

Abstract
To develop an effective strategy to isolate and use cell-free fetal DNA (cffDNA) for the combined use of next-generation sequencing (NGS) for diagnosing choroideremia and non-invasive prenatal testing (NIPT) for Y chromosome determination, a large Chinese family with an X-linked recessive disease, choroideremia, was recruited. Cell-free DNA was extracted from maternal plasma, and SRY polymerase chain reaction amplification was performed using NIPT. Sanger sequencing was subsequently used for fetal amniotic fluid DNA verification. A nonsense mutation (c.C799T:p.R267X) of the CHM gene on the X chromosome of the proband (IV:7) and another 5 males with choroideremia were detected, while 3 female carriers with no symptoms were also identified. The fetus (VI:7) was identified as female from the cffDNA, and the same heterozygous nonsense mutation present in her mother was also confirmed. At one and a half years of age, the female baby did not present with any associated symptoms of choroideremia. Therefore, cffDNA was successfully used for the combined use of NGS for diagnosing choroideremia in a large Chinese pedigree, and NIPT for Y chromosome determination. This approach should result in a markedly increased use of prenatal diagnosis and improvement, and more sophisticated clinical management of diseases in China and other developing countries. The establishment of a highly accurate method for prenatal gene diagnosis will allow for more reliable gene diagnosis, improved genetic counseling, and personalized clinical management of our patients.
Keywords: choroideremia, DNA mutation, next-generation sequencing, non-invasive prenatal testing, cell-free fetal DNA, SRY gene
Introduction
Choroideremia (CHM, OMIM: 303,100) is an X-linked recessive chorioretinal dystrophy characterized by progressive degeneration of the choroids, the retinal pigment epithelium, and the retinal photoreceptors. Its incidence is estimated to be between 1 in 50,000 and 1 in 100,000 people (1). However, it is likely that choroideremia is under-diagnosed due to its similarities to other eye disorders, such as retinitis pigmentosa (RP) (1,2). Affected males initially exhibit symptoms of night blindness by 10–20 years of age, followed by progressive peripheral visual field loss, with complete blindness occurring by age 40 in certain patients (3–5). Carrier females are usually asymptomatic; however, certain individuals do develop night blindness and/or visual field loss (6).
Choroideremia is caused by mutations in the CHM gene (OMIM: 300,390), which consists of 15 exons spanning at least 150 kb and is located at Xq21.2-q21.3 (7). The CHM gene encodes Rab escort protein 1 (REP1), a subunit of the dimeric holoenzyme, Rab geranylgeranyl transferase, which attaches 20-carbon isoprenoid groups to the cysteine residues of Rab proteins, a family of GTP-binding proteins that regulate vesicular traffic. Rab geranylgeranyl transferase is an enzyme complex that mediates correct intracellular vesicular transport (8). REP1 is composed of 653 amino acids and a single 95-kDa polypeptide (9). Cremers et al (10,11) first identified that the CHM gene is the disease-causing gene that, when partially deleted or disrupted in male patients with tapetochoroidal dystrophy (choroideremia, OMIM: 303,100), leads to choroideremia. Thus far, there are at least 195 CHM gene mutations reported in the Human Genome Mutation database (HGMD), and various methods were used to detect these CHM gene variants. Direct sequence analysis for identifying choroideremia-causing variants results in a detection rate of ~60–95%; however, this is recommended as the first investigative option (6,12,13), even though next-generation sequencing (NGS) will result in an almost 100% detection rate (14).
Human gene replacement clinical trials for choroideremia are currently under way. Initially, these phase 1/2 trials used an adeno-associated viral vector (AAV2) to deliver the REP1 gene directly to the retina, which improved visual outcomes in six individuals (15). With the increased likelihood that individuals affected with choroideremia may benefit from gene therapy trials or gene-specific treatments in the near future, the genetic confirmation of this disease, and identification of the specific variants in clinically prenatal diagnosed individuals, is increasingly a high priority for reproductive health in China and other developing countries.
Prenatal diagnosis is used to detect whether the fetus will suffer from a genetic disease after birth. Traditional invasive methods of sampling fetal materials include amniocentesis, chorionic villus sampling, umbilical cord blood sampling, and so forth (16–18), whereas non-invasive prenatal testing (NIPT) involves testing fetal genetic material through isolation of cell-free fetal DNA (cffDNA) from maternal peripheral blood, plasma, or serum. Using cffDNA for prenatal diagnosis to replace or complement existing invasive methods may greatly reduce the pain and the risk of a miscarriage; therefore, there is increased interest in using NIPT.
Herzenberg et al (19) first isolated fetal nucleated red blood cells using flow cytometry, but no efficient prenatal diagnosis was available at the time due to the limited amount of usable sample recovered. The use of NIPT did not become promising until Lo et al (20) confirmed that cffDNA exists in maternal peripheral blood. Although the concentration of cffDNA is extremely low in maternal peripheral blood (3–6%) (21), the goal of the present authors was to establish an effective method to measure the concentrations of cffDNA at different stages of gestation, and to optimize NIPT (22).
The cffDNA in NIPT is mainly used in the clinic for prenatal identification of X-linked disorders, including hemophilia, Huntington's disease, Duchenne muscular dystrophy, congenital adrenal cortical hyperplasia prenatal screening of fetal chromosomal aneuploidy, and detection of the Rhesus (Rh) factor (21,23–27). Maternal plasma cell-free DNA is mixed with maternal and fetal DNA, of which fetal DNA represents a minor portion of the total. The SRY gene is a sex-determining gene on the Y chromosome, which only exists in males. Using cffDNA from maternal plasma for fetal gender determination is mainly limited to Y chromosome sequences absent in the maternal genome, such as the SRY. Thus, the only way to identify those sequences is through male-bearing pregnancies (21).
Prenatal genetic diagnosis is often used when there is a family history of sex-linked diseases. Most sex-linked diseases are recessive X-linked and caused by a particular gene mutation on the X chromosome, such as choroideremia. The disease is normally manifested only in males, who carry a single X chromosome, whereas, in females, the normal allele on the second X chromosome compensates for the diseased allele (28). Using cffDNA for the combined use of NGS for detecting choroideremia mutations and NIPT for Y chromosome determination is novel.
The aim of the present study was to genetically identify variants of the choroideremia-causing gene by NGS, and to analyze the SRY gene using NIPT for the presence of X-linked recessive disease in a large family cohort diagnosed with choroideremia.
Materials and methods
Collection for samples
The present research study was approved by the Southwest Medical University review board, and was conducted according to the principles of the Declaration of Helsinki. Informed consent was obtained from all participants. Full medical and family histories were collected, a pedigree was formed, and an ophthalmologic examination that included wide-field fundus photography, wide-field autofluorescence, spectral domain optical coherence tomography, fundus photography, and eye/orbit ultrasound was performed.
Pregnant women signed informed consent forms for approval of the prenatal diagnosis. Methods for DNA extraction from the peripheral blood of associated family members and the amniotic fluid from the fetus were described previously (22,29,30). Briefly, 1X Blood Lysis buffer was added into peripheral blood on ice for 30 min, and centrifuged for 10 min at 1,600 × g. The filtrate was discarded, 1 ml Nucleic Lysis Buffer, 100 µl 20% SDS and 10 µl Protease K (20 mg/ml; Roche Diagnostics, Indianapolis, IN, USA) were added, and the mixture was incubated at 56°C for 4 h. An equivalent volume of phenol was added, mixed, and centrifuged for 10 min at 1,600 × g. Subsequently, a repeat step was performed using an equivalent volume of phenol and chloroform. The supernatant was retained, and 2.5 vols absolute ethyl alcohol was added to precipitate the DNA. Finally, the precipitated DNA was dissolved in 1X TE buffer. For DNA extraction from amniotic fluid, it was necessary to move directly to the Nucleic Lysis Buffer treatment step following centrifugation of the amniotic fluid, and then the identical procedure was followed with respect to DNA extraction from the peripheral blood.
cffDNA isolation from plasma and nested polymerase chain reaction (PCR)
Blood samples from the peripheral blood of pregnant women were collected in 2–3 ml EDTA-K2 anticoagulative tubes. Plasma separation from these blood samples was performed using a two-step centrifugation method, as previously described (22). Then, 20 µl Protease K (20 mg/ml) and 200 µl plasma were mixed, and centrifuged briefly after adding 200 µl Buffer AL (Qiagen, Dusseldorf, Germany). The solution was incubated at 56°C for 10 min, centrifuged briefly, 200 µl absolute ethyl alcohol was added, mixed, and subsequently transferred to the spin columns (Qiagen). The spin columns were centrifuged for 1 min at 6,000 × g, 500 µl Buffer AW2 (Qiagen) was added prior to centrifugation for 3 min at 20,000 × g, and then the filtrate was discarded. Next, the spin columns were centrifuged for 1 min at 20,000 × g, and the DNA was eluted with 60 µl Buffer AE (Qiagen), separated into aliquots and stored at −80°C prior to use.
The amplification of the SRY gene (Y chromosomal material) using nested PCR was performed as previously described (22). The primers used for this nested PCR were as follows: SRY-138 forward (F) (5′-TACAGGCCATGCACAGAGAG-3′) and SRY-138 reverse (R) (5′-TGTTGTCCAGTTGCACTTCG-3′) (first round); and SRY-116F (5′-GCACAGAGAGAAATACCCGAAT-3′) and SRY-116R (5′-GCACTTCGCTGCAGAGTACC-3′) (second round).
Amplification of DNA of the amniotic fluid
Amplification reactions were set up in a reaction volume of 10 µl (29,30). Each reaction contained 50 ng DNA, 300 nmol each gene specific primer for SRY or CHM, 5 µl 2X PCR MasterMix (Tiangen Biotech Co., Ltd., Beijing, China), and doubly distilled (dd)H2O. The following primer sets were used for these reactions: XES7 (5′-CCCGAATTCGACAATGCAATCATATGCCCC0CCC1-3′) and XES2 (5′-CTGTAGCGGTCCCTGTGCTGCGGTG-3′), or CHM7L (TGG GAG AAA AGG ATT TGT GTG) and CHM7R (ATG GAT CAG GTT TTG CTG CT), which produced a 609 or 637 base pair (bp) amplification product for the SRY or CHM gene, respectively. The following PCR program was used: An initial denaturation step at 95°C for 90 sec, followed by 34 cycles, each consisting of a denaturation step at 94°C for 30 sec, an annealing step at 65 or 62°C for 30 sec, and an extension step at 72°C for 25 sec, followed by an extension step at 72°C for 2 min.
Electrophoresis and silver staining
The PCR products were resolved on a 2% agarose gel or an 8% PAGE gel and detected using ultraviolet (UV) light or silver staining, respectively.
Real-time quantitative PCR (RT-qPCR)
RT-qPCR for the SRY gene and the control gene, β-ACTIN, were performed according to previously reported methods and using cffDNA as the sample template (22). Sequence-specific fluorescence-labeled probes and primers for TaqMan qPCR were matched by the Universal Probe Library Center software (Roche Applied Science, Penzberg, Germany) (31,32). The β-ACTIN gene was set up as a internal control using genomic DNA as the template. The amplification primers for the SRY gene were: SRY-71F (5′-CCAGCTAGGCCACTTACCG-3′), SRY-71R (5′-AGCTTTGTCCAGTGGCTGTAG-3′), and the Taqman probe was no. 71; and the primers for the β-ACTIN gene were: Q-β-actin55GF (5′-AAGTCCCTTGCCATCCTAAA-3′), and Q-β-actin55GR (5′-ATGCTATCACCTCCCCTGTG-3′), and the Taqman probe was no. 55. The following PCR program was used: An initial denaturation step at 95°C for 10 min, followed by 45 cycles, each consisting of a denaturation step at 94°C for 15 sec and an annealing step at 60°C for 1 min.
Capture panel design, library preparation, and targeted sequencing
A capture panel of retinal disease genes was used, as previously described (33,34). The probes on the panel covered 4,405 exons and corresponded to the splice junctions of 163 known retinal disease genes within a span of 1,176 Mbp.
Illumina paired-end libraries (Illumina, Inc., San Diego, CA, USA) were generated according to the manufacturer's sample preparation protocol for genomic DNA. Briefly, 1 µg each patient's genomic DNA (proband) was sheared into fragments of ~300-500 bp. The DNA fragments were end-repaired using polynucleotide kinase and Klenow fragment (large protein fragment). The 5′ ends of the DNA fragments were phosphorylated, and a single adenine base was added to the 3′ end. Illumina Y-shaped index adaptors were ligated to the repaired ends, and subsequently the DNA fragments were amplified by PCR for eight cycles. Fragments of 300–500 bp were isolated using bead purification. The pre-capture libraries were quantified, and their size distributions were determined using a commercial bio-analytical system (Agilent 2100 BioAnalyzer; Agilent Technologies, Santa Clara, CA, USA). For each capture reaction, 50 pre-capture libraries (60 ng/library) were pooled together. Hybridization and wash kits (Agilent Technologies, Inc., Santa Clara, CA, USA) were used for the washing and recovery of captured DNA, and performed according to the standard manufacturer's protocol. Captured libraries were quantified and sequenced (Illumina HiSeq 2000; Illumina, Inc.) as 100 bp paired-end reads, according to the manufacturer's protocol.
Bioinformatic analysis of sequenced data
Sequence reads were aligned to human genome reference version hg19 using the Burrows-Wheeler Aligner version 0.5.9 (35). After recalibration and local realignment using the Genome Analysis Toolkit (GATK, version 1.0.5974) (36), the refined sequencing results were subjected to variant calling using the Atlas2 toolkit (37). Several common variant databases, such as the 1,000 genome database [Build 20110521 and 20101123], the single nucleotide polymorphism database (dbSNP)137, the National Heart, Lung, and Blood Institute (NHLBI) GO Exome Sequencing Database, the National Institute of Environmental Health Sciences (NIEHS) Exome Sequencing Database, the YanHuang Project Database, and an internal control database of 997 exomes, were used to filter out common polymorphisms at an allele frequency >0.5% in all the above databases (35,38). To remove synonymous mutations, variant annotation was performed using ANNOVAR (39), and RefSeq genes were used as references to coordinate the mutations. SIFT, Polyphen2, LRT, MutationTaster, and MutationAssessor were used to make functional predictions of missense variants (40–42). The pathogenicity of novel missense mutations was predicted using the database, dbNSFP (43). The HGMD was used to search for known pathogenic mutations.
Mutation validation and segregation tests
The putative NGS mutations were validated using Sanger sequencing. Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) was used to design primers at least 100 bp upstream and downstream from the mutation, and the assigned names of the primers used for the CHM gene causative variation were CHM7L and CHM7R. After PCR amplification, the amplicons were sequenced on an ABI3500 sequencer. The DNA materials of other family members were also sequenced using Sanger sequencing in order to perform segregation tests.
The DNA from fetal amniotic fluid was PCR-amplified using the previously mentioned primer pairs, CHM7L and CHM7R, and Sanger sequencing was performed using primer CHM7 L for the prenatal gene diagnosis.
Results
Pedigree collection and clinical phenotypes
A six-generation pedigree of Chinese descent with 30 years of follow-up was recruited to the present study. The 60-year-old proband (IV:7, Fig. 1) presented with early clinical signs of nyctalopia or night blindness at around 1 year of age. This patient now presents with extreme ametropia (both eyes >1,000 degrees), severe retinal pigment degeneration, and coloboma of the choroid (Fig. 2). A progressive narrowing of the field of vision, or tunnel vision, and decreased visual acuity was also observed. The fundus photographs shown in Fig. 2 reveal that the retinal pigment epithelial layer was patchy, with shaded areas of scattered pigmentation; however, the layer appeared transparent in the central area called the macula (Fig. 2A and B, denoted by the black arrows). The pigment layer was extremely thin, possibly absent, or lacking pigment (Fig. 2A and B, denoted by the blue arrows), leading to its transparent nature, which allowed the sclera and deep choroidal blood vessels (Fig. 2A and B, blue arrows) in the areas outside the central macula to be visible (Fig. 2A and B, black arrows). The observed blood vessels were markedly smaller in size, and the boundary of the optic-papilla was clear. This observation was further confirmed in optical coherence tomography images, which revealed atrophy of the retina at the macular fovea, thinning of the fovea, and loss of choroids.
Figure 1.

Large pedigree of choroideremia. The 60-year-old proband (IV:7) is indicated by an arrow. Affected males are denoted by a filled square. Note that the choroideremia carrier (V:11) is a female from the proband's daughter and is pregnant with a fetus (VI:7) that will undergo prenatal diagnosis to demonstrate whether the baby will inherit a normal X chromosome.
Figure 2.

Fundus photograph and OCT images of choroideremia proband. (A) The left eye fundus photograph. (B) The right eye fundus photograph. Fundus photographs revealed that the retinal pigment epithelial layer was patchy with shaded areas of scattered pigmentation; however, the layer appeared transparent in the central area, called the macula (black arrow). The pigment layer was extremely thin, possibly absent, or lacking pigment (blue arrow), leading to its transparent nature, which allowed the sclera and deep choroidal blood vessels (blue arrow) in the areas outside the central macula to be visible (black arrow). The observed blood vessels were markedly smaller in size, and the boundary of the optic-papilla was clear. (C and D) Normal fundus photographs (control) are shown. (E) The OCT images reveal atrophy of the retina at the macular fovea, thinning of the fovea, and loss of choroids. OCT, optical coherence tomography.
A further 5 males were identified as having night blindness and severely restricted peripheral visual fields within the first decade of life, as is classically expected. The female carriers disclosed no noteworthy visual defects. The patients for III:5, III:8 and III:9 in Fig. 1 did not marry and produced no offspring, whereas the male of III:6 married and assisted in producing 3 girls, who were normal.
The pregnant woman (V:11, Fig. 1) with G2P1001 was recruited and signed the informed consents for prenatal diagnosis. DNAs from the peripheral blood of this pregnant woman and the amniotic fluid from the fetus (VI:7, Fig. 1) at 17 weeks of pregnancy were extracted for further analysis.
Capture sequencing, data processing, mutation screening and validation
To identify causative mutations in this family, targeted capture sequencing was performed. DNA from the proband (IV:7) was captured, and sequenced with at least ×20 depth coverage. An automatic variant calling, filtering, and annotation pipeline was used to process the capture sequencing data from the sample. The common polymorphisms with >0.5% frequency were filtered out, and non-pathogenic variations in any of the variant or internal control databases were queried. Sequence variants that were not annotated in any of the above public databases were prioritized for further analysis (data not shown).
A nonsense mutation (c.C799T:p.R267X) located in exon 6 of the CHM gene (GenBank accession number: NM_000,390, NP_000,381.1) on the X chromosome from the proband was detected. This nonsense mutation was subsequently confirmed using Sanger sequencing (Figs. 1 and 3A), while other known retinal disease-causing gene mutations, such as RP, were excluded. This mutation was not identified in 100 healthy controls, and introduced a termination codon into the open reading frame (ORF) of the CHM gene. This ORF disruption was predicted to result in a distinct truncated protein product, whereby 60% of the C-terminal amino acids of the REP1 protein were lost, leading to a functional defect in its protein escort capabilities. The identical mutation was subsequently identified in 5 males (III:5, 6, 8, 9, and IV:5) of this family (data not shown). The identical heterozygous mutation was also subsequently identified in two female carriers (III:2 and V:11) of this family (Fig. 3, and data not shown), which indicated that the mutation of the proband (IV:7) was inherited from the mother (III:2), leading to the pathogenic mutation in the male offspring. Ultimately, this mutation demonstrated perfect co-segregation with the disease in the family. The father, son, and grandson of the proband (III:1, V:12 and VI:6, respectively) and other members of the family were normal, with wild-type alleles of the CHM gene (Fig. 3 and data not shown).
Figure 3.

NGS and Sanger sequencing for the family with choroideremia. A nonsense mutation (c.C799T:p.R267X) located in exon 6 of the CHM gene (GenBank no: NM_000,390) on the X chromosome from the proband (IV:7) was detected by NGS. Sanger sequencing confirmed the gene panel result of NGS. (A) The proband (IV:7 in Fig. 1) exhibits a 799 C → T mutation at the first nucleotide position of codon 267 (R267X) on exon 6 of the CHM gene locus (cga-tga). (B) The carrier daughter of the proband, who exhibits heterozygosity. (C and D) The healthy males of the proband's son (V:12 in Fig. 1) and grandson (VI:6 in Fig. 1) harbor normal DNA sequences. Note that the arrows illustrate the position of the point mutation. NGS, next generation sequencing.
NIPT
cffDNA isolation from maternal peripheral blood plasma (V:11) was performed. PCR-based amplification of the SRY gene by nested PCR and subsequent silver staining revealed that the plasma DNA from both a pregnant woman with a known female fetus and no template were negative, whereas the plasma DNA from a pregnant woman carrying a male fetus were positive (Fig. 4A). The results of the SRY gene amplification from this woman's plasma DNA were verified by extracting the DNA from fetal amniocytes and using the amplification primer set, XES7 and XES2 (Fig. 4B). These results confirmed the successful identification of the fetus as female (VI: 7) using the cffDNA from the maternal peripheral blood plasma (V: 11).
Figure 4.

NIPT. (A) NIPT using cffDNA from the mother's blood (V:11 in Fig. 1). Lanes 1–6 are DNA PCR products determined using nested PCR for the mutational diagnosis from the maternal blood plasma (V: 11 in Fig. 1), a known male genomic DNA sample, a known female genomic DNA sample, a blank control without any DNA, a plasma DNA sample from a pregnant woman with a known female fetus, and a plasma DNA sample from a pregnant woman with a known male fetus, respectively. Lanes 2 and 6 show the positive amplification. Lane M indicates the DNA molecular-weight marker DL600, with fragment sizes of 600, 500, 400, 300, 200, and 100 bp. (B) Verification of the SRY gene from the fetal DNA of amniotic fluid in the pregnant woman. Lanes 1–5 represent the diagnosis from fetal amniotic fluid DNA (VI:7 in Fig. 1), a female genomic DNA sample (fetus's mother; V:11 in Fig. 1), a male genomic DNA sample (fetus's father; V:10 in Fig. 1), a male genomic DNA sample (fetus's brother; VI:6 in Fig. 1) and a blank control without any DNA, respectively. Lane M indicates the DNA molecular weight marker DL2000, with fragment sizes of 2000, 1000, 750, 500, 250 and 100 bp. (C and D) The real-time quantitative PCR profiles. (C) The profile for a plasma DNA sample from a pregnant woman and β-ACTIN as control using different dilution of genomic DNA. (D) The profile for a plasma DNA sample from a pregnant woman with the known baby boy and β-ACTIN as control using different dilution of genomic DNA. NIPT, non-invasive prenatal testing; bp, base pairs.
To further validate these cffDNA results, RT-qPCR was performed to determine the quantity of cffDNA in the maternal peripheral blood plasma. The results did not show any curve of amplification (Fig. 4C), whereas a positive control using male cffDNA did (Fig. 4D, denoted by the blue arrow). Thus, these data further supported the results obtained from the nested PCR experiment.
Verification and prenatal gene diagnosis by amniotic fluid DNA
DNA isolated from the amniotic fluid of the pregnant woman (V:11, Fig. 1) with the fetus (VI:7) was amplified, and Sanger sequencing was performed for prenatal gene diagnosis of the CHM gene mutation. The results shown in Fig. 5 illustrate that the heterozygous nonsense mutation (c.C799T:p.R267X) has been identified, indicating that this baby will inherit the pathogenic DNA mutation from her mother, since her father exhibited both a normal phenotype and genotype (data not shown). At the age of one and a half, this female baby exhibited no symptoms associated with this disease mutation.
Figure 5.
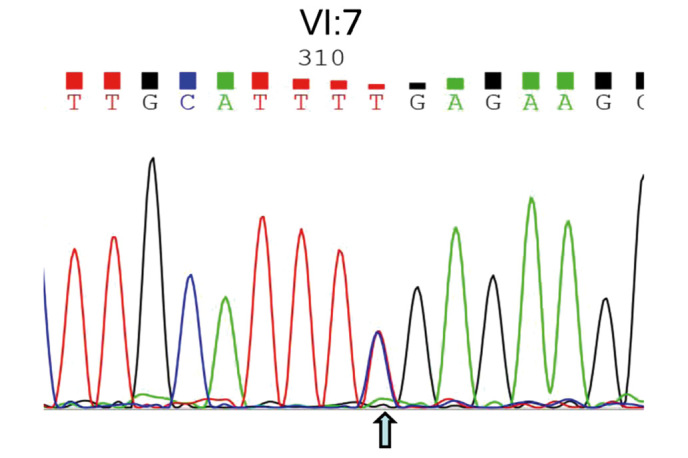
Sanger DNA sequencing of amniotic fluid DNA from the fetus. The heterozygous nonsense mutation (c.C799T:p.R267X) presenting in the fetus (VI:7) was identified, indicating that the mutation was inherited from her mother since her father possessed a wild type allele.
Discussion
Choroideremia is an X-linked recessive chorioretinal dystrophy caused by mutations in the CHM gene and characterized by progressive degeneration of the choroids, the retinal pigment epithelium, and the retinal photoreceptors (1,2,10,11). Detection of variants in the CHM gene were previously identified using a range of methods (6,12–14). Human gene replacement clinical trials are currently under way for treatment of choroideremia (15). With the increased likelihood that individuals affected with choroideremia may benefit from gene therapy or gene-specific treatments in the near future, the genetic confirmation of this disease and the identification of specific variants present prenatally are now a high priority for reproductive health in China and other developing countries.
In 1997, scientists identified cffDNA in maternal peripheral blood during pregnancy, opening the possibility for early NIPT for a variety of genetic conditions (20). Using cffDNA for prenatal testing may be able to replace or complement existing invasive methods, and greatly reduce the pain and risk of miscarriage compared with current methods. cffDNA has only been used for NIPT with a select number of single gene inheritance diseases and aneuploidy in chromosomes (23–25,44,45) due to the mixture of maternal DNA.
Due to the risk of miscarriage associated with traditional prenatal diagnostic methods or invasive methods, there is a growing interest for using NIPT (23–25,44,45). Currently, NIPT is mainly used to detect aneuploidy, the Rh antigen D (RhD) group, X-linked genetic disease, and certain single gene inheritance diseases (21,26,27). Although the gender of the fetus can often be determined using an ultrasound scan of the fetus in the second or third trimester, isolation of cffDNA in the first trimester would open up novel fields of NIPT with an earlier diagnosis and earlier, more precise management of medical care. Using isolated cffDNA present in the maternal plasma from women 6–10 weeks into their pregnancies will provide the required sensitivity, specificity, and accuracy required for NIPT. The present study has successfully managed to isolate cffDNA from maternal peripheral blood, and laid the foundation for a novel use of non-invasive prenatal diagnosis (NIPD) in medicine in the future.
In the present study, cffDNA was successfully used for the combined use of NGS for diagnosing choroideremia and NIPT for Y chromosome determination. First, a six generation pedigree of Chinese descent was recruited. The 60-year-old proband (IV:7) presented with early clinical signs of nyctalopia or night blindness at around 1 year of age. The proband's daughter became pregnant, and was included in our study. Secondly, a nonsense mutation (c.C799T:p.R267X) in the CHM gene on the X chromosome from the proband was detected by NGS, and confirmed by Sanger sequencing (Fig. 3). This mutation introduces a termination codon into the ORF of the CHM gene, thereby producing a distinct truncated protein product. The reduced quantity of fully functional REP1 prevents Rab proteins from reaching and attaching to targeted organelle membranes, which ultimately leads to premature cell death (8,46,47). Thirdly, NIPD was performed on cffDNA isolated from maternal peripheral blood plasma (V:11), and the fetus was successfully identified as a female baby (VI:7). Finally, verification of the prenatal gene diagnosis was performed successfully on amniotic fluid DNA. The heterozygous nonsense mutation (c.C799T:p.R267X) from the fetus (VI:7) was identified, indicating that the mutation in this baby was inherited from her mother (V:11). At 18 months' follow-up, the baby revealed no symptoms of choroideremia.
Thus, cffDNA has been successfully used for the combined use of NGS for diagnosing choroideremia and NIPD for Y chromosome determination. This approach should result in an increased use of prenatal diagnosis and an improved, more sophisticated clinical management of diseases in China and other developing countries. The establishment of a highly accurate method for prenatal gene diagnosis will allow more reliable gene diagnosis, improved genetic counseling, and personalized clinical management for our patients. Earlier identification of gene mutations responsible for diseases may aid in the diagnosis of retinal diseases, and assist in therapeutic research approaches. This will also afford patients the opportunity to make informed reproductive and pregnancy management decisions based on precise NIPD. Of course, more efficient approaches for detecting specific point mutations from cffDNA in different pedigrees should be developed in the future due to varied genetic mutational spectrums.
Acknowledgements
This study was supported in part by the National Natural Science Foundation of China (grant nos. 30371493, 81172049), the Science and Technology Innovation Team of Colleges and Universities of Sichuan Province (grant no. 13TD0032), the Research Foundation of the Science and Technology Department of Sichuan Province (grant nos. 14JC0797, 2015JY0038), and the Luzhou City Special Foundation (grant no. 2013LZLY-J10).
References
- 1.Coussa RG, Traboulsi EI. Choroideremia: A review of general findings and pathogenesis. Ophthalmic Genet. 2012;33:57–65. doi: 10.3109/13816810.2011.620056. [DOI] [PubMed] [Google Scholar]
- 2.Li S, Guan L, Fang S, Jiang H, Xiao X, Yang J, Wang P, Yin Y, Guo X, Wang J, et al. Exome sequencing reveals CHM mutations in six families with atypical choroideremia initially diagnosed as retinitis pigmentosa. Int J Mol Med. 2014;34:573–577. doi: 10.3892/ijmm.2014.1797. [DOI] [PubMed] [Google Scholar]
- 3.Esposito G, De Falco F, Tinto N, Testa F, Vitagliano L, Tandurella IC, Iannone L, Rossi S, Rinaldi E, Simonelli F, et al. Comprehensive mutation analysis (20 families) of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with Rab geranylgeranyl transferase. Hum Mutat. 2011;32:1460–1469. doi: 10.1002/humu.21591. [DOI] [PubMed] [Google Scholar]
- 4.Roberts MF, Fishman GA, Roberts DK, Heckenlively JR, Weleber RG, Anderson RJ, Grover S. Retrospective, longitudinal, and cross sectional study of visual acuity impairment in choroideremia. Br J Ophthalmol. 2002;86:658–662. doi: 10.1136/bjo.86.6.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Hoyos M, Lorda-Sanchez I, Gómez-Garre P, Villaverde C, Cantalapiedra D, Bustamante A, Diego-Alvarez D, Vallespin E, Gallego-Merlo J, Trujillo MJ, et al. New type of mutations in three Spanish families with choroideremia. Invest Ophthalmol Vis Sci. 2008;49:1315–1321. doi: 10.1167/iovs.07-1169. [DOI] [PubMed] [Google Scholar]
- 6.MacDonald IM, Smaoui N, Seabra MC. In: GeneReviews® [Internet] Seattle WA: University of Washington, Seattle; 2003. Choroideremia. [Updated 2010] 1993–2014; pp. 1993–2014. [Google Scholar]
- 7.van Bokhoven H, van den Hurk JA, Bogerd L, Philippe C, Gilgenkrantz S, de Jong P, Ropers HH, Cremers FP. Cloning and characterization of the human choroideremia gene. Hum Mol Genet. 1994;3:1041–1046. doi: 10.1093/hmg/3.7.1041. [DOI] [PubMed] [Google Scholar]
- 8.Strunnikova NV, Barb J, Sergeev YV, Thiagarajasubramanian A, Silvin C, Munson PJ, Macdonald IM. Loss-of-function mutations in Rab escort protein 1 (REP-1) affect intracellular transport in fibroblasts and monocytes of choroideremia patients. PLoS One. 2009;4:e8402. doi: 10.1371/journal.pone.0008402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Hurk JA, Schwartz M, van Bokhoven H, van de Pol TJ, Bogerd L, Pinckers AJ, Bleeker-Wagemakers EM, Pawlowitzki IH, Rüther K, Ropers HH, Cremers FP. Molecular basis of choroideremia (CHM): Mutations involving the rab escort protein-1 (REP-1) gene. Hum Mutat. 1997;9:110–117. doi: 10.1002/(SICI)1098-1004(1997)9:2<110::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 10.Cremers FP, Sankila EM, Brunsmann F, Jay M, Jay B, Wright A, Pinckers AJ, Schwartz M, van de Pol DJ, Wieringa B, et al. Deletions in patients with classical choroideremia vary in size from 45 kb to several megabases. Am J Hum Genet. 1990;47:622–628. [PMC free article] [PubMed] [Google Scholar]
- 11.Cremers FP, van de Pol DJ, van Kerkhoff LP, Wieringa B, Ropers HH. Cloning of a gene that is rearranged in patients with choroideraemia. Nature. 1990;347:674–677. doi: 10.1038/347674a0. [DOI] [PubMed] [Google Scholar]
- 12.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 13.McLaren TL, De Roach JN, Montgomery H, Hoffmann L, Kap C, Lamey TM. Genetic analysis of choroideremia families in the Australian population. Clin Experiment Ophthalmol. 2015;43:727–734. doi: 10.1111/ceo.12542. [DOI] [PubMed] [Google Scholar]
- 14.Gregg AR, Van den Veyver IB, Gross SJ, Madankumar R, Rink BD, Norton ME. Noninvasive prenatal screening by next-generation sequencing. Annu Rev Genomics Hum Genet. 2014;15:327–347. doi: 10.1146/annurev-genom-090413-025341. [DOI] [PubMed] [Google Scholar]
- 15.MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, Clark KR, During MJ, Cremers FP, Black GC, et al. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet. 2014;383:1129–1137. doi: 10.1016/S0140-6736(13)62117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simões M, Marques C, Gonçalves A, Pereira AP, Correia J, Castela J, Guerreiro C. Amniocentesis in HIV pregnant women: 16 years of experience. Infect Dis Obstet Gynecol. 2013;2013:914272. doi: 10.1155/2013/914272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cignini P, Dugo N, Giorlandino C, Gauci R, Spata A, Capriglione S, Cafà EV. Prenatal diagnosis of a fetus with a ring chromosome 20 characterized by array-CGH. J Prenat Med. 2012;6:72–73. [PMC free article] [PubMed] [Google Scholar]
- 18.Vora NL, Johnson KL, Peter I, Tighiouart H, Ralston SJ, Craigo SD, Bianchi DW. Circulating cell-free DNA levels increase variably following chorionic villus sampling. Prenat Diagn. 2010;30:325–328. doi: 10.1002/pd.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herzenberg LA, Bianchi DW, Schröder J, Cann HM, Iverson GM. Fetal cells in the blood of pregnant women: Detection and enrichment by fluorescence-activated cell sorting. Proc Natl Acad Sci USA. 1979;76:1453–1455. doi: 10.1073/pnas.76.3.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, Wainscoat JS. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–487. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- 21.Wright CF, Burton H. The use of cell-free fetal nucleic acids in maternal blood for non-invasive prenatal diagnosis. Hum Reprod Update. 2009;15:139–151. doi: 10.1093/humupd/dmn047. [DOI] [PubMed] [Google Scholar]
- 22.Yang WC, Zhu L, Qiu YM, Zhou BX, Cheng JL, Wei CL, Chen HC, Li LY, Fu XD, Fu JJ. Isolation and analysis of cell free fetal DNA from maternal peripheral blood in Chinese women. Genet Mol Res. 2015;14:18078–18089. doi: 10.4238/2015.December.22.34. [DOI] [PubMed] [Google Scholar]
- 23.Boon EM, Faas BH. Benefits and limitations of whole genome versus targeted approaches for noninvasive prenatal testing for fetal aneuploidies. Prenat Diagn. 2013;33:563–568. doi: 10.1002/pd.4111. [DOI] [PubMed] [Google Scholar]
- 24.Chiu RW, Lo YM. Clinical applications of maternal plasma fetal DNA analysis: Translating the fruits of 15 years of research. Clin Chem Lab Med. 2013;51:197–204. doi: 10.1515/cclm-2012-0601. [DOI] [PubMed] [Google Scholar]
- 25.Lv W, Wei X, Guo R, Liu Q, Zheng Y, Chang J, Bai T, Li H, Zhang J, Song Z, et al. Noninvasive prenatal testing for Wilson disease by use of circulating single-molecule amplification and resequencing technology (cSMART) Clin Chem. 2015;61:172–181. doi: 10.1373/clinchem.2014.229328. [DOI] [PubMed] [Google Scholar]
- 26.van den Oever JM, Bijlsma EK, Feenstra I, Muntjewerff N, Mathijssen IB, Bakker E, van Belzen MJ, Boon EM. Noninvasive prenatal diagnosis of Huntington disease: Detection of the paternally inherited expanded CAG repeat in maternal plasma. Prenat Diagn. 2015;35:945–949. doi: 10.1002/pd.4593. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Li X, Ge HJ, Xiao B, Zhang YY, Ying XM, Pan XY, Wang L, Xie WW, Ni L, et al. Haplotype-based approach for noninvasive prenatal tests of Duchenne muscular dystrophy using cell-free fetal DNA in maternal plasma. Genet Med. 2015;17:889–896. doi: 10.1038/gim.2014.207. [DOI] [PubMed] [Google Scholar]
- 28.Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: A population study. Am J Hum Genet. 1988;42:677–693. [PMC free article] [PubMed] [Google Scholar]
- 29.Fu JJ, Li LY, Li XR, Lu GX. Rapid prenatal gene diagnosis for β-thalassemia by amplification refractory mutation system (ARMS) Chin J Obstet Gynecol. 2000;35:359–360. [Google Scholar]
- 30.Fu JJ, Li LY, Lu GX. Prenatal gene diagnosis of spinal muscular atrophy by combined with the technique of PCR-SSCP, PCR followed by restriction enzyme digestion and linkage analysis. Chin J Neurol. 2001;34:74–78. [Google Scholar]
- 31.Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L, Xu J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275–289. doi: 10.1038/cr.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan MA, Tania M, Wei C, Mei Z, Fu S, Cheng J, Xu J, Fu J. Thymoquinone inhibits cancer metastasis by downregulating TWIST1 expression to reduce epithelial to mesenchymal transition. Oncotarget. 2015;6:19580–19591. doi: 10.18632/oncotarget.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, Bowne SJ, Sullivan LS, Luo H, Zhao L, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133:331–345. doi: 10.1007/s00439-013-1381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C, Milosavljevic A, Gibbs RA, Yu F. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. 1000 Genomes Project Consortium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Q, Cheng J, Yang W, Tania M, Wang H, Khan MA, Duan C, Zhu L, Chen R, Lv H, Fu J. Identification of a novel heterozygous missense mutation in the CACNA1F gene in a Chinese family with retinitis pigmentosa by next generation sequencing. Biomed Res Int. 2015;2015:907827. doi: 10.1155/2015/907827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia H, Huang X, Guo Y, Hu P, He G, Deng X, Xu H, Yang Z, Deng H. Identification of a novel MYO15A mutation in a Chinese family with autosomal recessive nonsyndromic hearing loss. PLoS One. 2015;10:e0136306. doi: 10.1371/journal.pone.0136306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Jian X, Boerwinkle E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jensen TJ, Zwiefelhofer T, Tim RC, Džakula Ž, Kim SK, Mazloom AR, Zhu Z, Tynan J, Lu T, McLennan G, et al. High-throughput massively parallel sequencing for fetal aneuploidy detection from maternal plasma. PLoS One. 2013;8:e57381. doi: 10.1371/journal.pone.0057381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Srinivasan A, Bianchi DW, Huang H, Sehnert AJ, Rava RP. Noninvasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. Am J Hum Genet. 2013;92:167–176. doi: 10.1016/j.ajhg.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonilha VL, Trzupek KM, Li Y, Francis PJ, Hollyfield JG, Rayborn ME, Smaoui N, Weleber RG. Choroideremia: Analysis of the retina from a female symptomatic carrier. Ophthalmic Genet. 2008;29:99–110. doi: 10.1080/13816810802206499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krock BL, Bilotta J, Perkins BD. Noncell-autonomous photoreceptor degeneration in a zebrafish model of choroideremia. Proc Natl Acad Sci USA. 2007;104:4600–4605. doi: 10.1073/pnas.0605818104. [DOI] [PMC free article] [PubMed] [Google Scholar]