs003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim YS, Song MY, Park JD, Song KS, Ryu HR, Chung YH, Chang HK, Lee JH, Oh KH, Kelman BJ, Hwang IK, Yu IJ. Part Fibre Toxicol. 2010;7:20. doi: 10.1186/1743-8977-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergin IL, Wilding LA, Morishita M, Walacavage K, Ault AP, Axson JL, Stark DI, Hashway SA, Capracotta SS, Leroueil PR, Maynard AD, Philbert MA. Nanotoxicology. 2015:1–9. doi: 10.3109/17435390.2015.1072588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Zande M, Vandebriel RJ, Van Doren E, Kramer E, Herrera Rivera Z, Serrano-Rojero CS, Gremmer ER, Mast J, Peters RJ, Hollman PC, Hendriksen PJ, Marvin HJ, Peijnenburg AA, Bouwmeester H. ACS Nano. 2012;6:7427–7442. doi: 10.1021/nn302649p. [DOI] [PubMed] [Google Scholar]
- 14.Axson JL, Stark DI, Bondy AL, Capracotta SS, Maynard AD, Philbert MA, Bergin IL, Ault AP. J Phys Chem C. 2015;119:20632–20641. doi: 10.1021/acs.jpcc.5b03634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mwilu SK, El Badawy AM, Bradham K, Nelson C, Thomas D, Scheckel KG, Tolaymat T, Ma LZ, Rogers KR. Sci Total Environ. 2013;447:90–98. doi: 10.1016/j.scitotenv.2012.12.036. [DOI] [PubMed] [Google Scholar]
- 16.Rogers KR, Bradham K, Tolaymat T, Thomas DJ, Hartmann T, Ma LZ, Williams A. Sci Total Environ. 2012;420:334–339. doi: 10.1016/j.scitotenv.2012.01.044. [DOI] [PubMed] [Google Scholar]
- 17.Walczak AP, Fokkink R, Peters R, Tromp P, Rivera ZEH, Rietjens I, Hendriksen PJM, Bouwmeester H. Nanotoxicology. 2013;7:1198–1210. doi: 10.3109/17435390.2012.726382. [DOI] [PubMed] [Google Scholar]
- 18.Stensberg MC, Wei Q, McLamore ES, Porterfield DM, Wei A, Sepúlveda MS. Nanomedicine (London, England) 2011;6:879–898. doi: 10.2217/nnm.11.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misra SK, Dybowska A, Berhanu D, Luoma SN, Valsami-Jones E. Sci Total Environ. 2012;438:225–232. doi: 10.1016/j.scitotenv.2012.08.066. [DOI] [PubMed] [Google Scholar]
- 20.Nel A, Xia T, Madler L, Li N. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- 21.Cedervall T, Lynch I, Lindman S, Berggard T, Thulin E, Nilsson H, Dawson KA, Linse S. Proc Natl Acad Sci U S A. 2007;104:2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch I, Cedervall T, Lundqvist M, Cabaleiro-Lago C, Linse S, Dawson KA. Advances in colloid and interface science. 2007;134–135:167–174. doi: 10.1016/j.cis.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 23.Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Nature Materials. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 24.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. P Natl Acad Sci USA. 2008;105:14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monopoli MP, Walczyk D, Campbell A, Elia G, Lynch I, Bombelli FB, Dawson KA. J Am Chem Soc. 2011;133:2525–2534. doi: 10.1021/ja107583h. [DOI] [PubMed] [Google Scholar]
- 26.Monopoli MP, Walczyk D, Campbell A, Elia G, Lynch I, Bombelli FB, Dawson KA. J Am Chem Soc. 2011;133:2525–2534. doi: 10.1021/ja107583h. [DOI] [PubMed] [Google Scholar]
- 27.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson K. Proc Natl Acad Sci. 2008;105:14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Docter D, Westmeier D, Markiewicz M, Stolte S, Knauer SK, Stauber RH. Chem Soc Rev. 2015;44:6094–6121. doi: 10.1039/c5cs00217f. [DOI] [PubMed] [Google Scholar]
- 29.Zhu RR, Wang WR, Sun XY, Liu H, Wang SL. Toxicology in vitro: an international journal published in association with BIBRA. 2010;24:1639–1647. doi: 10.1016/j.tiv.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Rahman MF. 2013 [Google Scholar]
- 31.Banerjee V, Das KP. Langmuir. 2014;30:4775–4783. doi: 10.1021/la5007007. [DOI] [PubMed] [Google Scholar]
- 32.Lundqvist M. Nat Nano. 2013;8:701–702. doi: 10.1038/nnano.2013.196. [DOI] [PubMed] [Google Scholar]
- 33.Albanese A, Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. ACS Nano. 2014;8:5515–5526. doi: 10.1021/nn4061012. [DOI] [PubMed] [Google Scholar]
- 34.Lartigue L, Wilhelm C, Servais J, Factor C, Dencausse A, Bacri JC, Luciani N, Gazeau F. ACS Nano. 2012;6:2665–2678. doi: 10.1021/nn300060u. [DOI] [PubMed] [Google Scholar]
- 35.Shannahan JH, Podila R, Aldossari AA, Emerson H, Powell BA, Ke PC, Rao AM, Brown JM. Toxicological sciences: an official journal of the Society of Toxicology. 2015;143:136–146. doi: 10.1093/toxsci/kfu217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elzey S, Grassian V. J Nanopart Res. 2010;12:1945–1958. [Google Scholar]
- 37.Stebounova L, Guio E, Grassian V. J Nanopart Res. 2011;13:233–244. [Google Scholar]
- 38.Rogers KR, Bradham K, Tolaymat T, Thomas DJ, Hartmann T, Ma L, Williams A. Sci Total Environ. 2012;420:334–339. doi: 10.1016/j.scitotenv.2012.01.044. [DOI] [PubMed] [Google Scholar]
- 39.Mwilu SK, El Badawy AM, Bradham K, Nelson C, Thomas D, Scheckel KG, Tolaymat T, Ma L, Rogers KR. Sci Total Environ. 2013;447:90–98. doi: 10.1016/j.scitotenv.2012.12.036. [DOI] [PubMed] [Google Scholar]
- 40.Walczak AP, Fokkink R, Peters R, Tromp P, Herrera Rivera ZE, Rietjens IM, Hendriksen PJ, Bouwmeester H. Nanotoxicology. 2013;7:1198–1210. doi: 10.3109/17435390.2012.726382. [DOI] [PubMed] [Google Scholar]
- 41.Axson JL, Stark DI, Bondy AL, Capracotta SS, Maynard AD, Philbert MA, Bergin IL, Ault AP. The Journal of Physical Chemistry C. 2015 doi: 10.1021/acs.jpcc.5b03634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frohlich E, Roblegg E. Toxicology. 2012;291:10–17. doi: 10.1016/j.tox.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frohlich E, Meindl C, Roblegg E, Ebner B, Absenger M, Pieber TR. Part Fibre Toxicol. 2012;9:26. doi: 10.1186/1743-8977-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bergin IL, Witzmann FA. Int J Biomed Nanosci Nanotechnol. 2013;3 doi: 10.1504/IJBNN.2013.054515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bohmert L, Girod M, Hansen U, Maul R, Knappe P, Niemann B, Weidner SM, Thunemann AF, Lampen A. Nanotoxicology. 2014;8:631–642. doi: 10.3109/17435390.2013.815284. [DOI] [PubMed] [Google Scholar]
- 46.Pharmacopeia U. USP The Pharmacopeia of the United States of America XVI. Mack Publishing Company; Easton: 1960. [Google Scholar]
- 47.Kalantzi L, Goumas K, Kalioras V, Abrahamsson B, Dressman JB, Reppas C. Pharmaceutical research. 2006;23:165–176. doi: 10.1007/s11095-005-8476-1. [DOI] [PubMed] [Google Scholar]
- 48.Culen M, Rezacova A, Jampilek J, Dohnal J. Journal of Pharmaceutical Sciences. 2013;102:2995–3017. doi: 10.1002/jps.23494. [DOI] [PubMed] [Google Scholar]
- 49.Steingoetter A, Fox M, Treier R, Weishaupt D, Marincek B, Boesiger P, Fried M, Schwizer W. Scandinavian journal of gastroenterology. 2006;41:1155–1164. doi: 10.1080/00365520600610451. [DOI] [PubMed] [Google Scholar]
- 50.Kwiatek MA, Menne D, Steingoetter A, Goetze O, Forras-Kaufman Z, Kaufman E, Fruehauf H, Boesiger P, Fried M, Schwizer W, Fox MR. American journal of physiology Gastrointestinal and liver physiology. 2009;297:G894–901. doi: 10.1152/ajpgi.00117.2009. [DOI] [PubMed] [Google Scholar]
- 51.Carr B, Wright M. Nanoparticle Tracking Analysis: A Review of Applications and Usage 2010–2012. NanoSight Ltd; Minton Park, London Road, Amesbury, Wiltshire, SP4 7RT: 2013. [Google Scholar]
- 52.Filipe V, Hawe A, Jiskoot W. Pharm Res. 2010;27:796–810. doi: 10.1007/s11095-010-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montes-Burgos I, Walczyk D, Hole P, Smith J, Lynch I, Dawson K. J Nanopart Res. 2010;12:47–53. [Google Scholar]
- 54.Malloy A. Mater Today. 2011;14:170–173. [Google Scholar]
- 55.Axson JL, Creamean JM, Bondy AL, Capracotta SS, Warner KY, Ault AP. Aerosol Sci Technol. 2014;49:24–34. doi: 10.1080/02786826.2014.991439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malloy A, Carr B. Particle & Particle Systems Characterization. 2006;23:197–204. [Google Scholar]
- 57.Anson ML, Mirsky AE. The Journal of General Physiology. 1932;16:59–63. doi: 10.1085/jgp.16.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rimon S, Perlmann GE. The Journal of biological chemistry. 1968;243:3566–3572. [PubMed] [Google Scholar]
- 59.Gray DM, Hung SH, Johnson KH. Methods Enzymol. 1995;246:19–34. doi: 10.1016/0076-6879(95)46005-5. [DOI] [PubMed] [Google Scholar]
- 60.Sreerama N, Woody RW. Protein science: a publication of the Protein Society. 2004;13:100–112. doi: 10.1110/ps.03258404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sönnichsen C, Franzl T, Wilk T, Von Plessen G, Feldmann J. New Journal of Physics. 2002;4:93. doi: 10.1103/PhysRevLett.88.077402. [DOI] [PubMed] [Google Scholar]
- 62.Manna A, Imae T, Aoi K, Okada M, Yogo T. Chemistry of Materials. 2001;13:1674–1681. [Google Scholar]
- 63.Ying M, Huang F, Ye H, Xu H, Shen L, Huan T, Huang S, Xie J, Tian S, Hu Z, He Z, Lu J, Zhou K. International journal of biological macromolecules. 2015;79:201–208. doi: 10.1016/j.ijbiomac.2015.04.057. [DOI] [PubMed] [Google Scholar]
- 64.Wang YQ, Zhang HM. Journal of agricultural and food chemistry. 2014;62:11303–11311. doi: 10.1021/jf504347w. [DOI] [PubMed] [Google Scholar]
- 65.Schmid F-X. eLS. John Wiley & Sons, Ltd; 2001. [DOI] [Google Scholar]
- 66.Wang YQ, Zhang HM. Spectrochimica acta Part A, Molecular and biomolecular spectroscopy. 2015;149:822–829. doi: 10.1016/j.saa.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 67.Konno T, Kamatari YO, Tanaka N, Kamikubo H, Dobson CM, Nagayama K. Biochemistry. 2000;39:4182–4190. doi: 10.1021/bi991923d. [DOI] [PubMed] [Google Scholar]
- 68.Kakinen A, Ding F, Chen P, Mortimer M, Kahru A, Ke PC. Nanotechnology. 2013;24:345101. doi: 10.1088/0957-4484/24/34/345101. [DOI] [PubMed] [Google Scholar]
- 69.Paula MMDS, Costa CSD, Baldin MC, Scaini G, Rezin GT, Segala K, Andrade VMD, Franco CV, Streck EL. Journal of the Brazilian Chemical Society. 2009;20:1556–1560. [Google Scholar]
- 70.Botasini S, Méndez E. J Nanopart Res. 2013;15:1–7. [Google Scholar]
- 71.Murdock RC, Braydich-Stolle L, Schrand AM, Schlager JJ, Hussain SM. Toxicological sciences: an official journal of the Society of Toxicology. 2008;101:239–253. doi: 10.1093/toxsci/kfm240. [DOI] [PubMed] [Google Scholar]
- 72.Kittler S, Greulich C, Gebauer JS, Diendorf J, Treuel L, Ruiz L, Gonzalez-Calbet JM, Vallet-Regi M, Zellner R, Koller M, Epple M. Journal of Materials Chemistry. 2010;20:512–518. [Google Scholar]
- 73.Greulich C, Kittler S, Epple M, Muhr G, Koller M. Langenbeck’s archives of surgery/Deutsche Gesellschaft fur Chirurgie. 2009;394:495–502. doi: 10.1007/s00423-009-0472-1. [DOI] [PubMed] [Google Scholar]
- 74.Meziani MJ, Sun YP. J Am Chem Soc. 2003;125:8015–8018. doi: 10.1021/ja030104k. [DOI] [PubMed] [Google Scholar]
- 75.Chen R, Choudhary P, Schurr RN, Bhattacharya P, Brown JM, Chun Ke P. Appl Phys Lett. 2012;100 doi: 10.1063/1.3672035. 013703-013703-013704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nel A, Madler L, Velegol D, Xia T, Hoek E, Somasundaran P, Klaessig F, Castranova V, Thompson M. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 77.Chakraborty S, Joshi P, Shanker V, Ansari ZA, Singh SP, Chakrabarti P. Langmuir. 2011;27:7722–7731. doi: 10.1021/la200787t. [DOI] [PubMed] [Google Scholar]
- 78.Kamatari YO, Dobson CM, Konno T. Protein science: a publication of the Protein Society. 2003;12:717–724. doi: 10.1110/ps.0219903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Richter C, Tanaka T, Yada RY. The Biochemical journal. 1998;335(Pt 3):481–490. doi: 10.1042/bj3350481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Y, Ni Y. Analyst. 2014;139:416–424. doi: 10.1039/c3an01818k. [DOI] [PubMed] [Google Scholar]
- 81.Di Silvio D, Rigby N, Bajka B, Mackie A, Bombelli FB. Int J Biochem Cell Biol. 2016;75:212–222. doi: 10.1016/j.biocel.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 82.Di Silvio D, Rigby N, Bajka B, Mayes A, Mackie A, Bombelli FB. Nanoscale. 2015;7:11980–11990. doi: 10.1039/c5nr02618k. [DOI] [PubMed] [Google Scholar]
- 83.Jeon S, Oberreitt DR, Van Schooneveld G, Hogan CJ. Anal Chem. 2016;88:7667–7674. doi: 10.1021/acs.analchem.6b01555. [DOI] [PubMed] [Google Scholar]
- 84.Miclaus T, Beer C, Chevallier J, Scavenius C, Bochenkov VE, Enghild JJ, Sutherland DS. Nat Commun. 2016;7:10. doi: 10.1038/ncomms11770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mao BH, Tsai JC, Chen CW, Yan SJ, Wang YJ. Nanotoxicology. 2016;10:1021–1040. doi: 10.1080/17435390.2016.1189614. [DOI] [PubMed] [Google Scholar]
- 86.Schaeffler M, Semmler-Behnke M, Sarioglu H, Takenaka S, Wenk A, Schleh C, Hauck SM, Johnston BD, Kreyling WG. Nanotechnology. 2013;24 doi: 10.1088/0957-4484/24/26/265103. [DOI] [PubMed] [Google Scholar]
- 87.Navarro E, Piccapietra F, Wagner B, Marconi F, Kaegi R, Odzak N, Sigg L, Behra R. Environ Sci Technol. 2008;42:8959–8964. doi: 10.1021/es801785m. [DOI] [PubMed] [Google Scholar]
- 88.Behra R, Sigg L, Clift MJD, Herzog F, Minghetti M, Johnston B, Petri-Fink A, Rothen-Rutishauser B. Journal of the Royal Society Interface. 2013;10:20130396. doi: 10.1098/rsif.2013.0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hsu-Kim H. Environ Sci Technol. 2007;41:2338–2342. doi: 10.1021/es062269+. [DOI] [PubMed] [Google Scholar]
- 90.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sielecki AR, Fujinaga M, Read RJ, James MNG. Journal of Molecular Biology. 1991;219:671–692. doi: 10.1016/0022-2836(91)90664-r. [DOI] [PubMed] [Google Scholar]
- 92.Cooper JB, Khan G, Taylor G, Tickle IJ, Blundell TL. J Mol Biol. 1990;214:199–222. doi: 10.1016/0022-2836(90)90156-G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.