

Abstract
BACKGROUND
Severe traumatic brain injury (TBI) may increase the risk of venous thromboembolic complications; however, early prevention with heparinoids is often withheld for its anticoagulant effect. New evidence suggests low molecular weight heparin reduces cerebral edema and improves neurological recovery after stroke and TBI, through blunting of cerebral leukocyte (LEU) recruitment. It remains unknown if unfractionated heparin (UFH) similarly affects brain inflammation and neurological recovery post-TBI. We hypothesized that UFH after TBI reduces cerebral edema by reducing LEU-mediated inflammation and improves neurological recovery.
METHODS
CD1 male mice underwent either TBI by controlled cortical impact (CCI) or sham craniotomy. UFH (75 U/kg or 225 U/kg) or vehicle (VEH, 0.9% saline) was administered 2, 11, 20, 27, and 34 hours after TBI. At 48 hours, pial intravital microscopy through a craniotomy was used to visualize live brain LEUs interacting with endothelium and microvascular fluorescein isothiocyanate–albumin leakage. Neurologic function (Garcia Neurological Test, GNT) and body weight loss ratios were evaluated 24 and 48 hours after TBI. Cerebral and lung wet-to-dry ratios were evaluated post mortem. ANOVA with Bonferroni correction was used to determine significance (p < 0.05).
RESULTS
Compared to positive controls (CCI), both UFH doses reduced post-TBI in vivo LEU rolling on endothelium, concurrent cerebrovascular albumin leakage, and ipsilateral cerebral water content after TBI. Additionally, only low dose UFH (75 U/kg) improved GNT at both 24 and 48 hours after TBI. High dose UFH (225 U/kg) significantly increased body weight loss above sham at 48 hours. Differences in lung water content and blood pressure between groups were not significant.
CONCLUSIONS
UFH after TBI reduces LEU recruitment, microvascular permeability, and brain edema to injured brain. Lower UFH doses concurrently improve neurological recovery whereas higher UFH may worsen functional recovery. Further study is needed to determine if this is caused by increased bleeding from injured brain with higher UFH doses.
Keywords: Heparin, traumatic brain injury, leukocyte
Traumatic brain injury (TBI) is the leading cause of death and disability in young Americans, with over 1.7 million cases occurring yearly.1 After severe TBI, the initial cerebral destruction often worsens with an evolving tissue injury and swelling that can progress to secondary brain injury. The development of secondary brain injury is a key determinant in the occurrence of post-TBI complications including the development of cerebral edema, intracranial hypertension, cerebral herniation, and brain death.2
After traumatic injury, circulating leukocytes (LEU) and endothelial cells (EC)3,4 interact in the microcirculation, and their subsequent activation leads to the release of toxic substances that cause progressive microvascular disruption of the blood-brain barrier (BBB) allowing extravascular leakage of plasma, LEU, and proteins.5–8
Intriguingly, heparins have emerged as capable of blunting leukocyte activation and the resultant tissue edema in systemic and central nervous system conditions.9–11 The pleiotropic effects of heparin have become manifest in over a century since its discovery in 1916.12 Irrespective of its anticoagulant effects, heparin administration in trauma and hemorrhagic shock seems to alter the microcirculation, blunting LEU and EC activation,13 which can be directly visualized in vivo using intravital microscopy.14 This has also been confirmed in cerebral insult models, concomitantly demonstrating reduced intracranial pressure and infarct size while resulting in improved neurological function.9,15 To our knowledge, no existing studies to date have evaluated in vivo unfractionated heparin (UFH) effects on LEU/ECs interactions in the cerebral microcirculation after TBI. We thus hypothesized that early, repeated administration of UFH after TBI reduces EC/LEU interactions while decreasing microvascular permeability and cerebral edema. We further postulated that UFH treatment would also result in improved neurological recovery.
MATERIALS AND METHODS
Experimental Design and Study Groups
All experimental procedures were conducted with approval of the Institutional Animal Care and Use Committee of the University of Pennsylvania. Adult male CD1 mice (25–30 g) (Charles River Laboratories, Wilmington, MA) were housed for 5 to 7 days in standard facilities before experiments with access to water and chow ad libitum. On day 1, mice were anesthetized with intraperitoneal ketamine (Hospira, Lake Forest, IL—100 mg/kg), xylazine (Akorn, Decatur, IL—10 mg/kg), and acepromazine (Boehringer Ingelheim, St. Joseph, MO—1 mg/kg). While maintained euthermic, a tunneled right external jugular vein line was placed for administration of rhodamine and FITC-labeled albumin for in vivo assessment of the microcirculation. Animals underwent CCI or sham craniotomy followed by repeated subcutaneous injections of UFH (100 usp Units/mL; Medefil Inc., Glendale Heights, IL) (75 U/kg or 225 U/kg) or an equal volume of 0.9% saline (NS, vehicle, VEH, 1 mL/kg) at 2, 11, 20,27, and 34 hours after CCI (Fig. 1). Forty-eight animals were randomized into four groups: negative control [No CCI] (VEH only, n = 6), positive control [CCI] (CCI + VEH, n = 14), low dose ufh [UFH75] (CCI + 75 U/kg UFH, n = 17), and high dose UFH [UFH225] (CCI + 225 U/kg UFH, n = 11). The above dosages of UFH were chosen to reflect the clinical VTE prophylaxis dose and triplication of this dose.
Figure 1.
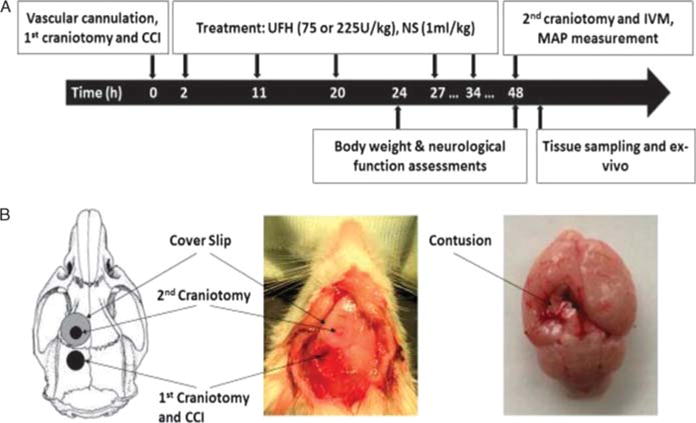
Experimental design and procedures. (A), Timeline of experimental procedures. (B), Schematic diagram showing the location of craniotomies and CCI-induced cortical contusion surrounded by penumbral area. CCI, controlled cortical impact; IVM, intravital microscopy; s.c., subcutaneously; UFH, unfractionated heparin;NS, normal saline.
Severe TBI Model
To recreate severe traumatic brain injury, a validated controlled cortical impact (CCI) model was used as described else-where.16,17 In brief, anesthetized animals were placed prone in a stereotactic device, their skull was exposed by an apical scalp incision and a left-sided, 4-mm craniotomy was created between bregma and lambda sutures using a trephine (Fig. 1). The exposed left, parietotemporal cortex covered with dura mater was then directly injured using a CCI device (AMS 201; AmScien Instruments, Richmond, VA). The device was armed with a 3-mm-diameter impactor tip, discharged with an impact velocity of 6 m/s that created a 1.0-mm cortical deformation.16 The resultant cerebral injury is reproducible and consistent with severe TBI.
Observation With Intravital Microscopy
In vivo pial intravital microscopy assessment of the cerebral microcirculation was conducted as described previously.18,19 Forty-eight hours after CCI, animals were anesthetized as earlier, placed in a stereotactic frame, and a second 2.5-mm craniotomy was created adjacent to the first using a dental drill (Henry Schein, Melville, NY). This craniotomy was then covered with a 5-mm coverslip (Fisher Scientific, Waltham, MA), and animals were then transferred to an intravital microscope (ECLIPSE FN1; Nikon Instruments, Melville, NY) after which they received 50 μL IV of 0.3% rhodamine 6G (Sigma-Aldrich, St. Louis, MO) to fluorescently label circulating leukocytes. Non-branching pial venules (25–50 μm diameter) were randomly selected and a 1-minute footage was recorded under a 590-nm epi-illumination emission filter using a digital camera (QuantEM; Photometrics, Tucson, AR). Intravenous bovine fluorescein isothiocyanate (FITC)–labeled albumin (Sigma-Aldrich, St. Louis, MO—100 mg/kg) was then administered intravenously for visualization of albumin leakage as a surrogate of microvascular permeability. The same pial regions were observed under a 488-nm fluorescent filter for 10 seconds, and images were captured for offline determination of microvascular leakage.
Microcirculatory Analysis
Video recordings and still images were imported into a digital analysis software (NIS-Elements, Nikon Instruments, Melville, NY), and leukocyte-endothelial interactions were quantified by a blinded observer documenting the following parameters1 : PMN rolling—mean number of labeled cells crossing a 100-μm venular segment2; PMN adhesion—labeled cells stationary for at least 30 seconds during the recording period. Fluorescently labeled spherical cells measuring 7 to 12 μm were counted as LEU and interactions were reported as number of cells per 100 μm per minute (Fig. 2A). FITC-labeled albumin fluorescence intensity was measured in three distinct regions within the vessel (venular intensity, Iv) and outside the vessel wall (perivenular intensity, Ip) (Fig. 2B). The ratio of venular intensity to perivenular intensity was averaged to determine the permeability index for a given vessel indicating the degree of leakage through the vessel wall at that time.
Figure 2.
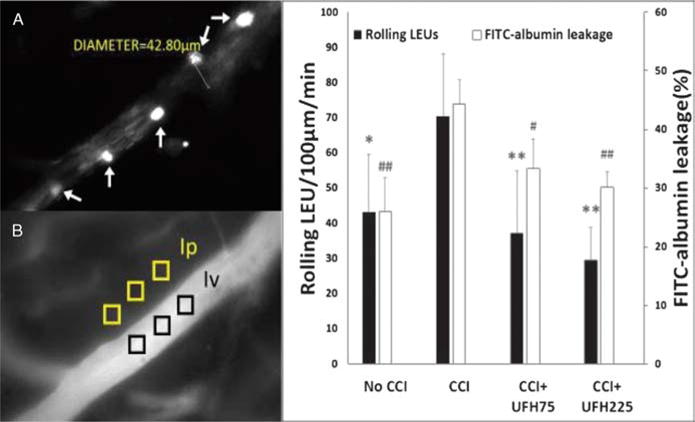
In vivo leukocyte/endothelial interactions and microvascular permeability. (A), Representative image showing LEU interacting with endothelium. White arrows indicate labeled LEU rolling on ECs. (B), Representative image showing FITC-albumin leakage in the cerebral microcirculation. The microvascular permeability index is expressed as the ratio of mean fluorescence of three separate locations outside the vessel wall (perivenular intensity, Ip) to mean fluorescence of three separate locations within the venule (venular intensity, Iv). (C), Leukocyte rolling and FITC-albumin leakage in the pial penumbral microcirculation 48 hours after CCI. Compared to positive controls (CCI), both UFH doses reduced post-TBI LEU rolling and cerebrovascular albumin leakage. Rolling LEU: *p <0.05, **p < 0.01 vs. CCI + VEH. FITC-albumin leakage: #p < 0.05, ##p < 0.01 vs. CCI + VEH.
Body Weight Loss, Neurological Recovery, and Systemic Hemodynamics
Body weight loss after injury is a widely used marker of illness and physical animal impairment. Animal body weights (W) were obtained before and 24 and 48 hours after CCI and extent of weight loss was expressed as weight loss ratio [(W0h − W24h or 48h)/W0h × 100%].
Neurologic function was assessed 24 and 48 hours after TBI using the validated modified Garcia Neurological Test (GNT),20,21 which scores motor, sensory, reflex, and balance ability to a maximum sum score of 18 points.
After completion of microscopy video recordings, the left common carotid artery was cannulated to measure mean arterial pressure (MAP). After MAP measurement, animals were sacrificed by ketamine overdose and cervical dislocation.
Brain and Lung Water Content
After sacrifice, brains were excised and divided along the midline into hemispheres. In the same setting, the left lung was excised via sternotomy. Both organs were weighed immediately (wet weight, WW) and again after 72 hours of drying at 70°C (dry weight, DW). Tissue water content in each organ was calculated using wet-to-dry ratios (% water content = [WW − DW]/WW × 100%).
Statistical Analysis
All data are presented as mean ± SD. Statistical analyses were performed using SPSS software (version 19; SPSS, Chicago, IL). Differences between groups were compared using analysis of variance with Bonferroni correction. A p value of less than 0.05 was considered statistically significant.
RESULTS
In Vivo LEU-EC Interactions and Microvascular Permeability
Forty-eight hours after CCI, the number of rolling leukocytes and degree of microvascular permeability was greatest in CCI animals and lowest in No CCI controls (Fig. 2).
Live leukocyte rolling in CCI animals (70.4 ± 17.9 LEUs/100 μm/min) was also significantly reduced with either low dose (37.2 ± 17.6 LEUs/100 μm/min, p < 0.01) or high dose (29.6 ± 9.3 LEUs/100 μm/min, p < 0.01) heparin to near No CCI levels (43.1 ± 16.6 LEUs/100 μm/min, p < 0.05 vs. CCI) (Fig. 2A, C). Differences in LEU rolling among both heparin groups or No CCI animals were not significant. Leukocyte adhesion was rare without any differences between groups (No CCI: 1.2 ± 1.8 LEUs/100μm/min, CCI: 1.7 ± 1.5 LEUs/100 μm/min, UFH75: 0.6 ± 0.7 LEUs/100 μm/min, UFH225: 0.8 ± 1.2 LEUs/100 μm/min, p > 0.05 for all comparisons).
Live cerebrovascular FITC-albumin leakage was greater in CCI animals (44.3 ± 4.2%) than either low dose (33.3 ± 5.1%, p < 0.05) or high dose (30.1 ± 2.7%, p < 0.01) heparin groups or No CCI animals (25.9 ± 5.8%, p < 0.01) (Fig. 2B, C). There were no significant differences in microvascular permeability between either heparin groups or No CCI groups.
Brain and Lung Edema
Forty-eight hours after injury, wet-to-dry ratios of the cerebral hemisphere ipsilateral to the injury were significantly greater in CCI animals (78.5 ± 0.6%) than in either UFH groups (UFH75: 77.2 ± 0.5%, UFH225: 77.3 ± 0.6%, p < 0.05 for both) or No CCI animals (76.7 ± 0.6%, p < 0.01) (Fig. 3A). Either heparin dosage reduced ipsilateral cerebral hemisphere edema to near No CCI levels (p > 0.05). Contralateral cerebral hemisphere edema was low and similar in all groups. There were no significant differences in lung water content among the different groups though there was great variability within each group (Fig. 3B).
Figure 3.
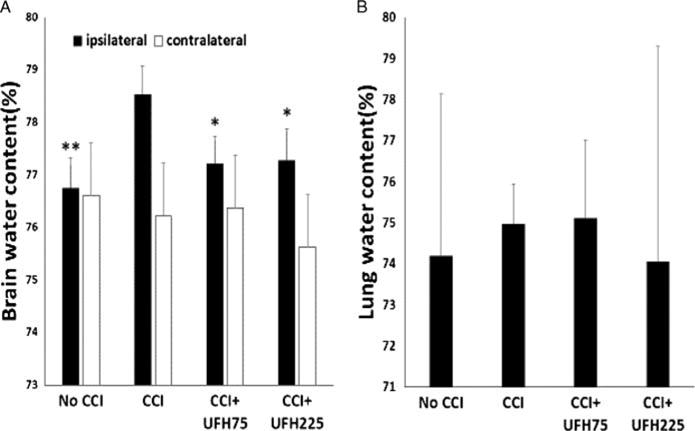
Animal outcomes. (A), Both UFH doses decreased brain water content in the hemisphere ipsilateral to the injury to levels nearing negative controls (No CCI). Ipsilateral hemisphere: *p < 0.05 or **p < 0.01 vs. CCI + VEH. Contralateral cerebral edema was similar in all groups. (B), There were no significant differences in lung water content among the different groups.
Body Weight Loss, Neurological Function, and Hemodynamics
Twenty-four hours after injury, body weight loss was lowest in No CCI animals, but this was not significantly different to that in other groups. Only high dose UFH animals (12.1 ±3.2%) demonstrated a significantly greater weight loss than No CCI controls (6.5 ± 1.4%, p < 0.05) 48 hours after injury (Fig. 4A).
Figure 4.
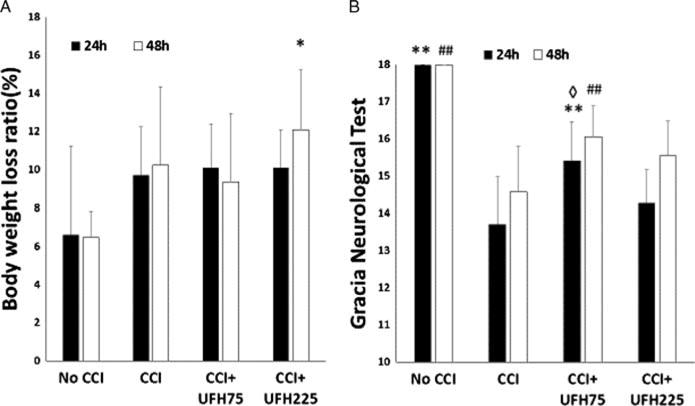
(A), No significant differences were found in body weight loss ratios 24 hours after injury among the different groups. Only UFH225 UFH significantly worsened body weight loss above negative controls (No CCI) at 48 hours. 48 hours: *p < 0.05 vs. No CCI. (B), Neurological function was normal in negative controls (No CCI) at both timeframes. Compared to positive controls (CCI), UFH75 but not UFH225 improved GNT both 24 and 48 hours after injury. Compared to UFH225 UFH75 significantly improved GNT at 24 hours. 24 hours: **p < 0.01 vs. CCI + VEH, ◊p < 0.05 vs. CCI + 225 U/kg UFH, 48 hours, ##p < 0.01 vs. CCI + VEH.
Neurologic functional recovery as determined by the Garcia Neurological Test was complete in No CCI animals at both 24 and 48 hours after injury (18 ± 0.0 for each) (Fig. 4B). Compared to GNT scores in CCI animals (24 h: 13.7 ± 1.3, 48 h: 14.6 ± 1.2), only low dose UFH significantly improved GNT at both time frames (24 h: 15.4 ± 1.1; 48 h: 16.1 ± 0.8, p < 0.01 for both). High dose UFH did not improve GNT scores over CCI animals 24 or 48 hours after injury. Moreover, low dose UFH significantly improved GNT over high dose UFH (14.3 ± 0.9, p < 0.05) 24 hours after injury (Fig. 4B).
No differences were found in mean arterial blood pressures at the completion of intravital microscopy (48 h) among any of the groups (No CCI: 75.8 ± 14.9 mm Hg, CCI: 68.3 ±14.5 mm Hg, UFH75: 81.2 ± 13.1 mm Hg, UFH225: 72.0 ± 16.0 mm Hg, p > 0.05 for all comparisons). There was no evidence of bleeding in any wound or body cavity that was grossly noted on inspection in any of the animals, though systematic evaluation for visible bleeding was not part of the protocol.
DISCUSSION
In the present study, we report how early repeated heparin administration reduced in vivo leukocyte rolling on endothelium after TBI, concurrently reducing live vascular permeability in the pericontusional cerebral vasculature. This was associated with reduced cerebral edema by either heparin doses, but only with concurrent neurological improvement by low dose heparin. Furthermore, high dose heparin resulted in a greater inability to regain lost body weight 48 hours after TBI.
Brain injury is a leading cause of death and disability after trauma, particularly affecting young patients.1 After severe TBI, many patients remain critically ill requiring intensive monitoring and management as the cerebral injury “blossoms” with ongoing tissue inflammation and neurologic compromise. In the hours to days after injury, pericontusional brain tissue continues to swell, often producing intracranial hypertension and placing the patient at risk for cerebral herniation. This ongoing post-injury inflammation is thought to follow disruption of the BBB and result from pervasive leakage of fluid and cells into the interstitial milieu.4 Such BBB disruption may be promoted by activation of the local host immune response through the local degranulation of sequestered LEUs and their release of toxic oxygen free radicals and proteinases that injure adjacent endothelium.22 Activation of LEU and ECs in the microcirculation stimulates their step-wise interaction. Activated leukocytes first slow out of circulatory flow as they roll on activated endothelium through weak interactions involving surface selectins.6,23 This initial contact leads to covalent bonding between surface CD11b leukocyte receptors interacting with endothelial receptors of the immunoglobulin superfamily including intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1).6 This strong interaction results in leukocyte arrest and firm adhesion to endothelium. LEU then transmigrate through the BBB into brain parenchyma activating resident astrocytes and microglia, which leads to further tissue injury and BBB disruption.24 This ongoing BBB breakdown allows leakage of plasma and proteins promoting regional edema formation and ongoing neuroinflammation, key elements in the progression to secondary brain injury. Secondary brain injury is thought to be the leading cause of poor outcomes after TBI including cognitive disability and paralysis, and coma and brain death.2,25 In the last decade, intense TBI research has sought to characterize neuroprotective agents acting in the neurovascular microcirculation and capable of slowing secondary brain injury26,27
Trauma, and in particular TBI, harbors a post-injury inflammatory state that also affects other host systems promoting a profound hypercoagulable state after a brief period of coagulopathy.28 TBI increases the general population risk of VTE three-to fourfold during that period.11 Heparins have been used to reduce VTE risk in brain injured patients.28,29 Heparin is a highly sulfated glycosaminoglycan most commonly stored in the secretory granules of mast cells.12 Intriguingly and irrespective of its anticoagulant properties, other effects of heparin have recently been revealed, most notably their ability to reduce inflammation after injury. In particular, animal and clinical studies have shown how heparins can reduce tissue oxidative pathways by reducing circulating oxygen free radicals while augmenting physiologic antioxidants.30 Additionally, heparins have been shown to possess anti-excitotoxic properties through their modulation of calcium homeostasis during inflammation.31 More than two decades ago, Chaudry et al. demonstrated how heparinization before hemorrhagic shock prevented endothelial cell dysfunction while improving microvascular flow and organ function without increasing bleeding complications.13,32 Dampening of both leukocyte and endothelial activation by heparins has also been shown in the microcirculation in different settings. In particular, UFH blunts upregulation of surface LEU selectins (L-, E-, and P-selectin), key adhesion molecules responsible for LEU rolling on endothelium in the systemic microcirculation in different animal models.33 UFH can also inhibit expression of LEU CD18/CD11b34 and its EC counter-ligands, ICAM-1 and VCAM-1,35,36 the receptors directly implicated in LEU adhesion to EC. In a cerebral ischemia rat model, heparin was found to inhibit leukocyte accumulation in ischemic tissue, reducing infarction size and improving neurological recovery.15 Likewise, in a bacterial meningitis rat model, heparin was shown to reduce in vivo leukocyte rolling and adhesion in cerebral venules, which was directly associated with greater regional cerebral blood flow and reduced intracranial pressure, brain edema, and leukocyte extravasation into cerebrospinal fluid.9
Specifically in TBI models, low molecular weight heparin (LMWH) has been shown to possess anti-inflammatory effects.37–40 Our group recently demonstrated in a TBI rodent model how the LMWH, enoxaparin, reduced LEU/EC interactions and concurrent microvascular permeability resulting in reduced cerebral edema and improved neurological recovery.19 However, despite multiple studies suggesting similar benefits of LMWH and UFH in TBI, the former regimen may have a higher propensity to cause expansion of intracranial hematomas,41 and as such, some centers caring for TBI patients prefer the use of UFH—at least in the first 24 hours after injury. In the current study using UFH, we found that leukocyte rolling on endothelium was similarly reduced by high and low dose UHF to levels similar to that found in uninjured animals. This was paralleled with reduced venular macromolecular leakage in the same vessels, again equally seen after treatment with either heparin regimen. This data concurs with that seen in other CNS conditions using LMWHs. Of note, LEU adhesion was rarely observed in any of the groups despite known effects directly affecting this step in EC/LEU interactions.9 This is likely because our epiluminecence exposure at the wavelength of Rhodamine needed to be extremely short to avoid thrombogenic effects on the pial surface. This short time frame was insufficient to allow the visualization of leukocyte adhesion. Nonetheless, only low dose UFH-treated animals demonstrated improved neurological recovery after injury, not those treated with high dose heparin. Additionally, high dose UFH treatment significantly prevented recovery of animal weight loss, suggesting a potentially harmful effect of this UFH dose beyond a simple lack of benefit as seen with the lower UFH dose. Taken together, this data may indicate a worsened functional recovery in animals treated with higher dose UFH. Though this cannot be implied from the current study, this may have occurred by hypoactivity or poor feeding in high dose-treated animals due to anemia or obtundation from increased brain hemorrhage. Though mean arterial pressure before animal sacrifice was similar in all groups, this finding may not have accounted for small degrees of worsened ongoing cerebral bleeding in high dose UFH animals negatively affecting intracranial dynamics and swelling.
Lung edema, on the other hand, did not differ in the different groups. Acute lung injury (ALI) is not uncommon after TBI42,43 and may be observed in up to 20% of severe head injury cases.44 In primary ALI animal models, UFH has been found to reduce lung edema.45,46 In a previous mouse TBI study, our group also found reduced lung water after treatment with LMWH 48 hours after CCI.18 However, in the current study, brain injured animals did not demonstrate significant reductions in pulmonary edema with either heparin regimen. These differing results may have occurred because our TBI model was insufficiently severe to cause detectable increases in pulmonary water using wet-to-dry methodology. Alternatively, the anti-inflammatory effects of heparin on the pulmonary microcirculation may be less potent than that of LMWHs. It may also be possible that high molecular weight FITC albumin injected systemically may have altered the rheology of the pulmonary circulation.
The current study is small and, as such, has important limitations. First, only two different doses of heparin were used, and we did not perform a dose-response curve of UFH administered both subcutaneously and intravenously. Such a dose-response evaluation would have better contrasted anti-inflammatory and anticoagulant properties encountered in the clinical setting. Furthermore, standard UFH dosing is usually every 8 hours, whereas the more convoluted dosage regimen used in our study occurred to allow the investigator the ability to leave the laboratory to rest in the 3 days that treatment was being administered. This notwithstanding, the dose chosen reflects typical human prophylactic doses of UFH employed in patients after injury and the high dose, a dose threefold higher. Instead of arbitrarily choosing this threefold increase, we could have chosen a therapeutic dose of UFH involving a bolus and continuous infusion, but this was not done to keep the animal model constant and avoid changing part of the study animals to an intravenous and continuous regimen. Second, we did not measure systemic white blood cell counts. Others have reported a leukocytosis that occurs after TBI and which persists for up to 48 hours after injury.47 Such an increase in circulating LEUs may have affected the observed numbers of rolling LEUs. Third, measurement of hemoglobin levels and histological cerebral hematoma size would have been optimal to determine if ongoing or increased bleeding occurred intracranially or elsewhere with higher UFH doses. This would have been useful to explain the persistent body weight loss and inferior neurological recovery in these animals. Instead, we measured mean arterial pressure, which, although it demonstrated no difference among the groups, remains a low-fidelity surrogate of ongoing or persistent bleeding. Finally, the differences found between groups in the studied outcomes were small, though statistically significant, and it remains unclear if this would translate into clinically relevant effects when tested in head injured humans as opposed to in a TBI rodent model.
In conclusion, the current study demonstrates that early repeated administration of UFH after TBI reduces live LEU interacting with endothelium in the pericontusional microcirculation, concurrently reducing local venular albumin leakage. This UFH-related reduction in observed cerebrovascular leakage was confirmed with reduced hemispheric cerebral water content indicating an overall reduced pericontusional cerebral edema by heparin. On the other hand, only low dose UFH—equivalent to standard prophylactic doses—concurrently improved neurological recovery at both 24 and 48 hours and did not result in persistent loss of functional recovery as reflected by persistent body weight loss. Early low dose prophylactic UFH administration after TBI may be beneficial beyond its ability to reduce VTE complications, slowing progression of secondary brain injury. Further study in larger animals and humans is needed to confirm that anti-inflammatory properties of heparin are not offset by the inherent risks of cerebral hemorrhage propagation after TBI.
Acknowledgments
The authors thank Ms. Robin Armstrong for her technical and organizational assistance.
Footnotes
This study was presented at the 46th Western Trauma Association annual meeting February 28–March 4, 2016, in Lake Tahoe, California.
AUTHORSHIP
K.N., K.K., S.L., J.M., V.E.J., and J.L.P. designed this study. K.N., S.L., and J.L.P. conducted the literature search. K.N., K.D.B., J.S.-P., J.C., and J.L.P. contributed to data collection. K.N., K.K., K.D.B., J.S.-P., J. C., J.M., V.E.J., and J.L.P. performed data analysis. All authors contributed to data interpretation. K.N. and J.L.P. wrote the manuscript, which all authors critically revised.
DISCLOSURE
The authors declare no conflicts of interest.
References
- 1.Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet. 2012;380(9847):1088–1098. doi: 10.1016/S0140-6736(12)60864-2. [DOI] [PubMed] [Google Scholar]
- 2.Pop V, Badaut J. A neurovascular perspective for long-term changes after brain trauma. Transl Stroke Res. 2011;2(4):533–545. doi: 10.1007/s12975-011-0126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hazeldine J, Lord JM, Belli A. Traumatic brain injury and peripheral immune suppression: primer and prospectus. Front Neurol. 2015;6:235. doi: 10.3389/fneur.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lukaszewicz AC, Soyer B, Payen D. Water, water, everywhere: sodium and water balance and the injured brain. Curr Opin Anaesthesiol. 2011;24(2):138–143. doi: 10.1097/ACO.0b013e32834458af. [DOI] [PubMed] [Google Scholar]
- 5.Vestweber D. Novel insights into leukocyte extravasation. Curr Opin Hematol. 2012;19(3):212–217. doi: 10.1097/MOH.0b013e3283523e78. [DOI] [PubMed] [Google Scholar]
- 6.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 7.Reglero-Real N, Marcos-Ramiro B, Millán J. Endothelial membrane reorganization during leukocyte extravasation. Cell Mol Life Sci. 2012;69(18):3079–3099. doi: 10.1007/s00018-012-0987-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thal SC, Neuhaus W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch Med Res. 2014;45(8):698–710. doi: 10.1016/j.arcmed.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Weber JR, Angstwurm K, Rosenkranz T, Lindauer U, Freyer D, Burger W, Busch C, Einhäupl KM, Dirnagl U. Heparin inhibits leukocyte rolling in pial vessels and attenuates inflammatory changes in a rat model of experimental bacterial meningitis. J Cereb Blood Flow Metab. 1997;17(11):1221–1229. doi: 10.1097/00004647-199711000-00011. [DOI] [PubMed] [Google Scholar]
- 10.Yanaka K, Spellman SR, McCarthy JB, Low WC, Camarata PJ. Reduction of brain injury using heparin to inhibit leukocyte accumulation in a rat model of transient focal cerebral ischemia. II. Dose-response effect and the therapeutic window. J Neurosurg. 1996;85(6):1108–1112. doi: 10.3171/jns.1996.85.6.1108. [DOI] [PubMed] [Google Scholar]
- 11.Reiff DA, Haricharan RN, Bullington NM, Griffin RL, McGwin G, Jr, Rue LW., 3rd Traumatic brain injury is associated with the development of deep vein thrombosis independent of pharmacological prophylaxis. J Trauma. 2009;66(5):1436–1440. doi: 10.1097/TA.0b013e31817fdf1c. [DOI] [PubMed] [Google Scholar]
- 12.Paschoa AF. Heparin: 100 years of pleiotropic effects. J Thromb Thrombolysis. 2016;41(4):636–643. doi: 10.1007/s11239-015-1261-z. [DOI] [PubMed] [Google Scholar]
- 13.Wang P, Singh G, Rana MW, Ba ZF, Chaudry IH. Preheparinization improves organ function after hemorrhage and resuscitation. Am J Physiol. 1990;259(3 Pt 2):R645–R650. doi: 10.1152/ajpregu.1990.259.3.R645. [DOI] [PubMed] [Google Scholar]
- 14.Xie X, Thorlacius H, Raud J, Hedqvist P, Lindbom L. Inhibitory effect of locally administered heparin on leukocyte rolling and chemoattractant-induced firm adhesion in rat mesenteric venules in vivo. Br J Pharmacol. 1997;122(5):906–910. doi: 10.1038/sj.bjp.0701454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yanaka K, Spellman SR, McCarthy JB, Oegema TR, Jr, Low WC, Camarata PJ. Reduction of brain injury using heparin to inhibit leukocyte accumulation in a rat model of transient focal cerebral ischemia. I. Protective mechanism. J Neurosurg. 1996;85(6):1102–1107. doi: 10.3171/jns.1996.85.6.1102. [DOI] [PubMed] [Google Scholar]
- 16.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12(2):169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- 17.Pascual JL, Murcy MA, Li S, Gong W, Eisenstadt R, Kumasaka K, Sims C, Smith DH, Browne K, Allen S, Baren J. Neuroprotective effects of progesterone in traumatic brain injury: blunted in vivo neutrophil activation at the blood-brain barrier. Am J Surg. 2013;206(6):840–845. doi: 10.1016/j.amjsurg.2013.07.016. discussion 845–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S, Eisenstadt R, Kumasaka K, Johnson VE, Marks J, Nagata K, Browne KD, Smith DH, Pascual JL. Does enoxaparin interfere with HMGB1 signaling after TBI? A potential mechanism for reduced cerebral edema and neurologic recovery. J Trauma Acute Care Surg. 2016;80(3):381–387. doi: 10.1097/TA.0000000000000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Marks JA, Eisenstadt R, Kumasaka K, Samadi D, Johnson VE, Holena DN, Allen SR, Browne KD, Smith DH, et al. Enoxaparin ameliorates posttraumatic brain injury edema and neurologic recovery, reducing cerebral leukocyte endothelial interactions and vessel permeability in vivo. J Trauma Acute Care Surg. 2015;79(1):78–84. doi: 10.1097/TA.0000000000000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26(4):627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- 21.Manaenko A, Lekic T, Ma Q, Ostrowski RP, Zhang JH, Tang J. Hydrogen inhalation is neuroprotective and improves functional outcomes in mice after intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:179–183. doi: 10.1007/978-3-7091-0693-8_30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiStasi MR, Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009;30(11):547–556. doi: 10.1016/j.it.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubes P, Ward PA. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 2000;10(1):127–135. doi: 10.1111/j.1750-3639.2000.tb00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moxon-Emre I, Schlichter LC. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropathol Exp Neurol. 2011;70(3):218–235. doi: 10.1097/NEN.0b013e31820d94a5. [DOI] [PubMed] [Google Scholar]
- 25.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6(7):393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Logsdon AF, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW. Role of microvascular disruption in brain damage from traumatic brain injury. Compr Physiol. 2015;5(3):1147–1160. doi: 10.1002/cphy.c140057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enzmann G, Mysiorek C, Gorina R, Cheng YJ, Ghavampour S, Hannocks MJ, Prinz V, Dirnagl U, Endres M, Prinz M, et al. The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol. 2013;125(3):395–412. doi: 10.1007/s00401-012-1076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim L, Schuster J, Holena DN, Sims CA, Levine J, Pascual JL. Early initiation of prophylactic heparin in severe traumatic brain injury is associated with accelerated improvement on brain imaging. J Emerg Trauma Shock. 2014;7(3):141–148. doi: 10.4103/0974-2700.136846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farooqui A, Hiser B, Barnes SL, Litofsky NS. Safety and efficacy of early thromboembolism chemoprophylaxis after intracranial hemorrhage from traumatic brain injury. J Neurosurg. 2013;119(6):1576–1582. doi: 10.3171/2013.8.JNS13424. [DOI] [PubMed] [Google Scholar]
- 30.Dzieciuchowicz Ł, Checinski P, Krauss H. Heparin reduces oxidative stress in the postoperative period. Med Sci Monit. 2002;8(9):CR657–CR660. [PubMed] [Google Scholar]
- 31.Hao L, Zhang Q, Yu T, Yu L, Cheng Y. Modulation of ultra-low-molecular-weight heparin on [Ca2+]i in nervous cells. Brain Res Bull. 2011;86(5–6):355–359. doi: 10.1016/j.brainresbull.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 32.Wang P, Ba ZF, Chaudry IH. Endothelial cell dysfunction occurs after hemorrhage in nonheparinized but not in preheparinized models. J Surg Res. 1993;54(5):499–506. doi: 10.1006/jsre.1993.1077. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Brown JR, Varki A, Esko JD. Heparin’s anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L-and P-selectins. J Clin Invest. 2002;110(1):127–136. doi: 10.1172/JCI14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diamond MS, Alon R, Parkos CA, Quinn MT, Springer TA. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD1) J Cell Biol. 1995;130(6):1473–1482. doi: 10.1083/jcb.130.6.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JH, Kim CH, Seo GH, Lee J, Kim JH, Kim DG, Ahn YS. Heparin attenuates the expression of TNFalpha-induced cerebral endothelial cell adhesion molecule. Korean J Physiol Pharmacol. 2008;12(5):231–236. doi: 10.4196/kjpp.2008.12.5.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller SJ, Hoggat AM, Faulk WP. Heparin regulates ICAM-1 expression in human endothelial cells: an example of non-cytokine-mediated endothelial activation. Thromb Haemost. 1998;80(3):481–487. [PubMed] [Google Scholar]
- 37.Sen O, Sonmez E, Cekinmez M, Ozen O, Caner H. Antithrombin III and enoxaparin treatment inhibit contusion-triggered cell death, inflammation, hemorrhage and apoptosis after severe traumatic brain injury in rats. Turk Neurosurg. 2011;21(2):203–209. doi: 10.5137/1019-5149.JTN.3646-10.1. [DOI] [PubMed] [Google Scholar]
- 38.Stutzmann JM, Mary V, Wahl F, Grosjean-Piot O, Uzan A, Pratt J. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Rev. 2002;8(1):1–30. doi: 10.1111/j.1527-3458.2002.tb00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wahl F, Grosjean-Piot O, Bareyre F, Uzan A, Stutzmann JM. Enoxaparin reduces brain edema, cerebral lesions, and improves motor and cognitive impairments induced by a traumatic brain injury in rats. J Neurotrauma. 2000;17(11):1055–1065. doi: 10.1089/neu.2000.17.1055. [DOI] [PubMed] [Google Scholar]
- 40.Župan Ž, Pilipović K, Dangubić B, Frković V, Šustic A, Župan G. Effects of enoxaparin in the rat hippocampus following traumatic brain injury. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(8):1846–1856. doi: 10.1016/j.pnpbp.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 41.Glassner S, Srivastava K, Cofnas P, Deegan B, Demaria P, Denis R, Ginzburg E. Prevention of venous thrombotic events in brain injury: review of current practices. Rambam Maimonides Med J. 2013;4(1):e0001. doi: 10.5041/RMMJ.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baumann A, Audibert G, McDonnell J, Mertes PM. Neurogenic pulmonary edema. Acta Anaesthesiol Scand. 2007;51(4):447–455. doi: 10.1111/j.1399-6576.2007.01276.x. [DOI] [PubMed] [Google Scholar]
- 43.Gajic O, Manno EM. Neurogenic pulmonary edema: another multiple-hit model of acute lung injury. Crit Care Med. 2007;35(8):1979–1980. doi: 10.1097/01.CCM.0000277254.12230.7D. [DOI] [PubMed] [Google Scholar]
- 44.Bratton SL, Davis RL. Acute lung injury in isolated traumatic brain injury. Neurosurgery. 1997;40(4):707–712. doi: 10.1097/00006123-199704000-00009. discussion 712. [DOI] [PubMed] [Google Scholar]
- 45.Li LF, Huang CC, Lin HC, Tsai YH, Quinn DA, Liao SK. Unfractionated heparin and enoxaparin reduce high-stretch ventilation augmented lung injury: a prospective, controlled animal experiment. Crit Care. 2009;13(4):R108. doi: 10.1186/cc7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X, Li Z, Zheng Z, Liu Y, Ma X. Unfractionated heparin ameliorates lipopolysaccharide-induced lung inflammation by downregulating nuclear factor-kB signaling pathway. Inflammation. 2013;36(6):1201–1208. doi: 10.1007/s10753-013-9656-5. [DOI] [PubMed] [Google Scholar]
- 47.Rhind SG, Crnko NT, Baker AJ, Morrison LJ, Shek PN, Scarpelini S, Rizoli SB. Prehospital resuscitation with hypertonic saline-dextran modulates inflammatory, coagulation and endothelial activation marker profiles in severe traumatic brain injured patients. J Neuroinflammation. 2010;7:5. doi: 10.1186/1742-2094-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]