

Abstract
Antimalarial drugs with the 4-aminoquinoline scaffold such as the important drugs, chloroquine (CQ) and amodiaquine (AQ), have been used to prevent and treat malaria for many years. The importance of these drugs is related to their simple usage, high efficacy, affordability, and cost-effectiveness of their synthesis. In recent years, with the spread of parasite resistance to CQ and cross-resistance to its other analogues have decreased their consumption in many geographical areas. On the other hand, AQ is an effective antimalarial drug which its usage has been restricted due to hepatic and hematological toxicities. The significance of the quinoline ring at quinoline-based antimalarial drugs has prompted research centers and pharmaceutical companies to focus on the design and synthesis of new analogues of these drugs, especially CQ and AQ analogues. Accordingly, various derivatives have been synthesized and evaluated in vitro and in vivo against the resistant strains of the malaria parasite to solve the problem of drug resistance. Also, the pharmacokinetic properties of these compounds have been evaluated to augment their efficacy and diminish their toxicity. Some of these analogues are currently in clinical and preclinical development. Consequently, the recent researches showed yet 4-aminoquinoline scaffold is active moiety in new compounds with antiplasmodial activity. Hence, the aim of this review article is to introduce of the novel synthetic analogues of CQ and AQ, which may constitute the next generation of antimalarial drugs with the 4-aminoquinoline scaffold.
Keywords: Malaria, Chloroquine, Amodiaquine, Drug resistance
What’s Known
Malaria is one of the most prevalent and deadly infectious diseases in the world. Advances in malaria research are often reviewed.
What’s New
The present review article introduces a new analogue of chloroquine to medicinal chemists and pharmacologists with a view to solving resistance and toxicity problems.
Introduction
Malaria is one of the most widespread parasitic diseases in the tropical and subtropical regions of the world and is transmitted by female Anopheles mosquitoes. Fives species of malaria parasites infect humans: Plasmodium falciparum (P. falciparum), P. vivax, P. malariae, P. ovale, and P. knowlesi. The most deadly form of malaria is P. falciparum, which has a shorter period of infection and more severe parasitemia. According to the latest report of the World Health Organization (WHO) in 2015, a total of 95 countries and territories had ongoing malaria transmission with an estimated 3.2 billion people at risk of malaria. Also, there were an estimated 214 million new cases of malaria and 438000 deaths annually.1 About 90% of all malaria deaths happen in the African region due to P. falciparum, especially among children younger than 5 years of age. During their life cycle, Plasmodium parasites are dependent on 2 hosts: The sexual cycle in the Anopheles mosquito as the main host and the asexual cycle in the human as the intermediate host.1 During a blood meal by the infected female Anopheles mosquito, sporozoites enter the human host and infect the liver cells. Then, sporozoites mature into tissue schizonts, which rupture and release merozoites (liver stage). In this stage, some species of Plasmodium can form hypnozoites, which can remain hidden in the liver for many weeks or years. Thereafter, merozoites move into blood, where they infect the red blood cells and form trophozoites. The ring stage trophozoites mature into blood schizonts (blood stage). These red blood cells rupture and infect the healthy red blood cells. In this stage, some trophozoites convert into gametocytes. If an Anopheles mosquito bites an infected person, the sexual cycle of the parasite in the mosquito begins (figure 1).2 The malaria symptoms typically develop within 10 days to 4 weeks after the infection.3 The common symptoms of malaria include high fevers, chills, sweats, headaches, nausea and vomiting, diarrhea, body pain, and bloody stool. Also, malaria may cause severe anemia because of the loss of red blood cells. If not quickly treated, malaria can become severe and may cause coma and death.4 Based on the different stages of the life cycle of the parasite, there are a variety of drugs that affect these stages and exert their therapeutic effects. For instance, quinine [1], chloroquine [CQ, 2], amodiaquine [AQ, 3] and mefloquine [MQ, 4] are fast-acting and highly effective blood schizonticidal drugs with a quinoline scaffold against malaria parasites-mainly P. falciparum. In general, CQ and AQ are called 4-aminoquinolones which are the most important and widely used class of antimalarial drugs. Nonetheless, the development of drug resistance has restricted their use for malaria prophylaxis and treatment. Recently, researchers have synthesized new compounds based on the 4-aminoquinoline scaffold and evaluated their antimalarial activities. The aim of this review article is introduction of new 4-aminoquinoline candidates (CQ and AQ analogues) with antiplasmodial activity against the CQ-sensitive and CQ-resistant strains of Plasmodium, especially P. falciparum.
Figure 1.
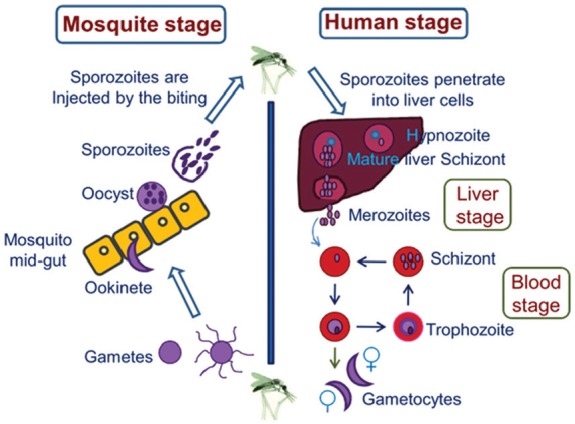
Life cycle of the malaria parasite: Humans and female Anopheles mosquitoes are two hosts of malaria parasites. In humans, the parasites grow and multiply in the liver cells (liver stage) and then in the red blood cells (blood stage). In mosquitoes, gametocytes are picked up during a blood meal and started sexual cycle.
Introduction of Antimalarial Drugs with the Quinoline Scaffold
For many years, the bark of the Cinchona tree was used to treat fever. Later years, its active ingredient, quinine [1], was isolated and identified as the oldest effective drug for malaria treatment. Quinine, as an alkaloid, is a fast-acting and highly effective blood schizonticidal drug against the species of human malaria, especially severe malaria. Quinine also has weak gametocide activity against P. vivax and P. malariae. However, there were concerns about the toxicity of quinine. Accordingly, quinine analogues were synthesized such as CQ [2], AQ [3], and MQ [4]. These analogues were inspired from the structure of the quinoline ring in quinine which is still used in the treatment of malaria (figure 2).3 CQ has been widely used since the1940s as the drug of choice for the treatment all types of malaria and has provided effective, affordable, and safe treatment for patients around the world.5 CQ is more effective and safer than quinine as a blood schizonticidal drug, but it is not active against the liver stage and mature gametocytes.
Figure 2.
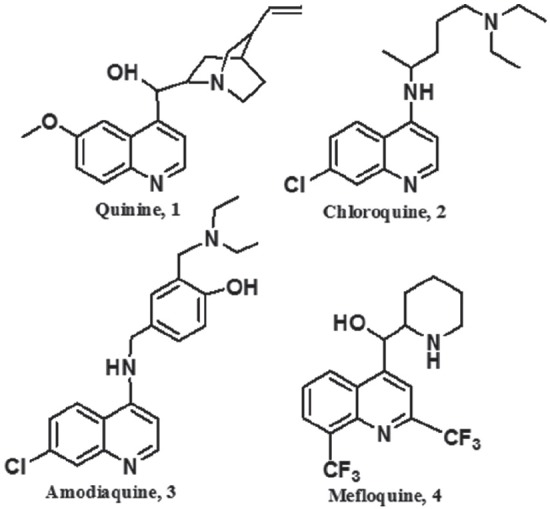
Chemical structure of antimalarial drugs containing the quinoline ring.
Today, the use of this drug has been restricted after widespread parasitic resistance to CQ in different regions, which was led to a major health concern in malaria-endemic areas. Therefore, researchers focused on the discovery of alternative antimalarial drugs that could be effective against CQ-resistant strains. As a result, other 4-aminoquinolines were developed as CQ analogous. AQ [3], a Mannich base 4-aminoquinoline, is a CQ analogue with a mechanism of action similar to CQ and is effective against CQ-resistant P. falciparum strains.3 Nevertheless, there is a cross-resistance between AQ and CQ.6 This is noticeable that AQ is associated with agranulocytosis and hepatotoxicity as side effects after its long-term administration; therefore, the clinical use of AQ has been severely limited.
MQ, which has a similar structure to quinine, is recommended for malaria prophylaxis and acute therapy in most malarious areas with CQ-resistant strains. The use of MQ was confirmed by the Center for Disease Control and Prevention (CDC). MQ is a potent blood schizonticidal drug with a long half-life, although the toxicity of this drug has limited its usage. Today, combination therapy MQ with artesunate is used strongly in the resistant regions.
The success of CQ in this group was due to its excellent clinical efficacy, low toxicity, easy usage, and cost-effective synthesis.7 Unfortunately, in today’s world, one of the most serious problems in the treatment, control, and elimination of malaria is drug resistance against affordable, safe, and readily available drugs such as CQ and pyrimethamine/sulfadoxine.8 Due to this serious health problem, researchers have focused on discovering new antimalarial agents. Further research on new synthetic analogues containing the quinoline ring has revealed that these compounds can be suitable alternative agents for old antimalarial drugs. Considering the importance of review articles,9 in this paper, we introduce a number of new CQ and AQ analogues containing the 4-aminoquinolone ring.
Mechanism of the Action of the Quinolone-Based Antimalarial Drugs
Different mechanisms of action have been reported for CQ and its analogues.10 CQ is a blood schizonticidal drug without activity against the liver stage.11 Accordingly, the parasite enters the host’s red blood cell and digests hemoglobin (Hb) in its acidic food vacuole (FV) as the dominant source of nutrition. Hb is a multi-subunit protein with an iron-containing heme group found in erythrocytes. After Hb digestion in the FV of the parasite, Hb is degraded to amino acids and heme (ferriprotoporphyrin ΙΧ) as a toxic by-product. The parasite needs these amino acids for its growth, and several malarial protease enzymes are involved in this degradation process. Plasmodium parasites do not have any enzymes to metabolize toxic heme, which is responsible for the parasite’s death. Nonetheless, the malaria parasite has developed a special process for the detoxification of heme. For this purpose, the malaria parasite converts heme to hemozoin as an insoluble crystalline form (malaria pigment) by heme polymerization. Hemozoin is not toxic for the parasite, and its formation is an essential mechanism for the detoxification and survival of the parasite (figure 3).12 Generally, small molecules with a 4-aminoquinoline moiety such as CQ and its analogues present antimalarial activity through the prevention of hemozoin formation which is resulted to the parasite’s death. The FV has a lysosomal structure with an approximate pH of 5–5.2, whereas CQ is a weak base with pKa of 8.1 and pKa’ of 10.2 which penetrates through the parasite’s membrane in its unprotonated form. In this situation, CQ is protonated and accumulated in the acidic FV of Plasmodium species, especially P. falciparum and is trapped in its acidic form (protonated form). CQ and its analogues can inhibit the formation of hemozoin and increase the intracellular heme (figure 3). In other words, it is inferred that CQ and its analogues somehow intervene with the parasite-feeding process.
Figure 3.
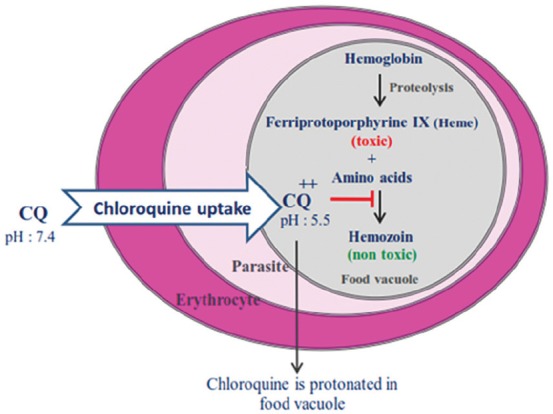
CQ is transferred to the food vacuole (FV) of the parasite and trapped in its acidic form. Then, Plasmodium parasite is killed by inhibiting haemozoin formation.
The Mechanism of Chloroquine Resistance in Plasmodium Falciparum
In 1957, the first parasite resistance to CQ was reported. Different factors were led to early resistance to the cheapest and safest antimalarial drug. These factors included frequent travels to malarious areas, uncontrolled treatment regimens with CQ, and finally the feeding of mosquitoes from different hosts.13 The weak accumulation of CQ into the FV of the parasite is the most important mechanism of resistance, accord with mutations in transporter proteins such as the P. falciparum CQ resistance transporter (PfCRT) and multi-drug resistant protein-1 (PfMDR1) which have located in the parasite’s FV membrane. Consequently, the decrease in drug accumulation is responsible for the loss of antiplasmodial activity. It has been reported that PfCRT mutation at amino acid 76 reduces the CQ uptake into the FV (figure 4).14,15 In other words, this mutation is resulted to CQ efflux out of the acidic FV,16,17 or the transporter (PfCRT) acts as a channel and allows the exit of CQ from the FV of the parasite.18,19 Furthermore, PfMDR1 transfer CQ into the vacuole.20 The mutation in PfMDR1 also reduces the transport capacity of the drug (figure 4).21
Figure 4.
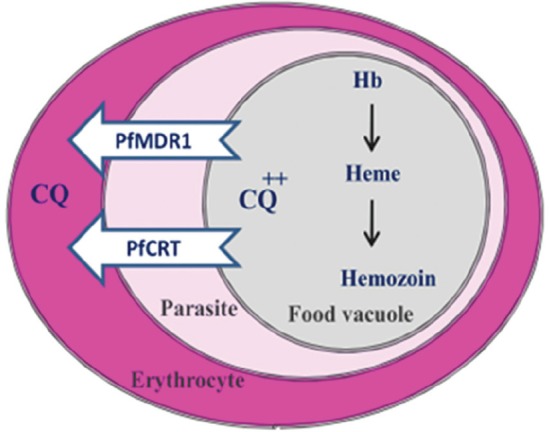
Mutations in transporter proteins (PfCRT and PfMDR1) decrease CQ accumulation in the parasite’s Food Vacuole.
Chloroquine and Amodiaquine Metabolites
CQ is swiftly metabolized into metabolites, N-desethyl chloroquine (DCQ) and N-bis-desethyl chloroquine (BDCQ), after oral or intravenous administration by the liver and kidney. BDCQ is obtained from the biotransformation of DCQ. The major active metabolite of CQ is DCQ with activity against CQ-sensitive strains, but its activity has decreased against CQ-resistant strains (figure 5). DCQ is rapidly found in blood or plasma with high concentrations.
Figure 5.
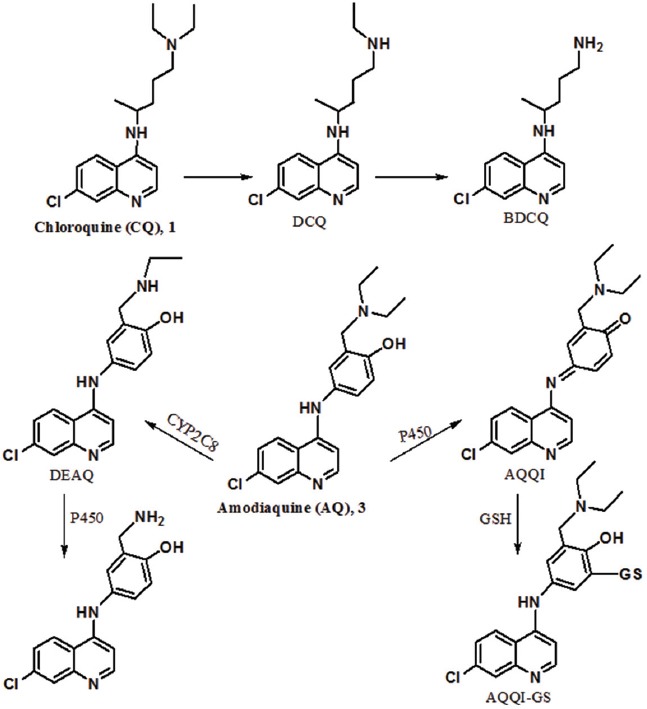
CQ is metabolized to active metabolite DCQ. AQ is metabolized to DEAQ as its major active metabolite and toxic metabolite (AQQI).
After oral administration, AQ is quickly absorbed from the gastrointestinal tract. AQ is extensively metabolized in the liver, and its major metabolite is desethyl amodiaquine (DEAQ). The metabolite is obtained from the N-deethylation of AQ by CYP2C8 (figure 5). While the formation of this metabolite is fast, its elimination is very slow with a half-life of 100 hours. AQ is more potent than its metabolite, whereas the concentration of DEAQ in blood is higher than AQ. In fact, DEAQ is responsible for antiplasmodial activity. Amodiaquine quinone-imine (AQQI) is another metabolite of AQ that is obtained in the presence of cytochrome P450 as a toxic metabolite. This metabolite is detoxified by glutathione and produces AQQI-GS. It is noteworthy that the high dose or long-term use of AQ is led to the depletion of glutathione levels and eventually, liver toxicity. The metabolite (AQQI) is also susceptible to nucleophilic attack by enzymes in addition to glutathione in the liver.6,22,23 Indeed, AQQI reacts with key human enzymes that have thiol groups and forms adducts with these enzymes. These adducts are also fatal for liver cells.
Importance of Chloroquine and Modifications in Its Structure
The spread of CQ resistance is a serious problem in malaria prevention and treatment that was first reported during the 1950s. This effective drug was the treatment of choice in many geographical areas, although with the development of CQ resistance, the combination of pyrimethamine/sulfadoxine was used for 20 years instead of CQ.24 Studies in this field demonstrated that, after a few years, sensitivity to CQ was recovered with limitation of mutation25-27 such as the deletion of PfCRT T76 mutation in Malawi after 8 years of its discontinuation.26 CQ [2] contains a quinoline ring and the pentamidine chain.2 In this drug, the presence of the flat heteroaromatic ring for connection to heme and the diethyl amino group for accumulation in the FV are the necessary factors in order to antimalarial activity (figure 6). Nowadays, due to the importance of CQ, researchers have focused on the design of new quinoline-based antimalarial compounds. In these derivatives, the quinoline ring has been often preserved and the pentamine chain has been replaced by other substituents.
Figure 6.
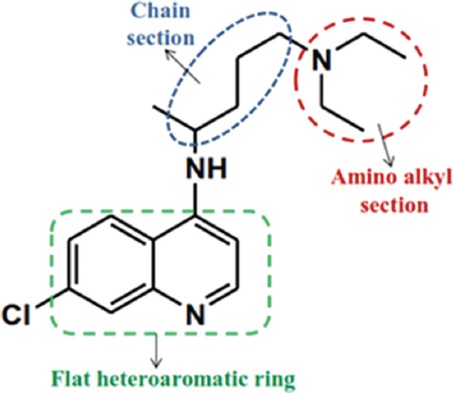
Important parts of CQ containing the quinoline ring, the pentamidine chain and amino alkyl head.
Accordingly, different derivatives have been designed and synthesized (figure 7). AQ-13 [5] is one of these analogues with a diamino-alkyl side chain shorter than the alkyl chain of CQ. AQ-13 present antimalarial activity against CQ-resistant strains (PfCRT). The compound has pharmacokinetic and mechanism of action, similar to those of CQ.28 In vivo studies showed that the N-deethylation of AQ-13 is led to specific changes in lipophilicity and increase the cross-resistance of this compound even more that of CQ.29
Figure 7.
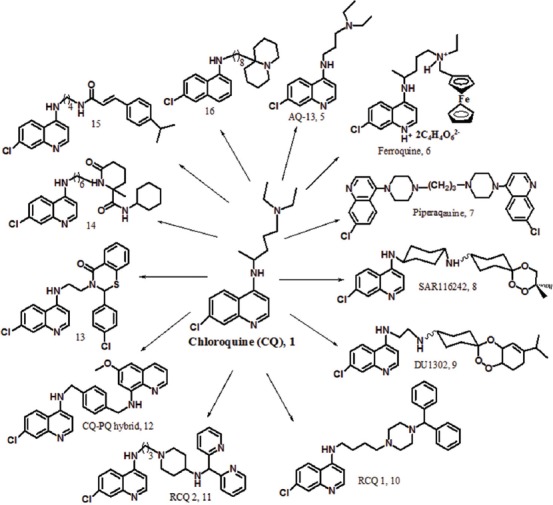
The synthetic analogues of CQ containing modified side chain with antimalarial activity.
Due to the influence of metal complexes in many pharmaceutical compounds, CQ complexes were also synthesized and evaluated on the blood stage of P. falciparum. Many of these compounds were more effective than CQ in both in vitro and in vivo tests.30,31 Currently, ferroquine [FQ, 6] has been introduced as a metal complex of CQ and a candidate for clinical studies (figure 7).32,33 In this compound, the ferrocene group alone does not have particular antimalarial activity; however, due to the tendency of the parasite to iron, the confrontation of the parasite with the compound increases.33,34 This compound is active against the CQ-resistant strains of P. falciparum and P. vivax. FQ in combination with artesunate is under investigation in phase II clinical trials.34-36
Piperaquine [7] is a dimer analogue of CQ that was widely used in the 1970s–1980s for the treatment of CQ-resistant P. falciparum strains in china (figure 7).37 The compound has two of the 4-aminoquinoline groups that have been join to each other by a dipiperazine-propyl linker. The appropriate activity of piperaquine against the resistant parasites is attributed to its bulky structure, which prevents its connection to the PfCRT site. Also, the compound is trapped powerfully into the FV of the parasite with its positive charges and prevented the heme detoxification system in the malaria parasite. The investigation of the pharmacokinetic properties of piperaquine demonstrated high lipid-solubility, long elimination half-life, good bioavailability, and fast clearance. These advantages, in addition to low cost and good tolerability, was resulted to the selection of the drug for children. Today, the prevalence of P. falciparum resistance to piperaquine has reduced monotherapy.38 Nevertheless, therapy with piperaquine and dihydroartemisinin (DHA) has exhibited high effectiveness, safety, and tolerability. The combination drug is called Duo-Cotecxin. The only problem with this combination therapy is the long half-life of piperaquine (16.5 d) and the short half-life of DHA (1/2 h).39,40 Recently, a combination of piperaquine with arterolane has been entered into phase ΙΙΙ clinical trials. This combination drug acts through 2 different mechanisms to overcome drug resistance.41
Considering the importance of combination therapy, research groups have focused on synthetic hybrid molecules containing 2 pharmacophores with a covalent linkage. For example, trioxaquine analogues exhibited strong activity against CQ and pyrimethamine-resistant strains in a single dose.42 In these hybrid molecules, drug-drug adverse interactions are minimized. Among the analogues of trioxaquine, compounds SAR116242 [8] and DU1302 [9] were selected as antimalarial candidates (figure 7). The excellent antimalarial activity of the hybrid [SAR116242, 8] against the CQ-sensitive and CQ-resistant strains of P. falciparum is attributed to the dual mechanism of action including hemozoin inhibition and heme alkylation. This compound is a racemic mixture, and investigations have demonstrated that both diastereoisomers have similar activity in vitro against Plasmodium. Also, the compound with the cyclohexyl linker has acceptable metabolic stability.42,43
AQ [3] is one of CQ analogues with a phenyl substituent. This is a Mannich base 4-aminoquinoline with a similar mechanism to that of CQ which was first introduced as a CQ alternative. Hence, AQ was used against CQ-resistant strains in P. falciparum parasites for prophylaxis and treatment. But its application was limited due to serious adverse effects such as agranulocytosis and hepatotoxicity with its long-term use.44 In addition, AQ has cross-resistance with CQ.6 Resistance to AQ is also attributed to mutation in the transporter PfCRT.6 In 1990, WHO recommended combination therapy of AQ especially with artemisinin derivatives for the treatment of uncomplicated malaria. Artemisinins are short-acting antimalarial drugs and their combination therapy with long-acting drugs is led to a delay in development of P. falciparum drug resistance.
Other CQ analogues [10] and [11] is named reversed CQ (RCQ). These compounds have a CQ-like moiety and a resistance reversal-like moiety.45 RCQs interact with different types of transporters (PfCRTs) and solve the problem of CQ-resistance in P. falciparum with inhibition hemozoin formation.45 In these compounds, the length of the linker between quinoline and nitrogen is an important factor to overcome CQ-resistance.
Also, the hybridization of CQ with primaquine (PQ) through different linkers displayed good activity against the different stages of P. falciparum.46 Indeed, these hybrid molecules can inhibit the transmission of the malaria parasite. The hybrid molecule [12] is active against the liver stage and the blood stage of the different strains of P. falciparum as well as the maturation of gametocytes. On the other hand, this hybrid compound present synergistic effect between PQ and CQ moieties which is responsible for increased activity. Accordingly, the compound [12] is active against CQ-resistance strains.
CQ analogues [13-14] with bulky side chains showed excellent activity against the CQ-resistant strain W2.47,48 These compounds do not have base moiety but inhibit hemozoin formation, similar to CQ. Previously, it was assumed that base side chain is a necessary part for antiplasmodial activity. The compound [13] exhibited superior in vitro activity in comparison with CQ and notable in vivo suppression assay. The compound [14] is more potent than CQ with a dual mechanism of action, inhibition of hemozoin formation, and inhibition of falcipain-2.
N-cinnamoylated CQ analogue [15], as a dual-stage antimalarial lead compound, displayed potent activity against the blood stage (CQ-resistant W2 and CQ-sensitive 3D7 strains) and the liver stage P. falciparum.49 The 4-amino-7-chloroquinoline moiety plays a serious role in these activities. On the other hand, the compound also had in vivo activity against the blood-stage rodent malaria parasites. A new analogue of CQ [16], containing a side chain with a bulky base head, showed excellent activity against the CQ-resistant (W2) strain of P. falciparum.50 In this compound, the spacer length between the bulky head and the quinolone ring plays an important role in antimalarial activity. The compound [16] exhibited low cytotoxicity on 2 human cell lines.
Synthesis of Amodiaquine Analogues to Reduce Its Toxicity
AQ [3] is a potent antimalarial drug but side effects of hepatotoxicity and agranulocytosis have limited its use. Due to the suitable activity of AQ in malaria treatment, researchers focused on the design and synthesis of new analogues of AQ to solve its toxicity problem (figure 8). In this regard, fluoro-amodiaquine [FAQ, 17] obtained to change the hydroxy group of the phenyl ring with a fluorine atom at the 4’-position of AQ.51 Fluorine is an important agent in medicinal chemistry, and researchers usually use fluorine in the drug design to block the metabolic sites. Therefore, FAQ is not metabolized to the toxic quinone-imine metabolite and keeps significant antimalarial activity against CQ-resistant strains. Tebuquine [TQ, 18] is another analogue of AQ which prepared by the substitution of 4-chloro phenyl moiety at the 5’-position of the hydroxyaniline side chain of AQ.52 This compound was more active than CQ and AQ in both in vitro and in vivo studies. TQ showed weak cross-resistance with CQ.53 The toxic metabolite of AQQI is formed after a long-term administration of TQ, similar to AQ. Subsequently, a synthetic compound [19] with 3’,5’- di(methylpyrrolidine) substituents, was designed with steric hindrance around the hydroxyl group at the 4’-position. As a result, the formation of the toxic metabolite of quinone-imine is restricted. Unfortunately, the compounds [18,19] have long half-lives, which is led to increased toxicity and spread of resistance.
Figure 8.
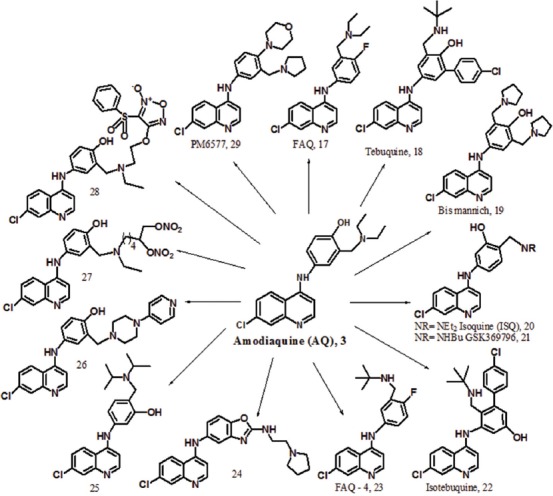
The synthetic analogues of AQ containing modified side chain with antimalarial activity.
Other non-toxic analogue of AQ [20-21] obtained to interchange the hydroxyl group at the 4’-position of AQ with dialkyl amino methylene at the 3’-position.54 With this change, the toxic quinone-imine metabolite is not formed. In addition, the compounds exhibited potent antimalarial activity against CQ- sensitive and CQ-resistant strains. Isoquine [ISQ, 20] is an isomer of AQ with a diethyl amino methylene substituent at the 4’-position which showed the strongest in vitro activity against P. falciparum and good in vivo activity after oral administration against P. Yoelii in comparison with AQ.55 The metabolic studies in rats confirmed the absence of glutathione metabolites in the bile, but they also presented severe hepatic first-pass metabolism, leading to the production of deethylated metabolites. This metabolic pathway reduces efficacy against CQ-resistant strains. In fact, ISO has less toxicity than AQ because of excretion glucuronide metabolites instead of glutathione conjugates.
GSK369796 [21] was synthesized as an N-tert-butyl analogue of ISQ to modify its low efficacy problem. Dealkylation of this compound is difficult because of steric hindrance around the N-dialkyl group. As a result, its effectiveness is preserved against the resistant strains. Pre-clinical evaluation of this compound [21] confirmed better pharmacodynamic and pharmacokinetic profile than that of ISQ.56 The advantages of GSK369796 include similar potency to AQ, low toxicity, and easy metabolism in comparison with AQ. With all of the listed advantages, the compound is not acceptable because of the lack of a suitable dose in compared with CQ. Other analogues of TQ were synthesized to avoid the quinone-imine formation. Mono-Mannich base [isotebuquine, 22] showed (IC50 = 0.3 ng/mL against D6, 0.4 ng/mL against W2, and 1.78 ng/mL against TM91C235) the best activity against P. falciparum.57 Isotebuquine has a hydroxyl group at the meta position of the aniline ring. A toxicity study in a murine monocyte-like macrophage presented low toxicity in comparison with TQ. The Thompson test in infected mice with P. berghei by oral administration did not show any significant activity, which may be the result of poor oral bioavailability.
In regard to the importance of halogen atoms in the drug design, 4’-fluoro-N-tert-butyl amodiaquine [FAQ-4, 23] was prepared with the displacement of the fluorine atom with 4’- hydroxy and 3’-N- tert-butyl with 3’-N-diethyl in AQ to block the metabolism site (N-dealkylating), which is resulted to the formation of the toxic metabolite of AQQI. This compound, with a suitable oral bioavailability and a good safety profile, is undergoing pre-clinical studies.51
The benzoxazole analogue of AQ [24] showed excellent activity against two CQ-resistant strains (K1 and W2) of P. falciparum with IC50 values of 8 and 2.2 nM, respectively.58 The low cytotoxicity of this compound against L6 (rat myoblast) cells and its high selectivity indices present a suitable alternative for AQ. The high potency to this analogue is related to the presence of an amine group. Additionally, the benzoxazole substituent attached to the 4-amino-7-chloroquinoline ring block the toxic metabolite formation, without loss of antiplasmodial activity.
The modification of ISQ as a non-toxic AQ analogue was led to the synthesis of a compound [25] with a diisopropylamine side chain.59 This compound presented good antimalarial activity with a MIC value of 10 µg/mL. However, the compound [25] do not have activity better than CQ. An in vitro evaluation of the compound [26] against the K1 strain of P. falciparum showed comparable activity with ISQ [20] and better activity than AQ with an IC50 value of 1.4 nM.60 Nonetheless, the toxicity of this compound is higher than AQ, with a narrow therapeutic window.
NO-AQ derivatives [27 and 28] were synthesized and displayed excellent in vitro activity against CQ-sensitive and CQ-resistant strains of P. falciparum.61 These compounds have either furoxan or nitrooxy moiety as a nitric oxide (NO)-donor group and are able to dilate a rat aorta strip with an NO-dependent mechanism. These hybrid molecules [27 and 28] displayed in vivo antiplasmodial activity similar to AQ. A new AQ analogue [PM6577, 29] with a morpholino group at 4’-position and a pyrrolidino group at 3’-position aniline ring showed antiplasmodial activity comparable to AQ when tested on the F32 and K1 strains of P. falciparum.62 Moreover, the cytotoxicity of this compound was evaluated on two human cell lines (a diploid embryonic lung and a neuroblastoma cell line) and showed low toxicity. PM6577 [29] presented in vivo efficiency comparable to AQ. The physiochemical (ADME) properties and permeability profile of the compound [29] is also evaluated in the pre-clinical phase.
Conclusion
The spread of resistance to current antimalarial drugs have prompted investigators and pharmaceutical companies to focus on the development of new antimalarial drug. Meanwhile, different organizations such as the Bill and Melinda Gates Foundation (BMGF) and Medicines for Malaria Venture (MMV) support malaria elimination programs around the world. Many new compounds have been synthesized by research groups and evaluated for antimalarial activity. Due to the appropriate efficacy of antimalarial drugs with a 4-aminoquinolone scaffold such as CQ and AQ, new analogues have been synthesized based on this scaffold and evaluated in both in vitro and in vivo tests against various CQ-sensitive and CQ-resistance strains of the malaria parasite, especially P. falciparum. This review article introduces new series of CQ analogues to overcome resistance and AQ analogues to reduce toxicity. All compounds have the active nucleus of the 7-chloro-4-aminoquinoline and present antimalarial activity against CQ-resistant strains. Some of these compounds have entered into pre-clinical and clinical developments. Therefore, further research on these analogues should be continued to achieve a new, efficient, cheap, and safe antimalarial drug with a 4-aminoquinoline structure as the next- generation of CQ/AQ analogues in clinical development.
Acknowledgment
The author acknowledges the management of Pasteur Institute of Iran.
Conflict of Interest: None declared.
References
- 1.World Health Organization. Antimicrobial resistance: Global report on surveillance 2014. Geneva: WHO; 2014. p. 257. [Google Scholar]
- 2.Winzeler EA. Malaria research in the post-genomic era. Nature. 2008;455:751–6. doi: 10.1038/nature07361. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flannery EL, Chatterjee AK, Winzeler EA. Antimalarial drug discovery - approaches and progress towards new medicines. Nat Rev Microbiol. 2013;11:849–62. doi: 10.1038/nrmicro3138. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM–5) Arlington: American Psychiatric Association publishing; 2013. p. 29. [Google Scholar]
- 5.Jensen M, Mehlhorn H. Seventy-five years of Resochin in the fight against malaria. Parasitol Res. 2009;105:609–27. doi: 10.1007/s00436-009-1524-8. [DOI] [PubMed] [Google Scholar]
- 6.Daily EB, Aquilante CL. Cytochrome P450 2C8 pharmacogenetics: A review of clinical studies. Pharmacogenomics. 2009;10:1489–510. doi: 10.2217/pgs.09.82. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells TN, Poll EM. When is enough enough? The need for a robust pipeline of high-quality antimalarials. Discov Med. 2010;9:389–98. [PubMed] [Google Scholar]
- 8.Winstanley PA, Ward SA, Snow RW. Clinical status and implications of antimalarial drug resistance. Microbes Infect. 2002;4:157–64. doi: 10.1016/S1286-4579(01)01523-4. [DOI] [PubMed] [Google Scholar]
- 9.Tahghighi A. Importance of metal complexes for development of potential leishmanicidal agents. J Organomet Chem. 2014;770:51–60. doi: 10.1016/j.jorganchem.2014.08.007. [DOI] [Google Scholar]
- 10.Rosenthal PJ. Antimalarial chemotherapy: Mechanisms of action, resistance, and new directions in drug discovery. New York: Springer Science & Business Media; 2001. p. 396. [DOI] [PubMed] [Google Scholar]
- 11.Hastings IM. The origins of antimalarial drug resistance. Trends Parasitol. 2004;20:512–8. doi: 10.1016/j.pt.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Golan DE, Tashjian AH, Armstrong EJ. Principles of pharmacology: The pathophysiologic basis of drug therapy. Philadelphia: Lippincott Williams & Wilkins; 2011. p. 1008. [Google Scholar]
- 13.D’Alessandro U, Buttiens H. History and importance of antimalarial drug resistance. Trop Med Int Health. 2001;6:845–8. doi: 10.1046/j.1365-3156.2001.00819.x. [DOI] [PubMed] [Google Scholar]
- 14.Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–3. doi: 10.1126/science.1074045. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–71. doi: 10.1016/S1097-2765(05)00077-8. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez CP, Stein WD, Lanzer M. Is PfCRT a channel or a carrier? Two competing models explaining chloroquine resistance in Plasmodium falciparum. Trends Parasitol. 2007;23:332–9. doi: 10.1016/j.pt.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 17.Martin RE, Marchetti RV, Cowan AI, Howitt SM, Broer S, Kirk K. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science. 2009;325:1680–2. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- 18.Bray PG, Mungthin M, Hastings IM, Biagini GA, Saidu DK, Lakshmanan V, et al. PfCRT and the trans-vacuolar proton electrochemical gradient: Regulating the access of chloroquine to ferriprotoporphyrin IX. Mol Microbiol. 2006;62:238–51. doi: 10.1111/j.1365-2958.2006.05368.x. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valderramos SG, Fidock DA. Transporters involved in resistance to antimalarial drugs. Trends Pharmacol Sci. 2006;27:594–601. doi: 10.1016/j.tips.2006.09.005. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson TJ, Nair S, Qin H, Singlam S, Brockman A, Paiphun L, et al. Are transporter genes other than the chloroquine resistance locus (pfcrt) and multidrug resistance gene (pfmdr) associated with antimalarial drug resistance? Antimicrob Agents Chemother. 2005;49:2180–8. doi: 10.1128/AAC.49.6.2180-2188.2005. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez CP, Rotmann A, Stein WD, Lanzer M. Polymorphisms within PfMDR1 alter the substrate specificity for anti-malarial drugs in Plasmodium falciparum. Mol Microbiol. 2008;70:786–98. doi: 10.1111/j.1365-2958.2008.06413.x. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu S, Atsumi R, Itokawa K, Iwasaki M, Aoki T, Ono C, et al. Metabolism-dependent hepatotoxicity of amodiaquine in glutathione-depleted mice. Arch Toxicol. 2009;83:701–7. doi: 10.1007/s00204-009-0436-9. [DOI] [PubMed] [Google Scholar]
- 23.Tafazoli S, O’Brien PJ. Amodiaquine-induced oxidative stress in a hepatocyte inflammation model. Toxicology. 2009;256:101–9. doi: 10.1016/j.tox.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Reyburn H. New WHO guidelines for the treatment of malaria. BMJ. 2010;340:c2637. doi: 10.1136/bmj.c2637. [DOI] [PubMed] [Google Scholar]
- 25.Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, et al. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med. 2006;355:1959–66. doi: 10.1056/NEJMoa062032. [DOI] [PubMed] [Google Scholar]
- 26.Djimde A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourte Y, et al. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344:257–63. doi: 10.1056/NEJM200101253440403. [DOI] [PubMed] [Google Scholar]
- 27.Read AF, Huijben S. Evolutionary biology and the avoidance of antimicrobial resistance. Evol Appl. 2009;2:40–51. doi: 10.1111/j.1752-4571.2008.00066.x. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saenz FE, Mutka T, Udenze K, Oduola AM, Kyle DE. Novel 4-aminoquinoline analogs highly active against the blood and sexual stages of Plasmodium in vivo and in vitro. Antimicrob Agents Chemother. 2012;56:4685–92. doi: 10.1128/AAC.01061-12. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramanathan-Girish S, Catz P, Creek MR, Wu B, Thomas D, Krogstad DJ, et al. Pharmacokinetics of the antimalarial drug, AQ-13, in rats and cynomolgus macaques. Int J Toxicol. 2004;23:179–89. doi: 10.1080/10915810490471352. [DOI] [PubMed] [Google Scholar]
- 30.Navarro M, Castro W, Martinez A, Sanchez Delgado RA. The mechanism of antimalarial action of [Au(CQ)(PPh(3))]PF(6): Structural effects and increased drug lipophilicity enhance heme aggregation inhibition at lipid/water interfaces. J Inorg Biochem. 2011;105:276–82. doi: 10.1016/j.jinorgbio.2010.11.005. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez A, Rajapakse CS, Naoulou B, Kopkalli Y, Davenport L, Sanchez-Delgado RA. The mechanism of antimalarial action of the ruthenium(II)-chloroquine complex [RuCl(2)(CQ)] (2) J Biol Inorg Chem. 2008;13:703–12. doi: 10.1007/s00775-008-0356-9. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pradines B, Fusai T, Daries W, Laloge V, Rogier C, Millet P, et al. Ferrocene-chloroquine analogues as antimalarial agents: In vitro activity of ferrochloroquine against 103 Gabonese isolates of Plasmodium falciparum. J Antimicrob Chemother. 2001;48:179–84. doi: 10.1093/jac/48.2.179. [DOI] [PubMed] [Google Scholar]
- 33.Biot C, Nosten F, Fraisse L, Ter-Minassian D, Khalife J, Dive D. The antimalarial ferroquine: From bench to clinic. Parasite. 2011;18:207–14. doi: 10.1051/parasite/2011183207. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubar F, Khalife J, Brocard J, Dive D, Biot C. Ferroquine, an ingenious antimalarial drug: Thoughts on the mechanism of action. Molecules. 2008;13:2900–7. doi: 10.3390/molecules13112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barends M, Jaidee A, Khaohirun N, Singhasivanon P, Nosten F. In vitro activity of ferroquine (SSR 97193) against Plasmodium falciparum isolates from the Thai-Burmese border. Malar J. 2007;6:81. doi: 10.1186/1475-2875-6-81. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leimanis ML, Jaidee A, Sriprawat K, Kaewpongsri S, Suwanarusk R, Barends M, et al. Plasmodium vivax susceptibility to ferroquine. Antimicrob Agents Chemother. 2010;54:2228–30. doi: 10.1128/AAC.01572-09. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis TM, Hung TY, Sim IK, Karunajeewa HA, Ilett KF. Piperaquine: A resurgent antimalarial drug. Drugs. 2005;65:75–87. doi: 10.2165/00003495-200565010-00004. [DOI] [PubMed] [Google Scholar]
- 38.Hung TY, Davis TM, Ilett KF, Karunajeewa H, Hewitt S, Denis MB, et al. Population pharmacokinetics of piperaquine in adults and children with uncomplicated falciparum or vivax malaria. Br J Clin Pharmacol. 2004;57:253–62. doi: 10.1046/j.1365-2125.2003.02004.x. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zwang J, Ashley EA, Karema C, D’Alessandro U, Smithuis F, Dorsey G, et al. Safety and efficacy of dihydroartemisinin-piperaquine in falciparum malaria: A prospective multi-centre individual patient data analysis. PLoS One. 2009;4:e6358. doi: 10.1371/journal.pone.0006358. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price RN, Hasugian AR, Ratcliff A, Siswantoro H, Purba HL, Kenangalem E, et al. Clinical and pharmacological determinants of the therapeutic response to dihydroartemisinin-piperaquine for drug-resistant malaria. Antimicrob Agents Chemother. 2007;51:4090–7. doi: 10.1128/AAC.00486-07. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snyder C, Chollet J, Santo-Tomas J, Scheurer C, Wittlin S. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp Parasitol. 2007;115:296–300. doi: 10.1016/j.exppara.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 42.Benoit-Vical F, Lelievre J, Berry A, Deymier C, Dechy-Cabaret O, Cazelles J, et al. Trioxaquines are new antimalarial agents active on all erythrocytic forms, including gametocytes. Antimicrob Agents Chemother. 2007;51:1463–72. doi: 10.1128/AAC.00967-06. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cosledan F, Fraisse L, Pellet A, Guillou F, Mordmuller B, Kremsner PG, et al. Selection of a trioxaquine as an antimalarial drug candidate. Proc Natl Acad Sci U S A. 2008;105:17579–84. doi: 10.1073/pnas.0804338105. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rudrapal M, Chetia D. Novel 4-aminoquinoline analogues as antimalarial agents: A review. Der Pharma Lett. 2011;3:29–36. [Google Scholar]
- 45.Burgess SJ, Kelly JX, Shomloo S, Wittlin S, Brun R, Liebmann K, et al. Synthesis, structure-activity relationship, and mode-of-action studies of antimalarial reversed chloroquine compounds. J Med Chem. 2010;53:6477–89. doi: 10.1021/jm1006484. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lodige M, Hiersch L. Design and Synthesis of Novel Hybrid Molecules against Malaria. Int J Med Chem. 2015;2015:458319. doi: 10.1155/2015/458319. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solomon VR, Haq W, Srivastava K, Puri SK, Katti SB. Synthesis and antimalarial activity of side chain modified 4-aminoquinoline derivatives. J Med Chem. 2007;50:394–8. doi: 10.1021/jm061002i. [DOI] [PubMed] [Google Scholar]
- 48.Musonda CC, Gut J, Rosenthal PJ, Yardley V, Carvalho de Souza RC, Chibale K. Application of multicomponent reactions to antimalarial drug discovery. Part 2: New antiplasmodial and antitrypanosomal 4-aminoquinoline gamma- and delta-lactams via a ‘catch and release’ protocol. Bioorg Med Chem. 2006;14:5605–15. doi: 10.1016/j.bmc.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 49.Perez BC, Teixeira C, Albuquerque IS, Gut J, Rosenthal PJ, Gomes JR, et al. N-cinnamoylated chloroquine analogues as dual-stage antimalarial leads. J Med Chem. 2013;56:556–67. doi: 10.1021/jm301654b. [DOI] [PubMed] [Google Scholar]
- 50.Tasso B, Novelli F, Tonelli M, Barteselli A, Basilico N, Parapini S, et al. Synthesis and Antiplasmodial Activity of Novel Chloroquine Analogues with Bulky Basic Side Chains. Chem Med Chem. 2015;10:1570–83. doi: 10.1002/cmdc.201500195. [DOI] [PubMed] [Google Scholar]
- 51.O’Neill PM, Shone AE, Stanford D, Nixon G, Asadollahy E, Park BK, et al. Synthesis, antimalarial activity, and preclinical pharmacology of a novel series of 4’-fluoro and 4’-chloro analogues of amodiaquine. Identification of a suitable “back-up” compound for N-tert-butyl isoquine. J Med Chem. 2009;52:1828–44. doi: 10.1021/jm8012757. [DOI] [PubMed] [Google Scholar]
- 52.Casagrande M, Basilico N, Parapini S, Romeo S, Taramelli D, Sparatore A. Novel amodiaquine congeners as potent antimalarial agents. Bioorg Med Chem. 2008;16:6813–23. doi: 10.1016/j.bmc.2008.05.068. [DOI] [PubMed] [Google Scholar]
- 53.Paunescu E, Susplugas S, Boll E, Varga RA, Mouray E, Grellier P, et al. Synthesis and antimalarial activity of new amino analogues of amodiaquine. Med Chem. 2008;4:407–25. doi: 10.2174/157340608785700153. [DOI] [PubMed] [Google Scholar]
- 54.O’Neill PM, Mukhtar A, Stocks PA, Randle LE, Hindley S, Ward SA, et al. Isoquine and related amodiaquine analogues: A new generation of improved 4-aminoquinoline antimalarials. J Med Chem. 2003;46:4933–45. doi: 10.1021/jm030796n. [DOI] [PubMed] [Google Scholar]
- 55.Delarue S, Girault S, Maes L, Debreu-Fontaine MA, Labaeid M, Grellier P, et al. Synthesis and in vitro and in vivo antimalarial activity of new 4-anilinoquinolines. J Med Chem. 2001;44:2827–33. doi: 10.1021/jm010842o. [DOI] [PubMed] [Google Scholar]
- 56.O’Neill PM, Park BK, Shone AE, Maggs JL, Roberts P, Stocks PA, et al. Candidate selection and preclinical evaluation of N-tert-butyl isoquine (GSK369796), an affordable and effective 4-aminoquinoline antimalarial for the 21st century. J Med Chem. 2009;52:1408–15. doi: 10.1021/jm8012618. [DOI] [PubMed] [Google Scholar]
- 57.Miroshnikova OV, Hudson TH, Gerena L, Kyle DE, Lin AJ. Synthesis and antimalarial activity of new isotebuquine analogues. J Med Chem. 2007;50:889–96. doi: 10.1021/jm061232x. [DOI] [PubMed] [Google Scholar]
- 58.Ongarora DS, Gut J, Rosenthal PJ, Masimirembwa CM, Chibale K. Benzoheterocyclic amodiaquine analogues with potent antiplasmodial activity: Synthesis and pharmacological evaluation. Bioorg Med Chem Lett. 2012;22:5046–50. doi: 10.1016/j.bmcl.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 59.Saha CN, Bhattacharya S, Chetia D. Synthesis and Antimalarial Screening of Some New Isoquine Analogues. International Journal of Chem Tech Research. 2009;1:322–8. [Google Scholar]
- 60.Guglielmo S, Bertinaria M, Rolando B, Crosetti M, Fruttero R, Yardley V, et al. A new series of amodiaquine analogues modified in the basic side chain with in vitro antileishmanial and antiplasmodial activity. Eur J Med Chem. 2009;44:5071–9. doi: 10.1016/j.ejmech.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 61.Bertinaria M, Guglielmo S, Rolando B, Giorgis M, Aragno C, Fruttero R, et al. Amodiaquine analogues containing NO-donor substructures: Synthesis and their preliminary evaluation as potential tools in the treatment of cerebral malaria. Eur J Med Chem. 2011;46:1757–67. doi: 10.1016/j.ejmech.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 62.Hochart G, Paunescu E, Boll E, Melnyk P. PM6280 and PM6577: ADME Study of Two Potent and Anti-malarial Amodiaquine Analogs with Improved Metabolic Stability. Research &Reviews: Journal of Medicinal & Organic Chemistry. 2015;2(1):1–14. [Google Scholar]