

Abstract
Perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS), chemicals present in a multitude of consumer products, are persistent organic pollutants. Both compounds induce hepatotoxic effects in rodents, including steatosis, hepatomegaly and liver cancer. The mechanisms of PFOA- and PFOS-induced hepatic dysfunction are not completely understood. We present evidence that PFOA and PFOS induce their hepatic effects via targeting hepatocyte nuclear factor 4-alpha (HNF4α). Human hepatocytes treated with PFOA and PFOS at a concentration relevant to occupational exposure caused a decrease in HNF4α protein without affecting HNF4α mRNA or causing cell death. RNA sequencing analysis combined with Ingenuity Pathway Analysis of global gene expression changes in human hepatocytes treated with PFOA or PFOS indicated alterations in the expression of genes involved in lipid metabolism and tumorigenesis, several of which are regulated by HNF4α. Further investigation of specific HNF4α target gene expression revealed that PFOA and PFOS could promote cellular dedifferentiation and increase cell proliferation by down regulating positive targets (differentiation genes such as CYP7A1) and inducing negative targets of HNF4α (pro-mitogenic genes such as CCND1). Furthermore, in silico docking simulations indicated that PFOA and PFOS could directly interact with HNF4α in a similar manner to endogenous fatty acids. Collectively, these results highlight HNF4α degradation as novel mechanism of PFOA and PFOS-mediated steatosis and tumorigenesis in human livers.
Keywords: Perfluorooctanoic acid, perfluorooctanesulfonic acid, hepatocyte nuclear factor 4-alpha, human hepatocyte, liver
Introduction
Perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) are structurally similar anthropogenic compounds that are used in various consumer products to provide soil, oil and water resistance to materials used in clothing, upholstery, and food packaging (Lindstrom et al., 2011, Suja et al., 2009). They are two of the most commonly used compounds in a larger class of chemicals known as perfluoroalkyl acids. The multitude of carbon-fluorine bonds present in both PFOA and PFOS make these compounds chemically stable, promoting resistance to environmental degradation and biotransformation (Giesy et al., 2010, Kudo and Kawashima, 2003). Both PFOA and PFOS are persistent organic pollutants and are present in detectable levels in humans and wildlife worldwide (Kudo and Kawashima, 2003, Suja et al., 2009). Excretion of these compounds is exceptionally slow, with an average half-life in humans on the order of 3–5 years (Kudo and Kawashima, 2003, Olsen et al., 2007). The stable chemical structures, as well as the slow rate of elimination, make PFOA and PFOS persistent organic pollutants with the potential to bioaccumulate and induce long-term health effects.
The primary sources of human exposure to PFOA and PFOS are consumption of food and water contaminated with either compound (D’Hollander et al., 2010, Domingo, 2012, Vestergren and Cousins, 2009). A recent study revealed that more than 98% of human serum samples examined in the United States contain detectable levels of PFOA/PFOS, which were dose dependently associated with positive liver function tests (Gleason et al., 2015). The general population of the United States has both PFOA and PFOS present in their blood at concentrations ranging from approximately 10–100 nM (Chang et al., 2014, Gleason et al., 2015, Lau, 2012) (Figure 1). As expected, residents of the areas surrounding cities including Decatur, Alabama and Cottage Grove, Minnesota, where the 3M company manufactured PFOA and PFOS, had higher concentrations in their blood, as a consequence of watershed contamination. Similar results were found for residents surrounding Parkersburg, West Virginia where DuPont manufactured the fluorinated compounds (Chang et al., 2014, Emmett et al., 2006, Landsteiner et al., 2014, Lau, 2012). Occupational workers that came in direct contact with PFOA or PFOS had the highest concentrations present in their blood, approaching 10 μM or higher (Chang et al., 2014, Lau, 2012, Olsen et al., 2007). Although concerns of significant public health issues induced by PFOA and PFOS have caused 3M and DuPont to cease production of these compounds in the United States (Kudo and Kawashima, 2003), they continue to be manufactured and utilized worldwide (Suja et al., 2009).
Figure 1. Human serum concentrations of PFOA and PFOS within different subpopulations of exposure in the United States.
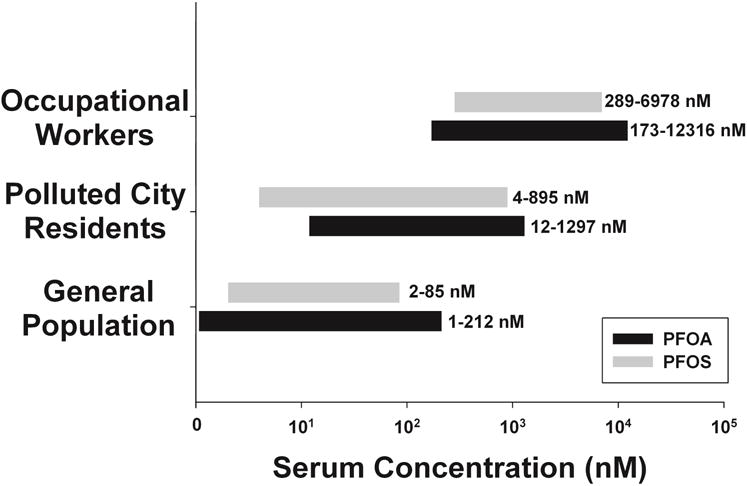
A literature review was conducted to determine concentrations of PFOA and PFOS in human serum of United States residents (Chang et al., 2014, Emmett et al., 2006, Gleason et al., 2015, Landsteiner et al., 2014, Lau, 2012, Olsen et al., 2007). Populations were categorized into three different groups based on the potential for exposure to either compound. General population data was collected from human blood banks across the United States and National Health and Nutrition Examination Survey (NHANES) data. Concentrations reported in the polluted city residents category included data collected from residents in the surrounding areas of the 3M facilities (Cottage Grove, MN and Decatur, AL) and the DuPont facility (Parkersburg, WV) responsible for the production of PFOA and PFOS. Concentrations reported in the occupational exposure group were observed in the serum samples collected from workers of the 3M and DuPont facilities.
In rodents, both compounds distribute primarily to the liver and plasma (Kennedy et al., 2004, Kudo and Kawashima, 2003). The phenotypic results of exposure to PFOA or PFOS include immunotoxicity, developmental toxicity and tumorigenesis (Lau et al., 2003, Lau et al., 2004, Lau et al., 2006, Qazi et al., 2010). A multitude of effects are observed in the liver, including hepatomegaly, steatosis and hepatocellular carcinoma (Butenhoff et al., 2012, Kennedy et al., 2004, Qazi et al., 2010). Initial studies identified nuclear receptor peroxisome proliferator-activated receptor alpha (PPARα) as a potential target for PFOA- and PFOS-induced liver dysfunction (Bjork et al., 2008, Elcombe et al., 2012). However, the hepatomegaly induced by both fluorocarbons was still observed in studies using PPARα null mice (Abbott et al., 2007, DeWitt et al., 2009, Qazi et al., 2009). These PPARα-independent effects are attributed to other nuclear receptors, including the constitutive androstane receptor (CAR) and PPARγ (Rosen et al., 2008). Furthermore, the expression of PPARα in rodents is much greater than it is in humans (Cohen et al., 2003, DeWitt et al., 2009, Klaunig et al., 2003). These findings suggest other factors are involved in the hepatic effects that occur in response to PFOA and PFOS.
Hepatocyte nuclear factor 4-alpha (HNF4α) is considered the master regulator of hepatic differentiation (Bonzo et al., 2012, Hwang-Verslues and Sladek, 2010, Parviz et al., 2003). It regulates various hepatocyte specific processes including liver development, transcriptional regulation of liver specific genes, regulation of lipid metabolism and maintaining hepatocellular quiescence and differentiation (Watt et al., 2003). In mice, conditional hepatocyte specific deletion of HNF4α results in hepatomegaly and hepatic steatosis, a liver phenotype similar to that observed in rodents administered PFOA or PFOS (Bonzo et al., 2012, Walesky et al., 2013b). A recent study indicated that PFOA exposure might reduce HNF4α expression in hepatocytes (Scharmach et al., 2012). These results suggest that HNF4α could be a relevant target of PFOA and PFOS in the liver. The goal of this study was to investigate the effects of occupationally relevant concentrations of PFOA and PFOS on HNF4α and its signaling network and to determine whether HNF4α down regulation could be a mechanism of PFOA- and PFOS-induced hepatic steatosis.
Materials and Methods
Materials
Unless otherwise noted, all materials were purchased from Sigma Aldrich (St. Louis, MO). Phenol red-free Williams’ Medium E and glutamine were purchased from Life Technologies (Grand Island, NY). For western blotting analysis, HNF4α antibody was purchased from R&D Systems (Minneapolis, MN). All other antibodies were purchased from Cell Signaling Technologies (Beverly, MA). Primers were ordered from Integrated DNA Technologies (Coralville, IA).
Isolation and culture of human hepatocytes
Primary human hepatocytes were isolated from liver explants by the Cell Isolation Core of the department of Pharmacology, Toxicology and Therapeutics at the University of Kansas Medical Center. All human tissues were obtained with informed consent from patients in accordance with ethical and institutional guidelines. The Institutional Review Board at the University of Kansas Medical Center approved this study. Hepatocytes were isolated using a standard multi-step collagenase procedure as described previously (Xie et al., 2014). At the time of isolation, cellular viability was 85% or greater. Cells were seeded to confluency in collagen-coated 6-well plates at a density of 1 × 106 cells per well, and were cultured with phenol red-free Williams’ Medium E supplemented with 2 mM L-glutamine, 10 mM HEPES buffer, 100 nM insulin, 100 nM dexamethasone, 100 U/mL penicillin, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin B. Cells were maintained in an incubator set to 37°C and a humidified atmosphere of 95% air and 5% CO2. Cells were treated following a brief period of cell attachment.
Hepatocellular PFOA and PFOS exposure protocol
PFOA (free acid) and PFOS (potassium salt) were dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 100 mM. Hepatocytes were exposed to concentrations ranging from 10 nM to 10 μM of either PFOA or PFOS, or their vehicle control (0.01% DMSO, referred to as Veh throughout). These concentrations were chosen based on the observed serum concentrations measured in humans (Figure 1). Culture medium was changed every 48 hours.
Assessment of PFOA- or PFOS-induced cytotoxicity
After exposure, cell death was assayed as described previously (Beggs et al., 2014). Briefly, culture supernatant was collected before 1% Triton in PBS was added for 30 minutes to lyse the cells. Both solutions were centrifuged at 600g for 5 minutes before ALT activity was measured as recommended by the manufacturer (Thermo Scientific, Marietta, OH). As a positive control for cell death, various concentrations of Triton dissolved in PBS were added to cells for 30 minutes before cytotoxicity was measured.
Protein isolation and Western blotting analysis
After treatment, protein was isolated using radio-immunoprecipitation assay (RIPA) buffer containing HALT protease and phosphatase inhibitors (Thermo Scientific) as described previously (Beggs et al., 2014, Walesky et al., 2013a). Briefly, protein concentration of cell extracts was determined using the bicinchoninic acid assay (Thermo Scientific). Protein expression of HNF4α and GAPDH was determined by loading 5–10 μg of protein onto NuPAGE 4–12% Bis-Tris gels (Life Technologies). Antibodies were diluted in 5% nonfat milk in Blotto to 1:2000 for HNF4α and NANOG, and 1:5000 for GAPDH. Secondary antibodies were diluted to 1:2000 in 5% nonfat milk in Blotto. Horseradish peroxidase (HRP) was visualized using SuperSignal® West Pico Chemiluminescent Substrate (Thermo Scientific).
RNA sequencing and Ingenuity Pathway Analysis
RNA was isolated from hepatocytes using TRI Reagent (Life Technologies). RNA Integrity was determined using an Agilent Bioanalyzer 2100 (Agilent Technologies; Santa Clara, CA) at the Genomics Core Facility of the University of Kansas Medical Center (Kansas City, KS). RNA sequencing and post sequencing bioinformatics analysis was conducted as described previously (Walesky et al., 2013a). Briefly, extracted RNA from cells treated for 48 hours with 10 μM of PFOA or PFOS, or Veh was sequenced using an Illumina HiSeq 2000 sequencer (Illumina, San Diego, CA). These genes were uploaded to Ingenuity Pathways Analysis (IPA, Ingenuity Systems) for gene set enrichment analysis.
Real-time PCR
A NanoDrop 1000 spectrophotometer (Thermo Scientific) was used to determine the quantity and quality of the RNA collected. Complementary DNA (cDNA) was prepared from 1 mg of RNA using deoxynucleotide triphosphate mix, random primers, RNase Inhibitor, reaction buffer and reverse transcriptase as recommended by the manufacturer (Promega, Madison, WI). RNA expression was determined using an Applied Biosystems StepOnePlus™ Real-Time PCR System with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). Fold change in gene expression was determined using the ddCt method (Livak and Schmittgen, 2001). The expression of genes of interest was normalized to the expression of GAPDH in humans and 18S ribosomal RNA in mice. Human primers for PCR experiments were designed as follows: ADH1B, 5′ -aagaagttttcactggatgcg- 3′ (forward) and 5′ -attgcctcaaaacgtcagga-3′ (reverse), AKR1B10, 5′ -gtaatgccatcggtggaaaa-3′ (forward) and 5′ -gcttctcgatctggaagtgg- 3′ (reverse), CCND1, 5′ -agaccttcgttgccctctgt- 3′ (forward) and 5′ -agttgttggggctcctcag- 3′ (reverse), CLDN1, 5′ -tttgactccttgctgaatctga- 3′ (forward) and 5′ -catacacttcatgccaacgg- 3′ (reverse), CYP7A1, 5′ -gagaaggcaaacgggtgaac- 3′ (forward) and 5′ -ggattggcaccaaattgcaga- 3′ (reverse), GAPDH, 5′ -tgatgacatcaagaaggtggtgaag-3′ (forward) and 5′ -tccttggaggccatgtgggccat- 3′ (reverse), HNF4α, 5′ -aagccgtccagaatgagc- 3′ (forward) and 5′ -aatgtcgccgttgatccc- 3′ (reverse), PLIN2, 5′ -gtgagatggcagagaacggt- 3′ (forward) and 5′ -agccccttacaggcataggt- 3′ (reverse), TAT, 5′ -caatgaaagatgccctggac- 3′ (forward) and 5′ -ccttagcttctaggggtgcc- 3′ (reverse). Murine primers for PCR experiments were designed as follows: 18S, 5′ -ttgacggaagggcaccaccag- 3′ (forward) and 5′ -gcaccaccacccacggaatcg- 3′ (reverse), AKR1B7, 5′ -ttctgattcggttccatgtcc- 3′ (forward) and 5′ -tccagttcctgttgaagctg- 3′ (reverse), CCND1, 5′ -gccctccgtatcttacttcaag- 3′ (forward) and 5′ -gcggtccaggtagttcatg- 3′ (reverse), and Egr1, 5′ -agcgccttcaatcctcaag- 3′ (forward) and 5′ -tttggctgggataactcgtc-3′ (reverse).
In silico docking
In order to identify the potential binding modes of PFOA and PFOS to HNF4α, an in silico molecular docking strategy was employed using the UCSF DOCK 6.0 software package (Allen et al., 2015). The crystal structure of HNF4α with myristic acid bound (1PZL) (Duda et al., 2004) was used as a receptor template. Myristic acid was removed and the HNF4α receptor structure was prepared using the DOCK PREP function in the UCSF CHIMERA software suite (Pettersen et al., 2004). All water molecules were removed from the solution structure as part of the docking preparations. Incomplete side chains with missing atoms in the original HNF4α structure were replaced using the Dunbrack rotamer library (Dunbrack, 2002). Hydrogens were added to the receptor, and hydrogen bonds were identified where present. Partial charges were calculated for the receptor with the AMBER module ANTECHAMBER, using the Gasteiger method. Once preparation of the receptor had been completed, it was saved in MOL2 format. This is the input file format for the grid-based scoring function from DOCK 6.0. The accessory module GRID uses this structure to generate the scoring grids for electrostatic and van der Waals interactions with the docked ligands. As an initial control, the myristic acid ligand was redocked to the empty HNF4α receptor template (data not shown). The PFOA and PFOS ligands were docked into a structural pocket characterized by the accessory program SPHGEN_CPP (Allen et al., 2015). Before docking, each ligand was energy minimized using the Driedling criterion (Pettersen et al., 2004). The docking parameters tested more than 1000 orientations of each ligand molecule (both PFOA and PFOS), using a rigid receptor/flexible ligand docking model. Upon completion, PFOA and PFOS were then ranked based on total energy score, consisting of the non-bonded terms of the AMBER force field. The Kd(app), or apparent dissociation constant, was calculated using the overall GRID energy score as a proxy for ligand binding free energy (ΔG°bind) and the equation Kd = eΔG°/RT. The Kd(app) is only provided for simplicity sake and should not be considered to represent an empirically determined dissociation constant.
Animal studies
Animal studies were conducted by Dr. Christopher Lau at the USEPS/NHEERL according to protocols approved by the U.S. EPA ORD/NHEERL Institutional Animal Care and Use Committee as previously described (Rosen et al., 2010). Ten week-old male CD-1 mice were obtained from Charles River Laboratories (Raleigh, NC). Animals were allowed one week to acclimate and provided pellet chow (LabDiet 5001, PMI Nutrition International) and tap water ad libitum. Animal facilities were controlled for temperature (20–24°C) and relative humidity (40–60%), and operated under a 12-h light-dark cycle. Mice were administered 3 mg/kg PFOA (ammonium salt) or 10 mg/kg PFOS (potassium salt) or their vehicle control (0.5% Tween-20) by oral gavage at a volume of 10 ml/kg, once daily for seven days. After treatment, mice were weighed and then sacrificed by exsanguination. The liver was removed and weighed. A section of the liver was fixed in 10% neutral buffered formalin for 48 hours, and further processed in paraffin. The blocks were sliced into 5-μM-thick sections and stained for proliferating cell nuclear antigen (PCNA). The rest of the liver was frozen in liquid nitrogen and stored at −80°C until it was used to prepare mRNA and RIPA extracts.
Statistics
All results are expressed as the mean ± the standard error of the mean. For all experiments not associated with RNA sequencing, two-tailed student’s t-test and when appropriate one-way ANOVA were applied with p < 0.05 being considered significant. The criterion for significance for RNA sequencing results was an absolute fold change of at least ± 1.5 relative to Veh control, and a q-value (the P-value adjusted for multiple hypothesis correction using the Benjamini-Hochberg procedure to control for false discovery rate) of ≤ 0.05.
Results
Neither PFOA nor PFOS cause cell death of hepatocytes
Cytotoxicity of hepatocytes was determined by measuring ALT activity in culture supernatant, and normalized to total ALT activity measured in lysed cells. Compared to Veh controls, cytotoxicity measured was not different in hepatocytes after 48 or 96 hours of exposure to either PFOA or PFOS at any of the concentrations tested, including the highest concentration of 10 μM (Figure 2).
Figure 2. PFOA- and PFOS-induced cytotoxicity.
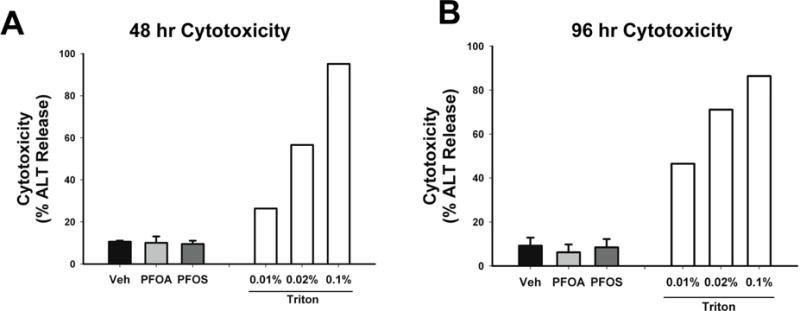
Hepatocytes were treated with Veh control (0.01% DMSO) or 10 μM of either PFOA or PFOS. Cell culture supernatant was collected after 48 (A) and 96 (B) hours of exposure. As a positive control of cytotoxicity, one set of hepatocytes was treated with various concentrations of Triton X-100 for 30 minutes (white bars). After treatment, cytotoxicity was measured as described in Materials and Methods. Data for Veh, PFOA and PFOS represent the mean ± SEM of 4 separate experiments performed in duplicate.
PFOA and PFOS decrease Hepatocellular HNF4α protein expression
No change in HNF4α protein expression was observed after 48 hours of exposure to PFOA (Figure 3A and B). After 96 hours of exposure to 10 μM PFOA, hepatocellular expression of HNF4α protein was significantly decreased (Figure 3D and E). However, lower concentrations of PFOA did not affect HNF4α expression. Exposure of cells to 10 μM PFOS for 48 hours caused a 30% decrease in HNF4α protein expression that was reduced by 40% at 96 hours (Figure 3A, C, D and F). Interestingly, neither PFOA nor PFOS at any concentration caused a change in HNF4α mRNA at 48 and 96 hours (Figure 4).
Figure 3. Concentration-response of HNF4α protein expression in hepatocytes exposed to PFOA or PFOS.

Hepatocytes were exposed to Veh control or various concentrations of either PFOA or PFOS (10–10,000 nM) for 48 or 96 hours. After treatment, the expression of HNF4α and GAPDH was determined by western blot analysis. Densitometry quantification was performed on HNF4α and GAPDH bands. The expression of HNF4α in each treatment is represented as a fold change compared to Veh control. Representative blots of HNF4α and GAPDH expression after 48 hours (A). Densitometry of HNF4α in PFOA-treated cells (B). Densitometry of HNF4α in PFOS-treated cells (C). Representative blots of HNF4α and GAPDH expression after 96 hours (D). Densitometry of HNF4α in PFOA-treated cells (E). Densitometry of HNF4α in PFOS-treated cells (F). * p < 0.05 compared to Veh-treated cells. Data represent the mean ± SEM of 3–5 separate samples.
Figure 4. HNF4α mRNA expression.
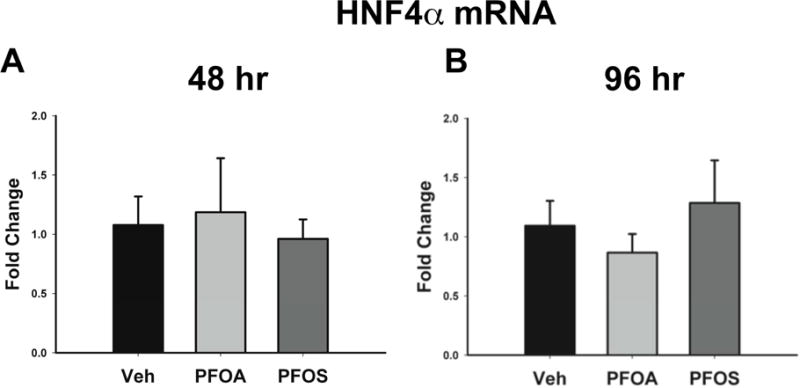
Hepatocytes were exposed to Veh control or 10 μM of either PFOA or PFOS. After exposure, mRNA was isolated. HNF4α expression was determined after 48 (A) and 96 (B) hours of exposure and normalized to GAPDH. Results are depicted as fold change relative to Veh treatment. Data represent the mean ± SEM of 6 separate samples.
PFOA alters the expression of genes involved in lipid metabolism
To identify the global gene expression changes induced by both chemicals, we performed RNA sequencing analysis of human hepatocytes treated with PFOA and PFOS (10μM concentration) for 48 hr. After 48 hours of exposure to PFOA, only 40 genes changed as compared to Veh control (20 upregulated and 20 downregulated). Table 1 depicts the top 10 genes that were either upregulated or downregulated in response to PFOA. These results were subjected to Ingenuity Pathway Analysis (IPA) to identify patterns associated with the changes in gene expression. PFOA-induced changes in gene expression were largely associated with lipid metabolism and hepatic steatosis (Table 2). Other notable functions associated with the gene changes include cholestasis and liver hyperplasia.
Table 1.
PFOA-induced changes in gene expression
| UPREGULATED | DOWNREGULATED | ||
|---|---|---|---|
|
| |||
| Gene Name | Fold Change | Gene Name | Fold Change |
| C2orf81 | 21.00 | RP5-890E16.2 | −7.11 |
| MUC4 | 3.91 | AC068580.5 | −6.82 |
| CIDEC | 3.26 | TRIM29 | −5.27 |
| CYP4A22 | 3.20 | CYP7A1 | −3.73 |
| PLIN2 | 3.11 | AP006285.1 | −3.41 |
| APOA4 | 2.99 | CXCL10 | −3.24 |
| RP11-701P16.4 | 2.92 | GFRA2 | −3.23 |
| ANGPTL4 | 2.79 | UNC13D | −2.62 |
| CYP4A11 | 2.73 | ADH1C | −2.60 |
| PCK1 | 2.64 | ELN | −2.55 |
Table 2.
Major biological and toxicological signaling pathways perturbed by PFOA
| Biological Function | Diseases or Function Annotation | p-value | Genes |
|---|---|---|---|
| Lipid Metabolism, Molecular Transport, Small Molecule Biochemistry | Concentration of Lipid | 2.20E-8 | ABCB11, ADH1C, ANGPTL4, APOA4, CIDEC, CPT1A, CREB3L3, PLIN2, TNFSF10 |
| Gastrointestinal Disease, Hepatic System Disease | Cholestasis | 4.06E-07 | ABCB11, ADH1C, CYP7A1, TNFSF10, UGT2B10 |
| Tissue Morphology | Abnormal morphology of white adipose tissue | 2.78E-06 | CIDEC, PCK1, PLIN2 |
|
| |||
| Toxicological Function | Diseases or Function Annotation | p-value | Genes |
| Liver steatosis | Hepatic steatosis | 4.51E-6 | ABCB11, CIDEC, CPT1A, CYP4A11, PCK1, PLIN2 |
| Liver cholestasis | cholestasis | 4.06E-07 | ABCB11, ADH1C, CYP7A1, TNFSF10, UGT2B10 |
| Liver hyperplasia | cholangiocarcinoma | 8.59E-5 | CPT1A, CYP4A11, PLIN2 |
PFOS alters the expression of genes involved in liver necrosis and carcinogenesis
Exposure to PFOS for 48 hours resulted in a greater number of gene changes compared to PFOA. PFOS exposure caused an upregulation of 89 genes and a down regulation in 592 genes. Table 3 depicts the top 10 genes that were either upregulated or downregulated in response to PFOS. IPA identified these gene changes associated most notably with two main functions, liver necrosis and carcinogenesis (Table 4).
Table 3.
PFOS-induced changes in gene expression
| UPREGULATED | DOWNREGULATED | ||
|---|---|---|---|
|
| |||
| Gene Name | Fold Change | Gene Name | Fold Change |
| RP11-367J11.3 | 6.60 | ALS2CR12 | −20.44 |
| NPAS1 | 6.31 | SLC26A7 | −17.42 |
| NPPB | 5.43 | LINC00939 | −8.38 |
| C1orf106 | 5.03 | ZNF540 | −8.27 |
| CTNND2 | 4.90 | RP11-1023L17.1 | −7.75 |
| EGR1 | 3.82 | CYP7A1 | −7.13 |
| KIF17 | 3.80 | ZNF84 | −6.88 |
| RP11-277L2.4 | 3.43 | CXCL10 | −6.53 |
| KRT8P32 | 3.37 | PEG3 | −6.24 |
| EPHA4 | 3.36 | HMGN5 | −6.14 |
Table 4.
Major biological and toxicological signaling pathways perturbed by PFOS
| Biological Function | Diseases or Function Annotation | p-value | Genes |
|---|---|---|---|
| Cancer | Adenocarcinoma | 1.56E-34 | 351 different genes |
| Cell Death and Survival, Gastrointestinal Disease, Hepatic System Disease | Death of liver cells | 4.50E-06 | ATF2, CXCL10, CYP7A1, DICER1, FAH, FGL2, FOS, HMOX1, IER3, IL33, IL6ST, IQGAP2, ITGAV, PIK3R1, RB1CC1, REL, SEPP1, TBK1, TNFSF10, YES1 |
| Lipid Metabolism, Small Molecule Biochemistry | Hydroxylation of lipid | 1.12E-05 | CYP2B6, CYP2C8, CYP3A4, CYP3A5, CYP4A11, CYP4A22, CYP7A1 |
|
| |||
| Toxicological Function | Diseases or Function Annotation | p-value | Genes |
| Liver necrosis/cell death | Necrosis of liver | 4.50E-6 | ATF2, CXCL10, CYP7A1, DICER1, FAH, FGL2, FOS, HMOX1, IER3, IL33, IL6ST, IQGAP2, ITGAV, PIK3R1, RB1CC1, REL, SEPP1, TBK1, TNFSF10, YES1 |
| Liver cholestasis | Secretion of taurocholic acid | 2.45E-03 | ABCB11, HMOX1 |
| Liver failure | Failure of liver | 1.21E-02 | ABCB11, ADRA1A, DUSP1, FAH, HMOX1 |
Predicted activation of upstream regulators in response to PFOA or PFOS
Further investigation into the gene changes caused by PFOA and PFOS revealed the potential for upstream regulators to be activated. In response to PFOA exposure, IPA suggests that the cells appear as though they have been exposed to PPAR agonists including rosiglitazone, WY-14,643 (pirinixic acid) and fenofibrate (Table 5). The software predicts that PPARα and PPARγ, the pharmacological targets of the aforementioned xenobiotics, might be activated by PFOA. In response to PFOS exposure, IPA suggests that the transcription factors NANOG and SOX11 are activated (Table 5).
Table 5.
Predicted activation of upstream regulators induced by PFOA and PFOS
| Upstream Regulator | Altered Target Molecules in Data Set | Z-score |
|---|---|---|
| PFOA | ||
|
| ||
| rosiglitazone | ANGPTL4, CIDEC, CPT1A, CXCL10, CYP2B6, PCK1, PLIN2 | 2.596 |
| WY-14,643 | ADH1C, ANGPTL4, APOA4, CIDEC, CPT1A, CREB3L3, CYP2B6, CYP4A11, CYP7A1, HMGCS2, PLIN2 | 2.579 |
| fenofibrate | ANGPTL4, CPT1A, CREB3L3, CYP4A11, CYP7A1 | 2.199 |
|
| ||
| PFOS | ||
|
| ||
| SOX11 | ADAM9, CD58, EGR1, IER2, IFIH1, IGIP, IL6ST | 2.646 |
| NANOG | EGR1, FOS, HNRNPH1, MEIS1, NRIP1 | 2.000 |
HNF4α regulated genes altered in response to PFOA and PFOS
We further determined the number of genes that are putative targets of HNF4α in the overall gene changes induced by PFOA and PFOS. The identification of these HNF4α target genes has been published previously. We conducted an exhaustive microarray and a separate RNA sequencing analysis in wild type and HNF4α -KO mice. The genes that were significantly different (both up and down) were then compared to published ChIP sequencing analysis to identify the genes with HNF4α binding sites upstream of the promoter region in mice (Gunewardena et al., 2015, Walesky et al., 2013a, Walesky et al., 2013b, Walesky and Apte, 2015). Specific filters were used to identify human orthologs, and we confirmed these genes are also targets in humans. These analyses led us to identify several new negative targets of HNF4α, most of which are involved in stimulation of cell proliferation. In the present study, the bona fide targets of HNF4α were compared to genes changed by PFOA and PFOS treatment. Of the 40 total genes that were altered in response to PFOA, 11 of them are regulated by HNF4α (Table 6). PFOS exposure changed the expression of 681 total genes in hepatocytes, and HNF4α is an upstream regulator of 90 of these genes (Table 6).
Table 6.
HNF4α-regulates a multitude of PFOA- and PFOS-altered genes
| HNF4α -regulated Genes | |
|---|---|
| PFOA | ABCB11, ADH1B, APOA4, CIDEC, CPT1A, CYP2B6, CYP7A1, HPS5, MUC4, PCK1, TAT |
| PFOS | AASS, ABCA6, ABCB10, ABCB11, ABCG5, ADH1B, ATF2, BLVRB, BLZF1, C6orf211, C8orf4, CAPZA2, CCDC82, CCP110, CD46, CEP83, CHST9, CIR1, CPED1, CPT1A, CRYZ, CYP2B6, CYP2C8, CYP3A4, CYP3A5, CYP7A1, DYNC2LI1, EDEM3, EGR1, EMC2, EPM2AIP1, EPPK1, EXO1, F11, F13B, GDF15, GOLGA4, GOLIM4, GRB14, IL6ST, JRKL, KRT7, LARP4, LIN7C, LRRC40, MIS18BP1, MLXIPL, NAMPT, NDUFA5, NEK7, NUAK2, PDCD10, PKM, PMS1, PRPF38B, RAD50, RBM41, RECQL, RMI1, ROCK1, RUVBL2, SCFD1, SCP2, SF3B1, SLC35A3, SLC38A4, SMC2, SRSF11, SSFA2, TAT, TDO2, TMEM123, TMF1, TPX2, TRAIP, TRAPPC8, TRIP11, TTC37, UFM1, UPF3B, USP15, ZBTB11, ZC3H15, ZNF146, ZNF224, ZNF277, ZNF300, ZNF345, ZNF644, ZRANB2 |
RT-PCR analysis of common PFOA- and PFOS-altered genes and putative HNF4α targets
The relative expression of some putative HNF4α target genes, as well as genes commonly altered by both PFOA and PFOS in the RNA sequencing analysis, was determined after 96-hour exposure. Both compounds significantly decreased the expression of positive targets of HNF4α (genes up regulated by HNF4α) including, CLDN1, CYP7A1, TAT and ADH1B at 96 hours (Figure 5A–D). Similarly, exposure to either compound resulted in a significant increase in the expression of negative targets of HNF4α (genes suppressed by HNF4α) including CCND1, AKR1B10 and PLIN2 (Figure 6A–C). NANOG mRNA increased following exposure to either PFOA or PFOS, but this was not statistically significant (Figure 6D). However, Western blot analysis confirmed a statistically significant increase in the protein expression of NANOG (Figure 6E and F).
Figure 5. Genes downregulated by PFOA or PFOS.
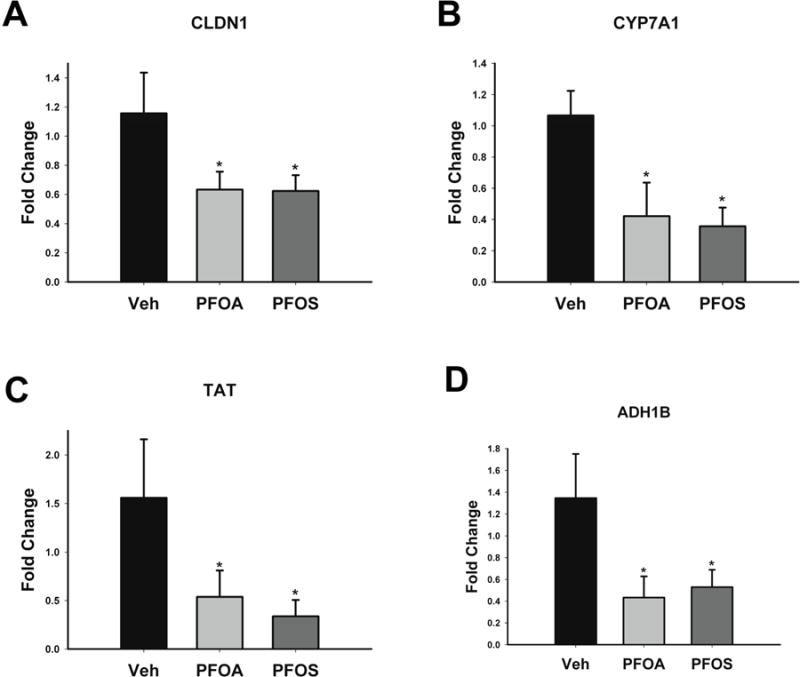
Hepatocytes were exposed to Veh control or 10 μM of either PFOA or PFOS for 96 hours. After exposure, mRNA was isolated. The expression of CLDN1 (A), CYP7A1 (B), TAT (C) and ADH1B (D) were determined, and normalized to GAPDH. Results are depicted as fold change relative to Veh treatment. * p < 0.05 compared to Veh-treated cells. Data represent the mean ± SEM of 6 separate samples performed in duplicate.
Figure 6. Genes upregulated by PFOA or PFOS and NANOG expression.
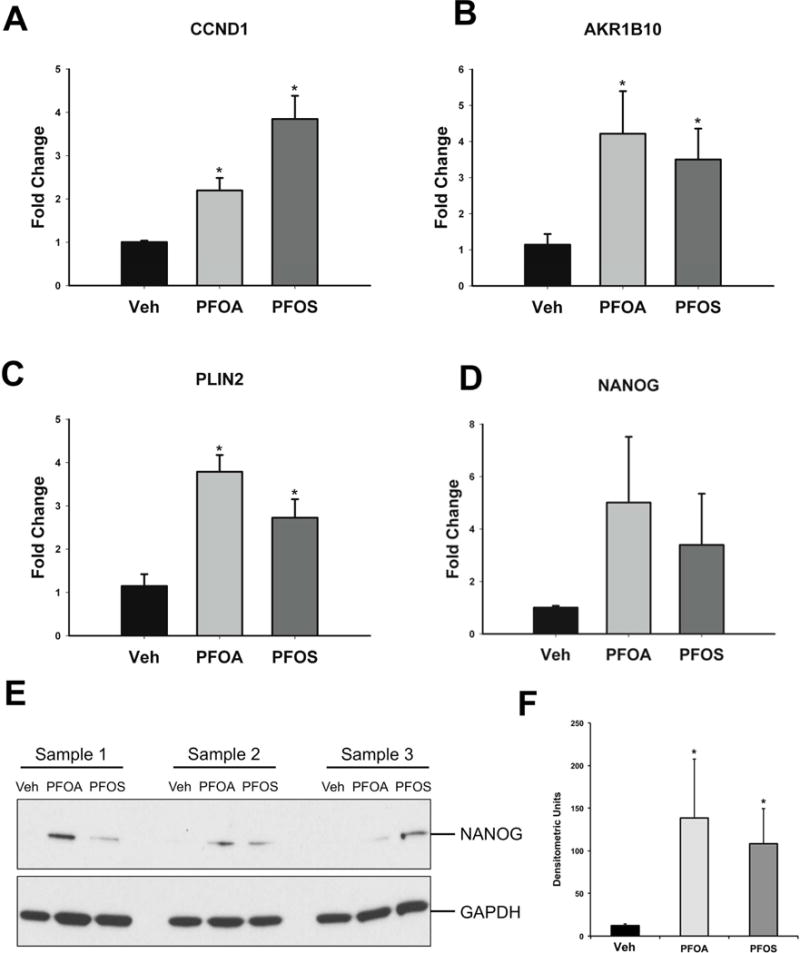
Hepatocytes were exposed to Veh control or 10 μM of either PFOA or PFOS for 96 hours. After exposure, mRNA was isolated. The expression of CCND1 (A), AKR1B10 (B), PLIN2 (C) and NANOG (D) were determined, and normalized to GAPDH. Results are depicted as fold change relative to Veh treatment. NANOG protein expression was also determined after 96 hours. (E) Representative blots of NANOG and GAPDH protein. (F) Densitometry of NANOG and GAPDH bands. * p < 0.05 compared to Veh-treated cells. Data represent the mean ± SEM of 3–6 separate samples performed in duplicate.
In silico docking studies of PFOA and PFOS to HNF4α
To further elucidate the mechanism of HNF4α protein destabilization, we performed in silico docking studies with PFOA and PFOS using the previously determined HNF4α crystal structure (Duda et al., 2004) as a receptor template. This particular structure (1PZL) was chosen due to the fact that HNF4α had been crystalized with an endogenous ligand bound. All docking simulations were performed using UCSF DOCK in flexible ligand mode (Allen et al., 2015). In the highest scoring docking pose for the PFOA simulation, PFOA bound at a similar location and adopted an almost identical conformation as the endogenous ligand, myristic acid (Figure 7A). PFOA was shown to interact with HNF4α hydrophobic residues A222, L234, L236, V242, L249, V255, I259, I346, and I349. This pattern of interaction was nearly identical to that observed with the fatty acid ligand myristic acid in the previously determined crystal structure (Duda et al., 2004). In contrast, PFOS bound in a different location and with a significantly altered conformation compared with that of either the myristic acid or PFOA ligands, with the ligand bound on the periphery of the active site (Figure 7B). While multiple protein-ligand interactions were observed in both docked structures, a substantially different pattern of interaction was observed with PFOS, with the ligand primarily contacting the polar residues S132, M252, Q345, E248, E251, and M354 of HNF4α (Figure 7B). In this case, the negatively charged sulfate moiety, which is not present in the PFOA structure, forms intermolecular contacts with the amino group of residue Q345 of HNF4α. While PFOA has a more favorable binding energy and Kd(app) compared to PFOS, the terms are comparable to that previously calculated for the myristic acid ligand (Table 7).
Figure 7. PFOA and PFOS ligands docked to HNF4α.
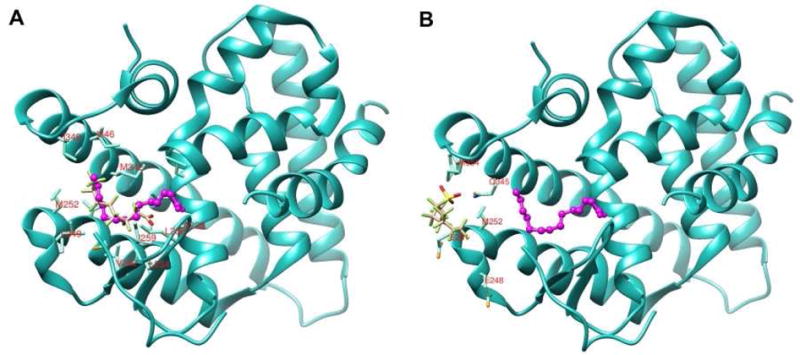
Ligands were docked into a vacant HNF4α receptor template (1PZL) using the program UCSF DOCK (Allen et al., 2015). PFOA (A) and PFOS (B) are represented as stick models, shown in tan, with individual functional groups color coded as per standard convention. The endogenous ligand (myristic acid) is overlaid in magenta in each structure, and depicted as a ball and stick model. Residues within 5 angstroms of each ligand binding site are labeled. Residues A173 through S369 in the HNF4α receptor have been cut away in each figure to make the ligand binding site more visible.
Table 7.
Kd(app) and binding energies of ligands interacting with HNF4α
| Ligand | Kd(app) | [GS] | [VDW] | [ES] | [IE] |
|---|---|---|---|---|---|
| Myristic Acid | 2.2 μM | −32.49 | −32.49 | −0.3051 | 25.92 |
| PFOA | 3.9 μM | −30.86 | −29.67 | −0.2697 | 20.49 |
| PFOS | 19.3 μM | −21.18 | −20.35 | −0.8317 | 17.67 |
[GS] = overall GRID energy score, consisting of the non-bonded terms of the AMBER molecular mechanics force field. [VDW] = Van der Waals contact energy score, [ES] = electrostatic surface potential score, [IE] = internal energy score
PFOA and PFOS decrease HNF4α protein expression in murine livers
Lastly, to determine if PFOA and PFOS cause a decrease in HNF4a protein expression in animals, mice were administered Veh and either PFOA or PFOS for seven days. Both compounds caused an increased liver weight to body weight ratio compared to Control (Veh-treated) mice (Figure 8A). Similar to results observed in human hepatocytes, both PFOA and PFOS caused a decrease in HNF4α protein expression in murine livers without affecting mRNA (Figure 8B). There was an increase in PCNA-positive cells observed in PFOA- and PFOS-treated mice compared to Control (Figure 8C), as well as an increase in the mRNA expression of a number of negatively-regulated HNF4α target genes (Figure 8D).
Figure 8. HNF4α down regulation in livers of mice treated with PFOA and PFOS.
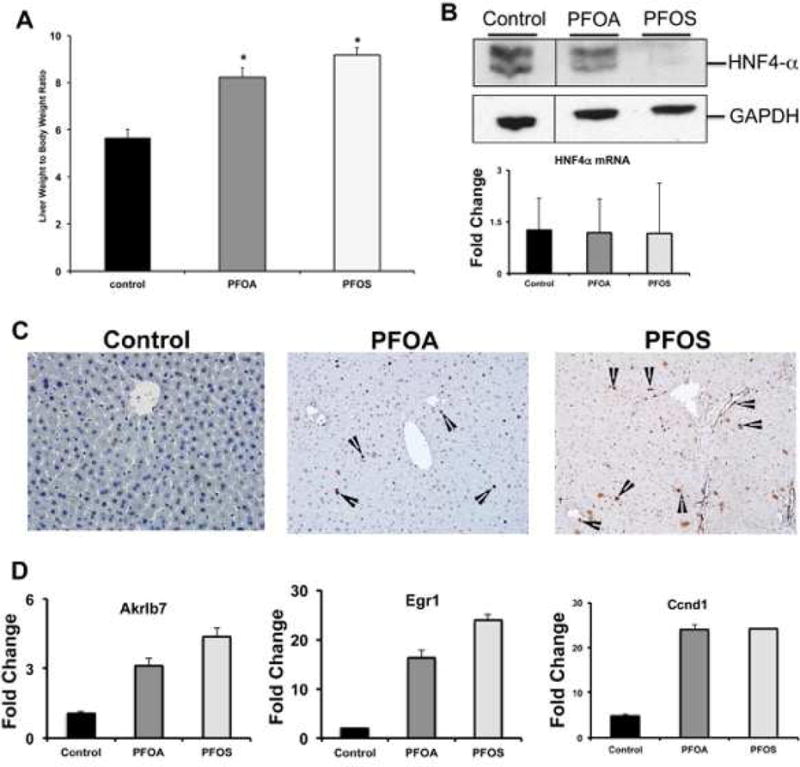
(A) Liver to bodyweight ratios of mice treated with PFOA and PFOS. (B) Hepatic HNF4α Western blot (upper panel) and HNF4α mRNA expression (lower panel) in PFOA and PFOS treated mice. (C) Representative photomicrographs of PCNA stained liver sections of (i) control, (ii) PFOA and (iii) PFOS treated mice. Arrowheads point to PCNA positive cells. (D) Bar graphs showing unregulated expression of some of the negative target genes of HNF4α associated with cell proliferation after PFOA and PFOS treatment. * p < 0.05 compared to Veh-treated mice. Data represent the mean ± SEM of 3–5 separate samples performed in duplicate.
Discussion
Whereas PFOA and PFOS have been known to induce steatosis and proliferation in the liver (Butenhoff et al., 2012, Klaunig et al., 2012, Qazi et al., 2010), the mechanisms have not been completely understood. Recent studies have implicated HNF4α as potentially accountable for PFOA-mediated effects in human hepatocytes and cell lines (Buhrke et al., 2015, Scharmach et al., 2012). Notably, conditional deletion of HNF4α in murine hepatocytes resulted in hepatomegaly and steatosis that mimic the hepatic phenotype observed after PFOA and PFOS administration (Walesky et al., 2013b). Our results corroborate that PFOA and PFOS induce hepatomegaly (Figure 8A) and suggest this might be the result of increased hepatocyte proliferation (Figure 8C). Based on these findings, we hypothesized that both PFOA and PFOS would cause HNF4α protein loss, and promote changes in the expression of genes associated with lipid metabolism and hepatocellular quiescence. Whereas HNF4α changes in PFOA and PFOS treated cells have been noted, this is a the first detailed analysis of HNF4α changes in human hepatocyte and mice after PFOA and PFOS treatment.
Previous studies have implicated PPARα in the hepatic effects of PFOA and PFOS. Interestingly, PPARα is transcriptionally regulated by HNF4α in mice (Martinez-Jimenez et al., 2010). This might explain the persisting phenotype observed PPARα null mice treated with PFOA and PFOS. Animal models focusing on PPARα signaling have received criticism for lacking translational relevance to humans that express less PPARα than rodents (Corton et al., 2014, Klaunig et al., 2003). We circumvented these issues by investigate the effect of PFOA and PFOS exposure on human hepatocytes.
Another important novelty of our study is the concentrations of PFOA and PFOS used in cell culture experiments. Our detailed literature survey demonstrated that 10μM concentration is most likely seen in occupational setting, which was used as the highest concentration in our studies. Majority of published studies have used significantly higher concentrations, which complicates data interpretation. Our studies conducted using human hepatocytes and occupationally relevant concentrations are is the closest to human situation. This was clear in the data as neither PFOA nor PFOS induced cytotoxicity (Figure 2), suggesting the observed results are not a consequence of cellular death, which is not observed in human exposure and validates our model.
RNA sequencing analysis is a powerful tool to determine global changes in gene expression. Our analysis revealed that PFOA predominantly changed genes involved in lipid metabolism and hepatic steatosis (Table 2). One of the novel findings is that the upstream regulator analysis suggested that PFOA treatment may activate PPARγ in addition to PPARα. This is interesting because PPARγ activation is known to promote adipogenesis and steatosis in the liver (Gavrilova et al., 2003, Moran-Salvador et al., 2011, Tailleux et al., 2012). PPARγ regulates the expression of PLIN2 (Okumura, 2011, Schadinger et al., 2005) that can promote fatty liver disease (Imai et al., 2007, Okumura, 2011) and is induced by both PFOA and PFOS (Figure 6C). However, there is also evidence that indicates PPARγ can protect against steatosis in certain contexts (Kawaguchi et al., 2004). Further studies are required to determine. If any other antagonistic mechanism is also activated after PFOA treatment, which may decrease the anti-steatotic effect of PFOA.
Another important finding is that both compounds promoted changes associated with cholestasis signaling (Table 2 and 4). Potentially, this occurs as a result of decreased CYP7A1 mRNA (Figure 5B). HNF4α is a transcriptional regulator of CYP7A1 (Kir et al., 2012, Sanyal et al., 2007), the rate-limiting enzyme involved in cholesterol catabolism and bile acid synthesis (Gilardi et al., 2007). In humans, CYP7A1 deficiency promotes hypercholesterolemia (Beigneux et al., 2002, Pullinger et al., 2002). Epidemiological evidence of individuals exposed to PFOA or PFOS suggest a positive correlation between exposure and increased serum cholesterol (Fu et al., 2014, Geiger et al., 2014, Kerger et al., 2011). Thus, the observed increase in cholesterol in humans exposed to PFOA or PFOS potentially results from a loss of HNF4α function and decreased CYP7A1 transcription.
PFOS predominantly induced changes in the expression of genes involved in carcinogenesis and cell death signaling (Table 4). HNF4α is critical for maintaining hepatocellular differentiation (DeLaForest et al., 2011), and loss of HNF4α functionality can promote the development of hepatocellular carcinoma (Lazarevich et al., 2004, Ning et al., 2010). The PFOS-induced increase in NANOG and down regulation of TAT and ADH1B genes, markers of hepatocellular differentiation (Hamazaki et al., 2001, Parent and Beretta, 2008) are significant findings, which suggest a novel de-differentiation-based mechanism of hepatic effects induced by PFOS.
PFOA and PFOS caused a decrease in CLDN1 mRNA (Figure 5A). The claudin-1 protein is involved in maintaining cellular adhesion and the formation of cell junctions (Gunzel and Yu, 2013, Morita et al., 1999), and loss of claudin-1 in the liver can promote metastasis (Georges et al., 2012, Holczbauer et al., 2013). PFOA and PFOS also caused an induction of the AKR1B10 gene (Figure 6B) that is associated with the progression of hepatocellular carcinoma (Matkowskyj et al., 2014). Lastly, hepatocellular deletion of HNF4α in mice resulted in hepatomegaly and increased cell division, as well as an increase in the mRNA of promitogenic CCND1 and Egr1, and the AKR1B10 murine homolog, AKR1B7. These genes are all negatively regulated by HNF4α (Walesky et al., 2013b). Both PFOA and PFOS caused an increase in CCND1 mRNA in hepatocytes (Figure 6A) and murine livers (Figure 8D). Collectively, these results suggest that PFOA and PFOS could promote hepatocellular dedifferentiation to a hepatoblast-type bipotential progenitor cell and might promote carcinogenesis of the human liver via HNF4α protein degradation.
HNF4α is considered an orphan receptor, and various fatty acids have been implicated as the endogenous ligands (Duda et al., 2004, Wisely et al., 2002, Yuan et al., 2009). However, these fatty acids do not act as classical agonists for HNF4α-mediated transcription. Rather, the fatty acids are purported to maintain the structural integrity of the HNF4α homodimer (Wisely et al., 2002). The chemical structures of PFOA and PFOS are analogous to these fatty acids, which suggests that either compound might competitively interact with the ligand-binding domain of HNF4α and displace the endogenous fatty acids, potentially signaling for protein degradation. Our in silico docking results support this hypothesis. PFOA binds with similar affinity and conformation to the putative endogenous ligand myristic acid (Figure 7A). PFOS binds in a significantly different location, and with an altered conformation, on the HNF4α receptor (Figure 7B). Interestingly, the binding between PFOA and HNF4α tends to be governed by hydrophobic interactions, whereas there is a strong electrostatic component to the interactions between PFOS and HNF4α. Although the computationally determined apparent binding affinity (Kd(app)) for PFOS is lower than PFOA, the alternative binding location at the entrance to the endogenous ligand binding site might block endogenous fatty acid ligand binding and “lock” HNF4α into an inactive conformation, one that would not be capable of DNA binding or receptor dimerization. Given that the gene modulation effects seen in cell culture were noticeably stronger with PFOS over PFOA, this suggests that the altered binding arrangement of PFOS could promote a conformation of HNF4α that might be targeted for degradation.
Collectively, these results indicate that at occupationally relevant serum concentrations PFOA and PFOS cause a decrease in HNF4α protein expression in human hepatocytes, and promote changes in the expression of genes involved in lipid metabolism and carcinogenesis. The steatosis, hepatomegaly and hepatocellular carcinoma that occur in rodents might be a consequence of a loss of HNF4α protein function promoted by these fluorinated compounds, perhaps by interacting with the ligand binding domain of HNF4α and interfering with protein stability. This study has revealed a novel potential mechanism for PFOA- and PFOS-induced hepatic effects in humans, including fatty liver disease and liver cancer. These data also suggest that exposure to PFOA or PFOS might sensitize the liver to injury from a secondary insult such as obesity or chronic liver injury.
Supplementary Material
Highlights.
PFOA and PFOS cause decreased HNF4α protein expression in human hepatocytes
PFOA and PFOS promote changes associated with lipid metabolism and carcinogenesis
PFOA and PFOS induced changes in gene expression associated with cellular dedifferentiation
PFOA and PFOS induce expression of Nanog, a transcription factor involved in stem cell development
Acknowledgments
This work was funded by The National Institutes of Health, grant numbers 1R01DK098414, P20 RR021940 and T32ES007079-34, and partially funded by the U.S. Environmental Protection Agency. The National Institutes of Health had no involvement in the design of this study or the interpretation of its results. The information in this document has been subjected to review by the National Health and Environmental Effects Research Laboratory of US EPA and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use. Ken Dorko and Hongmin Ni of the Cell Isolation Core at the University of Kansas Medical Center isolated human hepatocytes for experimental use.
Abbreviations
- ADH1B
alcohol dehydrogenase 1B
- AKR1B10
aldo-keto reductase 1B10
- ALT
alanine aminotransferase
- CCND1
cyclin D1
- CLDN1
claudin 1
- CYP7A1
cytochrome P450 7A1
- DMSO
dimethyl sulfoxide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HNF4α
hepatocyte nuclear factor 4-alpha
- IPA
Ingenuity Pathway Analysis
- PFOA
perfluorooctanoic acid
- PFOS
perfluorooctanesulfonic acid
- PLIN2
perilipin 2
- PPAR
peroxisome proliferator-activated receptor
- TAT
tyrosine aminotransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott BD, Wolf CJ, Schmid JE, Das KP, Zehr RD, Helfant L, Nakayama S, Lindstrom AB, Strynar MJ, Lau C. Perfluorooctanoic acid induced developmental toxicity in the mouse is dependent on expression of peroxisome proliferator activated receptor-alpha. Toxicol Sci. 2007;98:571–81. doi: 10.1093/toxsci/kfm110. [DOI] [PubMed] [Google Scholar]
- Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC. DOCK 6: Impact of new features and current docking performance. J Comput Chem. 2015;36:1132–56. doi: 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs KM, Fullerton AM, Miyakawa K, Ganey PE, Roth RA. Molecular Mechanisms of Hepatocellular Apoptosis Induced by Trovafloxacin-Tumor Necrosis Factor-alpha Interaction. Toxicol Sci. 2014;137:91–101. doi: 10.1093/toxsci/kft226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux A, Hofmann AF, Young SG. Human CYP7A1 deficiency: progress and enigmas. J Clin Invest. 2002;110:29–31. doi: 10.1172/JCI16076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork JA, Lau C, Chang SC, Butenhoff JL, Wallace KB. Perfluorooctane sulfonate-induced changes in fetal rat liver gene expression. Toxicology. 2008;251:8–20. doi: 10.1016/j.tox.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Bonzo JA, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J Biol Chem. 2012;287:7345–56. doi: 10.1074/jbc.M111.334599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhrke T, Kruger E, Pevny S, Rossler M, Bitter K, Lampen A. Perfluorooctanoic acid (PFOA) affects distinct molecular signalling pathways in human primary hepatocytes. Toxicology. 2015;333:53–62. doi: 10.1016/j.tox.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Butenhoff JL, Chang SC, Olsen GW, Thomford PJ. Chronic dietary toxicity and carcinogenicity study with potassium perfluorooctanesulfonate in Sprague Dawley rats. Toxicology. 2012;293:1–15. doi: 10.1016/j.tox.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Chang ET, Adami HO, Boffetta P, Cole P, Starr TB, Mandel JS. A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and cancer risk in humans. Crit Rev Toxicol. 2014;44(Suppl 1):1–81. doi: 10.3109/10408444.2014.905767. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Meek ME, Klaunig JE, Patton DE, Fenner-Crisp PA. The human relevance of information on carcinogenic modes of action: overview. Crit Rev Toxicol. 2003;33:581–9. doi: 10.1080/713608371. [DOI] [PubMed] [Google Scholar]
- Corton JC, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, Popp JA, Rhomberg L, Seed J, Klaunig JE. Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor alpha (PPARalpha) as a case study. Crit Rev Toxicol. 2014;44:1–49. doi: 10.3109/10408444.2013.835784. [DOI] [PubMed] [Google Scholar]
- D’Hollander W, de Voogt P, De Coen W, Bervoets L. Perfluorinated substances in human food and other sources of human exposure. Rev Environ Contam Toxicol. 2010;208:179–215. doi: 10.1007/978-1-4419-6880-7_4. [DOI] [PubMed] [Google Scholar]
- DeLaForest A, Nagaoka M, Si-Tayeb K, Noto FK, Konopka G, Battle MA, Duncan SA. HNF4A is essential for specification of hepatic progenitors from human pluripotent stem cells. Development. 2011;138:4143–53. doi: 10.1242/dev.062547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt JC, Shnyra A, Badr MZ, Loveless SE, Hoban D, Frame SR, Cunard R, Anderson SE, Meade BJ, Peden-Adams MM, Luebke RW, Luster MI. Immunotoxicity of perfluorooctanoic acid and perfluorooctane sulfonate and the role of peroxisome proliferator-activated receptor alpha. Crit Rev Toxicol. 2009;39:76–94. doi: 10.1080/10408440802209804. [DOI] [PubMed] [Google Scholar]
- Domingo JL. Health risks of dietary exposure to perfluorinated compounds. Environ Int. 2012;40:187–95. doi: 10.1016/j.envint.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Duda K, Chi YI, Shoelson SE. Structural basis for HNF-4alpha activation by ligand and coactivator binding. J Biol Chem. 2004;279:23311–6. doi: 10.1074/jbc.M400864200. [DOI] [PubMed] [Google Scholar]
- Dunbrack RL., Jr Rotamer libraries in the 21st century. Curr Opin Struct Biol. 2002;12:431–40. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Elcombe CR, Elcombe BM, Foster JR, Chang SC, Ehresman DJ, Butenhoff JL. Hepatocellular hypertrophy and cell proliferation in Sprague-Dawley rats from dietary exposure to potassium perfluorooctanesulfonate results from increased expression of xenosensor nuclear receptors PPARalpha and CAR/PXR. Toxicology. 2012;293:16–29. doi: 10.1016/j.tox.2011.12.014. [DOI] [PubMed] [Google Scholar]
- Emmett EA, Shofer FS, Zhang H, Freeman D, Desai C, Shaw LM. Community exposure to perfluorooctanoate: relationships between serum concentrations and exposure sources. J Occup Environ Med. 2006;48:759–70. doi: 10.1097/01.jom.0000232486.07658.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Wang T, Fu Q, Wang P, Lu Y. Associations between serum concentrations of perfluoroalkyl acids and serum lipid levels in a Chinese population. Ecotoxicol Environ Saf. 2014;106:246–52. doi: 10.1016/j.ecoenv.2014.04.039. [DOI] [PubMed] [Google Scholar]
- Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–76. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- Geiger SD, Xiao J, Ducatman A, Frisbee S, Innes K, Shankar A. The association between PFOA, PFOS and serum lipid levels in adolescents. Chemosphere. 2014;98:78–83. doi: 10.1016/j.chemosphere.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Georges R, Bergmann F, Hamdi H, Zepp M, Eyol E, Hielscher T, Berger MR, Adwan H. Sequential biphasic changes in claudin1 and claudin4 expression are correlated to colorectal cancer progression and liver metastasis. J Cell Mol Med. 2012;16:260–72. doi: 10.1111/j.1582-4934.2011.01289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesy JP, Naile JE, Khim JS, Jones PD, Newsted JL. Aquatic toxicology of perfluorinated chemicals. Rev Environ Contam Toxicol. 2010;202:1–52. doi: 10.1007/978-1-4419-1157-5_1. [DOI] [PubMed] [Google Scholar]
- Gilardi F, Mitro N, Godio C, Scotti E, Caruso D, Crestani M, De Fabiani E. The pharmacological exploitation of cholesterol 7alpha-hydroxylase, the key enzyme in bile acid synthesis: from binding resins to chromatin remodelling to reduce plasma cholesterol. Pharmacol Ther. 2007;116:449–72. doi: 10.1016/j.pharmthera.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Gleason JA, Post GB, Fagliano JA. Associations of perfluorinated chemical serum concentrations and biomarkers of liver function and uric acid in the US population (NHANES), 2007–2010. Environ Res. 2015;136:8–14. doi: 10.1016/j.envres.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Gunewardena S, Walesky C, Apte U. Global Gene Expression Changes in Liver Following Hepatocyte Nuclear Factor 4 alpha deletion in Adult Mice. Genom Data. 2015;5:126–128. doi: 10.1016/j.gdata.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013;93:525–69. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki T, Iiboshi Y, Oka M, Papst PJ, Meacham AM, Zon LI, Terada N. Hepatic maturation in differentiating embryonic stem cells in vitro. FEBS Lett. 2001;497:15–9. doi: 10.1016/s0014-5793(01)02423-1. [DOI] [PubMed] [Google Scholar]
- Holczbauer A, Gyongyosi B, Lotz G, Szijarto A, Kupcsulik P, Schaff Z, Kiss A. Distinct claudin expression profiles of hepatocellular carcinoma and metastatic colorectal and pancreatic carcinomas. J Histochem Cytochem. 2013;61:294–305. doi: 10.1369/0022155413479123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang-Verslues WW, Sladek FM. HNF4alpha–role in drug metabolism and potential drug target? Curr Opin Pharmacol. 2010;10:698–705. doi: 10.1016/j.coph.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Varela GM, Jackson MB, Graham MJ, Crooke RM, Ahima RS. Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology. 2007;132:1947–54. doi: 10.1053/j.gastro.2007.02.046. [DOI] [PubMed] [Google Scholar]
- Kawaguchi K, Sakaida I, Tsuchiya M, Omori K, Takami T, Okita K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient L-amino acid-defined diet. Biochem Biophys Res Commun. 2004;315:187–95. doi: 10.1016/j.bbrc.2004.01.038. [DOI] [PubMed] [Google Scholar]
- Kennedy GL, Jr, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, Biegel LB, Murphy SR, Farrar DG. The toxicology of perfluorooctanoate. Crit Rev Toxicol. 2004;34:351–84. doi: 10.1080/10408440490464705. [DOI] [PubMed] [Google Scholar]
- Kerger BD, Copeland TL, DeCaprio AP. Tenuous dose-response correlations for common disease states: case study of cholesterol and perfluorooctanoate/sulfonate (PFOA/PFOS) in the C8 Health Project. Drug Chem Toxicol. 2011;34:396–404. doi: 10.3109/01480545.2011.582502. [DOI] [PubMed] [Google Scholar]
- Kir S, Zhang Y, Gerard RD, Kliewer SA, Mangelsdorf DJ. Nuclear receptors HNF4alpha and LRH-1 cooperate in regulating Cyp7a1 in vivo. J Biol Chem. 2012;287:41334–41. doi: 10.1074/jbc.M112.421834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, DeLuca JG, Lai DY, McKee RH, Peters JM, Roberts RA, Fenner-Crisp PA. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Hocevar BA, Kamendulis LM. Mode of Action analysis of perfluorooctanoic acid (PFOA) tumorigenicity and Human Relevance. Reprod Toxicol. 2012;33:410–8. doi: 10.1016/j.reprotox.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Kudo N, Kawashima Y. Toxicity and toxicokinetics of perfluorooctanoic acid in humans and animals. J Toxicol Sci. 2003;28:49–57. doi: 10.2131/jts.28.49. [DOI] [PubMed] [Google Scholar]
- Landsteiner A, Huset C, Johnson J, Williams A. Biomonitoring for perfluorochemicals in a Minnesota community with known drinking water contamination. J Environ Health. 2014;77:14–9. [PubMed] [Google Scholar]
- Lau C, Thibodeaux JR, Hanson RG, Rogers JM, Grey BE, Stanton ME, Butenhoff JL, Stevenson LA. Exposure to perfluorooctane sulfonate during pregnancy in rat and mouse. II: postnatal evaluation. Toxicol Sci. 2003;74:382–92. doi: 10.1093/toxsci/kfg122. [DOI] [PubMed] [Google Scholar]
- Lau C, Butenhoff JL, Rogers JM. The developmental toxicity of perfluoroalkyl acids and their derivatives. Toxicol Appl Pharmacol. 2004;198:231–41. doi: 10.1016/j.taap.2003.11.031. [DOI] [PubMed] [Google Scholar]
- Lau C, Thibodeaux JR, Hanson RG, Narotsky MG, Rogers JM, Lindstrom AB, Strynar MJ. Effects of perfluorooctanoic acid exposure during pregnancy in the mouse. Toxicol Sci. 2006;90:510–8. doi: 10.1093/toxsci/kfj105. [DOI] [PubMed] [Google Scholar]
- Lau C. Perfluoroalkyl acids: recent research highlights. Reprod Toxicol. 2012;33:405–9. doi: 10.1016/j.reprotox.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, Fleishman DI, Engelhardt NV, Duncan SA. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology. 2004;39:1038–47. doi: 10.1002/hep.20155. [DOI] [PubMed] [Google Scholar]
- Lindstrom AB, Strynar MJ, Libelo EL. Polyfluorinated compounds: past, present, and future. Environ Sci Technol. 2011;45:7954–61. doi: 10.1021/es2011622. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martinez-Jimenez CP, Kyrmizi I, Cardot P, Gonzalez FJ, Talianidis I. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol Cell Biol. 2010;30:565–77. doi: 10.1128/MCB.00927-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkowskyj KA, Bai H, Liao J, Zhang W, Li H, Rao S, Omary R, Yang GY. Aldoketoreductase family 1B10 (AKR1B10) as a biomarker to distinguish hepatocellular carcinoma from benign liver lesions. Hum Pathol. 2014;45:834–43. doi: 10.1016/j.humpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, Gonzalez-Periz A, Lopez-Vicario C, Barak Y, Arroyo V, Claria J. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–50. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A. 1999;96:511–6. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning BF, Ding J, Yin C, Zhong W, Wu K, Zeng X, Yang W, Chen YX, Zhang JP, Zhang X, Wang HY, Xie WF. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res. 2010;70:7640–51. doi: 10.1158/0008-5472.CAN-10-0824. [DOI] [PubMed] [Google Scholar]
- Okumura T. Role of lipid droplet proteins in liver steatosis. J Physiol Biochem. 2011;67:629–36. doi: 10.1007/s13105-011-0110-6. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, Zobel LR. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect. 2007;115:1298–305. doi: 10.1289/ehp.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent R, Beretta L. Translational control plays a prominent role in the hepatocytic differentiation of HepaRG liver progenitor cells. Genome Biol. 2008;9:R19. doi: 10.1186/gb-2008-9-1-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, Rossi JM, Zaret KS, Duncan SA. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet. 2003;34:292–6. doi: 10.1038/ng1175. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, Verhagen A, Rivera CR, Mulvihill SJ, Malloy MJ, Kane JP. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109–17. doi: 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qazi MR, Xia Z, Bogdanska J, Chang SC, Ehresman DJ, Butenhoff JL, Nelson BD, DePierre JW, Abedi-Valugerdi M. The atrophy and changes in the cellular compositions of the thymus and spleen observed in mice subjected to short-term exposure to perfluorooctanesulfonate are high-dose phenomena mediated in part by peroxisome proliferator-activated receptor-alpha (PPARalpha) Toxicology. 2009;260:68–76. doi: 10.1016/j.tox.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Qazi MR, Abedi MR, Nelson BD, DePierre JW, Abedi-Valugerdi M. Dietary exposure to perfluorooctanoate or perfluorooctane sulfonate induces hypertrophy in centrilobular hepatocytes and alters the hepatic immune status in mice. Int Immunopharmacol. 2010;10:1420–7. doi: 10.1016/j.intimp.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Lee JS, Ren H, Vallanat B, Liu J, Waalkes MP, Abbott BD, Lau C, Corton JC. Toxicogenomic dissection of the perfluorooctanoic acid transcript profile in mouse liver: evidence for the involvement of nuclear receptors PPAR alpha and CAR. Toxicol Sci. 2008;103:46–56. doi: 10.1093/toxsci/kfn025. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Schmid JR, Corton JC, Zehr RD, Das KP, Abbott BD, Lau C. Gene Expression Profiling in Wild-Type and PPARalpha-Null Mice Exposed to Perfluorooctane Sulfonate Reveals PPARalpha-Independent Effects. PPAR Res. 2010;2010 doi: 10.1155/2010/794739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S, Bavner A, Haroniti A, Nilsson LM, Lundasen T, Rehnmark S, Witt MR, Einarsson C, Talianidis I, Gustafsson JA, Treuter E. Involvement of corepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proc Natl Acad Sci U S A. 2007;104:15665–70. doi: 10.1073/pnas.0706736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am J Physiol Endocrinol Metab. 2005;288:E1195–205. doi: 10.1152/ajpendo.00513.2004. [DOI] [PubMed] [Google Scholar]
- Scharmach E, Buhrke T, Lichtenstein D, Lampen A. Perfluorooctanoic acid affects the activity of the hepatocyte nuclear factor 4 alpha (HNF4alpha) Toxicol Lett. 2012;212:106–12. doi: 10.1016/j.toxlet.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Suja F, Pramanik BK, Zain SM. Contamination, bioaccumulation and toxic effects of perfluorinated chemicals (PFCs) in the water environment: a review paper. Water Sci Technol. 2009;60:1533–44. doi: 10.2166/wst.2009.504. [DOI] [PubMed] [Google Scholar]
- Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821:809–18. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Vestergren R, Cousins IT. Tracking the pathways of human exposure to perfluorocarboxylates. Environ Sci Technol. 2009;43:5565–75. doi: 10.1021/es900228k. [DOI] [PubMed] [Google Scholar]
- Walesky C, Edwards G, Borude P, Gunewardena S, O’Neil M, Yoo B, Apte U. Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology. 2013a;57:2480–90. doi: 10.1002/hep.26251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4alpha in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol. 2013b;304:G26–37. doi: 10.1152/ajpgi.00064.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walesky C, Apte U. Role of hepatocyte nuclear factor 4alpha (HNF4alpha) in cell proliferation and cancer. Gene Expr. 2015;16:101–8. doi: 10.3727/105221615X14181438356292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Garrison WD, Duncan SA. HNF4: a central regulator of hepatocyte differentiation and function. Hepatology. 2003;37:1249–53. doi: 10.1053/jhep.2003.50273. [DOI] [PubMed] [Google Scholar]
- Wisely GB, Miller AB, Davis RG, Thornquest AD, Jr, Johnson R, Spitzer T, Sefler A, Shearer B, Moore JT, Miller AB, Willson TM, Williams SP. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure. 2002;10:1225–34. doi: 10.1016/s0969-2126(02)00829-8. [DOI] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 2014;279:266–74. doi: 10.1016/j.taap.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Ta TC, Lin M, Evans JR, Dong Y, Bolotin E, Sherman MA, Forman BM, Sladek FM. Identification of an endogenous ligand bound to a native orphan nuclear receptor. PLoS One. 2009;4:e5609. doi: 10.1371/journal.pone.0005609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.