

Abstract
Almost a century ago, the first clinical account of the punch-drunk syndrome emerged, describing chronic neurological and neuropsychiatric sequelae occurring in former boxers. Thereafter, throughout the twentieth century, further reports added to our understanding of the neuropathological consequences of a career in boxing, leading to descriptions of a distinct neurodegenerative pathology, termed dementia pugilistica. During the past decade, growing recognition of this pathology in autopsy studies of non-boxers who were exposed to repetitive, mild traumatic brain injury, or to a single, moderate or severe traumatic brain injury, has led to an awareness that it is exposure to traumatic brain injury that carries with it a risk of this neurodegenerative disease, not the sport or the circumstance in which the injury is sustained. Furthermore, the neuropathology of the neurodegeneration that occurs after traumatic brain injury, now termed chronic traumatic encephalopathy, is acknowledged as being a complex, mixed, but distinctive pathology, the detail of which is reviewed in this article.
Keywords: traumatic brain injury, CTE, neurodegeneration, axons, tau, amyloid
INTRODUCTION
Traumatic brain injury (TBI) represents a leading cause of morbidity and mortality internationally. In the United States alone, there are upwards of 2.5 million TBIs annually, with just less than 300,000 requiring hospitalization, and approximately 2% of the US population (approximately 5 million people) living with long-term disabilities as a consequence of a TBI (1–4). As a result, the estimated annual cost for TBI-associated health care in the United States exceeds $60 billion (5). Nevertheless, the evolving neuropathologies of TBI survival remain poorly described. In particular, the association of a history of TBI with neurodegenerative disease has, thus far, been based on observations from limited case-series, which often have incomplete clinicopathological correlations. As such, recognition of TBI-associated neurodegeneration in clinics and at autopsy is comparatively rare, with the consequence that the true prevalence and socioeconomic burden of these late TBI outcomes remain unknown.
In his observations on the punch-drunk syndrome in former boxers almost 90 years ago, the New Jersey Medical Examiner, Harrison S. Martland, described a constellation of neuropsychiatric and motor symptoms, together with limited autopsy data, observing
the condition can no longer be ignored by the medical profession or the public. It is the duty of our profession to establish the existence or nonexistence of punch drunk by preparing accurate statistical data as to its incidence, careful neurologic examinations of fighters thought to be punch drunk, and careful histologic examinations of the brains of those who have died with symptoms. (6)
Thereafter, throughout the twentieth century, reports of further isolated case descriptions and short case-series (7–10) reinforced Martland’s observations on the link between exposure to repetitive TBI in boxing and late neurodegenerative disease associated with a distinct neuropathology later recognized and termed dementia pugilistica (DP) (7). Perhaps because this condition was thought restricted to the uniquely brutal sport of boxing, DP garnered remarkably little interest, certainly not the formal studies called for by Martland to establish the incidence and describe the detailed neuropathology. However, during the past decade or so, with growing recognition of the neuropathology of DP in athletes who were not boxers (11–20), and in survivors of a single, moderate or severe TBI (sTBI) (21–25), there has been increasing acknowledgment that it is brain injury per se that is associated with the risk of late neurodegenerative disease, rather than it being a unique consequence of participation in a single sport. Hence, the current, more encompassing term chronic traumatic encephalopathy (CTE) replaces DP.
Inevitably, as reports of CTE attributed to a growing list of circumstances, in particular sports, have emerged in the media and in peer reviewed manuscripts, there has been growing public concern. However, although it is almost nine decades on from Martland’s first descriptions, we remain little further forward in understanding the true prevalence of CTE, and remarkably few cases have been described. Further, no robust and validated operational criteria for the clinical diagnosis of CTE have been defined. At present, the diagnosis of CTE rests with neuropathological assessment conducted at autopsy. However, validated operational criteria are in their infancy, and understanding of the complex of pathologies contributing to CTE continues to be expanded. This review describes our current understanding of the range of pathologies observed in survivors of TBI, and the potential pathological substrates linking TBI to this late neurodegeneration.
TRAUMATIC BRAIN INJURY AND DEMENTIA
TBI is widely accepted as the strongest environmental risk factor for dementia (26–35). Indeed, not only is there evidence of a link between TBI and dementia, there is also evidence to support a dose–response relationship, with the risk of dementia increased in severe TBI versus moderate or mild TBI (34). However, much of the evidence supporting an association between TBI and the later development of neurodegenerative disease comes from observational and retrospective studies, and thus it may have been influenced by recall bias. The best current estimates suggest that between 5% and 15% of current cases of dementia may be TBI related (36), representing in excess of 675,000 dementia patients in the United States. However, as our appreciation of this link grows, and appropriately designed prospective studies report their findings, these figures undoubtedly will be revised.
Both historically and currently, TBI-associated dementia has been subdivided based on whether it follows an sTBI or repetitive, mild TBI (rTBI), with the associated clinical syndromes considered distinct. In particular, despite compelling literature to the contrary, there is a general presumption that CTE is limited to patients exposed to rTBI, most often athletes. This misperception may be due in part to a complete absence of comparative research or clinicopathological studies looking at material from both sTBI and rTBI patients in parallel. As such, considering late neurodegenerative outcomes from an sTBI and rTBI as different syndromes is, at best, premature and, arguably, flawed.
Following Martland’s seminal account of the punch-drunk syndrome, describing chronic neuropsychiatric sequelae and limited associated pathology in former boxers (6), multiple other reports followed and expanded on his findings. Of these, perhaps most enlightening were the observations by Roberts (37) on a cohort of 224 randomly selected professional boxers in which he reported that 17% displayed a “relatively stereotyped” clinical picture, comprising emotional lability, personality change, memory impairment and dementia, as well as pyramidal and extrapyramidal dysfunction, and cerebellar impairment. More recent descriptions of former nonboxer athletes (including from American football, ice hockey, and rugby) and military personnel have reported similar neuropsychiatric and behavioral problems, while expanding the descriptions to include aggression, poor judgment, depression, suicidal ideation, and, in some instances, suicide (17, 18, 39–41). Of note, these more recent case-series in nonboxer athletes report less prevalent motor symptoms than earlier reports of former boxers.
Studies reporting on dementia occurring following a moderate or severe sTBI typically describe the associated clinical syndrome as Alzheimer’s disease (AD) in type (27–35), with only occasional studies formally recording a clinical diagnosis based on validated consensus criteria (28, 30–32, 34, 35). Notably, neuropathological confirmation that the pathology after an sTBI conforms to AD is lacking in these reports. Certainly, there have been no suitably designed, robust studies in the current era of understanding of the neuropathology of CTE that confirm the dementia occurring in late survivors of an sTBI is AD. In that regard, in contrast to patients with AD and no history of TBI, dementia in association with an sTBI is reported to feature prominent motor and neuropsychiatric symptoms, including depression and suicide (42, 43). As such, in cases where it has been evaluated in detail, the clinical syndrome of neurodegeneration following an sTBI echoes that of rTBI, and appears distinct from AD. Thus, the traditional view permeating the literature that an sTBI is specifically associated with the development of AD (44, 45) appears speculative, at best, and without confirmatory neuropathology.
CHRONIC TRAUMATIC ENCEPHALOPATHY: A SPECTRUM OF NEUROPATHOLOGY OCCURRING WITH SURVIVAL AFTER TRAUMATIC BRAIN INJURY
Although Martland called for neuropathological assessment of DP in his original publication in 1928, it took until 1954 for the first formal account of the neuropathology of the brain of a former boxer to be described (46). Thereafter, several isolated case reports appeared during the following decades (9, 47–51), preceding the landmark observations of Corsellis et al. (52) on their neuropathological findings in a series of 15 former amateur and professional boxers. Although further studies on the material from this unique cohort followed, added to by many more studies both in boxers and, more recently, nonboxer athletes, this original publication remains informative. Of note, the picture that emerged from these original studies, and the observations made since, is of a complex of pathologies arising following exposure to TBI, whether repetitive or single, that might best be regarded as a polypathology. However, although increasing reports on the pathology of CTE are emerging, the often limited case numbers, biases in case selection, differences in methods, and the lack of matched-control observations create challenges in identifying informative pathology and in permitting comparative observations between rTBI and sTBI. The following account reviews the current understanding of the pathology of CTE and offers observations on priority areas requiring attention in future studies.
Macroscopic Neuropathology
A common observation in the early case reports of boxers was a degree of atrophy occurring in the cerebral hemispheres (9, 17, 46–49, 51–57) and cerebellum (51, 58), the former often showing preferential involvement of the temporal (59, 60) and frontal (9, 48, 50, 57, 60) lobes. In contrast, more recent reports on CTE in nonboxer athletes have documented relatively mild global atrophy, with the observation of a reduction in brain weight being made less consistently (11, 13–17, 39). Nevertheless, a pattern of macroscopic features, albeit relatively nonspecific, is emerging in studies on CTE in individuals exposed to rTBI, which encompasses atrophy of the mammillary bodies, a mild degree of ventricular enlargement (perhaps preferentially of the third ventricle), pallor of the substantia nigra, and thinning of the corpus callosum (46–49, 51–53, 56, 57, 59). In contrast to these observations in rTBI, formal reports of brain atrophy from autopsy examinations following survival after an sTBI are lacking. Notwithstanding this, generalized brain atrophy following an sTBI has been recognized, both at autopsy and in imaging studies, with the latter suggesting this atrophy continues beyond the acute phase and into longer-term survival (61–65). Further, as in rTBI, autopsy studies have reported notable thinning of the corpus callosum in a proportion of sTBI survivors at 1 year or more from injury (23).
Beyond evidence of cerebral atrophy, a feature that has been described in a majority of cases following exposure to rTBI is abnormalities of the septum pellucidum, in particular cavum septum pellucidum (CSP), septal fenestration, or, in occasional cases, complete absence of the septum (9, 11, 13, 14, 16, 17, 46–51, 53–59, 66–69). Although CSP has been reported in up to one-third of participants in population studies as being a normal finding (52, 70–73), evidence has suggested that there is both a higher prevalence and a greater extent of CSP in those exposed to rTBI (52). Further, imaging studies have confirmed CSP in vivo in boxers, with evidence in longitudinal studies supporting CSP as an acquired and evolving pathology (9, 50, 74, 75). In contrast to experiences in rTBI, to date CSP has not been documented as a specific feature in late survivors of an sTBI.
Microscopic Neuropathology
To date, some 150 or so cases have been reported describing the neuropathology of those exposed to rTBI from a range of sports—including boxing (22, 46, 52), American football, ice hockey, wrestling, soccer, and rugby union (11, 13–20, 40, 41)—and former military personnel (12, 19, 20), together with sporadic cases exposed to rTBI in other circumstances, including domestic abuse (58, 69, 76). Approximately 40 further cases reporting the pathology of survival after an sTBI have been described (21, 23, 77). However, although relatively small numbers of cases have been recorded in the literature, it should be noted this does not represent the entire world’s incidence of CTE during the period from the first clinical description by Martland or since the first pathology account in the 1950s. Instead, this represents recognized cases coming to autopsy, undoubtedly with considerably higher numbers either clinically diagnosed as an alternate dementia, such as AD, or not recognized as CTE during neuropathology assessment at autopsy. Notably, the pathology of CTE has been recognized only in circumstances where there has been exposure to brain injury, with this pathology featuring a range of abnormalities, including pathologies in tau, amyloid β (Aβ), and transactive response (TAR) DNA-binding protein 43 (TDP-43); neuroinflammation; axonal degeneration; degradation of white matter; and neuronal loss (21).
Tau
Under the high strain rates encountered in TBI, mechanical breaking of axonal microtubules occurs, leading to transport interruption as a component of diffuse axonal injury (DAI) (78–80) (Figure 1). This vulnerability of axons to mechanical loading arises as a result of their unique viscoelastic properties, in turn thought to derive from the biophysical properties of the microtubule-associated protein tau (80). Nevertheless, examination of tissue acquired at autopsy from patients dying in the acute phase after an sTBI (with up to 4 weeks’ survival) has revealed no evidence of immediate tau pathology (81). In contrast, where sought, abnormal accumulation of hyperphosphorylated tau is a constant in reported cases of CTE (22) and in a proportion of patients dying 1 year or more after an sTBI (21). Although the deposition of tau is a feature of a number of neurodegenerative pathologies and can be observed in normal aging, the pattern and distribution of tau pathology in CTE is sufficiently distinct to be proposed as pathognomonic. Specifically, whether in material from survivors of rTBI or an sTBI, this pathology is characterized by the accumulation of abnormal, hyperphosphorylated tau in both neurons and glia, showing a distinct perivascular accentuation and preferential involvement toward the depths of sulci in the neocortical gray matter (11, 17–19, 39, 40, 67, 82) (Figure 2). Typically, there is an irregular, patchy involvement within and between the involved cortical areas (17, 39, 40, 82). This pattern, originally observed by Geddes and colleagues (67, 82) in their description of tau pathology in boxers, has since been confirmed and refined in subsequent case-series, both in rTBI and sTBI, such that its appearance might be considered as defining CTE, when observed in the correct clinical context.
Figure 1.
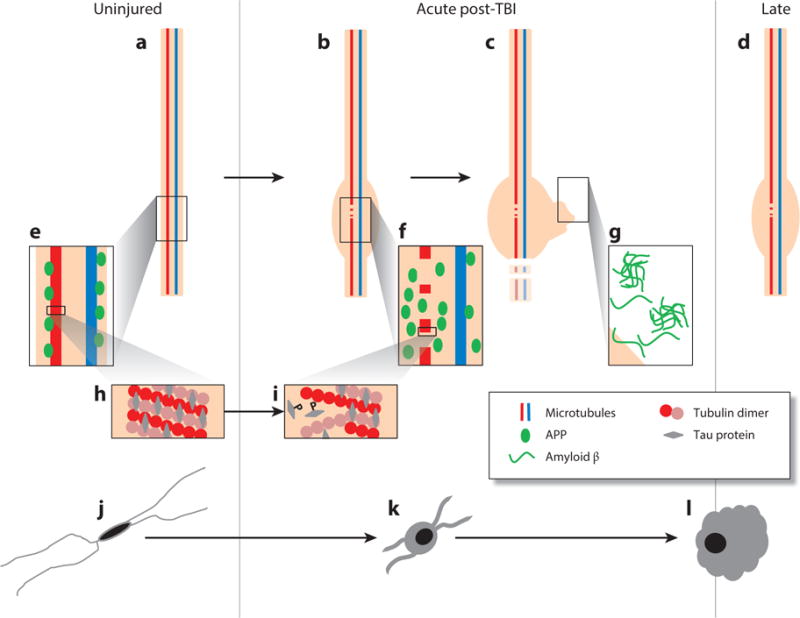
Proposed evolution of axonal and microglial pathologies contributing to late neurodegeneration following traumatic brain injury (TBI). In the intact, uninjured axon (a) microtubules composed of tubulin dimers bound by tau protein (h) transport cargo along the axon, including amyloid precursor protein (APP) and the enzymes responsible for its cleavage to amyloid β (Aβ) (e). As a consequence of dynamic stretch during injury, there is a change in the physical properties of tau protein, resulting in the mechanical breaking of microtubules, tau liberation, and its subsequent phosphorylation (i). At these sites of microtubule breakage, transport interruption follows, with the accumulation of transported cargo (f) resulting in axonal swelling (b), degeneration (c), and the liberation of large pools of Aβ, leading to plaque formation (g), a process that continues beyond the acute phase in a proportion of survivors (d). In parallel with this axonal pathology, there is a notable neuroinflammatory response marked by quiescent, ramified microglia (j) becoming activated (k); these activated and amoeboid microglia (l) persist beyond the acute phase in a proportion of survivors.
Figure 2.
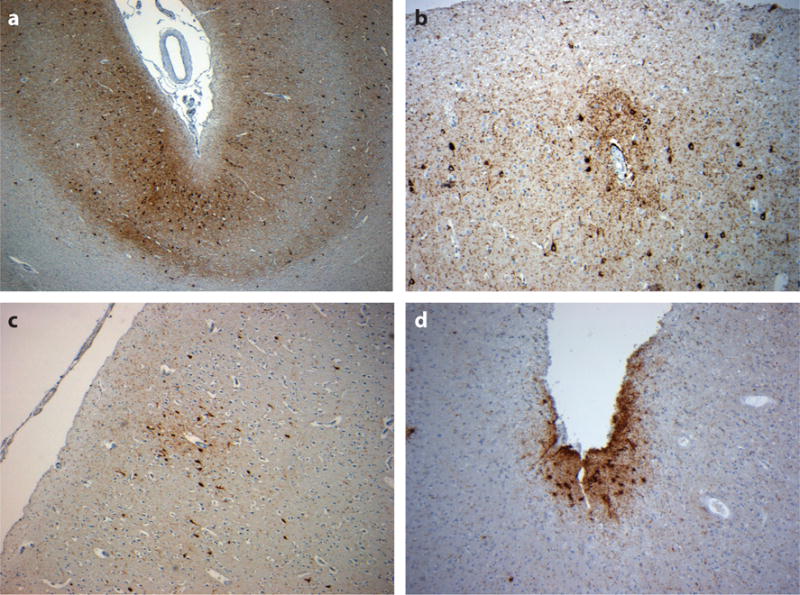
Neocortical tau pathology in chronic traumatic encephalopathy. Tau immunoreactive profiles are distributed throughout the neocortex, although they typically show a preferential distribution toward the superficial neocortical layers and depths of sulci [a, 49-year-old male 12 months following single, severe traumatic brain injury (TBI)], with a distinctive and characteristic perivascular accentuation of immunoreactive neurons and glia, whether exposed to repetitive, mild TBI (b, 56-year-old male, former rugby player) or a single, moderate or severe TBI (c, 48-year-old male 3 years following a single, severe TBI). The accumulations of subpial thorn-shaped astrocytes may also be observed (d, 59-year-old male, former soccer player). All sections stained for phosphorylated tau using antibody CP-13 (courtesy of Dr. P. Davies).
Remarkably, there remain just two cases, both former boxers, in which biochemical characterization of tau in CTE has been performed (55). In both cases, the hyperphosphorylated tau of CTE was indistinguishable from that observed in AD. When optimally assessed using immunostains for hyperphosphorylated tau (such as AT8, CP-13, or PHF-1), the neuronal tau pathologies appear as typical neurofibrillary tangles (NFTs) and pretangles that, beyond the perivascular accentuation and preferential involvement toward the depths of sulci, often show a distribution to superficial cortical layers (layers II and III) (39) in contrast to the involvement of deeper cortical layers that is more typical of AD. In the hippocampus, CTE is noted for the preferential involvement of sector CA2 by NFTs and extracellular tangles (Figure 3a), with NFTs and astroglial tau pathology also a feature in the hypothalamic region, in particular the mammillary bodies (39). Beyond these regions, there may be frequent involvement of deep gray nuclei and brainstem in the nucleus basalis of Meynert, substantia nigra (Figure 3b), locus coeruleus, raphe nuclei, and tectum. In contrast, the dorsal striatum (the caudate and putamen) appears relatively spared, as too are the cerebellar dentate nuclei. In addition to neuronal tangle and pretangle pathologies, frequent, and perhaps distinctive, grain-like tau-immunoreactive profiles and neurites are often present in affected neuropil. Further, tau-immunoreactive axonal profiles are often observed in subcortical and midline white matter (Figure 3c,d). Finally, glial pathology is commonly observed as tau-immunoreactive thorn-shaped astrocytes located in subpial and perivascular locations in the neocortex and subependymally (39).
Figure 3.
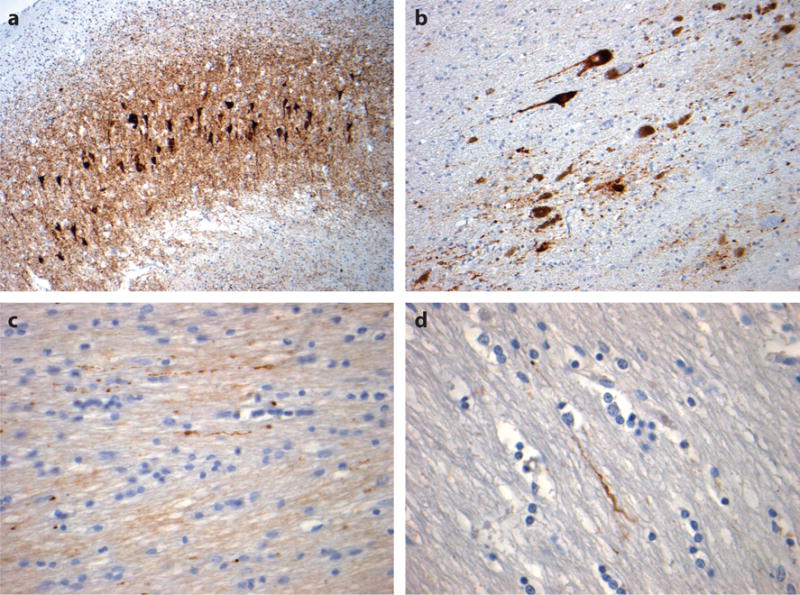
Tau pathology in chronic traumatic encephalopathy (CTE). In addition to neocortical tau pathology, tau-immunoreactive profiles are common in the hippocampus in CTE, with preferential involvement of sector CA2 by neurofibrillary tangles and extracellular tangles, as well as astroglial tau pathology (a, 59-year-old male, former soccer player). Elsewhere, tau pathologies are described in the deep gray nuclei and brainstem, where tau-immunoreactive substantia nigra neurons and neurites may be present, together with a degree of pigment incontinence (b, same case as in a). Scattered tau-immunoreactive axonal profiles are also common in subcortical and midline white matter (c, same case as a; d, 60-year-old male, former boxer). All sections stained for phosphorylated tau using antibody CP-13 (courtesy of Dr. P. Davies).
In relation to rTBI, these tau pathologies have been documented in virtually all cases. However, specifically for rTBI, there has been an unavoidable case-selection bias and a virtual absence of control material from uninjured cases in reports of CTE, rendering interpretations of the incidence of this pathology meaningless in the context of exposure to repetitive mild injury. In contrast, in the single study that looked at survivors of an sTBI, tau pathology was observed in up to 30% of patients at 1 year or more following injury, and this pathology was observed in greater density and in a wider distribution than in age-matched controls (21). However, as a retrospective archive-based study, no information was provided on the clinical status of these post-TBI tau-positive patients, hence, the clinical significance of the pathology identified at autopsy remains uncertain.
Regarding the hierarchical evolution of tau pathologies in CTE, putative staging protocols have been suggested by authors reviewing material from athletes after rTBI, and these range from restricted, focal, cortical pathology (stage I) to more extensive, widespread, cortical, hippocampal, and brainstem involvement (stage IV) (39). However, validation of this proposed staging, in particular clinicopathological correlation, has yet to be completed. Of note, the single study of a limited number of sTBI cases reported a hierarchy similar to that for AD (21), although this might reflect more limited anatomical sampling of the cohort.
Amyloid β
An early and consistent event in all severities of TBI, diffuse axonal injury results in cytoskeletal disruption with associated interruption of axonal transport (61, 83–85). In common medicolegal practice, DAI is sought by examining for evidence of transport interruption through the immunocytochemical demonstration of the abnormal accumulation of β-amyloid precursor protein (APP) in injured axons (83, 86, 87). Normally transported by fast axonal transport, following TBI, APP may be observed in damaged axons within hours of injury (Figures 1f and 4a). In addition, colocalizing with APP at these sites of injury are the enzymes presenilin-1 and β-site–APP-cleaving enzyme, which is required to cleave APP to Aβ (88–93). Thus, DAI creates an environment in which high concentrations of Aβ protein may be generated following TBI (93) (Figure 1g).
Figure 4.
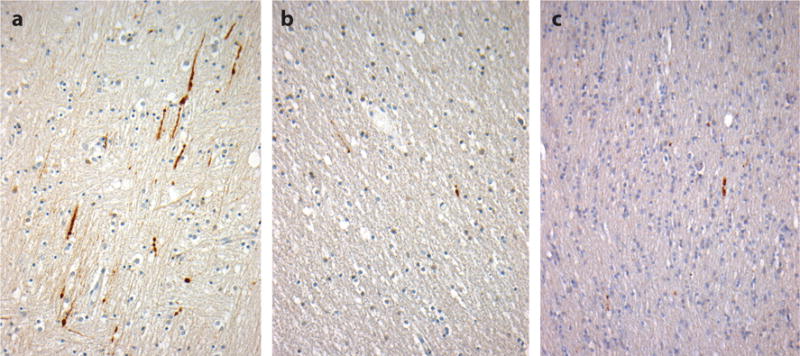
Axonal pathology in the corpus callosum with varying durations of survival after traumatic brain injury (TBI). A constant in all severities of TBI is diffuse axonal injury (DAI) with associated interruption of axonal transport. In tissue sections, DAI is typically revealed by staining for amyloid precursor protein (APP) and is detectable in under an hour following injury as immunoreactive axonal profiles with varying abnormal morphologies (a, 18-year-old male 11 h after severe TBI). Beyond this acute-phase axonal injury, evidence of ongoing interruption of axonal transport is marked by scattered, morphologically abnormal axons; staining for APP remains present in survivors 1 year or more after a single, moderate or severe TBI (b, 24-year-old male 8 years after a single, severe TBI) and in material from individuals exposed to repetitive, mild TBI (c, 59-year-old male, former soccer player). All sections stained for an antibody to the N-terminal amino acids 66–81 of APP (EMD Millipore).
A hallmark pathology of AD, Aβ plaques are identified in autopsy material from up to 30% of patients dying acutely following an sTBI, and at higher density than observed in equivalent material from uninjured age-matched controls (Figure 5a) (24, 25, 94). Further, Aβ plaques can also be demonstrated in pericontusional, surgically excised tissue from sTBI survivors (94). Typically, these acute plaques are diffuse in nature and similar to those of early-stage AD. However, whereas plaques in AD are thought to develop slowly and occur predominantly in the elderly, TBI-associated plaques are detectable within hours of injury and across a range of ages, including in young adults. Notably, the appearance of these Aβ plaques in the acute phase might represent not only excessive Aβ genesis as a consequence of DAI but also the failure of normal Aβ-clearance pathways, which become overwhelmed by excess parenchymal Aβ production.
Figure 5.
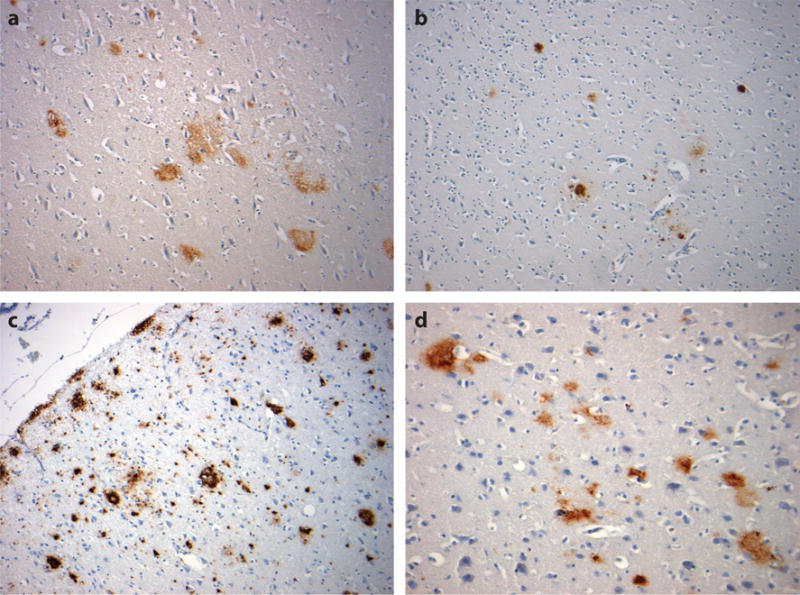
Amyloid β (Aβ) plaque pathologies following traumatic brain injury (TBI). Diffuse Aβ plaques can be identified in autopsy and surgical material from approximately 30% of TBI patients during the acute phase post-injury (a, 51-year-old male 24 h following severe TBI). In the following weeks to months, these diffuse plaques resolve, only to reemerge in approximately 30% of survivors 1 year or more after a single, moderate or severe TBI as both neuritic and diffuse Aβ plaques (b, 55-year-old female 47 years after a single, severe TBI). Aβ plaques are also present in a majority of cases of chronic traumatic encephalopathy following exposure to repetitive, mild TBI; these are typically, although not exclusively, diffuse in subtype (c, 60-year-old male, former boxer; d, 59-year-old male, former soccer player). All sections stained using antibody 6F/3D, specific for the N-terminal epitope of Aβ (Dako, Agilent Technologies).
In the months following injury, evidence suggests that the normal order in amyloid cycling might be restored, with these acute TBI plaques clearing (92). However, in observations of material acquired at autopsy from people who survived 1 year or more after an sTBI, amyloid plaques were again observed in considerably greater density and in wider distribution than they were in material from age-matched uninjured controls (21). Furthermore, although the acute plaques following TBI are typically diffuse in nature, those observed in survivors of an sTBI are more typically neuritic in type (21), thus more reminiscent of plaques in established AD (Figure 5b).
In studies reviewing cases from the Corsellis Archive, Roberts and colleagues (53) observed the “occult aftermath of boxing,” referring to the often-abundant Aβ plaques identified in material acquired at autopsy from a cohort of 20 retired amateur and professional boxers. In this series, samples from all but one case contained diffuse amyloid plaque pathology; the exception was a 22-year-old former professional boxer who had had a 3-year career and only 3 professional fights. Of note, however, the remaining 19 cases ranged in age from 53 to 83 (median 65) years, thus arguably representing an older cohort. All NFT-positive cases in this cohort contained plaque. In contemporary descriptions of CTE in former athletes, Aβ plaques are often reported as a less consistent finding (39). However, where documented, Aβ plaque pathology is present in a majority of cases, the proportion increasing with advancing age (95). Hence, the current recognition of tau pathologies leading to putative CTE diagnoses in comparably younger athletes might underlie the occasionally misreported assertion that amyloid pathologies are absent or rare in CTE, when indeed they appear to be frequently observed in older and, perhaps crucially, clinically confirmed cases. In studies of material from patients with histories of exposure to rTBI, these Aβ plaques are almost invariably described as diffuse in nature (Figure 5c,d), in contrast to the neuritic plaque more often observed in long-term survivors of an sTBI. However, although much research attention is focused on tau pathology in CTE, comparatively little attention is directed toward amyloid pathologies.
Transactive Response DNA-Binding Protein 43
The 43 kDa transactive response (TAR) DNA-binding protein 43 (TDP-43) is a nuclear protein widely expressed throughout the body. However, under certain conditions TDP-43 may translocate from the nucleus to the cytoplasm, forming polyubiquitinated and hyperphosphorylated pathological inclusion bodies; this abnormal TDP-43 is recognized as a major disease-associated protein in a number of neurodegenerative conditions, including frontotemporal lobar dementia and amyotrophic lateral sclerosis (96), and as a minor component of a variety of other conditions, including AD, Parkinson’s disease, and a small proportion of normal, aged individuals (97–99). Of relevance, animal modeling studies of TBI have suggested that axonal injury might upregulate TDP-43, leading to its translocation from the nuclear to cytoplasmic compartment (100–102). With recovery, TDP-43 is restored to the nucleus, suggesting this redistribution represents some form of acute-phase response (100, 101). Thus, there is the potential that TBI-induced axonal injury might influence neuronal TDP-43 processing.
In limited observations in CTE following exposure to rTBI, which included material from boxers and nonboxer athletes, neuronal cytoplasmic TDP-43 inclusions and distinctive grain-like profiles in the surrounding neuropil, where sought, have been documented in the hippocampus, temporal neocortex, and amygdala in a majority of cases (18, 39, 77, 103). In a small proportion of these cases, this TDP-43 pathology coincided with a clinical diagnosis of amyotrophic lateral sclerosis, with the autopsy examination revealing features consistent with CTE (18). Based on these few observations, a diagnosis of chronic traumatic encephalomyopathy has been proposed (18), although it remains to be seen whether with further case experience this putative diagnosis will survive rigorous scrutiny.
In contrast to the similarities in pathologies in tau and Aβ in survivors of rTBI and sTBI, there is a notable difference in TDP-43 pathology. Specifically, following survival after an sTBI, although increased cytoplasmic immunoreactivity to physiological, nonphosphorylated TDP-43 has been observed in acute and late survivors (1 year or more after injury), abnormally phosphorylated TDP-43 cytoplasmic inclusions have not been reported to the extent or distribution beyond that seen in material from uninjured controls (77). This intriguing apparent difference in pathologies between survivors of rTBI and sTBI might suggest a physiological role for TDP-43 in the response to injury, which is somehow modified after exposure to repetitive brain injury, and thus precipitates cytoplasmic accumulation of hyperphosphorylated TDP-43.
Neuroinflammation
Neuroinflammation is increasingly regarded as an important facet of the pathology of a range of neurodegenerative disorders (104–106), with neuroinflammation in AD recognized as an early event in disease pathogenesis (107–110). Of note, as regards TBI, studies exploring serum or cerebrospinal fluid from patients surviving an sTBI have reported elevated proinflammatory cytokine levels that correlated with poor neuropsychiatric outcome, including suicidal ideation (111–113).
Immediately following injury, TBI induces a complex neuroinflammatory response in both humans and animal models, a consequence of blood–brain barrier compromise, and an acute-phase inflammatory cell reaction featuring polymorphonuclear leukocytes, T lymphocytes, macrophages, and natural killer cells, together with activation of resident microglia (23, 114). Under normal circumstances it might be anticipated that this acute-phase inflammation would resolve. However, in a proportion of survivors there is accumulating evidence that neuroinflammation persists beyond the acute phase (Figure 1j–l). Examination of material acquired at autopsy from patients surviving an sTBI has revealed a remarkable and ongoing neuroinflammatory response in a proportion of survivors, even decades after survival following injury (Figure 6) (23). Intriguingly, although these observations noted florid neuroinflammation, with numerous activated microglia in the corpus callosum, in vivo positron emission tomography studies in surviving patients after an sTBI using the ligand [11C]PK-11195 have reported no significant differences in binding in this region, although they have reported a difference in binding in the thalamic region compared with control patients (115). As such, the possibility that imaging studies might considerably underestimate or misrepresent neuroinflammation following TBI could be considered. Of note, evidence of persistent inflammation in these imaging studies has been correlated with clinical evidence of impaired cognition.
Figure 6.
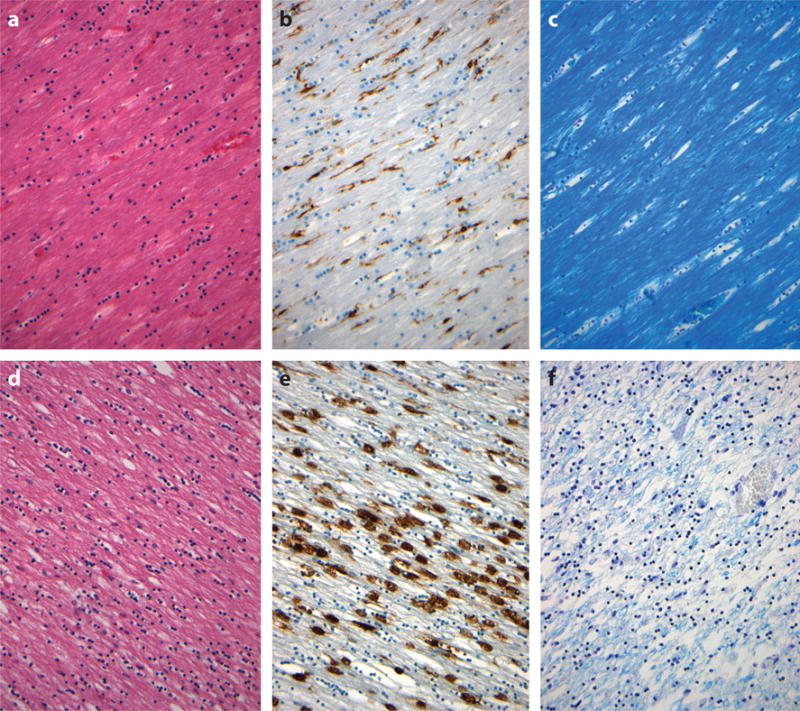
Neuroinflammation and degradation of white matter in the corpus callosum occurring with survival following traumatic brain injury (TBI). Material from approximately 30% of survivors at 1 year or more after a single, moderate or severe TBI shows evidence of white matter degradation as rarefaction in staining with Hematoxylin and eosin (d) when compared with material from uninjured controls (a). Accompanying this is evidence of ongoing neuroinflammation in the form of numerous amoeboid, activated microglia (e) in contrast to the quiescent, ramified microglia in uninjured controls (b). Staining for myelin with Luxol fast blue–Cresyl violet demonstrates an associated loss of myelin with evidence of continued myelin degradation after trauma (f). (a–c) Sections from the corpus callosum of a 38-year-old male non-TBI control whose cause of death was sudden, unexpected death in epilepsy. (d–f) Sections from the corpus callosum of a 56-year-old male with 3-year survival after a single, severe TBI. (b) and (e) stained for HLA-DP, -DQ, and -DR using antibody CR3/43 (Dako, Agilent Technologies) to reveal activated microglia.
In contrast to the limited, although detailed, studies of neuroinflammation following an sTBI, there has been comparatively little characterization of the extent and distribution of the neuroinflammatory response following rTBI, although neuroinflammation has been mentioned in several reports (47, 52, 56, 116). Further, numerous experimental models of rTBI have demonstrated persistent microglial activation after repetitive injury that has been associated with ongoing, degenerative pathology and evidence of cognitive deficit at up to 12 months postinjury (117, 118).
Although neuroinflammation after TBI has been increasingly documented, it remains unclear whether this neuroinflammation is primary—representing dysregulated inflammation driving late TBI pathology—or secondary—responding to ongoing post-TBI pathologies. However, data from experimental models have suggested that the immediate microglial inflammatory response arising following TBI is of a mixed reparative M2a (alternative) and neurotoxic M1 (classic) phenotype that might then be switched to an M1-predominant phenotype during the chronic phase (114, 119, 120). Whether the microglial responses in human TBI, in particular the phenotypic profiles in early-versus-late survivors, mirror observations from animal studies has yet to be confirmed.
Neuronal Loss
In studies on tissue acquired at autopsy from patients who had sustained an sTBI, a degree of acute-phase neuronal loss has been documented (121, 122). Given the multiple acute pathologies of TBI, including diffuse axonal injury and the acute-phase neuroinflammatory response, this neuronal loss is to be anticipated. However, there is evidence suggesting that this neuronal loss might continue beyond the acute phase in a proportion of patients, with active degeneration of neurons via programmed cell death being observed in material from patients in a persistent vegetative state up to 1 year following a single, severe TBI, and this is accompanied by evidence of a continued reduction in hippocampal and thalamic neuronal densities (122–124).
Neuronal loss has been described in the majority of studies of boxers and nonboxer athletes with CTE, involving the neocortex, substantia nigra, locus coeruleus, and cerebellum (12–16, 46, 48, 49, 51, 55, 57, 58, 68, 125, 126). Varying degrees of cell loss have been described, from widespread and diffuse to more patchy, or selective, changes. Of note, given reports of motor symptoms in CTE (in particular, Parkinsonian symptoms), although there have been reports of neuronal loss from the substantia nigra, this is typically accompanied by nigral NFTs, but synuclein-associated pathologies, including Lewy bodies, have not been a feature (13, 14, 46, 49, 52, 59). In contrast to rTBI, little is known about the vulnerability of the substantia nigra with survival from an sTBI.
White Matter Degradation and Continued Axonal Degeneration
As noted, a degree of DAI is regarded as a constant in all severities of TBI and is associated with cytoskeletal disruption and interruption of axonal transport, thus creating a milieu favoring rapid Aβ genesis in the acute setting (83–85, 88, 90, 91, 93). Since the earliest accounts of axonal injury after TBI, there has been an awareness that these white matter pathologies might continue beyond the immediate acute phase following injury (Figure 6). Specifically, using the Marchi technique, Strich (127) demonstrated evidence of ongoing myelin degeneration at autopsy up to 15 months after survival from a single, severe TBI.
As for progressive axonopathy after an sTBI, a swine model of DAI demonstrated that axons continue to degenerate months after injury (91), an observation subsequently supported in studies of patients surviving an sTBI, which show evidence of ongoing axonal pathology in a proportion of survivors, even decades after injury (23, 92) (Figure 1d). This progressive axonal pathology is characterized as morphologically abnormal APP-immunoreactive swollen axonal profiles in multiple regions (Figure 4). These profiles have been observed in isolation and as small clusters of somewhat granular axonal bulbs, indicative of disconnected axon terminals and providing evidence of ongoing axonal degeneration occurring with long-term survival. In addition, in long-term survival in humans, this axonal loss in the corpus callosum was accompanied by a remarkable thinning of the white matter and diminished or abnormal myelin staining, which coincided with amoeboid microglia that contained myelin breakdown products, in keeping with ongoing myelin phagocytosis (23).
In contrast to current experience in sTBI, accounts of white matter pathology after rTBI are limited. While early reports did not seek specific evidence of axonal pathology, subsequent reports have described morphologically abnormal axonal profiles accumulating transport-interrupted proteins, including phosphorylated tau (19, 39), although these reports have not characterized the extent and distribution, or made reference to controls or agonal state. As such, although these limited observations are noteworthy, the formal characterization of axonal pathology in rTBI remains to be pursued. Similarly, somewhat limited reports have described mixed observations regarding white matter pathology after rTBI, from foci of degeneration or rarefaction (12, 47, 56, 125), with reduced or patchy myelin staining (17, 47, 48, 52), to reports suggesting no evidence of demyelination (49).
GENETICS
Despite axonal pathology and linked Aβ production appearing ubiquitous after TBI, only 30% of patients develop Aβ plaques in the acute-phase following an sTBI (24, 25, 94), with a similar proportion of long-term sTBI survivors (21) and a majority of rTBI survivors (22, 95) depositing plaques. Among the possible explanations for this variable response to injury, there may potentially be a genetic predisposition whereby some individuals cannot sufficiently clear Aβ during the acute phase, and others may slowly lose the battle to keep Aβ deposition at bay over a period of years after TBI.
APOE and Traumatic Brain Injury
Possession of the ε4 allele of the APOE gene has long been associated with an increased risk of AD (128, 129). Specifically, possession of the ε4 allele is associated with the incidence of Aβ pathologies in AD and also with age-related Aβ deposition in cognitively intact individuals (130). In TBI there is considerable evidence supporting an association between the APOE genotype and prognosis following an sTBI, with ε4 carriers demonstrating worse immediate and 6-month outcomes than non-ε4 carriers in a variety of clinical indices (131–140). Further, with a longer duration of survival, the influence of the ε4 allele appears to be synergistic with TBI, with individuals possessing this allele at greater risk of dementia following injury than non-ε4 carriers (30, 32, 34, 141–143). This contrasts with longitudinal studies on outcome in late survivors of an sTBI which, while confirming deterioration in measures of functional and cognitive outcomes over time, have reported no particular association with the APOE genotype; although the age at follow-up in these studies might be argued to be relatively young in comparison with studies reporting an association between TBI and dementia (47).
Regarding mild TBI, thus far there has been no convincing evidence of an association between the APOE genotype and injury risk (144, 145). However, it should be noted that the number of injury episodes in these studies has been small. In contrast, studies of longer-term outcomes after rTBI have reported that possession of the ε4 allele is associated with greater neurological impairment in high-exposure boxers (that is, those with more than 12 professional bouts) (146) and poorer cognitive performance in older, professional American footballers (147). However, once again, the number of participants in these studies was small.
In relation to neuropathology, following an sTBI, individuals possessing the ε4 allele have demonstrated more severe contusional injury and show a trend toward more marked, diffuse, hypoxic brain injury (138). In addition, there is an increased incidence of amyloid plaque pathology in autopsy material from patients dying during the acute-phase after an sTBI compared with those who do not carry the ε4 allele (148). However, the association between APOE and late neuropathologies after an sTBI remains unknown. Although there are no data on the influence of the APOE genotype on acute pathology after exposure to rTBI, studies of its influence on late pathology after exposure to rTBI are informative. Original reports suggested that there is no particular association between the APOE genotype and CTE pathology as defined by characteristic tau deposition (11, 39). However, in an appraisal of a cohort of neuropathologically confirmed CTE cases, specifically those with Aβ pathologies, McKee and colleagues (95) reported a clear association between possession of the APOE ε4 allele, Aβ pathologies, and more advanced disease.
Intriguingly, although the pathognomonic lesion of CTE has been proposed to be tau, and considerable attention in the literature has focused on CTE as a tauopathy, the limited clinical reports of outcomes after rTBI and of dementia after an sTBI suggest that possession of the APOE ε4 allele is associated with an increased risk of poor outcome. Further, in emerging pathology studies in CTE after rTBI, the APOE ε4 allele appears to be more closely associated with Aβ pathologies than with tau. Together these observations suggest that clinically relevant CTE might extend beyond being a pure tauopathy, with Aβ pathologies undoubtedly meriting closer attention.
Neprilysin and Traumatic Brain Injury
Neprilysin is recognized as the principal Aβ-degrading enzyme, with microsatellite polymorphism in the promoter region of the neprilysin gene (NEP) linked to amyloid pathologies, including AD and cerebral amyloid angiopathy (149–151). In TBI, a relationship between this GT repeat polymorphism and Aβ plaque pathology has been demonstrated at up to 1 month following an sTBI (152). Specifically, individuals carrying a longer repeat (greater than 41 total GT repeats) were at considerably greater risk of Aβ plaque pathology after TBI than those with shorter repeat sequences. These findings implicate neprilysin as having an important role in post-traumatic Aβ metabolism and plaque formation. However, although an increase in intra-axonal neprilysin immunoreactivity has been observed in association with Aβ accumulation at up to 3 years following an sTBI (92), the longer-term association between NEP and neurodegenerative pathologies after TBI remains unknown. To date, no studies of neprilysin and rTBI outcome or pathology have been conducted.
CONCLUSIONS
Almost a century on from the first clinical descriptions of the punch-drunk syndrome (6), several decades after the first comprehensive descriptions of an associated neuropathology (52), and with growing recognition during the past decade of the range of circumstances in which the clinical syndrome and pathology arise, there can remain no doubt that in some individuals exposure to brain injury is associated with later development of a distinct neurodegenerative pathology. Thus far, although remarkably few cases have come to attention, this neuropathology has been described only in autopsy material from individuals exposed to TBI, either as rTBI or as an sTBI. Regarding the former, exposure to rTBI is most often described in relation to sports-associated mild TBI, with the list of sports in which autopsy-proven CTE has been documented continuing to grow. As such, it is clear that it is exposure to TBI that is associated with the development of CTE, and not the sport or environment in which TBI is encountered.
The association between exposure to TBI and an increased risk of dementia is widely acknowledged. However, over the decades there has evolved an unsubstantiated view that dementia associated with exposure to rTBI represents CTE and is clinically and pathologically distinct from dementia that arises following exposure to an sTBI, traditionally regarded as AD (44, 45). Although studies in those exposed to rTBI have distinguished their cohort from the outset as former boxers, American footballers, or other athletes, and often are supported by neuropathological confirmation at autopsy, studies of dementia following an sTBI are typically retrospective clinical reports with no autopsy confirmation of diagnosis. As a consequence, the diagnosis of AD in these studies on dementia associated with an sTBI is subject to the same errors in reporting as dementia diagnosed without autopsy validation. Of note, in cases where careful clinical review of patients with AD has been performed, the clinical syndrome in those exposed to an sTBI has been reported as distinct from that in patients with no exposure to TBI (42, 43). Further, although hampered by small case numbers, varied methods, and the limited number of cases reported, when suitable data are available, the pathologies of survival from sTBI and rTBI appear comparable, and distinct from descriptions of existing neurodegenerative pathologies, including AD (Table 1).
Table 1.
Clinicopathological characteristics of chronic traumatic encephalopathy in patients exposed to repetitive, mild traumatic brain injury (rTBI) or surviving 1 year or more after a single, moderate or severe TBI (sTBI)
| Characteristics | rTBI | sTBI |
|---|---|---|
| Clinical history | ||
| Personality change | + | + |
| Impulsivity or disinhibition | + | + |
| Depression, suicidality | + | + |
| Cognitive impairment | + | + |
| Gait disturbance | + | + |
| Parkinsonism | + | ? |
| Macroscopic appearances | ||
| Cerebral atrophy | + | (+) |
| Cerebellar atrophy | + | ? |
| Ventricular enlargement | + | + |
| Thinning of the corpus callosum | + | + |
| Abnormalities of the septum pellucidum, including cavum septum | + | − |
| Evidence of previous TBI | +/− | +/− |
| Microscopic appearances | ||
| Tau | ||
| Perivascular accentuation | + | + |
| Neurofibrillary tangles | + | + |
| Glial profiles | + | + |
| Grain-like profiles | + | + |
| Sulcal depths | + | + |
| Superficial neocortical layers | + | + |
| Patchy distribution | + | ? |
| Hippocampal CA2 involvement | + | ? |
| Hypothalamic region | + | ? |
| Brainstem | + | ? |
| Axonal | (+) | (+) |
| Amyloid β | ||
| Diffuse plaques | + | + |
| Neuritic plaques | + | + |
| TDP-43 | ||
| Hyperphosphorylated inclusions | + | − |
| Translocated from nucleus to cytoplasm | − | + |
| Neuroinflammation | (+) | + |
| Axonal degeneration | (+) | + |
| Neuronal loss | + | + |
| Myelin degradation | ? | + |
+, present; −, absent; +/−, may be either present or absent; (+), reported present but not formally assessed; ?, unknown.
Regarding this pathology, as more robust assessments emerge, it is becoming clearer that the neuropathology of survival from TBI is complex and multifaceted. An apparent constant is pathologies in tau, with a relatively stereotyped pattern and distribution proposed as sufficiently distinctive as to be pathognomonic. However, although tau pathologies have attracted attention, almost to the exclusion of wider pathologies, the complex of pathologies after TBI extends to include at least pathologies in Aβ and TDP-43, axonal degeneration, white matter degradation, neuronal loss, and persistent neuroinflammation. The significance of this complex of mixed pathologies to the clinical presentations of CTE remains to be established. Further, although there is emerging evidence on the potential contribution of genotype to the development of pathology after TBI, undoubtedly, greater efforts are required to establish risk factors for developing CTE including, but not limited to, the contribution of the severity and number of injuries, genotype, and the influence of comorbid pathologies.
Undoubtedly, to advance understanding of the part that TBI might play in precipitating this distinct, complex, neurodegenerative pathology, and to gain insight into the incidence, risk factors, and risk modifiers, concerted efforts are required to establish collaborative programs of research to document outcomes and, critically, neuropathology in survivors of TBI, regardless of the source of injury. Through these studies insight may be provided to inform strategies for prevention and much-needed robust, in vivo diagnostics. Until then, CTE remains an autopsy-defined diagnosis. However, despite remarkably few cases having been thoroughly evaluated neuropathologically, there can be no question that CTE is intimately associated with exposure to TBI.
SUMMARY POINTS.
Exposure to traumatic brain injury (TBI) is associated with an increased risk of neurodegenerative disease.
Chronic traumatic encephalopathy (CTE), the pathology of neurodegeneration occurring after TBI, is a complex polypathology featuring tau as a constant, with abnormalities in amyloid β and TDP-43, axonal degeneration, neuroinflammation, neuronal loss, and white matter degradation also being described.
It is exposure to TBI per se that is associated with the risk of CTE, rather than the environment or circumstances of exposure.
The clinical syndrome and neuropathology are comparable for survival after a single, moderate or severe TBI (sTBI) and after repetitive, mild TBI. Importantly, there are no neuropathological data to support neurodegeneration after an sTBI in Alzheimer’s disease.
Global initiatives are required to support programs of research in survivors of TBI that are directed at documenting outcomes and, critically, the associated neuropathology.
Acknowledgments
This work was supported by National Institutes of Health grants NS038104 (D.H.S. and W.S.), NS056202, NS092389, and AG038911 (D.H.S.); United States Department of Defense grant PT110785 (D.H.S. and W.S.); a National Health Service Research Scotland Career Researcher Fellowship (W.S.); and the Sackler Institute (J.H.).
Glossary
- TBI
traumatic brain injury
- DP
dementia pugilistica
- sTBI
single, moderate or severe traumatic brain injury
- CTE
chronic traumatic encephalopathy
- rTBI
repetitive, mild traumatic brain injury
- AD
Alzheimer’s disease
- CSP
cavum septum pellucidum
- Aβ
amyloid β
- DAI
diffuse axonal injury
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Coronado VG, McGuire LC, Faul M, Sugerman DE, Pearson WS. Epidemiology and public health issues. In: Zasler ND, Katz D, Zafonte RD, Arciniegas DB, Bullock MR, Kreutzer JS, editors. Brain Injury Medicine: Principles and Practice. 2nd New York: Demos Med; 2012. pp. 84–100. [Google Scholar]
- 2.Coronado VG, McGuire LC, Sarmiento K, Bell J, Lionbarger MR, et al. Trends in traumatic brain injury in the U.S. and the public health response: 1995–2009. J Saf Res. 2012;43:299–307. doi: 10.1016/j.jsr.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Faul M, Xu L, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths. Atlanta: CDC, Natl. Cent. Inj. Prev. Control; 2010. [Google Scholar]
- 4.CDC (Cent. Dis. Control Prev.) Traumatic Brain Injury in the United States: A Report to Congress. Atlanta: CDC; 1999. [Google Scholar]
- 5.Finklestein E, Corso P, Miller T. The Incidence and Economic Burden of Injuries in the United States. New York: Oxford Univ. Press; 2006. [Google Scholar]
- 6.Martland H. Punch drunk. JAMA. 1928;91:1103–7. [Google Scholar]
- 7.Millspaugh J. Dementia pugilistica. U S Navy Med Bull. 1937;35:297–303. [Google Scholar]
- 8.Critchley M. Medical aspects of boxing, particularly from a neurological standpoint. BMJ. 1957;1:357–62. doi: 10.1136/bmj.1.5015.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mawdsley C, Ferguson FR. Neurological disease in boxers. Lancet. 1963;282:795–801. doi: 10.1016/s0140-6736(63)90498-7. [DOI] [PubMed] [Google Scholar]
- 10.Spillane JD. Five boxers. BMJ. 1962;2:1205–10. doi: 10.1136/bmj.2.5314.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. 2011;69:173–83. doi: 10.1227/NEU.0b013e318212bc7b. [DOI] [PubMed] [Google Scholar]
- 12.Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus. 2011;31:E3. doi: 10.3171/2011.9.FOCUS11178. [DOI] [PubMed] [Google Scholar]
- 13.Omalu BI, DeKosky ST, Hamilton RL, Minster RL, Kamboh MI, et al. Chronic traumatic encephalopathy in a National Football League player: part II. Neurosurgery. 2006;59:1086–93. doi: 10.1227/01.NEU.0000245601.69451.27. [DOI] [PubMed] [Google Scholar]
- 14.Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–34. doi: 10.1227/01.neu.0000163407.92769.ed. [DOI] [PubMed] [Google Scholar]
- 15.Omalu BI, Fitzsimmons RP, Hammers J, Bailes J. Chronic traumatic encephalopathy in a professional American wrestler. J Forensic Nurs. 2010;6:130–36. doi: 10.1111/j.1939-3938.2010.01078.x. [DOI] [PubMed] [Google Scholar]
- 16.Omalu BI, Hamilton RL, Kamboh MI, DeKosky ST, Bailes J. Chronic traumatic encephalopathy (CTE) in a National Football League player: case report and emerging medicolegal practice questions. J Forensic Nurs. 2010;6:40–46. doi: 10.1111/j.1939-3938.2009.01064.x. [DOI] [PubMed] [Google Scholar]
- 17.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–35. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–29. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra60. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKee AC, Stein TD, Nowinski CJ, Stern RA, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–49. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol. 2013;9:211–21. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts GW, Gentleman SM, Lynch A, Graham DI. β A4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–23. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- 25.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57:419–25. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Molgaard CA, Stanford EP, Morton DJ, Ryden LA, Schubert KR, Golbeck AL. Epidemiology of head trauma and neurocognitive impairment in a multi-ethnic population. Neuroepidemiology. 1990;9:233–42. doi: 10.1159/000110778. [DOI] [PubMed] [Google Scholar]
- 27.Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer’s disease. Neurology. 1985;35:264–67. doi: 10.1212/wnl.35.2.264. [DOI] [PubMed] [Google Scholar]
- 28.Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. Int J Epidemiol. 1991;20(Suppl. 2):S28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- 29.Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, et al. The association between head trauma and Alzheimer’s disease. Am J Epidemiol. 1990;131:491–501. doi: 10.1093/oxfordjournals.aje.a115523. [DOI] [PubMed] [Google Scholar]
- 30.O’Meara ES, Kukull WA, Sheppard L, Bowen JD, McCormick WC, et al. Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. Am J Epidemiol. 1997;146:373–84. doi: 10.1093/oxfordjournals.aje.a009290. [DOI] [PubMed] [Google Scholar]
- 31.Salib E, Hillier V. Head injury and the risk of Alzheimer’s disease: a case control study. Int J Geriatr Psychiatry. 1997;12:363–68. doi: 10.1002/(sici)1099-1166(199703)12:3<363::aid-gps515>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 32.Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–23. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 33.Schofield PW, Tang M, Marder K, Bell K, Dooneief G, et al. Alzheimer’s disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry. 1997;62:119–24. doi: 10.1136/jnnp.62.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–66. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 35.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74:857–62. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shively S, Scher AI, Perl DP, Diaz-Arrastia R. Dementia resulting from traumatic brain injury: What is the pathology? Arch Neurol. 2012;69:1245–51. doi: 10.1001/archneurol.2011.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts G. Brain Damage in Boxers: A Study of the Prevalence of Traumatic Encephalopathy Among Ex-Professional Boxers. London: Pitman; 1969. [Google Scholar]
- 38.Deleted in proof
- 39.McKee AC, Stein TD, Nowinski CJ, Stern RA, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stewart W, McNamara PH, Lawlor B, Hutchinson S, Farrell M. Chronic traumatic encephalopathy: a potential late and under recognized consequence of rugby union? QJM. 2016;109:11–15. doi: 10.1093/qjmed/hcv070. [DOI] [PubMed] [Google Scholar]
- 41.McKee AC, Daneshvar DH, Alvarez VE, Stein TD. The neuropathology of sport. Acta Neuropathol. 2014;127:29–51. doi: 10.1007/s00401-013-1230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dams-O’Connor K, Spielman L, Hammond FM, Sayed N, Culver C, Diaz-Arrastia R. An exploration of clinical dementia phenotypes among individuals with and without traumatic brain injury. NeuroRehabilitation. 2013;32:199–209. doi: 10.3233/NRE-130838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sayed N, Culver C, Dams-O’Connor K, Hammond F, Diaz-Arrastia R. Clinical phenotype of dementia after traumatic brain injury. J Neurotrauma. 2013;30:1117–22. doi: 10.1089/neu.2012.2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeKosky ST, Blennow K, Ikonomovic MD, Gandy S. Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nat Rev Neurol. 2013;9:192–200. doi: 10.1038/nrneurol.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeKosky ST, Ikonomovic MD, Gandy S. Traumatic brain injury—football, warfare, and long-term effects. N Engl J Med. 2010;363:1293–96. doi: 10.1056/NEJMp1007051. [DOI] [PubMed] [Google Scholar]
- 46.Brandenburg W, Hallervorden J. Dementia pugilistica with anatomical findings. Virchows Arch. 1954;325:680–709. doi: 10.1007/BF00955101. In German. [DOI] [PubMed] [Google Scholar]
- 47.Payne EE. Brains of boxers. Neurochirurgia. 1968;11:173–88. doi: 10.1055/s-0028-1095326. [DOI] [PubMed] [Google Scholar]
- 48.Neubuerger KT, Sinton DW, Denst J. Cerebral atrophy associated with boxing. AMA Arch Neurol Psychiatry. 1959;81:403–8. doi: 10.1001/archneurpsyc.1959.02340160001001. [DOI] [PubMed] [Google Scholar]
- 49.Constantinidis J, Tissot R. Generalized Alzheimer’s neurofibrillary lesions without senile plaques. (Presentation of one anatomo-clinical case) Schweiz Arch Neurol Neurochir Psychiatr. 1967;100:117–30. In French. [PubMed] [Google Scholar]
- 50.Ferguson FR, Mawdsley C. Chronic Encephalopathy in Boxers: 8th International Congress of Neurology, Vienna. Vienna: Wiener Med. Akad.; 1965. [Google Scholar]
- 51.Grahmann H, Ule G. Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica & traumatic encephalopathy of boxers) Psychiatr Neurol. 1957;134:261–83. In German. [PubMed] [Google Scholar]
- 52.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 53.Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–78. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jordan BD, Kanik AB, Horwich MS, Sweeney D, Relkin NR, et al. Apolipoprotein E ε4 and fatal cerebral amyloid angiopathy associated with dementia pugilistica. Ann Neurol. 1995;38:698–99. doi: 10.1002/ana.410380429. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001;101:518–24. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- 56.Saing T, Dick M, Nelson PT, Kim RC, Cribbs DH, Head E. Frontal cortex neuropathology in dementia pugilistica. J Neurotrauma. 2012;29:1054–70. doi: 10.1089/neu.2011.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nowak LA, Smith GG, Reyes PF. Dementia in a retired world boxing champion: case report and literature review. Clin Neuropathol. 2009;28:275–80. [PubMed] [Google Scholar]
- 58.Williams DJ, Tannenberg AE. Dementia pugilistica in an alcoholic achondroplastic dwarf. Pathology. 1996;28:102–4. doi: 10.1080/00313029600169653. [DOI] [PubMed] [Google Scholar]
- 59.Drachman D, Newall K. Case 12-1999—a 67-year-old man with three years of dementia. N Engl J Med. 1999;340:1269–77. doi: 10.1056/NEJM199904223401609. [DOI] [PubMed] [Google Scholar]
- 60.Areza-Fegyveres R, Rosemberg S, Castro RM, Porto CS, Bahia VS, et al. Dementia pugilistica with clinical features of Alzheimer’s disease. Arq Neuropsiquiatr. 2007;65:830–33. doi: 10.1590/s0004-282x2007000500019. [DOI] [PubMed] [Google Scholar]
- 61.Farbota KD, Sodhi A, Bendlin BB, McLaren DG, Xu G, et al. Longitudinal volumetric changes following traumatic brain injury: a tensor-based morphometry study. J Int Neuropsychol Soc. 2012;18:1006–18. doi: 10.1017/S1355617712000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ross DE, Ochs AL, Seabaugh JM, Demark MF, Shrader CR, et al. Progressive brain atrophy in patients with chronic neuropsychiatric symptoms after mild traumatic brain injury: a preliminary study. Brain Inj. 2012;26:1500–9. doi: 10.3109/02699052.2012.694570. [DOI] [PubMed] [Google Scholar]
- 63.Ross DE. Review of longitudinal studies of MRI brain volumetry in patients with traumatic brain injury. Brain Inj. 2011;25:1271–78. doi: 10.3109/02699052.2011.624568. [DOI] [PubMed] [Google Scholar]
- 64.Tomaiuolo F, Carlesimo GA, Di Paola M, Petrides M, Fera F, et al. Gross morphology and morphometric sequelae in the hippocampus, fornix, and corpus callosum of patients with severe nonmissile traumatic brain injury without macroscopically detectable lesions: a T1 weighted MRI study. J Neurol Neurosurg Psychiatry. 2004;75:1314–22. doi: 10.1136/jnnp.2003.017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Warner MA, Marquez de la Plata C, Spence J, Wang JY, Harper C, et al. Assessing spatial relationships between axonal integrity, regional brain volumes, and neuropsychological outcomes after traumatic axonal injury. J Neurotrauma. 2010;27:2121–30. doi: 10.1089/neu.2010.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allsop D, Haga S, Bruton C, Ishii T, Roberts GW. Neurofibrillary tangles in some cases of dementia pugilistica share antigens with amyloid β-protein of Alzheimer’s disease. Am J Pathol. 1990;136:255–60. [PMC free article] [PubMed] [Google Scholar]
- 67.Geddes JF, Vowles GH, Nicoll JA, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–78. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 68.Hof PR, Bouras C, Buee L, Delacourte A, Perl DP, Morrison JH. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol. 1992;85:23–30. doi: 10.1007/BF00304630. [DOI] [PubMed] [Google Scholar]
- 69.Hof PR, Knabe R, Bovier P, Bouras C. Neuropathological observations in a case of autism presenting with self-injury behavior. Acta Neuropathol. 1991;82:321–26. doi: 10.1007/BF00308819. [DOI] [PubMed] [Google Scholar]
- 70.Bogdanoff B, Natter HM. Incidence of cavum septum pellucidum in adults: a sign of boxer’s encephalopathy. Neurology. 1989;39:991–92. doi: 10.1212/wnl.39.7.991. [DOI] [PubMed] [Google Scholar]
- 71.Bodensteiner JB, Schaefer GB. Dementia pugilistica and cavum septi pellucidi: born to box? Sports Med. 1997;24:361–65. doi: 10.2165/00007256-199724060-00002. [DOI] [PubMed] [Google Scholar]
- 72.Macpherson P, Teasdale E. CT demonstration of a 5th ventricle—a finding to KO boxers? Neuroradiology. 1988;30:506–10. doi: 10.1007/BF00339691. [DOI] [PubMed] [Google Scholar]
- 73.Schwidde JT. Incidence of cavum septi pellucidi and cavum Vergae in 1,032 human brains. AMA Arch Neurol Psychiatry. 1952;67:625–32. doi: 10.1001/archneurpsyc.1952.02320170043006. [DOI] [PubMed] [Google Scholar]
- 74.Casson IR, Siegel O, Sham R, Campbell EA, Tarlau M, DiDomenico A. Brain damage in modern boxers. JAMA. 1984;251:2663–67. [PubMed] [Google Scholar]
- 75.Jordan BD, Jahre C, Hauser WA, Zimmerman RD, Zarrelli M, et al. CT of 338 active professional boxers. Radiology. 1992;185:509–12. doi: 10.1148/radiology.185.2.1410364. [DOI] [PubMed] [Google Scholar]
- 76.Roberts GW, Whitwell HL, Acland PR, Bruton CJ. Dementia in a punch-drunk wife. Lancet. 1990;335:918–19. doi: 10.1016/0140-6736(90)90520-f. [DOI] [PubMed] [Google Scholar]
- 77.Johnson VE, Stewart W, Trojanowski JQ, Smith DH. Acute and chronically increased immunore-activity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol. 2011;122:715–26. doi: 10.1007/s00401-011-0909-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tang-Schomer MD, Patel AR, Baas PW, Smith DH. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 2010;24:1401–10. doi: 10.1096/fj.09-142844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol. 2012;233:364–72. doi: 10.1016/j.expneurol.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahmadzadeh H, Smith DH, Shenoy VB. Viscoelasticity of tau proteins leads to strain rate-dependent breaking of microtubules during axonal stretch injury: predictions from a mathematical model. Biophys J. 2014;106:1123–33. doi: 10.1016/j.bpj.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith C, Graham DI, Murray LS, Nicoll JA. Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. 2003;29:496–502. doi: 10.1046/j.1365-2990.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- 82.Geddes JF, Vowles GH, Robinson SF, Sutcliffe JC. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol. 1996;22:12–16. [PubMed] [Google Scholar]
- 83.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–63. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- 85.Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 86.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. β-amyloid precursor protein (βAPP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–44. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- 87.Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for β-amyloid precursor protein. Acta Neuropathol. 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- 88.Smith DH, Chen XH, Iwata A, Graham DI. Amyloid β accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003;98:1072–77. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- 89.Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. Neuromolecular Med. 2003;4:59–72. doi: 10.1385/NMM:4:1-2:59. [DOI] [PubMed] [Google Scholar]
- 90.Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, et al. Accumulation of amyloid β and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–92. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 91.Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-β, β-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–71. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid βplaques despite persistent accumulation of amyloid β in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19:214–23. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-β pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11:361–70. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 95.Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, et al. β-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. 2015;130:21–34. doi: 10.1007/s00401-015-1435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–33. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 97.Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–20. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol. 2009;256:1205–14. doi: 10.1007/s00415-009-5069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64:1388–94. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- 100.Sato T, Takeuchi S, Saito A, Ding W, Bamba H, et al. Axonal ligation induces transient redistribution of TDP-43 in brainstem motor neurons. Neuroscience. 2009;164:1565–78. doi: 10.1016/j.neuroscience.2009.09.050. [DOI] [PubMed] [Google Scholar]
- 101.Moisse K, Mepham J, Volkening K, Welch I, Hill T, Strong MJ. Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL−/− mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009;1296:176–86. doi: 10.1016/j.brainres.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 102.Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009;1249:202–11. doi: 10.1016/j.brainres.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 103.King A, Sweeney F, Bodi I, Troakes C, Maekawa S, Al-Sarraj S. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer’s disease. Neuropathology. 2010;30:408–19. doi: 10.1111/j.1440-1789.2009.01085.x. [DOI] [PubMed] [Google Scholar]
- 104.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 105.Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, McCluskey L, et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLOS ONE. 2012;7:e39216. doi: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brettschneider J, Libon DJ, Toledo JB, Xie SX, McCluskey L, et al. Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol. 2012;123:395–407. doi: 10.1007/s00401-011-0932-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, et al. Blocking TGF-β-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–87. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, et al. Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLOS ONE. 2013;8:e59586. doi: 10.1371/journal.pone.0059586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, et al. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–92. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 111.Juengst SB, Kumar RG, Arenth PM, Wagner AK. Exploratory associations with tumor necrosis factor-α, disinhibition and suicidal endorsement after traumatic brain injury. Brain Behav Immun. 2014;41:134–43. doi: 10.1016/j.bbi.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 112.Lindqvist D, Wolkowitz OM, Mellon S, Yehuda R, Flory JD, et al. Proinflammatory milieu in combat-related PTSD is independent of depression and early life stress. Brain Behav Immun. 2014;42:81–88. doi: 10.1016/j.bbi.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 113.Kumar RG, Boles JA, Wagner AK. Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months postinjury. J Head Trauma Rehabil. 2015;30:369–81. doi: 10.1097/HTR.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 114.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–77. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 116.Adams CW, Bruton CJ. The cerebral vasculature in dementia pugilistica. J Neurol Neurosurg Psychiatry. 1989;52:600–4. doi: 10.1136/jnnp.52.5.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75:241–54. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- 118.Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int. 2014;5:184. doi: 10.4103/2152-7806.147566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shaw K, MacKinnon MA, Raghupathi R, Saatman KE, McLntosh TK, Graham DI. TUNEL-positive staining in white and grey matter after fatal head injury in man. Clin Neuropathol. 2001;20:106–12. [PubMed] [Google Scholar]
- 122.Maxwell WL, Dhillon K, Harper L, Espin J, MacIntosh TK, et al. There is differential loss of pyramidal cells from the human hippocampus with survival after blunt head injury. J Neuropathol Exp Neurol. 2003;62:272–79. doi: 10.1093/jnen/62.3.272. [DOI] [PubMed] [Google Scholar]
- 123.Williams S, Raghupathi R, MacKinnon MA, McIntosh TK, Saatman KE, Graham DI. In situ DNA fragmentation occurs in white matter up to 12 months after head injury in man. Acta Neuropathol. 2001;102:581–90. doi: 10.1007/s004010100410. [DOI] [PubMed] [Google Scholar]
- 124.Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol. 2006;65:478–88. doi: 10.1097/01.jnen.0000229241.28619.75. [DOI] [PubMed] [Google Scholar]
- 125.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 126.Mann DM, Yates PO, Hawkes J. The pathology of the human locus ceruleus. Clin Neuropathol. 1983;2:1–7. [PubMed] [Google Scholar]
- 127.Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19:163–85. doi: 10.1136/jnnp.19.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–23. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 129.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, et al. Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 130.Jack CR, Jr, Wiste HJ, Weigand SD, Knopman DS, Vemuri P, et al. Age, sex, and APOE ε4 effects on memory, brain structure, and β-amyloid across the adult life span. JAMA Neurol. 2015;72:511–19. doi: 10.1001/jamaneurol.2014.4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Teasdale GM, Nicoll JA, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. 1997;350:1069–71. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- 132.Sorbi S, Nacmias B, Piacentini S, Repice A, Latorraca S, et al. ApoE as a prognostic factor for post-traumatic coma. Nat Med. 1995;1:852. doi: 10.1038/nm0995-852. [DOI] [PubMed] [Google Scholar]
- 133.Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, et al. Apolipoprotein E-ε4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–48. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 134.Liberman JN, Stewart WF, Wesnes K, Troncoso J. Apolipoprotein E ε4 and short-term recovery from predominantly mild brain injury. Neurology. 2002;58:1038–44. doi: 10.1212/wnl.58.7.1038. [DOI] [PubMed] [Google Scholar]
- 135.Sundstrom A, Marklund P, Nilsson LG, Cruts M, Adolfsson R, et al. APOE influences on neuropsychological function after mild head injury: within-person comparisons. Neurology. 2004;62:1963–66. doi: 10.1212/01.wnl.0000129268.83927.a8. [DOI] [PubMed] [Google Scholar]
- 136.Lichtman SW, Seliger G, Tycko B, Marder K. Apolipoprotein E and functional recovery from brain injury following postacute rehabilitation. Neurology. 2000;55:1536–39. doi: 10.1212/wnl.55.10.1536. [DOI] [PubMed] [Google Scholar]
- 137.Liaquat I, Dunn LT, Nicoll JA, Teasdale GM, Norrie JD. Effect of apolipoprotein E genotype on hematoma volume after trauma. J Neurosurg. 2002;96:90–96. doi: 10.3171/jns.2002.96.1.0090. [DOI] [PubMed] [Google Scholar]
- 138.Smith C, Graham DI, Murray LS, Stewart J, Nicoll JA. Association of APOE e4 and cerebrovascular pathology in traumatic brain injury. J Neurol Neurosurg Psychiatry. 2006;77:363–66. doi: 10.1136/jnnp.2005.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Diaz-Arrastia R, Gong Y, Fair S, Scott KD, Garcia MC, et al. Increased risk of late posttraumatic seizures associated with inheritance of APOE ε4 allele. Arch Neurol. 2003;60:818–22. doi: 10.1001/archneur.60.6.818. [DOI] [PubMed] [Google Scholar]
- 140.Teasdale GM, Murray GD, Nicoll JA. The association between APOE ε4, age and outcome after head injury: a prospective cohort study. Brain. 2005;128:2556–61. doi: 10.1093/brain/awh595. [DOI] [PubMed] [Google Scholar]
- 141.Mayeux R, Ottman R, Maestre G, Ngai C, Tang MX, et al. Synergistic effects of traumatic head injury and apolipoprotein-ε4 in patients with Alzheimer’s disease. Neurology. 1995;45:555–57. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 142.Katzman R, Aronson M, Fuld P, Kawas C, Brown T, et al. Development of dementing illnesses in an 80-year-old volunteer cohort. Ann Neurol. 1989;25:317–24. doi: 10.1002/ana.410250402. [DOI] [PubMed] [Google Scholar]
- 143.Mauri M, Sinforiani E, Bono G, Cittadella R, Quattrone A, et al. Interaction between apolipoprotein ε4 and traumatic brain injury in patients with Alzheimer’s disease and mild cognitive impairment. Funct Neurol. 2006;21:223–28. [PubMed] [Google Scholar]
- 144.Kristman VL, Tator CH, Kreiger N, Richards D, Mainwaring L, et al. Does the apolipoprotein ε4 allele predispose varsity athletes to concussion? A prospective cohort study. Clin J Sport Med. 2008;18:322–28. doi: 10.1097/JSM.0b013e31817e6f3e. [DOI] [PubMed] [Google Scholar]
- 145.Tierney RT, Mansell JL, Higgins M, McDevitt JK, Toone N, et al. Apolipoprotein E genotype and concussion in college athletes. Clin J Sport Med. 2010;20:464–68. doi: 10.1097/JSM.0b013e3181fc0a81. [DOI] [PubMed] [Google Scholar]
- 146.Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoprotein E ε4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278:136–40. [PubMed] [Google Scholar]
- 147.Kutner KC, Erlanger DM, Tsai J, Jordan B, Relkin NR. Lower cognitive performance of older football players possessing apolipoprotein E ε4. Neurosurgery. 2000;47:651–58. doi: 10.1097/00006123-200009000-00026. [DOI] [PubMed] [Google Scholar]
- 148.Nicoll JA, Roberts GW, Graham DI. Apolipoprotein E ε4 allele is associated with deposition of amyloid β-protein following head injury. Nat Med. 1995;1:135–37. doi: 10.1038/nm0295-135. [DOI] [PubMed] [Google Scholar]
- 149.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–50. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 150.Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. BioEssays. 2001;23:261–69. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 151.Yasojima K, Akiyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett. 2001;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- 152.Johnson VE, Stewart W, Stewart JE, Graham DI, Praestgaard AH, Smith DH. A neprilysin polymorphism and amyloid-β plaques following traumatic brain injury. J Neurotrauma. 2009;26:1197–202. doi: 10.1089/neu.2008.0843. [DOI] [PMC free article] [PubMed] [Google Scholar]