

Abstract
Loss of function mutations in Kelch Like ECH Associated Protein 1 (KEAP1) or gain-of-function mutations in nuclear factor erythroid 2-related factor 2 (NRF2) are common in non-small cell lung cancer (NSCLC) and is associated with therapeutic resistance. To discover novel NRF2 inhibitors for targeted therapy, we conducted a quantitative high-throughput screen using a diverse set of ~400,000 small molecules (Molecular Libraries Small Molecule Repository Library, MLSMR) at the National Center for Advancing Translational Sciences. We identified ML385 as a probe molecule that binds to NRF2 and inhibits its downstream target gene expression. Specifically, ML385 binds to the Neh1, the Cap ‘N’ Collar Basic Leucine Zipper (CNC-bZIP) domain of NRF2, and interferes with the binding of the V-Maf Avian Musculoaponeurotic Fibrosarcoma Oncogene Homolog G (MAFG)-NRF2 protein complex to regulatory DNA binding sequences. In clonogenic assays, when used in combination with platinum-based drugs such as doxorubicin or taxol, ML385 substantially enhances cytotoxicity in NSCLC cells compared to single agents alone. ML385 shows specificity and selectivity for NSCLC cells with KEAP1 mutation leading to gain of NRF2 function. In preclinical models of NSCLC with gain of NRF2 function, ML385 in combination with carboplatin showed significant anti-tumor activity. We demonstrate the discovery and validation of ML385 as a novel and specific NRF2 inhibitor and conclude that targeting NRF2 may represent a promising strategy for the treatment of advanced NSCLC.
Keywords: NRF2, KEAP1, tumor, therapeutic resistance, chemotherapy
Graphical abstract
Introduction
Lung cancer is the leading cause of cancer deaths in the US in both males and females and has a particularly poor prognosis. Survival rates have improved slightly since the 1990’s with 5-year relative survival rates of 49%, 16%, and 2% for patients with local, regional, and distant stage disease, respectively1. The standard treatment for advanced non-small cell lung cancer (NSCLC) typically includes platinum-based doublet chemotherapy. Other drugs used to treat NSCLC in combination with platinum therapy include paclitaxel, docetaxel, vinorelbine, gemcitabine, irinotecan, and pemetrexed2. Unfortunately, the major hurdle in the use of platinum-doublet chemotherapy regimens in patients with NSCLC is the emergence of primary or secondary drug resistance. To date, multiple mechanisms of resistance to platinum-doublet combination therapy are known and often these mechanisms are combinatorial.3
The redox sensitive bZIP transcription factor, nuclear factor erythroid 2-related factor 2 (NRF2), has emerged as the “master regulator” of cell survival through coordinated induction of cytoprotective antioxidant genes, phase-II detoxification enzymes, multidrug transporters, and central metabolic pathways4–6. Kelch-like ECH-associated protein 1 (KEAP1) negatively regulates NRF2 by directly binding and leading to its proteasomal degradation. Due to its cytoprotective functions, the activation of NRF2 signaling has long been a goal for chemoprevention studies. We discovered somatic loss-of-function mutations in KEAP1 leading to constitutive activation of NRF2 signaling in NSCLC, particularly in adenocarcinomas7. Activating mutations in NRF2, which impair its binding to KEAP1, were found in lung squamous cell lung carcinomas8–10. Overall, genome-sequencing studies using NSCLC tumors have revealed frequent mutations and somatic copy number alterations leading to an impaired KEAP1-NRF2 interaction (Supplementary Table 1)8–12. In addition to lung cancer, KEAP1 and NRF2 mutations have been reported in ovarian, esophageal, head and neck, gallbladder, prostate, and renal cancers7, 10, 12–15
Gain of NRF2 function in NSCLC confers resistance against cytotoxic chemotherapy as well as ionizing radiation therapy and accelerates tumor growth. RNA interference (RNAi)– mediated inhibition of NRF2 expression in KEAP1-mutant lung cancer cells suppresses tumor growth and results in increased sensitivity to chemotherapeutic drugs and ionizing radiation–induced cell death in vitro and in vivo10, 13, 16–20. Furthermore, somatic mutations in NRF2 or KEAP1 leading to constitutive NRF2 activation in lung cancers are associated with decreased recurrence-free survival21. A recent study by DiNicola et al (2011) showed that multiple oncogenes (K-RasG12D, B-RafV619E, and MycERT2) upregulate NRF2 signaling and NRF2 is essential for oncogenic transformation and senescence evasion22. In the last several years, data from a number of preclinical and clinical studies have established NRF2 as a novel regulator of therapeutic resistance and tumorigenesis and have provided a compelling rationale to explore the inhibition of NRF2 as a novel therapeutic approach in NSCLC and other highly recalcitrant tumors21, 23.
To discover novel, specific, first-in-class small molecule NRF2 inhibitors with the aim of reducing therapeutic resistance in lung cancer, we conducted a quantitative high-throughput screen (qHTS) of the Molecular Libraries Small Molecule Repository (MLSMR) and identified a thiazole-indoline compound series. Medicinal chemistry optimization led to ML385, a novel inhibitor of NRF2 signaling, in NSCLC cells. ML385 in combination with carboplatin demonstrated specificity, selectivity, and in vivo efficacy in NSCLC models. These promising results provide a strong rationale to further develop and test NRF2 inhibitors and potentially use these inhibitors in clinical trial settings in advanced lung cancer.
Methods
Cell Culture and reagents
A549, H1437, H838, H460, and BEAS2B cells were purchased from American Type Culture Collection (Manassas) and were cultured under recommended conditions.
Generation of NRF2 activity reporter cell lines
A549 NRF2-ARE-Fluc stable cell line–A549 cells were transfected with a firefly luciferase reporter (Fluc) construct driven by a minimal TATA promoter with upstream NRF2-specific antioxidant response element (ARE) enhancer sequence from human NQO1 promoter ARE and clones stably expressing ARE-FLuc7 were screened and validated. The HEK293 CMV-Fluc stable cell line constitutively expresses Fluc under the control of the CMV promoter. The H838 NRF2-ARE-Fluc7 and H1437 NRF2-ARE-Fluc cell lines were prepared and validated similar to that of A549 NRF2-ARE-Fluc stable cell line, and these two cell lines express a firefly luciferase reporter construct driven by a minimal promoter of NRF2-specific ARE.
High-throughput screening of the MLSMR library
We used the following assays to screen the MLSMR library to identify potential NRF2 inhibitors:
Primary assay: multiplexed NRF2 reporter gene and CellTiter-Fluor cell viability assays in A549 cells
Counter assay 1: Biochemical firefly luciferase assay
Counter assay 2: Multiplexed CMV driven luciferase reporter gene and CellTiter-Fluor cell viability assays
Counter assay 3: GR-beta lactamase reporter gene assay
Confirmation assay 1: multiplexed NRF2 reporter gene and CellTiter-Fluor cell viability assays in H838 cells
Confirmation assay 2: multiplexed NRF2 reporter gene and CellTiter-Fluor cell viability assays in H1437 cells
A detailed description of the primary screening assay, counter, and confirmation assays is included in the Supplementary Data Section. The screening data have been deposited into PubChem with AID 504444 (http://pubchem.ncbi.nlm.nih.gov/bioassay/504444).
Fluorescence polarization method
A fluorescence polarization (FP) assay was performed using a fluorescein-labeled ARE DNA duplex as a FP probe. The fluorescein-ARE probe was diluted to the appropriate concentration in phosphate-buffered saline (PBS), while Nrf2/MAFG (1:1) hetero-dimer was prepared by gel filtration using mixed samples of purified proteins. Protein complex was then mixed with the buffer containing serially-diluted chemicals or buffer and incubated at 4°C for 1 h. Then, the fluorescein-labeled ARE was mixed with the protein sample and incubated at 4°C for another 1 h. After pre-warming samples to 25°C for 2 to 3 min, fluorescence anisotropy and total intensity were measured for each dilution using a FlexStation-3 (Molecular Devices) in Basic Binding Assay-FP mode.
Nickel pull-down streptavidin-HRP assay
Full-length NRF2 (1-605 AA), Neh1, the Cap-n-collar (CNC) bZip domain of NRF2 (434–561 AA) and ΔNeh1 fragments were cloned in a pET14B expression vector. The excess amount of purified histidine-tagged NRF2 proteins was bound to the pre-charged and pre-equilibrated Ni-NTA beads (Novagen) and was incubated for 30 min on ice. After incubation, the NRF2-bound NTA-resin was washed (3×) with PBS. Subsequently, biotin-labeled ML385 or control compounds were added at a concentration of 10 μM. After 1 h incubation on ice, beads with protein were washed (3×) with PBS. For the competition assay, ML385 and compound 3 were added at a concentration of 10 μM, incubated on ice, and washed (3×) with PBS. Next, 5 μg of horseradish peroxidase (HRP)-conjugated streptavidin was added to the tube, followed by a 30-min incubation on ice, followed by an 8× wash with PBS. Lastly, bound protein-drug complex was eluted with PBS containing 10 mM EDTA, mixed 1:1 with SuperSignal West PICO solution, and the HRP activity was measured using well-scan mode in a Flexistation-3 (Molecular Devices).
Western blot analysis
Immunoblot analysis was performed as described by Singh and coworkers7. The following antibodies were used for immunoblotting: anti- β-ACTIN (Sigma), anti-KEAP1 (E-20) anti-NRF2 (H-300) and anti-GAPDH (FL-335) (SantaCruz Biotechnology), anti-GCLM (ab55436) and anti- GCLC (ab41463) (Abcam) and anti-NQO1 (A180) (Cell signaling technologies).
Real time RT-PCR
Total RNA was extracted from tumors and cells using the RNeasy kit (Qiagen). The reverse transcription reaction was performed using a high capacity cDNA synthesis kit (Applied Biosystems). Quantitative RT-PCR analyses of human NRF2, GCLc, GCLm, GSR, NQO1, HO-1, ABCC1, ABCC2, and ABCG2 were performed by using assay-on-demand primers and probe sets from Applied Biosystems. β-actin was used for normalization7, 18, 24. Biological replicates of tumors (n=4 or more) and cell culture samples (n=2–3) were used.
Clonogenic assays
Exponentially growing cells were counted, diluted, and seeded in triplicate at 800–1000 cells/well in a 6-well plate. Cells were incubated at 37°C for 24 h in a humidified CO2 incubator and then exposed to drugs or vehicle for 72 h. To assess clonogenic survival following drug exposure, cell cultures were incubated in complete growth medium at 37°C for 11–14 days and then stained with 50% methanol-crystal violet solution. Only colonies with more than 50 cells were counted20, 24.
NQO1 enzyme activity measurement
Cells were treated with vehicle or ML385 for 72 h. The enzyme activity in the total protein lysate was determined as described previously7, 25, 26.
Total antioxidant capacity and GSH measurement
Cells were treated with vehicle or ML385 for 72 h. The total antioxidant capacity and glutathione levels were measured using antioxidant and glutathione assay kits, respectively (Cayman Chemical Company).
Caspase activity assay
Caspase activity was measured using the Caspase-Glo® 3/7 Assay kit (Promega) as per the manufacturer’s instructions. The CellTiter-Blue assay (Promega) was utilized to quantify cell density and to normalize caspase activity. Briefly, cells were treated with ML385 for 36 h. An equal amount of CellTiter-Blue reagent (Promega) was added to the wells and the fluorescence was measured after 30 min. The CellTiter-Blue reagent was discarded and the Caspase-Glo (100 μL) reagent was added to the cells and incubated at 37°C for an additional 60–90 min. The resulting luminescence was recorded and the caspase activity was normalized to cell number.
Pharmacokinetic analysis of ML385 in CD-1 mice
For pharmacokinetic analysis, male CD-1 mice (n=3/time point) were administered a 30 mg/kg intraperitoneal (IP) dose of (vehicle: Solutol/Cremophor EL/polyethylene glycol 400/water [15/10/35/40,v/v/v/v]) of ML385. Blood samples were collected at pre-treatment, 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24 h and plasma samples were harvested. Plasma concentration of ML385 was determined using a qualified LC-MS/MS. A simulation was conducted to predict the in vivo exposure after a multiple-dose treatment based on the single-dose study results.
Determination of ML385 concentration in tumor samples
An UPLC-MS/MS method was developed to determine the concentration of ML385 in tumor samples. The details are included in the supplementary methods section.
Establishment of tumor xenografts and treatment
Tumor xenografts were established as described previously18. A549 cells (5.0×106) and H460 cells (1.0×106) were injected subcutaneously into the flank of athymic nude mice and the tumor dimensions were measured by caliper at an interval of 3–5 days18. The tumor volumes were calculated using the following formula: [length (mm) × width (mm) × width (mm) × 0.5]. Once the tumor volumes were approximately 50–100 mm3, mice were randomly allocated into 4 groups: vehicle, ML385, carboplatin, and ML385 in combination with carboplatin. Vehicle, carboplatin (5 mg/kg daily Monday to Friday)18, ML385 (30 mg/kg daily Monday to Friday), or ML385 in combination with carboplatin were administered intraperitoneally for 3 weeks. At the end of treatment period, mice were sacrificed and the tumor, blood, lung, and liver samples were collected.
For the orthotopic lung tumor model, A549 (1.0×106) and H460 cells (1.0×106) were diluted 1:1 in matrigel (30 μL) and were injected directly into the lungs. Ten days post-cell implantation, mice were imaged. Mice with visible localized lung tumor were randomly divided into 4 groups: vehicle, ML385, carboplatin, and ML385+carboplatin. Vehicle, carboplatin, ML385, or ML385 in combination with carboplatin were administered intraperitoneally for 2 weeks using the same regimen as described above. High-resolution lung micro-computed tomography (CT) images were acquired in 512 projections (270 μA, 75 kVp), and the data were reconstructed using the ordered subsets-expectation maximization algorithm. Volume-rendered whole lung images were generated using Amira 5.3.0 software (Visage Imaging Inc). For each mouse, pretreatment available lung volume was defined as 100% compared to post-treatment lung volumes. All experimental protocols conducted on the mice were performed in accordance with NIH guidelines and were approved by the Johns Hopkins University Animal Care and Use Committee.
Measurement of platinum levels in tumor
Platinum levels were determined using ICP-MS technique. The measurement and analysis was carried out at Actslab, Canada.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections were treated with anti-Ki-67 antibody at a dilution of 1:100 for 1 h and stained using LSAB+System-HRP kit (DakoCytomation) according to the manufacturer’s instructions. Non-immune rabbit IgG (Jackson ImmunoResearch Laboratories) was used as a negative control18.
Statistical analysis
Statistical significance between the control and the treatment groups was calculated by using a one-way ANOVA and post hoc test (Bonferroni’s Multiple Comparison Test) (GraphPad Prism 6). A p-value of 0.05 or less was considered statistically significant.
Results and Discussion
Screening for first-in-class small molecule inhibitors of NRF2
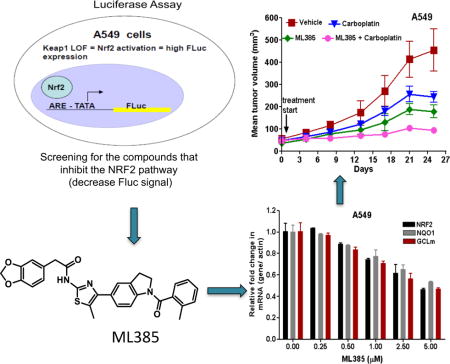
To identify specific, first-in-class small molecule inhibitors of NRF2, we performed a cell-based qHTS of the MLSMR library (~400,000 small molecules) using a novel NRF2 reporter gene assay multiplexed with a cytotoxicity readout. For the primary screen, we used A549 cells, with a point mutation in the Keap1 gene(G333C) and loss of heterozygosity at 19p13.2 that leads to the loss of KEAP1 activity and gain of NRF2 function7. These cells were stably transfected with a firefly luciferase reporter (Fluc) construct driven by a minimal TATA promoter with an upstream NRF2-specific ARE enhancer sequence from the human NQO1 promoter7, 27. Budesonide, a glucocorticoid, which was previously identified as an inhibitor of NRF2 signaling28, 29 with an IC50 value of 1 nM, was used as a control for NRF2 mediated ARE reporter activity. Staurosporine, a well-known apoptosis inducer, was used as an additional control. To select for compounds that inhibited NRF2 reporter activity, the screening of each compound was done at 6 concentrations in the MLSMR library (46 μM to 3 nM with 1:3 dilution factor; PubChem AID 504444), as described in the methods section. A multiplexed cytotoxicity readout was used to identify and to exclude potentially toxic compounds, whose effects could reduce luciferase reporter activity and thus mimic NRF2 inhibition. A summary of the qHTS activity outcomes is provided in Figure 1a. After the primary screen, 1712 putative NRF2 inhibitors were identified to be active in A549 cells and a clustering analysis was performed to identify the top chemical series30. Of these, 1552 compounds were reconfirmed as inhibitors of NRF2-dependent luciferase activity in A549 cells, yielding a 91% confirmation rate. These compounds were further counter-screened in a HEK293-CMV-luciferase reporter cell line and using an in vitro luciferase enzyme activity assay to filter out non-specific transcriptional inhibitors and luciferase enzyme inhibitors, respectively, (Figure 1b). Next, the putative NRF2 inhibitors were screened for activity in two alternative NSCLC cell lines with biallelic inactivation of KEAP1 (H838-ARE-luciferase and H1437-ARE-luciferase) (Figure 1c)7, 31. Forty-three compounds that showed NRF2 inhibitory activity in all 3 cell lines were selected for further characterization in a qRT-PCR assay, which measured the expression of the NRF2-dependent genes NAD(P)H dehydrogenase, quinone-1 (NQO1) and glutamate-cysteine ligase, modifier subunit (GCLm). These compounds were further tested using a glucocorticoid receptor (GR)-responsive beta-lactamase reporter cell line to filter out the molecules capable of inhibiting GR signaling, which have been shown to inhibit NRF2 signaling28, 29.
Figure 1. Summary of the NRF2 qHTS screening results.
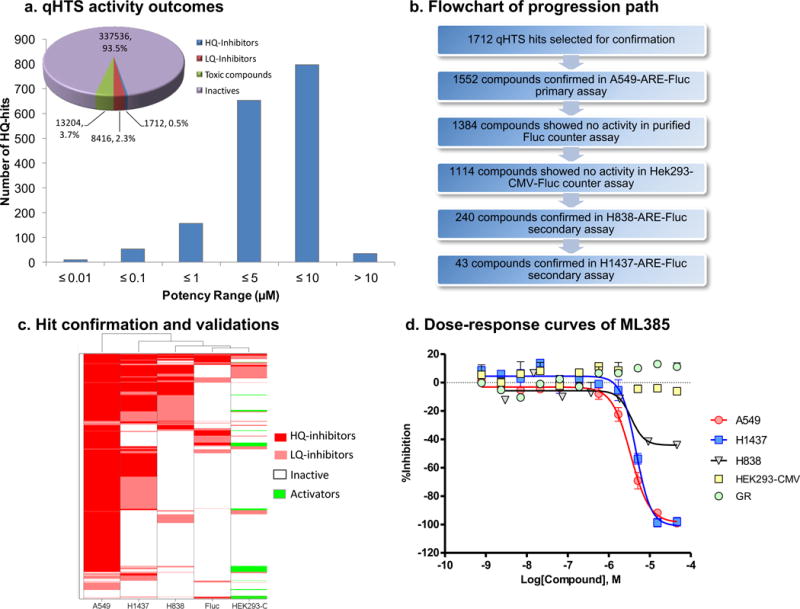
(a) Pie chart displaying the breakdown of the screening hit classes; high-quality (HQ) inhibitors are compounds in curve class −1.1, −1.2, −2.1, and −2.2 with efficacy higher than 50%; low-quality (LQ) inhibitors are compounds in curve class −3 with single point activity or those with shallow or poor-fitting curves; toxic compounds are those showing prominent cell killing effects from the cytotoxicity readout; inactives are compounds with class 4 curves. Histogram displaying the potency distribution of the HQ-inhibitors. (b) Assay flowchart for the validation of small molecule inhibitors of NRF2. (c) Heat map showing the activity profile of 1712 qHTS hits in 3 ARE NRF2 assays (A549, H1437, and H838 cells), purified firefly luciferase biochemical counter assay and HEK293-CMV counter assay. Compound IDs are given at right and the assay types are listed at the bottom of the heat map. Hierarchical clustering analysis was done using Spotfire DecisionSite 8.2. (d) Concentration-response curves of ML385 in three ARE NRF2-luc assays (A549, H1437, and H838 cells), HEK293-CMV counter assay and glucocorticoid receptor (GR)-responsive beta-lactamase reporter cell-based assay.
These data led to the identification of a thiazole-indoline structural series (shown as 1 in Figure 2a) that passed the aforementioned activity and selectivity criteria. Basic structure activity relationship studies uncovered the importance of an ortho-substituent on the benzoyl group, giving ML385 (2) as an active NRF2 inhibitor with desired preliminary biological activity (Figure 1d and 2a), while 3, bearing a piperazine in place of the benzodioxole, was a key inactive analog used in subsequent studies.
Figure 2. ML385 inhibits DNA binding activity of NRF2-MAFG complex.
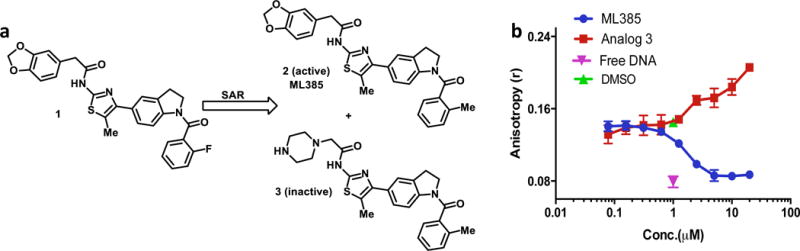
(a) Chemical structure of qHTS hit 1 and subsequent active NRF2 inhibitor ML385 and inactive analog 3. (b) ML385 inhibits the binding of NRF2-MAFG protein complex to fluorescein-labeled ARE DNA. Fluorescence intensity was measured to get anisotropy value and IC50 was calculated by fitting of sigmoid curve (R2>0.97).
A number of kinases have been reported to affect NRF2 activity32. Recently, it is reported that AEM1 inhibits NRF2 signaling in lung cancer cells33. To determine whether ML385 potentially inhibits NRF2 through direct interaction with a kinase, as well as to uncover the potential off-target kinase activity, we evaluated ML385 activity in A549 cells using an activity-based proteomics platform (KiNativ, ActivX Biosciences). This assay measures the ability of small molecules to inhibit the covalent labeling of protein kinases by a broadly reactive ATP acyl-phosphate probe directly in native cell lysates. Interestingly, ML385 (5 μM) did not exhibit significant inhibition of any of the ~170 kinases that were evaluated (Supplementary Table 2).
ML385 directly interacts with NRF2 protein
To test whether ML385 interacts with NRF2 and affects the DNA binding activity of the NRF2-MAFG protein complex, a fluorescence polarization assay was developed using fluorescein-labeled ARE-DNA duplex (Figure 2b; Supplementary Figure 1). The addition of ML385 decreased anisotropy in a dose-dependent manner, with an IC50 of 1.9 μM, suggesting that the NRF2-MAFG protein complex was dissociated from fluorescein-labeled ARE-DNA. Importantly, the inactive analog 3 increased anisotropy and did not inhibit the binding of the NRF2-MAFG complex to ARE-DNA duplex (Figure 2b).
To demonstrate the direct interaction between ML385 and NRF2 protein, we synthesized biotin-labeled analogs of ML385, including an active biotin analog (AB-ML385) with conjugation through the meta-position of phenyl acetamide and an inactive biotin (IB-ML385) analog with conjugation through the para-position of the same phenyl ring (Figure 3a). AB-ML385 retained NRF2 inhibitory activity, although with reduced potency, as measured by real time RT-PCR (Figure 3b), while IB-385 was inactive. We validated the interaction between AB-ML385 and NRF2 using histidine-tagged human NRF2 with a nickel (Ni+) pull-down assay, followed by the measurement of streptavidin-HRP activity. Only AB-ML385 showed interaction with NRF2 protein, while IB-ML385 showed no interaction with NRF2 protein (Figure 3c–d). Importantly, competition with ML385 reduced the interaction between NRF2 protein and AB-ML385 giving a reduced HRP signal. Again, competition with analog 3 had no effect on the NRF2 protein and AB-ML385 interaction suggesting that the interaction between NRF2 and ML385 is specific (Figure 3d).
Figure 3. ML385 directly interacts with purified NRF2 protein.
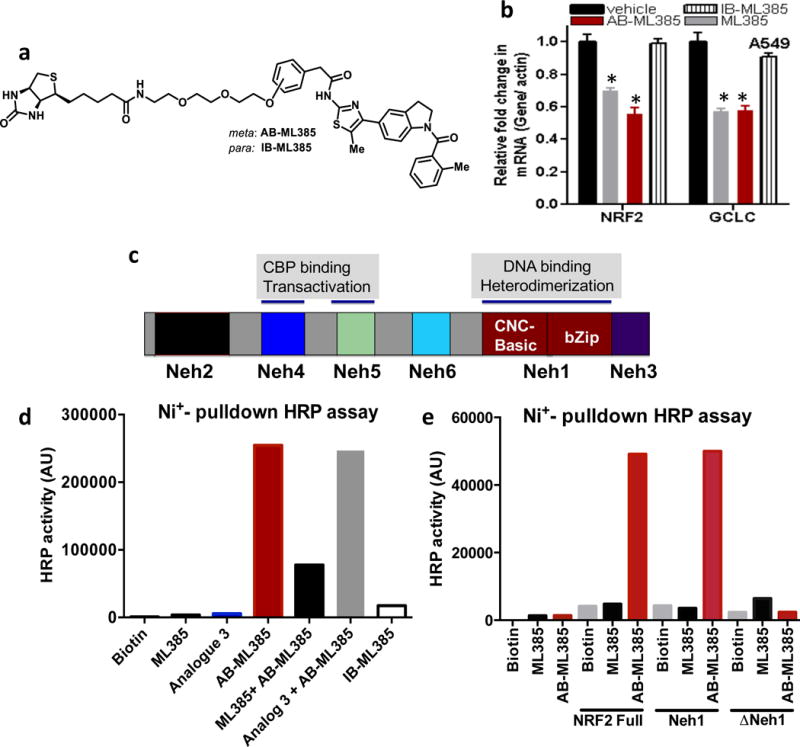
(a) Chemical structure of active Biotin conjugated ML385 (AB-ML385) and inactive biotin conjugated analog (IB-ML385). (b) AB-ML385 (10 μM) attenuates NRF2 and GCLc mRNA expression, while IB-ML385 does not. ML385 was used at a concentration of 5 μM. Error bars represent ±S.D. ‘*’P<0.05 relative to vehicle. (c) Domain architecture of NRF2 protein. (d) AB-ML385 binds to purified histidine-tagged full length NRF2 protein in Ni+ pull-down assay and is competed away by ML385. (e) AB-ML385 selectively binds to Neh1 domain of NRF2. NRF2 full-length NRF2 protein; Neh1, Neh1 domain of NRF2, ΔNeh1, full-length NRF2 protein lacking Neh1 domain.
To identify the specific domain of NRF2 protein that interacts with AB-ML385, we cloned individual domains of NRF2 and screened for potential binding with AB-ML385. The Ni+ affinity pull down assay revealed Neh1, the Cap-n-collar, b-Zip domain of NRF2, as the key domain interacting with AB-ML385 (Figure 3e). The Ni+ affinity pull-down assay using NRF2 protein lacking Neh1 domain (ΔNeh1) showed no interaction with AB-ML385. Taken together, these results suggest that ML385 binds to the Neh1 DNA binding domain of NRF2, blocks the binding of the NRF2-MAFG complex to the ARE sequence in the promoter and reduces transcriptional activity.
ML385 inhibits NRF2 signaling in lung cancer cells with KEAP1 mutations
To determine whether the interaction between NRF2 and ML385 inhibits NRF2-mediated transcription, we cultured A549 cells in the presence of ML385 for 72 h and measured the changes in the expression levels of NRF2 and its target genes. As shown in Figure 4a, we detected a dose-dependent reduction in the NRF2 transcriptional activity and the maximum inhibitory concentration was 5 μM ML385. To determine the time-dependent kinetics of this effect, we treated A549 cells with ML385 (5 μM) and measured the expression of the antioxidant target genes at various time points. Again, we noticed a time-dependent decrease in NRF2 signaling and the maximum decline was at 72 h (Figure 4b). In addition to reducing mRNA levels of NRF2 target genes, we observed a reduction in NRF2 mRNA levels. These results are in agreement with previous results, which showed that NRF2 auto-regulates its own transcription34. We then transfected A549 cells with a NRF2 gene promoter luciferase reporter construct and treated these cells with either ML385 or vehicle. Treatment with ML385 led to diminished NRF2 promoter activity, which was consistent with the activity of NRF2 protein regulating its own transcription (Supplementary Figure 2 and b). To determine whether ML385 causes global inhibition of NRF2 signaling in lung cancer cells with KEAP1 mutations, we treated A549 cells with ML385 for 48 and 72 h and measured the expression of several NRF2-dependent genes by real time RT-PCR. To confirm the specificity of ML385 for NRF2, we used inactive analog 3. As expected, we noticed a time-dependent reduction in transcript levels of glutathione synthesis and recycling enzymes, members of the thioredoxin family, and glucose metabolism-related genes in A549 cells treated with ML385. Compound 3 did not induce significant changes in NRF2-dependent gene expression (Figure 4c). As expected, A549 cells treated with ML385 demonstrated a dose-dependent reduction in NRF2 protein levels and its target genes (Figure 4d–e, Supplementary Figure 3a). Corroborating gene expression data, ML385 treatment significantly attenuated NQO1 enzyme activity and reduced GSH levels along with cellular antioxidant capacity (Figure 4f–h).
Figure 4. ML385 inhibits NRF2 signaling in lung cancer cells.
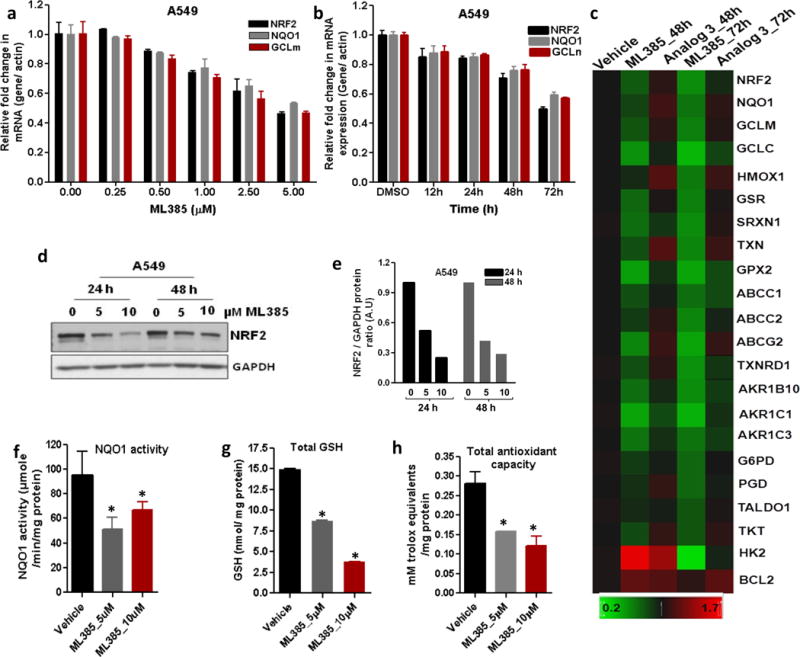
(a) Dose-dependent inhibition of NRF2-mediated transcription by ML385. Error bars represent ±S.D. (b) Time-dependent reduction in NRF2 and its target genes in A549 cells after treatment with ML385 (5μM). Error bars represent ±S.D. ‘*’P<0.05 relative to vehicle. (c) Heat map showing expression of NRF2-dependent antioxidant and drug detoxification genes in A549 cells treated with ML385 for 48 h or 72 h. Inactive analog 3 (5 μM) was used as a control. Gene expression was measured by real time RT-PCR. (d) Immunoblot showing relative levels of NRF2 protein at 24 h and 48 h post-treatment with ML385. (e) Densitometric quantification of NRF2 immunoblot data. (f–h) Treatment with ML385 attenuates antioxidant enzyme activity resulting in lower NQO1 enzyme activity, total antioxidant capacity, and reduced glutathione levels in A549 cells. Error bars represent ±S.D. ‘*’P<0.05 relative to vehicle.
Next, we selected H460, a large cell carcinoma cell line harboring a KEAP1 mutation, to further validate ML385 for NRF2 inhibitory activity7, 18. Similar to the results in the A549 cells, a treatment of H460 with ML385 led to a dose- and time-dependent reduction in the levels of mRNA of NRF2’s signature downstream genes and lowered GSH levels and antioxidant capacity (Supplementary Figures 3b and 4). Lastly, we validated NRF2 inhibitory activity of ML385 in a lung cancer cell line harboring an oncogenic NRF2 mutation. Using EBC1 cells, a squamous lung cancer cell line with a gain-of-function mutation in NRF2, we performed dose-response studies with ML385. Again, the treatment with ML385 led to a dose-dependent reduction in NRF2 and downstream glutathione-related gene expression (Supplementary Figure 5). Taken together, these results suggest that ML385 targets NRF2 signaling in lung cancer cells.
ML385 is selectively cytotoxic in NSCLC cells with KEAP1 mutations and further potentiates the toxicity chemotherapy drugs
Previously, we demonstrated that constitutive activation of NRF2 signaling in cancer cells confers chemoresistance and radioresistance and have established NRF2 as a primary therapeutic resistance factor in cancer cells7, 18, 24, 35. To determine whether ML385 is selectively toxic to cells with high NRF2 activity resulting from the loss of KEAP1 activity, we used a pair of isogenic H460 lung cancer lines with wild-type (WT) and mutant KEAP1. H460 parental cells harbor a point mutation (D262H) in KEAP1 leading to loss of KEAP1 activity7 and using the homologous recombination approach, we knocked in a WT KEAP1 allele. The expression of WT KEAP1 protein in H460-cells led to reduced NRF2 protein levels and attenuated downstream NRF2 signaling (Supplementary Figure 6a–c). Treatment with ML385 lead to a significant reduction in NRF2 and downstream target gene expression selectively in KEAP1 mutant H460 cells (Supplementary Figure 6d). Thus, H460 KEAP1 knock-in cells were found to be resistant to ML385-mediated growth inhibition as determined by clonogenic assay (Figure 5a). Next, we evaluated the activity of ML385 in a non-tumorigenic lung epithelial cell line (BEAS2B). As expected, BEAS2B cells, which have wild-type NRF2 and KEAP1 signaling, were resistant to the growth inhibitory effects of ML385 (Figure 5b). Collectively, these results demonstrate that ML385 selectively affects the colony forming ability or growth of lung cancer cells with gain of NRF2 function.
Figure 5. ML385 is selectively toxic to cells with KEAP1 mutations and potentiates the toxicity of chemotherapy drugs in NSCLC cells with KEAP1 mutations.
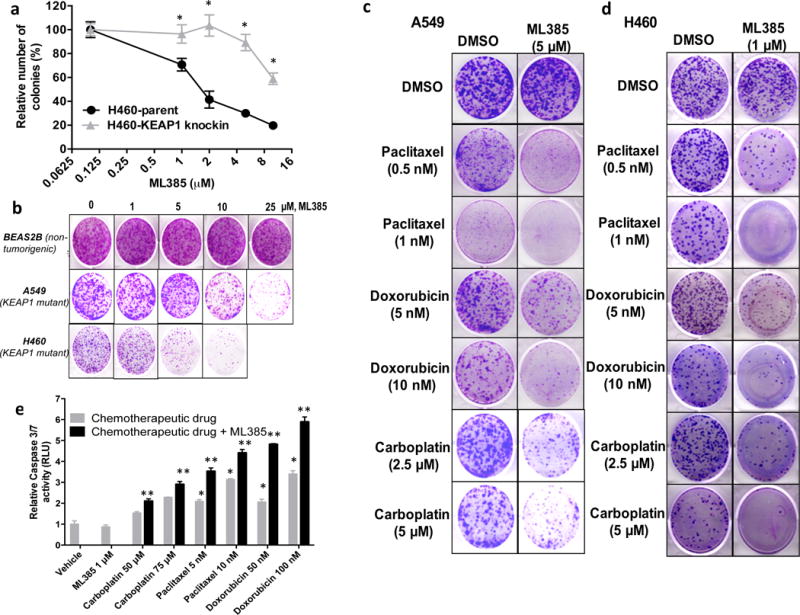
(a) H460, a NSCLC line with a point mutation in KEAP1, is more sensitive to ML385 than H460-KEAP1 Knock-in H460 cells expressing WT KEAP1. ‘*’P<0.05 relative to H460-KEAP1 Knock-in cells. (b) Treatment with ML385 selectively inhibits the colony forming ability of lung cancer cells but has no effect on the growth of non-tumorigenic BEAS2B cells. (c–d) H460 and A549 cells were treated with different concentrations of paclitaxel, doxorubicin, or carboplatin alone or in combination with ML385 for 72 h. At the end of treatment, regular growth medium was added and cells were further incubated for 8–10 days and stained with crystal violet. (e) H460 cells treated with ML385 in combination with chemotherapy drug showed increased caspase 3/7 activity, a marker of apoptosis. Cells treated with chemotherapy drug alone or ML385 in combination with chemotherapy drug were incubated with luminogenic caspase substrate and change in luminescence was measured. Caspase activity was normalized to the number of viable cells using CellTiter-Blue assay. Error bars represent ±S.D. ‘*’P<0.05 relative to vehicle or ML385; ‘**’ P<0.05 relative to chemotherapy drug alone.
To study whether ML385-mediated attenuation of NRF2 signaling enhances the sensitivity of lung cancer cells to chemotherapy drugs such as carboplatin, paclitaxel, or doxorubicin, we treated H460 and A549 cells with ML385 alone or in combination with chemotherapy drugs for 72 h. Co-treatment with ML385 and chemotherapy drugs for 72 h resulted in a higher toxicity and reduced numbers of colonies as compared to single agents alone in both the H460 and A549 cell line models (Figures 5c–d). To determine whether the reduced number of colonies in cells treated with ML385 in combination with chemotherapy were due to increased caspase-3-mediated apoptosis, we analyzed caspase-3/7 activity using a luminescence-based assay. As expected, cells treated with ML385 in combination with paclitaxel or ML385 in combination with carboplatin demonstrated elevated levels of caspase-3/7 activity (Figure 5e). These results indicate that ML385 enhances the cytotoxic activity of chemotherapy by blocking NRF2 signaling.
ML385 shows anti-tumor activity in NSCLC both as a single agent and in combination with carboplatin
To determine whether ML385 has an appropriate pharmacokinetic (PK) profile for in vivo studies, we dosed CD-1 mice at 30 mg/kg IP. The PK profile showed that ML385 has a half-life (t1/2 = 2.82 h) after IP injection supporting its use in in vivo efficacy studies (Supplementary Figure 7). To determine whether the combination of ML385 and carboplatin observed in cell culture could be recapitulated in vivo, we performed subcutaneous xenograft experiments using A549 and H460 cells. Mice were dosed with ML385, carboplatin, or ML385 in combination with carboplatin for 3–4 weeks and the tumor volumes were measured biweekly. A549 and H460 tumors treated with ML385 in combination with carboplatin showed a significant reduction in tumor growth in both cell lines compared to vehicle. Although the treatment with a single agent (either ML385 or carboplatin) led to a reduction in tumor growth, the magnitude of these effects was variable between cell lines and did not reach statistical significance (Figure 6a–d, Supplementary Figure 8a–b). These results are consistent with prior findings that were obtained with NRF2 siRNA18 in combination with chemotherapeutic drugs. Tumor samples were analyzed for exposure to ML385 4–6 h post last treatment with ML385. We detected ML385 at intratumoral concentrations of ~1 μM in both single agent and combination treatment cohorts. Mice tolerated the combination treatment and the analysis of serum samples for liver and toxicity-related markers revealed no evident signs of toxicity (Supplementary Table 3). ML385 in combination with carboplatin led to a significant reduction in tumor cell proliferation, demonstrated by fewer Ki-67 positive cells (Figure 6e, Supplementary Figure 8c). The RT-PCR and immunoblot analyses of tumor samples were used to determine whether antitumor activity correlated with modulation of the pharmacodynamic markers of NRF2 signaling by ML385. Tumor samples treated with ML385 showed a significant reduction in NRF2 protein level and its downstream target genes (Figure 6f–g). Determination of platinum levels in A549 and H460 tumors treated with carboplatin alone or ML385 in combination with carboplatin by inductively-coupled plasma mass spectrometry (ICP-MS) revealed ~2-fold higher platinum levels in tumors treated with combination therapy (Figure 6h). Collectively, these results suggest that ML385 potentiates the cytotoxic activity of carboplatin partly by blocking the NRF2-dependent drug detoxification pathway leading to increased drug retention in the tumor. The anti-tumor activity of ML385 in combination with carboplatin was replicated in an independent investigator’s laboratory using H460 xenografts (Supplementary Figure 9a, b).
Figure 6. Therapeutic efficacy of ML385 as a single agent and in combination with carboplatin in subcutaneous lung tumor xenografts.
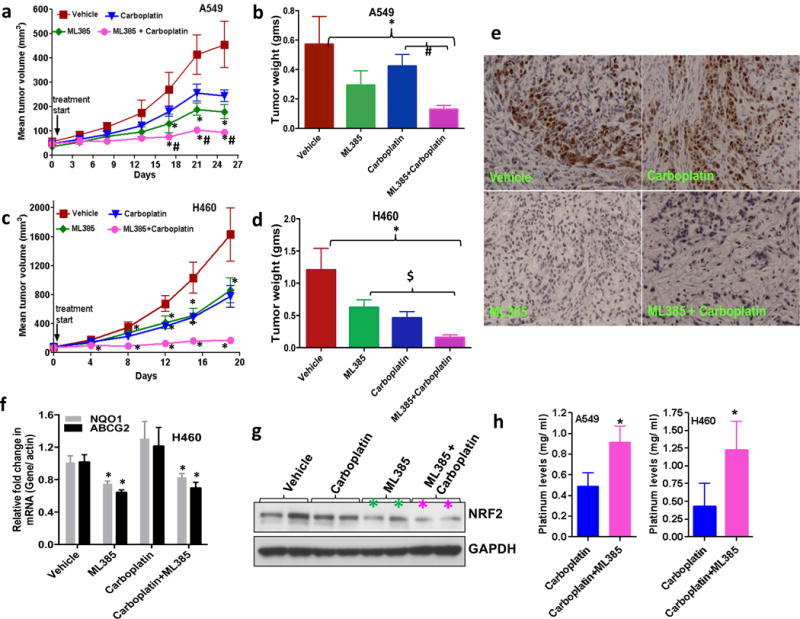
(a) ML385 shows anti-tumor activity as a single agent in sensitized A549 xenograft tumors to carboplatin therapy. Values represent tumor volume ± S.E.M. for all groups. n = 7 mice/group; ‘*’P<0.05 relative to vehicle; ‘#’ P<0.05 relative to carboplatin. (b) Treatment with ML385 or ML385 in combination with carboplatin significantly reduced tumor weight as compared to vehicle group. Efficacy of ML385 alone was comparable to carboplatin. ‘*’P<0.05 relative to vehicle; ‘#’ P<0.05 relative to carboplatin. (c-d) ML385 sensitized H460 lung tumors to carboplatin treatment. Groups of H460 tumors treated with ML385 or ML385 in combination with carboplatin showed a significant reduction in tumor volume and weight as compared to the vehicle group. Efficacy of ML385 alone was comparable to carboplatin. n = 5–7 mice/group; ‘*’P<0.05 relative to vehicle; ‘$’ P<0.05 relative to ML385. (e) The proliferative index based on Ki-67 immuno-reactivity in A549 subcutaneous tumors. (f) Treatment with ML385 attenuated the expression of NRF2-dependent genes in H460 tumors. ‘*’P<0.05 relative to vehicle or carboplatin (g) Immunoblot showing reduction in NRF2 protein in H460 tumors treated with ML385. (h) Bar graph showing platinum levels in A549 and H460 tumors treated with carboplatin alone or ML385 in combination with carboplatin. ‘*’P<0.05, relative to carboplatin alone.
ML385 and carboplatin combination therapy blocks orthotopic human lung tumor growth
We evaluated the therapeutic efficacy of ML385, alone and in combination with carboplatin in orthotopic models of human NSCLC that closely recapitulate clinical patterns of lung cancer progression. In our model, we sought to establish a single tumor within the left or right lung. Animals underwent micro-CT imaging, and the mice with approximately 3–7 mm nodules were randomly allocated into treatment groups. In the A549 lung cancer orthotopic model, mice treated with vehicle had 34% lung volume at 3 weeks compared to their pre-treatment volume. Single agent carboplatin or ML385-treated groups had 42% and 57% of their pretreatment lung volume at 3 weeks, respectively (Figure 7a–b). Although the differences in lung volume between the vehicle and the single agent (carboplatin and ML385)-treated groups did not reach a statistical significance, mice in the vehicle-treated group died immediately after 3 weeks, while those in the ML385 or carboplatin-treated group survived. The antitumor and anti-metastatic effect of carboplatin and ML385 combination treatment, as determined by tumor free lung volume, was significantly higher than vehicle or carboplatin monotherapy with 74% lung volume retention at 3 weeks post initiation of the combination treatment (Figure 7a–b, Supplementary Figure 10a).
Figure 7. ML385 shows significant anti-tumor efficacy in combination with carboplatin in an orthotopic lung tumor model.
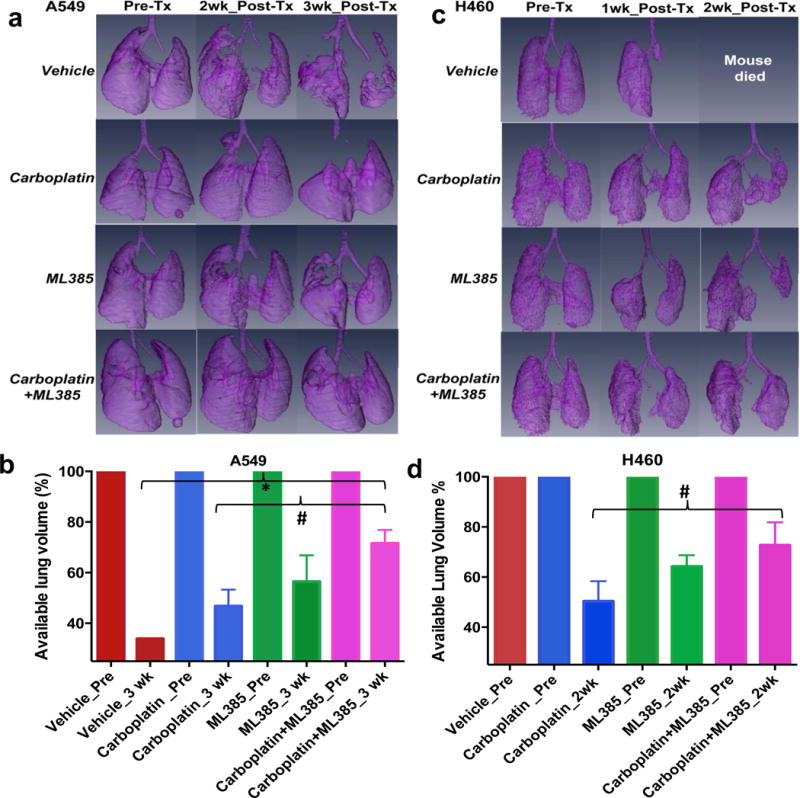
(a) Volume rendered 3-D lung images demonstrate the effectiveness of combination therapy in A549 tumors. Representative images show the available lung volume that is not taken over by the lung tumor pre -and post-treatment with vehicle, ML385, carboplatin or ML385 in combination with carboplatin. (n=2–5 mice/group). ‘Tx’-treatment. (b) Bar graph showing average tumor free lung volume in different groups at 3 wks post-treatment suggesting that combination therapy is more effective in reducing tumor growth. Mice in the vehicle-treated group died after 3 wks. For each group, pretreatment available lung volume was defined as 100% and was compared with post-treatment lung volumes. Values represent lung volume ± S.E.M. ‘*’P<0.05 relative to vehicle; ‘#’ P<0.05 relative to carboplatin. (c) Combination therapy shows a significant growth reduction of H460 tumors. Representative images show the available lung volume that is not taken over by the lung tumor pre- and post-treatment with vehicle, ML385, carboplatin or ML385 in combination with carboplatin. Mice in the vehicle-treated group did not survive for 2 wks post treatment. (n=2–4 mice/group). (d) Bar graph showing H460 tumor-free lung volume in different groups at 2 wks post-treatment suggesting that combination therapy is more effective in reducing tumor growth compared to carboplatin monotherapy. Values represent lung volume ± S.E.M ‘*’P<0.05, relative to carboplatin.
Next, we performed a similar experiment using an H460 tumor model with a well-described aggressive growth pattern, which spreads across the entire lung to the mediastinum if not treated at an early stage. The vehicle-treated animals died prior to the end of the two-week treatment period. A micro-CT analysis of the lungs of H460 tumor-bearing mice showed that those treated with ML385 monotherapy had 64% of the pre-treatment lung volume at 2 weeks post-treatment. Treatment with carboplatin yielded 50% lung volume compared to pre-treatment volume, which was not significantly different from that noted in the ML385 group (Figure 7c–d). Again, when ML385 and carboplatin were combined, the antitumor effects were significantly enhanced over carboplatin monotherapy, with mice showing 73% of pre-treatment lung volume at 2 weeks post-treatment (Figure 7d; Supplementary Figure 10b). Overall, these data indicate that ML385 in combination with carboplatin has a substantial in vivo efficacy in orthotopic NSCLC models.
Although aberrant gene expression is considered a hallmark of cancer, attempts to pharmacologically target transcription or transcription factors have not been very successful leading some scientists to consider that transcription factors are undruggable. Certainly, specificity of activity for a particular transcription factor represents a challenge; for example, in the case of NRF2, brusatol, a quassinoid, has been reported to inhibit NRF2 signaling36, yet it has also has been shown to inhibit DNA, RNA, and protein synthesis as well as oxidative phosphorylation and c-myc activity in leukemia cells37. In addition, recently, a high-throughput screen of 30,000 compounds has led to the identification of NRF2 inhibitor, which has a selective response in cell lines with gain of NRF2 function33.
In summary, our large, quantitative high-throughput screen led to the identification of specific small molecule inhibitor of NRF2, ML385. We have demonstrated that ML385 specifically and directly interacts with NRF2 protein, blocks NRF2 transcriptional activity, and enhances the efficacy of carboplatin and other chemotherapeutic drugs in lung cancer cells. The marked tumor growth inhibition observed in subcutaneous and orthotopic NSCLC models supports development of ML385 as a novel, potent selective inhibitor of NRF2 and as a potential anti-cancer agent for the treatment of NSCLC patients harboring KEAP1 mutations. However, a potential limitation of the current study is that we did not evaluate the effects of ML385 on the activity of other transcription factors including Cap-N-Collar bZIP and AP-1 members. Additional studies are needed to confirm if ML385 is selective for NRF2 or modulates activity of additional transcription factors and to evaluate the comprehensive role of NRF2 inhibitors in NSCLC patients in clinical trial settings.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R03 MH092170 and R01 CA140492 (S.B), R01 CA206155 (S.B.), National Cancer Institute SPORE grants P50 CA058184 (S.B.) and Flight Attendant Medical Research Institute (A.S). We thank Dr. Kensler for providing NRF2-promoter construct; Chris McNees for help with immunoblotting experiments and Ellen Tully for performing immunohistochemistry experiments. We thank S. Michael and R. Jones for automation support; P. Shinn, M. Itkin, and D. Van Leer for assistance with compound management, and W. Leister, H. Baker, C. LeClair, and E. Fernandez for analytical chemistry and compound purification support. Y-Q.Z., J.M.R., L.L., K.R.B., A.W., X.X., S.S., M.X., M.F., Z.L., M.S., and M.B.B. were supported by the intramural research program of the National Center for Advancing Translational Sciences and the Molecular Libraries Initiative of the NIH Roadmap for Medical Research (U54MH084681).
References
- 1.American Cancer Society. Cancer Facts and Figures 2013. Atlanta, Ga: American Cancer Society; 2013. [Google Scholar]
- 2.Hirsh V. Systemic therapies in metastatic non-small-cell lung cancer with emphasis on targeted therapies: the rational approach. Curr Oncol. 2010;17:13–23. doi: 10.3747/co.v17i2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsvetkova E, Goss GD. Drug resistance and its significance for treatment decisions in non-small-cell lung cancer. Curr Oncol. 2012;19:S45–51. doi: 10.3747/co.19.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 5.Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, Nasipuri P, Krausz KW, Wakabayashi N, Dewi R, Boros LG, Gonzalez FJ, Gabrielson E, Wong KK, Girnun G, Biswal S. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest. 2013;123:2921–2934. doi: 10.1172/JCI66353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, de Waal L, Sharifnia T, Brooks A, Greulich H, Banerji S, Zander T, Seidel D, Leenders F, Ansen S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Janne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, Meyerson M. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, Chu A, Collisson EA, Cope L, Creighton CJ, Getz G, Herman JG, Johnson BE, Kucherlapati R, Ladanyi M, Maher CA, Robertson G, Sander C, Shen R, Sinha R, Sivachenko A, Thomas RK, Travis WD, Tsao MS, Weinstein JN, Wigle DA, Baylin SB, Govindan R, Meyerson M. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muscarella LA, Barbano R, D’Angelo V, Copetti M, Coco M, Balsamo T, la Torre A, Notarangelo A, Troiano M, Parisi S, Icolaro N, Catapano D, Valori VM, Pellegrini F, Merla G, Carella M, Fazio VM, Parrella P. Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient’s outcome. Epigenetics. 2011;6:317–325. doi: 10.4161/epi.6.3.14408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, Shibata T, Yamamoto M, Hirohashi S. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 13.Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, Hirohashi S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–1368. 1368 e1351–1354. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 14.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BW, Tan MH, Zhang Z, Yang XJ, Zhou M, Gardie B, Molinie V, Richard S, Tan PH, Teh BT, Furge KA. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer cell. 2011;20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 15.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 16.Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W, Zhang DD. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010;70:5486–5496. doi: 10.1158/0008-5472.CAN-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK, Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, Blackford A, Goodman SN, Bunz F, Watson WH, Gabrielson E, Feinstein E, Biswal S. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008;68:7975–7984. doi: 10.1158/0008-5472.CAN-08-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, Yoshimatsu Y, Tachimori Y, Kushima R, Kiyono T, Yamamoto M. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia. 2011;13:864–873. doi: 10.1593/neo.11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, Wu H, Bova SG, Biswal S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Molecular cancer therapeutics. 2010;9:336–346. doi: 10.1158/1535-7163.MCT-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN, Minna JD, Stewart DJ, Wistuba II. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16:3743–3753. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konstantinopoulos PA, Spentzos D, Fountzilas E, Francoeur N, Sanisetty S, Grammatikos AP, Hecht JL, Cannistra SA. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011;71:5081–5089. doi: 10.1158/0008-5472.CAN-10-4668. [DOI] [PubMed] [Google Scholar]
- 24.Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid Redox Signal. 2010;13:1627–1637. doi: 10.1089/ars.2010.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prochaska HJ, Santamaria AB. Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: a screening assay for anticarcinogenic enzyme inducers. Anal Biochem. 1988;169:328–336. doi: 10.1016/0003-2697(88)90292-8. [DOI] [PubMed] [Google Scholar]
- 26.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 27.Lee JM, Moehlenkamp JD, Hanson JM, Johnson JA. Nrf2-dependent activation of the antioxidant responsive element by tert-butylhydroquinone is independent of oxidative stress in IMR-32 human neuroblastoma cells. Biochem Biophys Res Commun. 2001;280:286–292. doi: 10.1006/bbrc.2000.4106. [DOI] [PubMed] [Google Scholar]
- 28.Kratschmar DV, Calabrese D, Walsh J, Lister A, Birk J, Appenzeller-Herzog C, Moulin P, Goldring CE, Odermatt A. Suppression of the Nrf2-dependent antioxidant response by glucocorticoids and 11beta-HSD1-mediated glucocorticoid activation in hepatic cells. PLoS One. 2012;7:e36774. doi: 10.1371/journal.pone.0036774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ki SH, Cho IJ, Choi DW, Kim SG. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol Cell Biol. 2005;25:4150–4165. doi: 10.1128/MCB.25.10.4150-4165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh AHC, Manna SK, Acquaah-Mensah G, Carratero J, Kumar S, Nasipuri P, Krausz KW, Wakabayashi N, Dewi R, Boros LG, Gonzalez F, Gabrielson E, Wong KK, Girnun G, Biswal S. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest. 2013;123 doi: 10.1172/JCI66353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abazeed ME, Adams DJ, Hurov KE, Tamayo P, Creighton CJ, Sonkin D, Giacomelli AO, Du C, Fries DF, Wong KK, Mesirov JP, Loeffler JS, Schreiber SL, Hammerman PS, Meyerson M. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. 2013;73:6289–6298. doi: 10.1158/0008-5472.CAN-13-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bollong MJ, Yun H, Sherwood L, Woods AK, Lairson LL, Schultz PG. A Small Molecule Inhibits Deregulated NRF2 Transcriptional Activity in Cancer. ACS chemical biology. 2015;10:2193–2198. doi: 10.1021/acschembio.5b00448. [DOI] [PubMed] [Google Scholar]
- 34.Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–2892. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh A, Wu H, Zhang P, Happel C, Ma J, Biswal S. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol Cancer Ther. 2010;9:2365–2376. doi: 10.1158/1535-7163.MCT-10-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, Zhang DD. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108:1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall IH, Lee KH, Eigebaly SA, Imakura Y, Sumida Y, Wu RY. Antitumor agents. XXXIV: Mechanism of action of bruceoside A and brusatol on nucleic acid metabolism of P-388 lymphocytic leukemia cells. Journal of pharmaceutical sciences. 1979;68:883–887. doi: 10.1002/jps.2600680726. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.