

Abstract
In vitro, some RNAs can form stable four-stranded structures known as G-quadruplexes. Although RNA G-quadruplexes have been implicated in post-transcriptional gene regulation and diseases, direct evidence for their formation in cells has been lacking. Here, we identified thousands of mammalian RNA regions that can fold into G-quadruplexes in vitro, but in contrast to previous assumptions, these regions were overwhelmingly unfolded in cells. Model RNA G-quadruplexes that were unfolded in eukaryotic cells were folded when ectopically expressed in Escherichia coli; however, they impaired translation and growth, which helps explain why we detected few G-quadruplex–forming regions in bacterial transcriptomes. Our results suggest that eukaryotes have a robust machinery that globally unfolds RNA G-quadruplexes, whereas some bacteria have instead undergone evolutionary depletion of G-quadruplex–forming sequences.
MAIN TEXT
Many cellular RNAs contain regions that fold into stable structures required for function (1, 2). These structures can be studied using chemical probes that modify accessible or flexible nucleotides (3–5). For example, dimethyl sulfate (DMS) methylates A and C residues that are not protected by Watson–Crick pairing or other interactions, and because these modifications stall reverse transcriptase, primer-extension reactions can detect modification and thereby report on the folding state of these nucleotides. DMS also penetrates living cells and modifies RNAs within these cells, and with high-throughput sequencing of global primer-extension products, the intracellular folding of numerous RNAs can be simultaneously monitored in a procedure called DMS-seq (6, 7). Analogous high-throughput methods have also been developed using cell-permeable SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) reagents (8, 9). These methods reveal important differences between RNA structures formed in vivo and those formed in vitro (7, 9). However, these high-throughput methods are designed to detect Watson–Crick pairing, which leaves the folding states of noncanonical structures difficult to assess.
One such noncanonical structure is the RNA G-quadruplex (RG4), in which four strands of RNA interact, either intramolecularly or intermolecularly, through the formation of two or more layers of G-quartets, in which each of four G residues pairs to two neighboring G residues (Fig. 1A) (10, 11). Due to the extensive hydrogen-bonding and base-stacking interactions, RG4 structures can be very stable, with in vitro melting temperatures well exceeding physiological temperatures. This stability typically depends on the presence of K+, which is the optimal size to bind at the center of two stacked G-quartets and thereby counter the otherwise repulsive partial negative charges that converge at the quadruplex core (Fig. 1A).
Fig. 1.
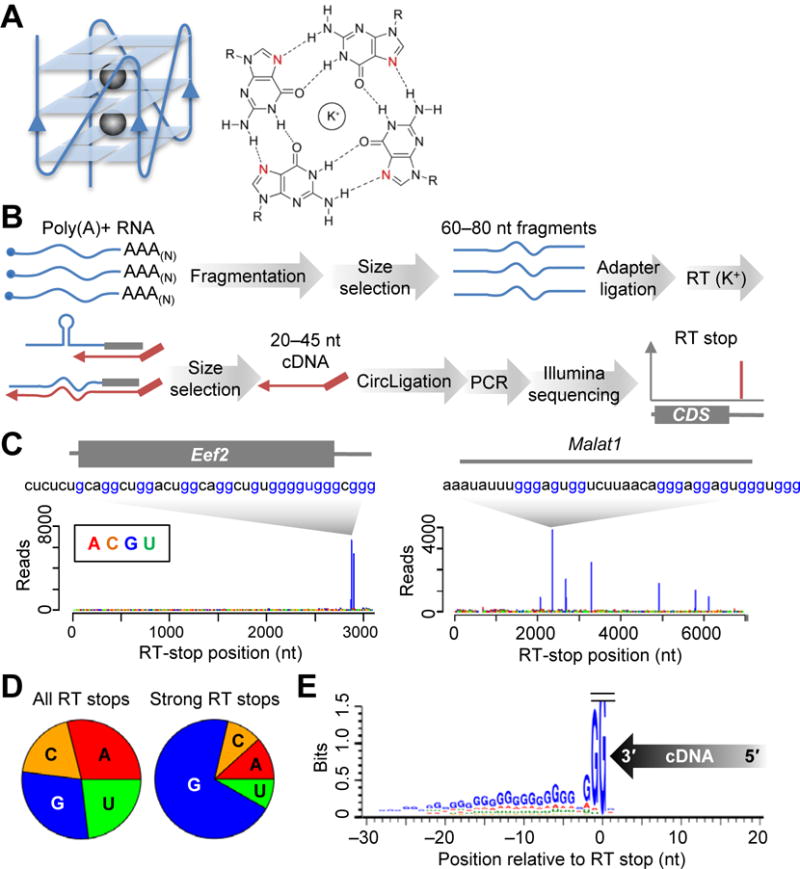
Strong RT stops at G-rich regions in the mESC transcriptome. (A) The RNA G-quadruplex. The schematic (left) depicts a three-tiered RG4, with a parallel RNA-backbone orientation (solid line) and three G-quartets stabilized by two K+ ions (spheres). The chemical structure of a G-quartet (right) highlights hydrogen bonding (dashed lines) to the N7 positions (red) and K+-facilitated convergence of the exocyclic oxygens of the four G residues (R, ribose). (B) Schematic of RT-stop profiling. See text for explanation. (C) RT-stop profiles for Eef2 mRNA (box, coding sequence) and Malat1. Bars representing each RT stop are colored according to the identity of the template nucleotide at the stall (position 0). Also shown for each transcript is the 40-nt RNA segment ending at the strongest stop. (D) Fraction of RT stops observed at each nucleotide, comparing all RT stops with only strong RT stops. (E) Nucleotide composition of the flanking sequences of strong RT stops at G. The direction of cDNA synthesis is indicated (arrow), with the 3′-terminal cDNA nucleotide assigned position +1. Template nucleotides are plotted at heights indicating the information content of their enrichment (bits).
Because of the high stability of RG4 structures in vitro and the high concentration of K+ in cells (typically >100 mM, well above that required for quadruplex formation), regions that fold into RG4 structures in vitro are generally assumed to fold into these structures in cells. Indeed, RG4s are implicated in control of mRNA processing and translation, with recently proposed roles in human diseases, such as cancer (12) and neurodegeneration (13). Supporting the idea that RG4s are folded in cells, immunostaining with G4-specific antibodies yields a detectable, albeit weak, RNase-sensitive signal in the cytoplasm (14). However, these immunostaining results leave open the possibility of folding during the processes of fixing, permeabilizing or staining cells, and even if this signal represented quadruplex formation in cells, it could not speak to either the sequence identities or the overall fraction of RG4 regions that fold in vivo.
Many RNAs with quadruplex-forming capacity
To systematically search for structure-forming potential in mammalian cellular RNAs, we exploited the ability of stable structures to stall reverse transcription. Poly(A)-selected mRNAs from mouse embryonic stem cells (mESCs) were randomly fragmented, and 60–80-nt fragments were ligated to a common 3′ adapter used for global primer extension. Complementary DNAs (cDNAs) resulting from reverse transcription that stalled after only 20–45 nt of extension were purified and sequenced to identify the RT stops (Fig. 1B), using a procedure resembling that developed for DMS-seq (7). As illustrated for the Eef2 (Eukaryotic elongation factor 2) mRNA and the Malat1 (Metastasis-associated lung-adenocarcinoma transcript 1) noncoding RNA, most of the strong RT stops (65%) were at G nucleotides (Fig. 1, C and D and fig. S1; p < 10−15, χ2 test). Analysis of the flanking sequences of these strong RT stops at G nucleotides showed that the 30 nucleotides upstream of the RT stops were also enriched in G (and depleted in C), particularly at positions −1 and −2 (92 and 66% G, respectively; Fig. 1E). In contrast, no enrichment was detected downstream, except for weak G enrichment at position +1 (38% G; Fig. 1E).
The upstream G enrichment, together with recent studies of individual transcripts (15), suggested that formation of intramolecular RG4 structures caused these strong RT stops. To test this possibility, we examined whether these RT stops were sensitive to the identity of the monovalent counter ion, and found that substituting K+ in the RT reaction with either Na+ or Li+ greatly diminished the RT stops at G residues (Fig. 2, A and B, and fig. S2A). Another diagnostic feature of RG4s is their sensitivity to modification of the N7 position of G (Fig. 1A). Methylating this position by using DMS (16) under denaturing conditions (95°C, 0 mM K+) also substantially diminished the RT stops at G residues, despite the presence of K+ during RT (Fig. 2, A and B, and fig. S2B). Most strong RT stops that were K+-dependent were also DMS-sensitive (Fig. 2C and table S1; p <10−15, χ2 test), and vice versa. Moreover, 6,140 (90%) of the 6,812 RT stops that exhibited ≥ 2 fold decrease in Na+/Li+ reactions and ≥ 2 fold decrease after 95°C DMS treatment were at G nucleotides (fig. S2C and table S1). In contrast, the 2,120 DMS-sensitive but K+-independent RT stops did not exhibit strong nucleotide enrichment at position 0 (fig. S2C), as would be expected for RT stops caused by other types of stable structures. Analysis of the remaining 672 RT stops that were K+-dependent and DMS-sensitive but not at G nucleotides showed that their upstream sequences were also somewhat enriched in G (fig. S2D), suggesting that at least some of these RT stops also involved RG4 structures that caused RT to stall before reaching the 3′-terminal G nucleotides. Collectively, these results indicated that most G-rich regions that caused strong RT stops did so by forming RG4 structures in vitro.
Fig. 2.
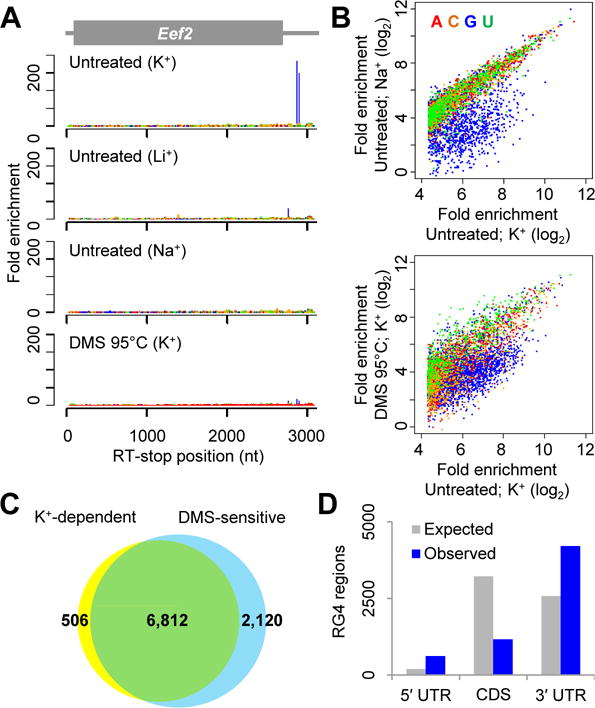
Folded RG4 structures cause strong RT stops. (A) RT-stop profiles of Eef2, showing enrichment observed in the original conditions (K+) and that observed when either substituting the monovalent cation used during RT (Li+ or Na+) or treating the RNA with DMS under denaturing conditions prior to RT (DMS 95°C). At each position within the mRNA, the fold enrichment was calculated as the number of RT-stop reads observed at that position, divided by the average number of RT-stop reads observed for all the mRNA positions with the same nucleotide identity. (B) Global analyses of strong RT stops, comparing the enrichment of stops observed in the original conditions (untreated; K+) to those observed when either substituting K+ with Na+ (top) or pretreating RNA with DMS at 95°C (bottom). RT-stop values are colored according to the template nucleotide at position 0. The distributions are truncated at the left because stops with <20 fold enrichment in the untreated K+ sample were not classified as strong stops. (C) Overlap between K+-dependent and DMS-sensitive strong RT stops (yellow and blue, respectively). (D) Abundance of RG4 regions within mRNA translated and untranslated regions. The expected abundances were estimated based on the relative number of G nucleotides within these three regions of detected mRNAs.
The four strands of RG4 structures typically assume a parallel orientation (17) (Fig. 1A). The circular dichroism spectra of the 60-nt regions upstream of K+-dependent strong RT stops in Eef2 and Malat1, as well as that of a canonical RG4 sequence G3A2G3A2G3A2G3 (hereafter referred to as the G3A2 quadruplex), exhibited a K+-dependent increase at 263 nm, diagnostic of parallel RG4 structures (17) (fig. S3).
Features of mammalian RG4 regions
Of the many endogenous RNA sequences with predicted RG4-forming potential (18), only ~100 have been experimentally tested (11, 19). Therefore, the 6,140 RG4 regions in the mESC transcriptome, 4,034 of which were non-overlapping, considerably expanded the repertoire of endogenous RNA sequences with experimentally supported RG4-forming capacity. Nonetheless, cellular transcripts presumably contain additional regions with intrinsic RG4-forming potential not detected in our experiment. For example, our strategy would miss 1) structures with stabilities insufficient to block RT, 2) structures spanning more than ~60 nt, which would be too large to reside within the RNA fragments assayed for RT stops, or 3) regions within transcripts that were not expressed in mESCs at levels sufficient to be detected in our sequencing.
To benchmark our method using previously supported examples, nearly all of which are in human transcripts (19), we performed RT-stop profiling on mRNA from human cell lines. 12,009 and 12,035 RG4 regions (6,506 and 6,281 non-overlapping regions) were identified in the HEK293T and HeLa transcriptome, respectively, with 7,852 non-overlapping regions identified in at least one of the two cell lines, and 4,935 identified in both cell lines (table S2). Of the known RG4 regions within detected mRNAs, approximately half were detected as K+-dependent strong RT stops (fig. S4A and table S2).
Recently, a high-throughput method has been developed to identify genomic sequences that can fold into DNA G-quadruplexes in vitro (20). Because DNA and RNA of the same sequence often have distinct three-dimensional structures and some regions of DNA are either not expressed as RNA or are expressed as spliced transcripts that do not match the DNA, we expected that many DNA- or RNA-specific G4 regions would exist. Indeed, only 0.16% of the recently identified DNA G4 regions corresponded to RG4 regions found in HeLa and HEK293T cells, and of the non-overlapping HeLa/HEK293T RG4 regions that uniquely mapped to the human genome, only 19% mapped to identified DNA G4 regions (fig. S4B).
Compared to control regions with matched nucleotide composition, the identified RG4 regions were more likely to have the four G-triplets needed to match the canonical RG4 motif (fig. S3C), G≥3NxG≥3NxG≥3NxG≥3, in which each Nx represents a linker of any sequence ranging from 1 to ~7 nt in length (18). However, 37% of these regions had fewer than four G-triplets within 60 nt upstream of the RT stop (fig. S4C) and thus would be missed by most G4-searching algorithms (18). The 6,140 RG4 regions from mESCs were found in 2,792 transcripts (table S1), which included both mRNAs and noncoding RNAs, such as Malat1, which was sufficiently abundant to be analyzed despite its lack of a poly(A) tail. As previously predicted (18), RG4 regions were enriched within untranslated regions (UTRs) relative to mRNA coding sequences (CDSs) (Fig. 2E; p < 10−15, χ2 test), as might be expected if some of these regions have regulatory functions. However, G nucleotides within the 60-nt regions upstream of RT stops were not more conserved than G nucleotides within flanking regions (fig. S5), suggesting that the RG4 structure-forming capacity of most RG4 regions was not evolutionarily conserved.
In sum, RT-stop profiling identified thousands of RG4 regions in the mammalian transcriptomes, thereby expanding the catalog of experimentally supported endogenous RG4 regions by >100 fold. As predicted computationally (18), regions that form RG4 structures in vitro are not an esoteric feature of dozens of mRNAs but rather ubiquitous within mammalian transcriptomes, bringing to the fore the question of their in vivo folding status.
Globally unfolded RG4 regions in mESCs
To identify RG4 regions that are folded in cells, we combined RT-stop profiling with elements of DMS-seq (7) to develop to a method that measures, transcriptome-wide, the in vivo folding states of endogenous sequences with RG4-forming potential (Fig. 3A). In this method, cells are first treated with DMS, which rapidly enters and randomly methylates accessible N7 positions of G residues (4). RNA isolated from these cells is then subjected to RT-stop profiling. Although DMS modifies the N7 position of G more efficiently than it modifies the N1 and N3 positions of A and C residues, respectively (21), modification at N7 does not prevent Watson–Crick pairing and thus does not cause an RT stop. Nonetheless, RT-stop profiling can distinguish between RG4 regions that are folded in cells from those that are not because those that are folded in vivo are protected from modification at positions participating in the RG4 structure, enabling them to later refold during RT to generate RT stops, whereas those that are unfolded in cells can be irreversibly modified at residues that would otherwise participate in quadruplex formation in vitro, resulting in RT read-through and correspondingly attenuated RT stops (Fig. 3A).
Fig. 3.
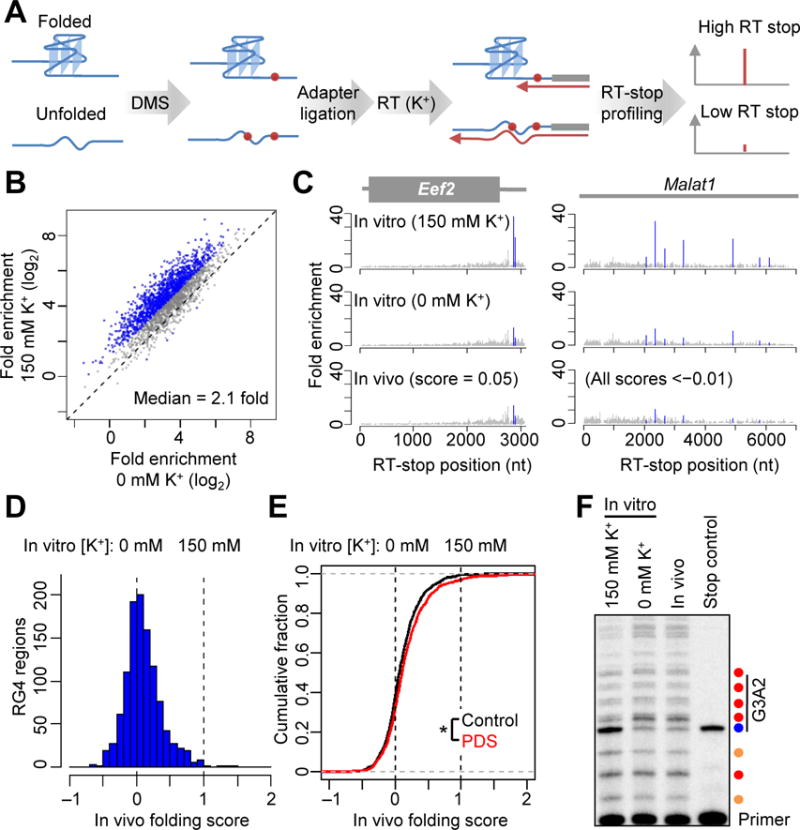
RG4 regions are unfolded in mESCs. (A) Schematic of transcriptome-wide probing of RG4 folding. (B) Probing RG4 folding in vitro. The RT-stop enrichment observed after DMS treatment of RNA folded in K+ (150 mM K+) was compared to that observed after DMS treatment of RNA folded without K+ (0 mM K+). Values for regions with differences of ≥2 fold are indicated (blue). (C) RT-stop profiles of Eef2 and Malat1, showing results observed after DMS treatment in vitro, either with or without K+, and those observed after DMS treatment in vivo. RT-stop enrichment values corresponding to RG4 regions are in blue; values for other RT stops at G nucleotides are in gray. In vivo folding scores for the RG4 regions are shown. (D) The distribution of in vivo folding scores of the 1,141 mESCs RG4 regions that were examined. The 0 and 1 reference values, which represent the signal observed when treating with DMS in vitro after folding with or without K+ (panel B), are marked (dashed lines). (E) Distribution of in vivo folding scores observed for RG4 regions after either treating the cells with 2 μM PDS for 24 hrs (PDS, red) or mock treatment (control, black). *, p < 10−8, paired t-test. (F) Gene-specific primer extension of the ectopically expressed G3A2 quadruplex probed in vitro (after folding in either 0 or 150 mM K+) or in vivo. Shown is a phosphorimage of a denaturing gel that resolved the extension products of a P33-radiolabeled primer. In the stop control, the β-mercaptoethanol quench was added to cells before DMS. The stronger RT stops are colored according to the nucleotide at the stall (A, red; C, orange; G, blue). The RG4 region is indicated (vertical line). For additional controls and sequencing ladders, see fig. S6E.
Reasoning that the RT-stop signals of different RG4 regions might have different sensitivities to DMS treatment, we first determined, for each RG4 region, the difference in RT-stop signal observed when mRNAs were modified in vitro either with or without K+. On average, the mESC RG4 regions refolded and DMS-treated in the presence of K+ had RT stops that were 2.5-fold stronger than those observed when refolding and treating in the absence of K+ (median 2.1 fold), and 1,342 regions had a difference of ≥2 fold (Fig. 3B and table S3). These in vitro results confirmed that DMS accessibility with readout from RT-stop profiling could indeed report on the folding states of many RG4 regions.
To probe the intracellular folding state of these regions, we treated mESCs with DMS and extracted poly(A)-selected RNAs for RT-stop profiling. As a positive control, results within the 5.8S rRNA were analyzed as a DMS-seq experiment (monitoring RT stops at A and C nucleotides), which showed that, as expected (7), DMS probing in vivo captured known Watson–Crick pairing within the 5.8S rRNA, as well as the intermolecular pairing between the 5.8S and 28S rRNAs (fig. S6A). Moreover, the RT-stop signals for RG4s were highly correlated between biological replicates (fig. S6B, Pearson’s r = 0.88). Inspection of the RG4 regions in both Eef2 mRNA and Malat1 indicated that these RG4 regions were accessible to DMS modification in vivo, as revealed by greatly reduced RT-stop signals (Fig. 3C). The signals observed for the in vivo-modified sample resembled those observed when omitting K+ from the in vitro folding and modification reaction, which indicted that these RG4 regions were unfolded in mESCs (Fig. 3C).
To infer the folding state, DMS-probing assays must be performed within their dynamic range; beyond this range, a transiently unfolded region might instead appear to be mostly unfolded, as most the molecules eventually become modified. The RT-stop signal at RG4 regions diminished in the unfolded reference (0 mM K+) but did not reach baseline (Fig. 3B and C), which indicated that our in vitro treatment left a fraction of these molecules unmodified and thus showed that our in vitro modification was within its dynamic range. Moreover, DMS modification of A’s and C’s in vivo resembled that observed for our in vitro references (fig. S6C), which indicated that our in vivo probing was also within the dynamic range of the assay.
We next expanded the analysis to 1,141 regions that retained a strong RT-stop signal (10 fold above background) when treated with DMS in the presence of K+ in vitro and had at least a 50% reduction in that signal when K+ was excluded. For each of these regions, an in vivo folding score was calculated in which the RT-stop signal observed in vivo was expressed relative to the range of signal observed in vitro, assigning scores of 1 and 0 to the signals observed in vitro with and without K+, respectively. In vivo folding scores for the 1,141 RG4 regions centered near 0 (median = 0.06) (Fig. 3D and table S3), which indicated that in mESCs, the folding of most RG4 regions resembled the unfolded state observed in vitro without K+. RG4 regions in 5′ UTRs, CDSs and 3′ UTRs, as well as those in noncoding RNAs, were similarly unfolded (fig. S6D). Treating mESCs with pyridostatin (PDS), a G4-stabilizing reagent (14, 15), induced a detectable but modest increase in global RG4 folding (0.04 increase in median folding score) (Fig. 3E; p < 10−8, paired t-test).
Although most RG4 regions are unfolded in mESCs, we cannot rule out the possibility that a few RG4 structures form in cells but could not be distinguished from experimental variability, or escaped our detection for other reasons, such as stable folding even in the absence of K+. An inability of DMS to penetrate the cell and modify the regions cannot be a source of false-negatives, as the decrease in the RG4-specific RT stops observed for RNA isolated from DMS-treated cells confirmed that DMS was indeed able to access and efficiently modify these regions in vivo. To confirm the unfolded state of RG4 regions with strong canonical motifs, we inserted the G3A2 quadruplex into an mRNA 3′ UTR, ectopically expressed the mRNA in HEK293T cells, and performed DMS modification followed by gene-specific primer extension. Again, the RT-stop pattern observed after DMS modification in vivo strongly resembled that observed after modifying in vitro without K+ (Fig. 3F and fig. S6E), further supporting the conclusion that RG4 regions are mostly unfolded in mammalian cells.
Globally unfolded RG4 regions in yeast cells
To determine whether the globally unfolded state of RG4 regions extends beyond mammalian cells, we applied our methods to the budding yeast Saccharomyces cerevisiae. We identified 744 strong RT stops within RNA isolated from exponentially growing yeast (table S4A), 133 of which were K+-dependent stops at G nucleotides. Among them, 47 showed ≥ 2-fold difference in RT-stop signal when comparing samples probed after folding with and without K+ (Fig. 4A and table S4B). The folding scores of endogenous RG4 regions centered near 0 (median = −0.15) (Fig. 4, B and C), again indicating a globally unfolded state. As observed in HEK293 cells, the ectopically expressed G3A2 quadruplex was also highly accessible to DMS, as indicated by an RT stop matching that observed for RNA modified in vitro without K+ (Fig. 4D). These results indicate that the globally unfolded state of RG4 regions is a broadly conserved feature of eukaryotic cells.
Fig. 4.
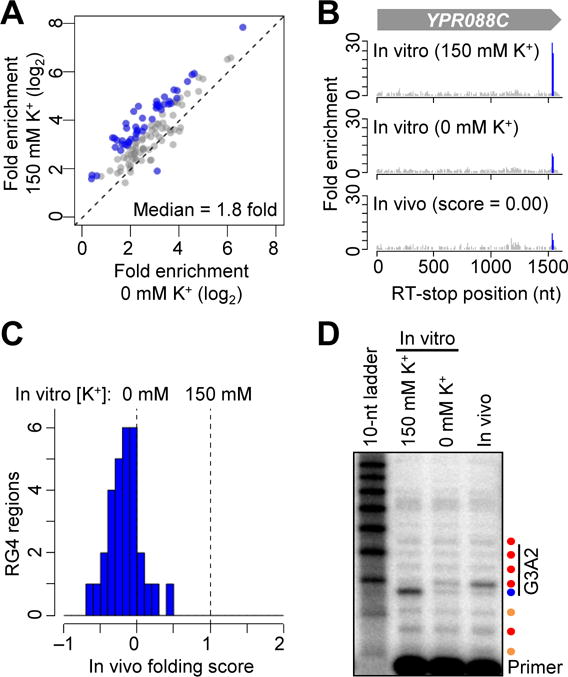
RG4 regions are unfolded in S. cerevisiae. (A) Probing in vitro folding of yeast RG4s. Otherwise, as in Fig. 3B. (B) RT-stop profiles of YPR088C. Otherwise, as in Fig. 3C. (C) The distribution of in vivo folding scores of the 31 RG4 regions that were examined in yeast. Otherwise, as in Fig. 3D. (D) Gene-specific primer extension of the ectopically expressed G3A2 quadruplex probed in vitro (after folding in either 0 or 150 mM K+) or in vivo. The primer was labeled with P32; otherwise, as in Fig. 3F.
SHAPE probing of RG4 regions
In addition to the chemical probes that modify the bases, such as DMS, probes that modify ribose 2′-hydroxyl groups with efficiency depending on the local chemical environment, known as SHAPE reagents, can provide useful tools for studying RNA structures (3, 5). Among these reagents, 2-methylnicotinic acid imidazolide (NAI) has been used to probe Watson–Crick RNA structures in cells (9, 22). To test whether NAI can also distinguish the folding states of RG4 regions, we used it to treat the G3A2 quadruplex folded in vitro with or without K+ and quantified its reactivity at each nucleotide using gene-specific primer extension, substituting Na+ for K+ in the RT reaction, so that modifications within RG4 regions could be detected (Fig. 5A). Whereas the formation of Watson–Crick structure typically decreases SHAPE reactivity, formation of the G3A2 quadruplex in the presence of K+ increased NAI reactivity (Fig. 5A). Furthermore, the enhanced reactivity occurred at the last G residue of each of the first three G tracts of the G3A2 quadruplex (Fig. 5A), consistent with a recent report describing in vitro NAI probing of two other G-quadruplexes (23). Perhaps the transition between a G-tract and a short loop in a parallel RG4 structure bends the RNA backbone to expose the 2′-hydroxyl of the last residue of the G tract (fig. S7A) (24). In vivo NAI treatment of the G3A2 quadruplex ectopically expressed in S. cerevisiae generated a modification pattern resembling that observed for this region folded in vitro without K+ (Fig. 5A), supporting the conclusion that this quadruplex is unfolded in yeast cells. Analogous results were observed for another model RG4, which had single-nucleotide U loops linking the G tracts (the G3U quadruplex) (fig. S7B).
Fig. 5.
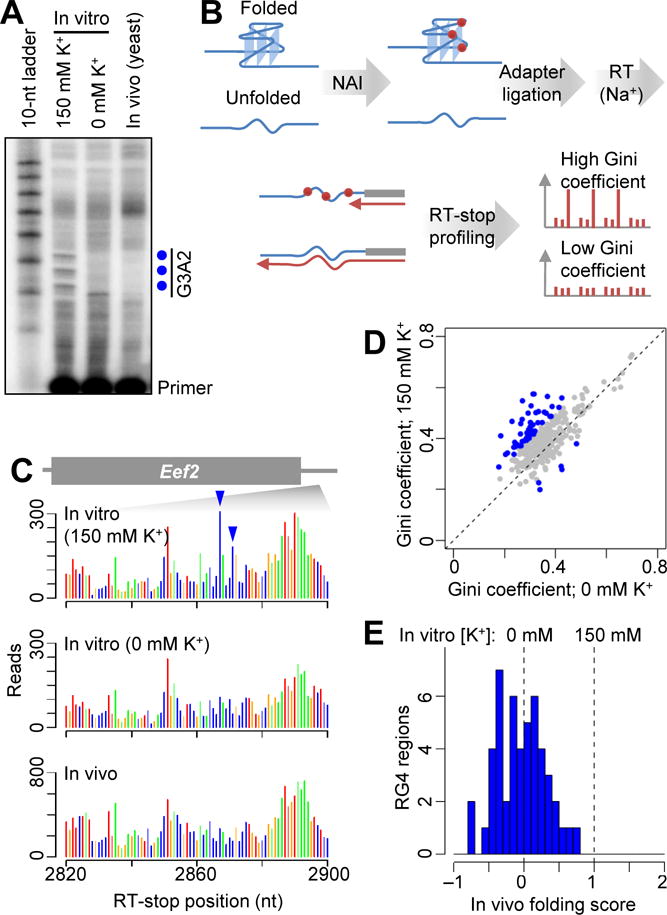
Probing RG4 folding using NAI. (A) Gene-specific primer extension of the ectopically expressed G3A2 quadruplex probed with NAI in vitro (after folding in either 0 or 150 mM K+) or in yeast. Shown is a phosphorimage of a denaturing gel that resolved the extension products of a P32-radiolabeled primer. RT stops corresponding to the preferential modifications of NAI within the G3A2 quadruplex are indicated (blue dots). (B) Schematic of transcriptome-wide probing of RG4 folding using NAI. (C) RT-stop profiles of an RG4 and its flanking regions within the Eef2 3′ UTR, showing raw read counts colored according to the identity of the template nucleotide at the stall (position 0). G residues preferentially modified after folding in K+ are indicated (blue arrowheads). (D) Comparison of Gini coefficients of the 310 RG4 regions with ≥100 RT stop reads in each of the two samples probed with NAI in vitro after folding in either 0 or 150 mM K+. Values for regions with differences of ≥0.1 between the two samples are indicated (blue). (E) Distribution of in vivo folding scores of the 49 RG4 regions that were examined using NAI probing. Otherwise, as in Fig. 3D.
To probe endogenous RG4 regions, we treated mESC RNA with NAI either in vitro (refolded with or without K+) or in vivo, and used RT-stop profiling with Na+ to determine the modification patterns (Fig. 5B). As with the G3A2 quadruplex, when folding endogenous RG4 regions in the presence of K+ in vitro, we observed preferential modification of the last G residue in G-tracts followed by short loops (Fig. 5C and fig. S7C). This pattern generated greater unevenness of modifications among G nucleotides within the RG4 region, which we quantified by calculating the Gini coefficient (7) for each of the 310 non-overlapping endogenous RG4 regions that had sufficient read coverage (≥100 RT-stop reads at G nucleotides in each sample). Among these, 49 had a ≥0.1 increase in Gini coefficient when comparing the modification observed in vitro after folding with K+ compared to that observed after folding without K+ (Fig. 5D). For these 49 regions, we calculated in vivo folding scores calibrated on the Gini-coefficient differences observed in vitro (table S5). As observed with the DMS probing, the distribution of in vivo folding scores centered near 0 (median = −0.02) (Fig. 5E), indicating that the in vivo NAI modification patterns of most RG4 regions resembled those of the unfolded state.
NAI probing complements DMS probing in three respects. First, NAI preferentially modifies specific residues of folded RG4s, whereas DMS modifies residues of unfolded RG4s. Second, NAI modification generates RT stops without requiring RG4 refolding, whereas DMS probing requires the refolding of RG4 structures in the presence of K+ to generate an RT stop. Third, NAI probing might detect less stable RG4 structures that do not stall RT in vitro and thereby escape identification using RT-stop profiling. However, unlike DMS probing of RG4 regions, NAI probing does not focus the signal onto a single RT-stop nucleotide, and it requires specific RG4 configurations, such as G-tracts followed by short loops, which also reduced the number of quantifiable RG4 regions. Nevertheless, the results from these two complementary chemical-probing methods both indicated that, despite the high intracellular K+ concentration, RG4 regions are overwhelmingly unfolded in eukaryotic cells.
Robust RG4 unfolding in eukaryotic cells
Our results in eukaryotic cells resembled those of recent high-throughput studies showing that Watson–Crick secondary structures that form in vitro are frequently unfolded in cells (7, 9), except the intracellular unfolding of RG4 regions was more pervasive. Whereas the previous studies identify many instances in which Watson–Crick structures do form in vivo, as expected from the known Watson–Crick pairing within ribosomal RNAs, tRNAs, pri-microRNAs, mRNAs, etc., we found no compelling evidence for the folding of an RG4 region in eukaryotic cells, which implies that these cells have a very effective molecular machinery that specifically remodels RG4s and maintains them in their unfolded state.
This remodeling presumably involves ATP-dependent processes. ATP depletion in yeast causes a global increase in Watson–Crick structures, suggesting that ATP-dependent processes, in particular ATP-dependent RNA helicases, play a major role in the cellular remodeling of these structures (7). Among the characterized ATP-dependent RNA helicases, DEAH box-containing helicase 36 (DHX36) accounts for most RG4-unfolding activity in HeLa cell extracts (25). To test whether DHX36 contributes to the globally unfolded state of RG4 regions in vivo, we applied DMS probing to mouse embryonic fibroblasts (MEFs) in which DHX36 was inducibly deleted through Cre-mediated recombination (26). The global distribution of folding scores was largely unchanged after DHX36 deletion, and values for individual RG4 regions were highly correlated before and after DHX36 deletion (fig. S8A–C), indicating that DHX36 was dispensable for the global unfolding of endogenous RG4 regions. We also tested whether ATP depletion affected RG4 folding and found that the ectopically expressed G3A2 quadruplex remained largely unfolded (fig. S8D). Although redundant functions with other helicases and the inability to completely deplete ATP might explain the negative results of these experiments, our results show that the mechanism responsible for remodeling RG4s and maintaining their unfolded state is robust to either the deletion of a key helicase known to unfold RG4 structures or the substantial depletion of ATP.
Folding of RG4 structures in bacteria
We next applied our methods to bacterial transcriptomes. Compared to the mammalian transcriptome, the E. coli transcriptome was substantially depleted in regions with K+-dependent strong RT stops (Fig. 6A and table S6). Only 35 K+-dependent strong RT stops were identified in E. coli, of which only 14 (40%) were at G nucleotides. Among these 14, none had differential DMS accessibility when comparing the results of in vitro modification with and without K+ (table S6). Similar depletion was observed within the transcriptomes of the other two bacteria we examined, Pseudomonas putida and Synechococcus sp WH8102 (Fig. 6A and table S6), even though their genomes are more G-rich than mammalian genomes. Only one region within the transcriptomes of these two species passed our cutoffs for calculating an in vivo folding score (a P. putida region with a folding score of 0.5, table S6).
Fig. 6.
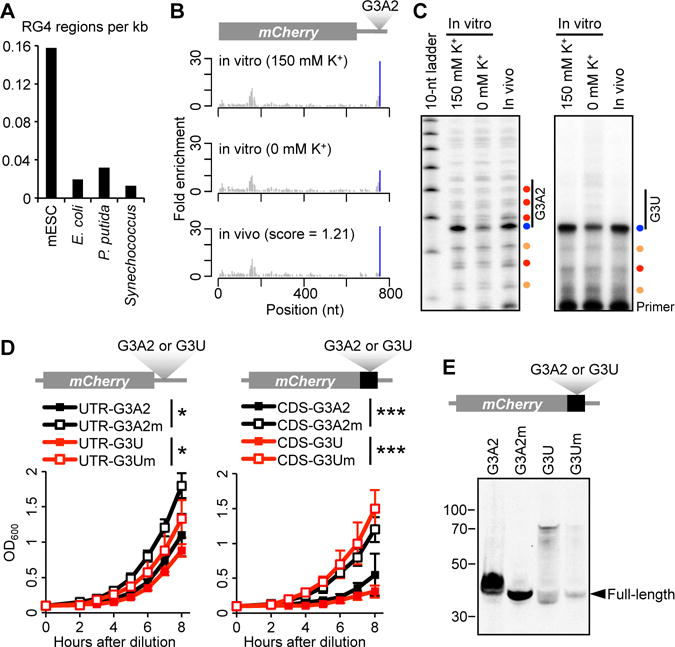
RG4 folding and interference with growth and translation in E. coli. (A) Density of RG4 regions in bacterial and mESC transcriptomes. For each species, the number of RG4 regions, as identified from K+-dependent strong RT stops at G nucleotides, was normalized to the total length of all detected transcripts. (B) RT-stop profiles of an ectopically expressed mCherry mRNA with an G3A2 quadruplex inserted into its 3′ UTR, showing results observed after DMS treatment in vitro, either with or without K+, and in vivo. Otherwise, as in Fig. 3C. (C) Gene-specific primer extension of ectopically expressed G3A2 (left, P33-labeled primer) and G3U (right, P32-labeled primer) quadruplexes probed with DMS. Otherwise, as in Fig. 3F. (D) Growth curves of strains expressing mCherry transcripts with the indicated RG4 (G3A2 or G3U) or RG4 mutant (G3A2m or G3Um) in either the 3′ UTR (left) or the CDS (right). Growth was monitored using optical density at a wavelength of 600 nm (OD600). Plotted are mean values ± SD (n = 6). *, p < 0.05; *** p < 0.001; Student’s t tests using measurements at the last time point. (E) Immunoblot probed for the translation products of the mCherry constructs with either the indicted RG4 or its respective mutant inserted between the mCherry sequence and the stop codon. Mobilities of molecular-weight markers and the full-length products are shown.
Having acquired evidence for only a single, weak RG4 region in endogenously expressed bacterial RNA, we ectopically expressed the G3A2 quadruplex within the 3′ UTR of an mCherry transcript and probed its folding state in E. coli. In contrast to our results in eukaryotic cells, the strong RT stop corresponding to the G3A2 quadruplex was resistant to in vivo DMS modification, indicating that this region was folded in E. coli cells (Fig. 6B and C). Likewise, the G3U quadruplex, was also folded in E.coli (Fig. 6C). Although intracellular NAI probing of the G3A2 quadruplex was inconclusive, intracellular NAI probing of the G3U quadruplex generated the modification pattern specific to that of the folded G3U quadruplex (fig. S9), confirming that RG4 folding rather than protein binding protected the region from DMS modification in vivo. Thus, RG4 regions are permitted to fold in E.coli but are strongly depleted among endogenous E.coli RNAs.
To understand this depletion, we compared the growth of strains that expressed G3A2 or G3U quadruplexes to those of strains that expressed the corresponding quadruplex mutants in which point substitutions abolished RG4-forming capacity and found that the RG4-expressing strains grew more slowly than the corresponding mutant-expressing strains (Fig. 6D). Moreover, these growth defects were exacerbated after introducing stop-codon mutations that caused the mCherry coding sequence to extend through the RG4 regions (Fig. 6D). Although effects from the RG4 regions in UTRs might be attributable to either RNA or DNA quadruplex formation, the enhanced growth defects observed after introducing stop-codon mutations were attributable to only RG4 structures.
To determine the influence of folded RG4 structures on translation, we examined the translation products from each of the strains. Consistent with a previous study (27), RG4 regions downstream of the stop codon did not substantially influence mCherry production. In contrast, the G3A2 quadruplex upstream of the stop codon caused read-through of the stop codon and/or frame-shifting, generating polypeptides that were longer than expected (Fig. 6E). The G3U quadruplex also perturbed translation, causing the production of both longer and shorter polypeptides (Fig. 6E). The products of the expected size dominated when mutant RG4 regions were placed upstream of the stop codon, which indicated that the aberrant translation products were primarily the consequence of stable RG4 structures.
Discussion
The mammalian, yeast, and bacterial cells that we studied all strongly avoid the presence of folded RG4 structures in their transcriptomes but do so through different mechanisms. Based on our in vivo probing, the eukaryotic cells appear to have a robust and effective molecular machinery that specifically unfolds and maintains the thousands of RG4 regions in an unfolded state, whereas bacteria lack this machinery and have instead eliminated sequences with RG4-forming potential over the course of evolution. When considering the impaired growth rates observed for strains ectopically expressing RG4 regions, the bacterial mechanism is easy to understand, but how might the eukaryotic mechanism act? Although the critical factors remain to be identified, this mechanism differs from that which unfolds Watson–Crick structure in two key aspects. First, it is less sensitive to ATP depletion, and second, it is more pervasive, unfolding essentially every RG4 that could be monitored in mESCs, MEFs and yeast, whereas the activities that unfold Watson–Crick structure allow many RNAs to remain folded.
We suspect that single-stranded RNA-binding proteins lie at the center of the mechanism that unfolds most eukaryotic RG4 regions. A wide variety of abundant RNA-binding proteins bind to G-rich RNA, including the hnRNP F/H family (28, 29), hnRNP D0 (30), hnRNP M (31), hnRNP A/B (32), hnRNP A1 (33, 34), hnRNP A2 (35), CBF-A (35), and SRSF1/2 (36). The solution structures of the three quasi-RNA recognition motifs (qRRMs) of hnRNP F in complex with G-tract RNA show how qRRMs could maintain G-tracts in a single-stranded conformation without blocking solvent accessibility to the N7 positions (37), which is consistent with our DMS probing results. Regardless of the identity of the machinery that operates in eukaryotic cells, it must be acting broadly throughout the transcriptome, including on untranslated RNAs and nuclear RNAs, as illustrated by the unfolding of RG4 regions within Malat1 (Fig. 3C), a nuclear noncoding RNA.
The evolutionary depletion of RG4 regions might be more tenable for bacteria than for eukaryotes for two reasons. First, maintaining machinery dedicated to the remodeling of RG4s would be more costly for species under greater selective pressure to minimize their genomes. Second, species with smaller genomes would face less frequent de novo emergence of new RG4 regions. On the other hand, the eukaryotic mechanism provides opportunities for regulation. Indeed, the relative enrichment of RG4-forming regions in untranslated regions hints at the possibility that RG4 regions might be allowed to fold and impart regulatory functions in certain cell types/states or subcellular compartments (38). Alternatively, these regions might impart function through transient folding that cannot be detected in our steady-state measurements. Another possibility is that the previously reported regulatory roles of RG4 regions, such as translational repression by RG4 regions within 5′ UTRs (10), might result from the stable association of the RNA-binding proteins that maintain the RG4 regions in the unfolded state. In this scenario, the bound proteins rather than a folded RG4 would inhibit translation initiation. Clearly, more needs to be learned about this RNA structure in its native cellular contexts, and our results and methods provide the framework for doing so.
Materials and Methods
In vivo DMS modification
DMS (Sigma-Aldrich; 50% diluted with ethanol) was added to mESCs or HEK293T cells cultured in 15 cm dishes to a final concentration of 8%, and evenly distributed by slow swirling. After incubating at 37°C for 5 minutes, the media and excess DMS were decanted, and cells were washed twice with 25% β-mercaptoethanol (Sigma-Aldrich) in PBS to quench any residual DMS. After washing, cells were lysed in 10 mL TRIzol reagent (Invitrogen) supplemented with 5% β-mercaptoethanol, and lysates were stored at −80°C. DMS was added to 10 ml of yeast culture to a final concentration of 8%. After incubating at 30°C with continuous shaking for 5 minutes, two volumes of 25% β-mercaptoethanol were added to the culture to stop the modification. Cells were harvested by centrifugation at 4,000 rpm for 5 minutes and washed with 25% β-mercaptoethanol until no residual DMS was observed at the bottom of tubes. After the final centrifugation, cells were resuspended in RNAlater solution (Invitrogen) and stored at −80°C. DMS treatment of the E. coli culture was similar to that of the yeast culture, except it was performed at 37°C. For additional details on culture of and transfection of mammalian cells, culture and induction of yeast cells, culture and induction of bacteria, and construction of RG4 expression constructs, see the supplementary materials (SM).
In vitro folding and DMS modification
Poly(A)-selected RNA in 1 mM Mg2+ and 50 mM Tris-Cl (pH 7.0), either with or without 150 mM K+, was heated to 80°C for 2 minutes and then rapidly cooled to 0°C for 1 minute. DMS was added to a final concentration of 8% and the mixture was incubated at either 37°C (mammalian and E. coli RNA) or 30°C (yeast RNA) for 5 minutes with constant mixing. Two volumes of 25% β-mercaptoethanol were added to stop the reaction before RNA was phenol-chloroform extracted and precipitated. For details on RNA purification, see SM.
RT-stop profiling
The DMS-seq protocol (7) was adapted to detect RT stops in unmodified RNA. 1 μg poly(A)-selected RNA in 10 mM Tris-Cl (pH 7.5) was denatured at 95°C for 2 minutes, supplemented with RNA-fragmentation reagent (Ambion) and incubated at 95°C for additional 1 minute before adding EDTA stop solution (Ambion). After ethanol precipitation, RNA fragments were dephosphorylated at their 3′ ends using T4 polynucleotide kinase (New England BioLabs). 60–80-nt RNA fragments were gel-purified and ligated to a pre-adenylated 3′ DNA adapter (AppTCGTATGCCGTCTTCTGCTTGddC) using T4 RNA ligase 1 (New England BioLabs) without ATP. Products of the expected size (82–102 nt) were gel-purified and resuspended in 6 μl water. For reverse transcription, 1 μl 0.2 M Tris-Cl (pH 7.5), 1 μl 1.5 M KCl (or NaCl or LiCl), 0.5 μl 60 mM MgCl2, 0.5 μl 10 mM dNTP mix and 0.5 μl 1 μM 5′-radiolabeled primer (32p-NNNNNNGATCGTCGGACTGTAGAACTCTGAACCTGTCG/iSp18/CAAGCAGAAGACGGCATACG, in which N is any nucleotide, and iSp18 is an 18-atom hexa-ethyleneglycol spacer, IDT) were added to the RNA template. The mixture was incubated at 80°C for 2 minutes then cooled down to 42°C and incubated for additional 2 minutes before adding 100 U SuperScript III reverse transcriptase (Invitrogen). After incubation at 42°C for 10 minutes, the reaction was stopped with addition of 1 μl 1 M NaOH, and the mixture was heated at 98°C for 15 minutes to hydrolyze the RNA. cDNAs from extension that stalled after addition of 20–45 nt were separated from primers and full-length cDNAs on a 10% urea gel, eluted and precipitated. Purified cDNA fragments were circularized using 50 U CircLigase (Epicentre) at 60°C for 4 hours before inactivation at 80°C for 10 minutes. Circularized cDNAs were amplified using a 5′ indexed primer (AATGATACGGCGACCACCGACAGGTTGGAATTCTCGGGTGCCAAGGAACTCCAGTCACxxxxxxATCCGACAGGTTCAGAGTTCTACAGTCCGA, in which xxxxxx is the multiplexing index), a common 3′ primer (CAAGCAGAAGACGGCATACGA), and Platinum Taq DNA Polymerase High Fidelity (Invitrogen) for 10–13 cycles of PCR. Libraries were purified on an 8% formamide gel and sequenced on a HiSeq 2000 sequencing machine (Illumina; 40 cycles, single-end mode). For details on transcript-specific analyses of model RG4 regions using primer-extension assays, see SM.
Analysis of sequencing reads
For each read that uniquely mapped to the cognate transcriptome, the nucleotide immediately upstream of the first aligned position was annotated as an RT stop. At each position of the transcriptome with ≥ 3 RT-stop reads, a fold-enrichment value (f) for RT stops was calculated as the ratio between the number of reads stalled at that position and the background read density, which was the average number of reads over all positions of the same nucleotide within the same transcript (e.g., all G nucleotides). RT stops with ≥10 reads and fold enrichment values ≥20 were designated strong stops (fig. S1). When calculating the fold enrichment values for negative-control samples (Li+, Na+, and 95°C DMS), the position under consideration was assigned a pseudo read count of 1 if it had no RT-stop reads (with no change to the background read density). Strong RT stops for which enrichment decreased by more than 50% in 150 mM Na+ compared to 150 mM K+ were designated K+-dependent. For details on read mapping, see SM.
NAI probing
NAI was synthesized as described (22) and stored as a 1M solution in DMSO at −80°C. For treatment in vivo, mESCs and yeast cells were treated with 80 mM NAI for 15 minutes at 37°C and 30°C, respectively, and washed three times with PBS before RNA extraction and poly(A) selection. For treatment in vitro, poly(A)-selected RNA in 1 mM Mg2+ and 50 mM Tris-Cl (pH 7.0), either with or without 150 mM K+, and was heated to 80°C for 2 minutes and then rapidly cooled to 0°C for 1 minute. This refolded RNA was treated with 80 mM NAI for 5 minutes at either 37°C (mammalian RNA) or 30°C (yeast RNA). After treatment, RNA was phenol-chloroform extracted and precipitated. NAI-treated RNA was subjected to RT-stop profiling, using 150 mM Na+ instead of 150 mM K+ during primer extension. Gini coefficients were calculated for each non-overlapping RG4-containing region (identified as 60-nt regions upstream of K+-dependent strong RT stops) as
where n denotes the number of G residues in the RG4 region, and ri denotes the RT-stop read number at position i.
Calculation of in vivo folding scores
For each RG4 region that retained a strong RT-stop signal (10 fold above background) when treated with DMS in the presence of K+ in vitro and had at least a 50% reduction in that signal when K+ was excluded, an in vivo folding score (s) was calculated as
For the regions that had ≥100 RT-stop reads at G nucleotides after NAI probing and a difference of ≥ 0.1 in Gini coefficients when comparing results of RNA folded in vitro with K+ to those of RNA folded in vitro without K+, an in vivo folding score (s) was calculated as
Although folding scores were calculated using linear functions, the conclusions of this study were not dependent on a linear relationship between the fraction of folded molecules and the extent of DMS or NAI modification.
Supplementary Material
Acknowledgments
We thank S. Rouskin and members of the Bartel lab for helpful discussions, C. Kayatekin, G. Johnson, and K. Heindl for experimental assistance, J. S. Yoo, T. Fujita and Y. Nagamine for the Dhx36 cell lines, S. Chisholm, S. Biller, and K. Dooley for the Synechococcus culture. This work was supported by NIH grant GM118135 (D.P.B.). J.U.G. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-2152-13). D.P.B. is an investigator of the Howard Hughes Medical Institute. Sequencing data were deposited in GEO (accession number GSE83617).
Footnotes
References and Notes
- 1.Mortimer SA, Kidwell MA, Doudna JA. Insights into RNA structure and function from genome-wide studies. Nat Rev Genet. 2014 Jul;15:469. doi: 10.1038/nrg3681. [DOI] [PubMed] [Google Scholar]
- 2.Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011 Sep;12:641. doi: 10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weeks KM. Advances in RNA structure analysis by chemical probing. Curr Opin Struct Biol. 2010 Jun;20:295. doi: 10.1016/j.sbi.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wells SE, Hughes JM, Igel AH, Ares M., Jr Use of dimethyl sulfate to probe RNA structure in vivo. Methods Enzymol. 2000;318:479. doi: 10.1016/s0076-6879(00)18071-1. [DOI] [PubMed] [Google Scholar]
- 5.Kubota M, Tran C, Spitale RC. Progress and challenges for chemical probing of RNA structure inside living cells. Nat Chem Biol. 2015 Dec;11:933. doi: 10.1038/nchembio.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding Y, et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014 Jan 30;505:696. doi: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- 7.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014 Jan 30;505:701. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loughrey D, Watters KE, Settle AH, Lucks JB. SHAPE-Seq 2.0: systematic optimization and extension of high-throughput chemical probing of RNA secondary structure with next generation sequencing. Nucleic Acids Res. 2014 Dec 1;42 doi: 10.1093/nar/gku909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spitale RC, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015 Mar 26;519:486. doi: 10.1038/nature14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bugaut A, Balasubramanian S. 5′-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res. 2012 Jun;40:4727. doi: 10.1093/nar/gks068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Millevoi S, Moine H, Vagner S. G-quadruplexes in RNA biology. Wiley Interdiscip Rev RNA. 2012 Jul-Aug;3:495. doi: 10.1002/wrna.1113. [DOI] [PubMed] [Google Scholar]
- 12.Wolfe AL, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014 Sep 4;513:65. doi: 10.1038/nature13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haeusler AR, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014 Mar 13;507:195. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biffi G, Di Antonio M, Tannahill D, Balasubramanian S. Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat Chem. 2014 Jan;6:75. doi: 10.1038/nchem.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwok CK, Balasubramanian S. Targeted Detection of G-Quadruplexes in Cellular RNAs. Angew Chem Int Ed Engl. 2015 Jun 1;54:6751. doi: 10.1002/anie.201500891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peattie DA. Direct chemical method for sequencing RNA. Proc Natl Acad Sci U S A. 1979 Apr;76:1760. doi: 10.1073/pnas.76.4.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vorlickova M, et al. Circular dichroism and guanine quadruplexes. Methods. 2012 May;57:64. doi: 10.1016/j.ymeth.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 18.Huppert JL, Bugaut A, Kumari S, Balasubramanian S. G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res. 2008 Nov;36:6260. doi: 10.1093/nar/gkn511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garant JM, Luce MJ, Scott MS, Perreault JP. G4RNA: an RNA G-quadruplex database. Database (Oxford) 2015;2015 doi: 10.1093/database/bav059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chambers VS, et al. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat Biotechnol. 2015 Aug;33:877. doi: 10.1038/nbt.3295. [DOI] [PubMed] [Google Scholar]
- 21.Lawley PD, Brookes P. Further Studies on the Alkylation of Nucleic Acids and Their Constituent Nucleotides. Biochem J. 1963 Oct;89:127. doi: 10.1042/bj0890127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spitale RC, et al. RNA SHAPE analysis in living cells. Nat Chem Biol. 2013 Jan;9:18. doi: 10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwok CK, Sahakyan AB, Balasubramanian S. Structural Analysis using SHALiPE to Reveal RNA G-Quadruplex Formation in Human Precursor MicroRNA. Angew Chem Int Ed Engl. 2016 Jul 25;55:8958. doi: 10.1002/anie.201603562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collie GW, Haider SM, Neidle S, Parkinson GN. A crystallographic and modelling study of a human telomeric RNA (TERRA) quadruplex. Nucleic Acids Res. 2010 Sep;38:5569. doi: 10.1093/nar/gkq259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Creacy SD, et al. G4 resolvase 1 binds both DNA and RNA tetramolecular quadruplex with high affinity and is the major source of tetramolecular quadruplex G4-DNA and G4-RNA resolving activity in HeLa cell lysates. J Biol Chem. 2008 Dec 12;283:34626. doi: 10.1074/jbc.M806277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo JS, et al. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog. 2014 Mar;10:e1004012. doi: 10.1371/journal.ppat.1004012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holder IT, Hartig JS. A matter of location: influence of G-quadruplexes on Escherichia coli gene expression. Chem Biol. 2014 Nov 20;21:1511. doi: 10.1016/j.chembiol.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Matunis MJ, Xing J, Dreyfuss G. The hnRNP F protein: unique primary structure, nucleic acid-binding properties, and subcellular localization. Nucleic Acids Res. 1994 Mar 25;22:1059. doi: 10.1093/nar/22.6.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caputi M, Zahler AM. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H′/F/2H9 family. J Biol Chem. 2001 Nov 23;276:43850. doi: 10.1074/jbc.M102861200. [DOI] [PubMed] [Google Scholar]
- 30.Nagata T, et al. Structure and interactions with RNA of the N-terminal UUAG-specific RNA-binding domain of hnRNP D0. J Mol Biol. 1999 Mar 26;287:221. doi: 10.1006/jmbi.1999.2616. [DOI] [PubMed] [Google Scholar]
- 31.Datar KV, Dreyfuss G, Swanson MS. The human hnRNP M proteins: identification of a methionine/arginine-rich repeat motif in ribonucleoproteins. Nucleic Acids Res. 1993 Feb 11;21:439. doi: 10.1093/nar/21.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar A, Sierakowska H, Szer W. Purification and RNA binding properties of a C-type hnRNP protein from HeLa cells. J Biol Chem. 1987 Dec 15;262:17126. [PubMed] [Google Scholar]
- 33.Zhang QS, Manche L, Xu RM, Krainer AR. hnRNP A1 associates with telomere ends and stimulates telomerase activity. Rna. 2006 Jun;12:1116. doi: 10.1261/rna.58806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burd CG, Dreyfuss G. RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. Embo J. 1994 Mar 1;13:1197. doi: 10.1002/j.1460-2075.1994.tb06369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khateb S, Weisman-Shomer P, Hershco-Shani I, Ludwig AL, Fry M. The tetraplex (CGG)n destabilizing proteins hnRNP A2 and CBF-A enhance the in vivo translation of fragile X premutation mRNA. Nucleic Acids Res. 2007;35:5775. doi: 10.1093/nar/gkm636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Expert-Bezancon A, et al. hnRNP A1 and the SR proteins ASF/SF2 and SC35 have antagonistic functions in splicing of beta-tropomyosin exon 6B. J Biol Chem. 2004 Sep 10;279:38249. doi: 10.1074/jbc.M405377200. [DOI] [PubMed] [Google Scholar]
- 37.Dominguez C, Fisette JF, Chabot B, Allain FH. Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. Nat Struct Mol Biol. 2010 Jul;17:853. doi: 10.1038/nsmb.1814. [DOI] [PubMed] [Google Scholar]
- 38.Subramanian M, et al. G-quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO Rep. 2011 Jul;12:697. doi: 10.1038/embor.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.