

Abstract
Herpesviruses are unusual among enveloped viruses because they bud twice yet acquire a single envelope. Furthermore, unlike other DNA viruses that replicate in the nucleus, herpesviruses do not exit it by passing through the nuclear pores or by rupturing the nuclear envelope. Instead, herpesviruses have a complex mechanism of nuclear escape whereby nascent capsids bud at the inner nuclear membrane to form perinuclear virions that subsequently fuse with the outer nuclear membrane, releasing capsids into the cytosol. This makes them some of the very few known viruses that bud into the nuclear envelope. The envelope acquired during nuclear budding does not end up in the mature viral particle but instead allows the capsid to translocate from the nucleus into the cytosol. The viral nuclear egress complex (NEC) is a critical player in the nuclear egress, yet its function and mechanism have remained enigmatic. Recent studies have demonstrated that the NEC buds membranes without the help of other proteins by forming a honeycomb coat, which established the NEC as the first virally encoded budding machine that operates at the nuclear, as opposed to cytoplasmic, membrane. This review discusses our current understanding of the NEC budding mechanism, with the emphasis on studies that illuminated the structure of the NEC coat and its role in capsid budding during herpesvirus nuclear escape.
1. Introduction
Replication in eukaryotic host cells, which separate their compartments by multiple membranes, presents viruses with a number of challenges. During infection, viruses interact with cellular membranes in many ways. Viruses must breach host membranes to deliver their genomes inside the cells. Once infection is underway, host membranes are used as envelope sources during budding of enveloped viruses or for building replication compartments by some RNA viruses. To accomplish this, viruses have had to become experts in membrane manipulation, yet we are only beginning to understand the mechanisms by which these processes are accomplished.
Herpesviruses are double-stranded DNA, enveloped viruses that infect nearly all vertebrates, from mice to elephants, and even invertebrates such as oysters, scallops, and snails (Davison et al., 2009). The hallmark of herpesviruses is their ability to establish lifelong latent infections in the infected hosts from which they periodically reactivate. Reactivations result not only in a substantial disease burden but also in a high rate of new infections. Herpesviruses that infect mammals and birds belong to the family of Herpesviridae and are divided into three subfamilies, α-, β-, and γ-herpesviruses. Eight human herpesviruses, which contain members of all three subfamilies, are ubiquitous; yet, most infections are asymptomatic as the immune system controls the virus, a testament to the sophisticated mechanism of coexistence with the host. However, disruption of this coexistence results in viral reactivation and a range of ailments from skin lesions and ocular diseases to encephalitis, cancers, congenital infections, and disseminated disease in immunocompromised people, e.g., organ transplant recipients or AIDS patients. Understanding the mechanisms by which herpesviruses manipulate their hosts, including host membranes, is necessary to develop better ways to prevent and control infections.
Herpesviruses are challenging to study because of their inherent complexity: they encode nearly a hundred genes, many of which are unique; despite such large coding capacity, herpesviral proteins are nearly always multifunctional; and herpesviral processes often require multiple proteins where other viruses use only one. One of the most complicated stages in herpesviral replication is viral exit out of the cell, termed egress, which is coupled to viral morphogenesis.
During egress, herpesviruses have to get across several membranes at different cellular locations (Fig. 1). Herpesviruses are also enveloped, and while egressing the cell must gain a lipid envelope as well as other viral components. Enveloped viruses typically acquire their lipid envelope by budding at a cytoplasmic membrane, either the plasma membrane or other cellular membranes. Herpesviruses are unusual in that while they have a single-bilayer envelope, they bud twice. Only the second, and final, budding event at cytoplasmic membranes results in the formation and release of the mature infectious virus while the envelope acquired during the first budding event does not end up in the mature viral particle. The initial budding event is also unusual, because it occurs in the nucleus at the nuclear envelope and serves to allow the viral capsids to escape from the nucleus. Herpesviruses are dsDNA viruses, and their genomes are replicated and packaged into capsids inside the nucleus. Most traffic in and out of the nucleus, which is surrounded by the nuclear envelope, occurs through the nuclear pores. Herpesvirus capsids are too large to fit through the nuclear pores, and to exit the nucleus, capsids bud into the inner nuclear membrane (INM) forming immature viral particles in the perinuclear space (Fig. 1) (Johnson and Baines, 2011). This process is often referred to as the primary envelopment, to distinguish it from the secondary envelopment, which occurs in the cytosol. Perinuclear viral particles then fuse with the outer nuclear membrane (ONM) thereby releasing naked capsids into the cytoplasm in a process termed deenvelopment. As the result, capsids are translocated from the nucleus to the cytosol. Herpesviruses then bud again, this time into cytoplasmic membranes derived from Trans-Golgi Network or early endosomes (Hollinshead et al., 2012; Johnson and Baines, 2011; Owen et al., 2015) to be released from the cell by exocytosis (Hogue et al., 2014) (Fig. 1). Currently, there are only few examples for viruses that bud at the nuclear membrane. It has been reported that insect viruses use nuclear budding (Shen and Chen, 2012; Yuan et al., 2011), but of all known viruses that infect vertebrates, herpesviruses are unique in their nuclear exit strategy.
Fig. 1.
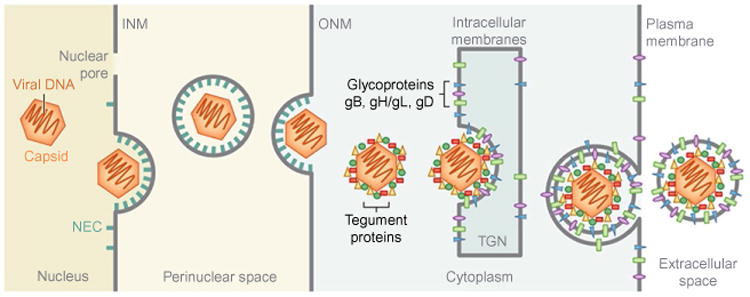
Herpesvirus egress. Herpesviruses assemble their capsids and package their DNA genome in the nucleus. Nucleocapsids bud at the inner nuclear membrane (INM), with the help of the nuclear egress complex (NEC), to form the perinuclear viral particles, which fuse with the outer nuclear membrane (ONM). As the result, the capsids are released into the cytoplasm where they undergo further maturation steps, e.g., assembly of a tegument layer around the capsids. During the second budding event, at the cytoplasmic membranes derived from the Trans-Golgi Network or early endosomes, the capsids acquire their final lipid envelope, which contains glycoproteins required for cell entry. The mature capsids are released from the cell through the secretory pathway. Reprinted from Bigalke, J.M., Heldwein, E.E., 2016. Nuclear exodus: herpesviruses lead the way. Annu. Rev. Virol., 3.
Efficient nuclear egress requires several viral and cellular proteins, but only two viral proteins are essential for the initial budding event at the INM (Bubeck et al., 2004; Chang and Roizman, 1993; Farina et al., 2005; Fuchs et al., 2002; Muranyi et al., 2002; Reynolds et al., 2001; Roller et al., 2000). These two conserved proteins, termed UL31 and UL34 in α-herpesviruses and known by other names in β- and γ-herpesviruses, form the nuclear egress complex (NEC). While the importance of the NEC in nuclear budding has been appreciated for a number of years, its specific function had remained enigmatic until very recently when the NEC was discovered to have an intrinsic ability to vesiculate membranes in vitro in the absence of any other proteins or chemical energy (Bigalke et al., 2014; Lorenz et al., 2015). The NEC drives membrane budding by oligomerizing on the membrane and forming a hexagonal scaffold, or a coat, inside the bud (Bigalke and Heldwein, 2015a; Bigalke et al., 2014; Hagen et al., 2015). The NEC is also capable of membrane scission, which makes it a virally encoded budding nanomachine that can operate independently of other viral or host factors. Furthermore, the crystal structures of NEC from several different herpesviruses (Bigalke and Heldwein, 2015b; Lye et al., 2015; Walzer et al., 2015; Zeev-Ben-Mordehai et al., 2015) have illuminated critical mechanistic and structural features of NEC-mediated budding. The NEC structures provide a three-dimensional roadmap to enable the dissection of its budding mechanism and the design of inhibitors to block it.
In this review, we describe the recent breakthroughs in our understanding of the NEC-mediated budding mechanism, with the emphasis on structures of the NEC heterodimer and the honeycomb lattice it forms in vitro and inside cells. These groundbreaking insights are transforming the way we think about how herpesviruses manipulate host membranes.
2. The Role of Nec in Membrane Budding
2.1 The NEC Is Composed of UL31 and UL34
The NEC is a heterodimer (Bigalke et al., 2014) composed of UL31 and UL34 (Liang and Baines, 2005; Lotzerich et al., 2006; Roller et al., 2010) that is located at the INM (Gonnella et al., 2005; Lotzerich et al., 2006; Reynolds et al., 2001; Sam et al., 2009) and face the nucleoplasm (Reynolds et al., 2001). UL31 and UL34 genes are conserved among all herpesviruses but are known by other names in β-herpesviruses (CMV: UL53 and UL50) and γ-herpesviruses (EBV: BFLF2 and BFRF1). HSV-1 UL34 is a 275-aa protein with a single C-terminal transmembrane (TM) region, while HSV-1 UL31 is a 306-aa protein that is not membrane anchored (Fig. 2). UL31 homologs have nuclear localization signals (Funk et al., 2015; Li et al., 2015; Passvogel et al., 2015), and in the absence of UL34, they are diffusely distributed throughout the nucleoplasm (Zhu et al., 1999). Although some UL34 homologs have predicted nuclear localization signals, most do not. Regardless, in the absence of UL31, UL34 localizes to the perinuclear region. UL34 is presumably anchored in the ONM and diffuses to INM and back yet is only retained in the INM when it binds UL31, in agreement with the currently favored model of diffusion-retention model of membrane protein sorting to the INM (Ungricht and Kutay, 2015; Ungricht et al., 2015). UL31 and UL34 thus appear to be synthesized and trafficked to the nucleus separately (UL31: cytosolic ribosomes and nuclear import machinery; UL34: rough ER and ONM/INM migrating) and only encounter each other at the INM. Formation of the NEC is thus a prerequisite for proper localization of both UL31 and UL34 to the INM (Fuchs et al., 2002; Funk et al., 2015; Johnson and Baines, 2011; Mettenleiter et al., 2009; Reynolds et al., 2001).
Fig. 2.
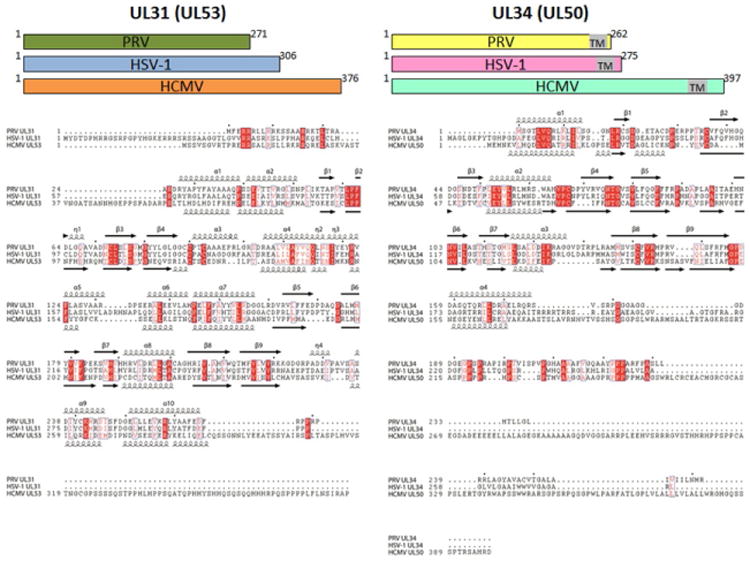
Sequence alignment of UL31 and UL34 proteins from PRV, HSV-1, and HCMV. Secondary structure for PRV and HSV-1 proteins is indicated above the sequences and for HCMV proteins, below the sequence alignment. HCMV bears additional residues at the C-termini of UL31 and UL34, which were absent from the crystallized constructs. Sequence alignment was done with Clustal Omega (Sievers and Higgins, 2014) and visualized with ESPript 3 (Robert and Gouet, 2014).
2.2 The NEC Is Necessary for Nuclear Capsid Budding and Sufficient for INM Vesiculation
The NEC is required for nuclear egress. In the absence of either UL31 or UL34, viral replication is strongly impaired and most capsids accumulate in the nucleus (Bubeck et al., 2004; Chang and Roizman, 1993; Farina et al., 2005; Fuchs et al., 2002a,b; Klupp et al., 2000; Muranyi et al., 2002; Reynolds et al., 2001; Roller et al., 2000). Expression of the NEC from pseudorabies virus (PRV), KSHV, or EBV in transfected cells is sufficient to drive formation of perinuclear vesicles (Desai et al., 2012; Klupp et al., 2007; Luitweiler et al., 2013) or multilayered ruffles in the nuclear envelope (Gonnella et al., 2005). These experiments showed that no other viral proteins besides UL31 and UL34 were necessary for vesiculation and highlighted a key role for the NEC in membrane budding.
2.3 The NEC Is a Complete Membrane-Budding Machine
Although the above experiments made clear that in cells, the NEC can drive membrane vesiculation in the absence of any other viral proteins, they left unanswered the question of the specific function of the NEC in membrane budding. Did it enlist host membrane-deforming machinery or was the NEC capable of mediating membrane deformation and budding directly?
This question was answered when purified recombinant HSV-1 NEC was shown to vesiculate synthetic lipid membranes in vitro in the absence of any other factors (Bigalke et al., 2014). This was first demonstrated with the HSV-1 NEC lacking the UL34 TM region (aka soluble NEC) in vitro (Bigalke et al., 2014) and later also shown with the membrane-anchored NEC from the related PRV (Lorenz et al., 2015).
Although the NEC is normally anchored in the INM by means of the UL34 TM segment, the fact that soluble NEC could mediate membrane budding and scission (Bigalke et al., 2014) suggests that the UL34 TM does not play an active role in budding beyond anchoring. Membrane interaction by soluble HSV-1 NEC required the presence of acidic lipids in membranes, which means that the soluble NEC uses electrostatic interactions between basic residues and acidic lipid headgroups for membrane recruitment (Bigalke et al., 2014). The requirement for acidic lipids became less stringent once the HSV-1 or PRV NEC was recruited to membranes by means of an artificial membrane anchor in the form of a C-terminal His-tag that anchored NEC to membranes containing Ni-chelating lipid (Bigalke et al., 2014; Lorenz et al., 2015). Thus, acidic lipids do not appear necessary for budding. Nevertheless, lipid composition may play an important role in budding because efficient in vitro budding mediated by PRV NEC was dependent on the presence of cholesterol and sphingomyelin (Lorenz et al., 2015).
In vitro, NEC-mediated budding occurs rapidly, within minutes, and requires no chemical energy in the form of ATP. Moreover, NEC carries out the budding process to completion by accomplishing not only formation of the bud but also the scission of its neck. The ability of the purified recombinant NEC to mediate budding and scission of the synthetic lipid vesicles in vitro clearly demonstrated for the first time that the NEC represents a complete virus-encoded membrane-budding machine that, at least in vitro, does not require any additional viral or host factors (Bigalke et al., 2014; Lorenz et al., 2015). Although so far demonstrated only in HSV-1 and PRV, members of the α-herpesvirus subfamily, the intrinsic ability of the NEC to vesiculate membranes is likely conserved among other herpesvirus subfamilies given the conservation of the UL31 and UL34 homologs and their general requirement for nuclear budding.
Budding of many other enveloped viruses requires cellular ESCRT proteins (Votteler and Sundquist, 2013). Nuclear egress of HSV-1 is insensitive to Vps4 dominant-negative mutation (Crump et al., 2007), which would exclude ESCRT involvement. The ability of the NEC to vesiculate membranes in vitro explains why nuclear budding by herpesviruses may not require ESCRTs. Additional experimental evidence is needed; however, before ESCRT involvement can be completely ruled out. Although the NEC alone is sufficient for membrane budding in vitro, efficient budding in cells, for example, efficient neck scission, could potentially benefit from involvement of host factors such as nuclear ESCRT proteins recently found to participate in reformation of nuclear envelope following mitosis (Olmos et al., 2015).
Many enveloped viruses encode their own membrane scaffolding, or matrix, proteins but most require host ESCRT machinery for membrane scission (Adell et al., 2016; Chen and Lamb, 2008; Hurley, 2015; Sundquist and Krausslich, 2012; Votteler and Sundquist, 2013). For example, HIV matrix protein Gag scaffolds the inner surface of the membrane bud and recruits cellular ESCRT-III proteins for membrane scission (Carlson and Hurley, 2012; Wollert et al., 2009) by assembling into a dome-like helical polymer on the inner surface of the neck and constricting it (Effantin et al., 2013). Budding of alphaviruses is thought to be driven entirely by the formation of a virus-encoded outer coat and its interactions with the nucleocapsid, without a role for the ESCRT machinery (Jose et al., 2009; Taylor et al., 2007). Several members of the Flaviviridae family, such as yellow fever virus and hepatitis C virus, are dependent on the ESCRT complex for scission but drive the membrane deformation process by forming a viral protein coat (Carpp et al., 2011; Corless et al., 2010; Mukhopadhyay et al., 2005). By contrast, influenza virus encodes complete membrane-budding machinery, in which two viral proteins, M1 and M2, mediate bud scaffolding and scission, respectively (Rossman and Lamb, 2011, 2013; Rossman et al., 2010). Influenza budding thus does not require the assistance of host proteins.
NEC-mediated nuclear budding by herpesviruses expands the repertoire of ESCRT-independent budding mechanisms. Moreover, the NEC-mediated budding mechanism may be unique because the NEC mediates both membrane scaffolding and scission. While scaffolding is achieved through the formation of a honeycomb coat, how NEC accomplishes scission is yet unclear. Rapid formation of the honeycomb lattice that scaffolds the membrane from the inside could, in principle, provide the driving force for scission by narrowing the neck of the bud. Alternatively, the NEC could change lipid line tension at the neck of the bud to generate membrane curvature conducive to scission, a mechanism that has been proposed to play a role in scission mediated by Influenza M2 protein (Rossman and Lamb, 2013; Rossman et al., 2010). Further work is necessary to determine the mechanism of membrane scission by NEC.
3. Nec Structures
3.1 The Overall Architecture of the NEC
Recently, five structures of NEC from three different herpesviruses, two α-herpesviruses HSV-1 and PRV and a β-herpesvirus HCMV, were determined (PDB IDs as follows: HSV-1: 4ZXS (2.8 Å resolution) (Bigalke and Heldwein, 2015b); PRV: 4Z3U (2.8 Å resolution) (Bigalke and Heldwein, 2015b) and 5E8C (2.9 Å resolution) (Zeev-Ben-Mordehai et al., 2015); and HCMV: 5DOB (2.5 Å resolution) (Lye et al., 2015) and 5D5N (2.4 Å resolution) (Walzer et al., 2015)). The overall NEC fold is very similar in all structures: an elongated, nearly cylindrical molecule of approximately 80 Å × 35 Å × 45 Å (Fig. 3). UL34 (HCMV UL50) has a globular core and forms a pedestal. UL31 (HCMV UL53) consists of a globular core positioned on top of the UL34 pedestal and an N-terminal hook-like extension, composed of two helices, that wraps around the base of UL34 (Fig. 3).
Fig. 3.
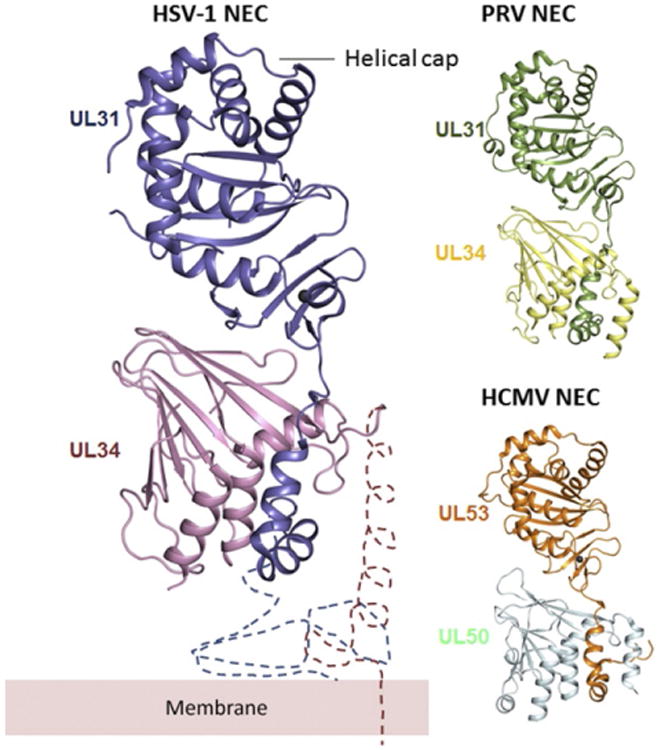
Comparison of HSV-1, PRV, and HCMV NEC crystal structures. All structures are shown in the same orientation. UL31 and UL34 form an elongated complex, with UL31 wrapping its N-terminal hook around UL34. The two molecules interact extensively, which implies high binding affinity. The membrane-proximal end is located at the bottom of the heterodimer in this orientation. The regions important for membrane interaction are missing from the structure and are indicated schematically, along with the membrane, only for HSV-1 but are expected to have a similar location in PRV and HCMV NEC. The C-terminal helix (α4) in HSV-1 UL34 was not resolved in the crystal structure. Overall, NEC structures from three different viruses are very similar, but the relative orientations of UL31 and UL34 are slightly different in HCMV (PDB ID: 4ZXS, 4Z3U, and 5DOB).
To obtain diffraction-quality crystals, the N-terminus of UL31 (17–50 residues) and the C-terminus of UL34 (85–222 residues), which include residues necessary for membrane interactions in UL31 and UL34 (Bigalke et al., 2014) and the TM anchor of UL34, were omitted in all cases (Fig. 4A). In all structures, the last resolved residues abutting the membrane-interacting regions in UL31 and UL34 are located near each other at the base of the NEC (Fig. 3), which places the missing membrane-proximal (MP) regions nearby. Therefore, the MP end of the NEC is located at the base of the UL34 pedestal while the helical cap in UL31 is its membrane-distal (MD) end (Fig. 3).
Fig. 4.
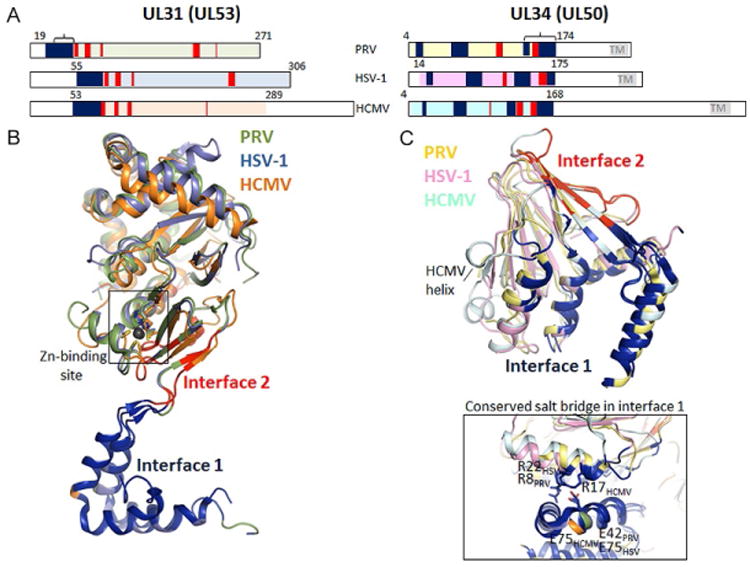
Detailed analysis of the UL31–UL34 interaction. The bar diagram of the crystallized constructs (numbered) and the regions of interactions separated into two interfaces: 1 (blue) and 2 (red). Interface 1 includes the UL31 (UL53) N-terminal hook and multiple regions throughout UL34 (UL50). Interface 2 is restricted to several residues within the C-terminal half of UL34 (UL50) and residues within the globular core in UL31 (UL53). Brackets indicate the binding sites within UL31 and UL34 predicted on the basis of deletion mutagenesis, prior to crystal structures (bar diagram). UL31 and UL34 from HSV-1, PRV, and CMV were superimposed to visualize the similarities in folds (PDB codes 4ZXS, 4Z3U, 5DOB). All UL31 (UL53) molecules contain a zinc-binding motif, with zinc coordinated by three strictly conserved cysteines and one histidine. This element is likely to be important for structural integrity of the complex. Interface 1 and 2 differ between the viruses with regard to salt bridges and hydrogen bonds, but one salt bridge at interface 1 is conserved in all three structures (inlet bottom right) and may be important for complex formation throughout all herpesviruses.
3.2 UL31 and UL34 Structures
UL34 (HCMV UL50) has a novel globular fold described as a β-taco (Bigalke and Heldwein, 2015b; Leigh et al., 2015): a loose β-sandwich is formed by two β-sheets composed of four and five antiparallel β-strands, respectively (Fig. 4C). Three helices (four in HCMV) are located at the top of the taco. In all structures except HSV-1 UL34, another C-terminal helix is located at the side of the taco.
Like UL34, UL31 has an unusual fold, although a portion of it can be aligned to proteins harboring the Bergerat fold, an α-β-β-α-β-β motif found among the ATP-binding members of the GHKL ATPase/kinase superfamily, which includes proteins such as histidine kinases, chaperones Hsp90, and DNA topoisomerase II (Bergerat et al., 1997; Dutta and Inouye, 2000). UL31 has an additional β strand between the second strand and the second helix of the classic Bergerat fold, which results in α-β-β-β-α-β-β topology. Due to the largely hydrophobic nature of the region that corresponds to the ATP-binding site in the GHKL family members, UL31 is unlikely to bind ATP.
All UL31 molecules bind a zinc ion, which is coordinated by three cysteines and one histidine (Fig. 4B). All four zinc-coordinating residues are strictly conserved among UL31 sequences from α-, β-, and γ-herpesvirus subfamilies, and thus zinc binding by UL31 is likely a common feature among herpesviruses. Only two other residues, P9531 and S11031, are conserved across all three subfamilies, underscoring the important role of zinc coordination. The zinc-coordinating residues come from distant regions of UL31, and the CCCH-type zinc-binding site in UL31 does not resemble a zinc finger. Instead, zinc coordination may stabilize UL31 structure by anchoring the surface-exposed helix α3 to the lower β sheet.
3.3 Comparison of the NEC Structures
Despite the overall resemblance, the NEC structures from α-herpesviruses HSV-1 and PRV are more similar to each other than to the NEC structures from HCMV, a β-herpesvirus (Figs. 3 and 4). The rmsd values range between 1.1 and 1.4 Å for HSV-1 vs PRV NECs, ∼3.1 Å for HSV-1 vs HCMV, and 4.3 Å for PRV vs HCMV alignments. UL34 structures are more similar (rmsd values range between 0.6 and 1.6 Å) than UL31 structures (rmsd values range between 1.1 and 3.1 Å). The main difference among UL34 structures lies in the absence of the C-terminal helix α4 from HSV-1 UL34, which is not resolved in the structure. Additionally, the loop between strands β1 and β2 in HSV-1 and PRV UL34 is a short helix in HCMV UL50 (Figs. 2 and 4C). The main differences in UL31 structures are in the conformation of several loops, the orientation of the N-terminal hook relative to the globular part of the molecule, and helices α3—α5 and α10 (Figs. 2 and 4C).
3.4 The Complex Interactions at the NEC Interface
UL31 and UL34 interaction buries a large accessible surface area, nearly 2000 Å2, which underlies the NEC stability. UL31 and UL34 have two distinct interfaces. Interface 1 is formed by the V-shaped hook of UL31, whereas interface 2 is formed by the globular core of UL31 (Fig. 4). Prior to structure determination, two regions necessary for complex formation, one within UL31 and the other within UL34, were proposed based on deletion analysis (Liang and Baines, 2005; Sam et al., 2009; Schnee et al., 2012) and designated as the respective binding sites (Fig. 4A). While both regions participate in UL31/UL34 interactions, they map to different interfaces and hardly contact one another, which underscores the complexity of UL31/ UL34 interactions that involve multiple regions throughout the protein sequence.
Interface 1 contributes approximately two-thirds of the contacts and is thus more important for the NEC stability. Interface 1 features a salt bridge present in all structures (HSV-1: E7531–R2234, PRV: E4231–R834, HCMV: E7553–R1750) (Fig. 4C). In HSV-1 NEC, interface 1 is stabilized by two additional salt bridges (R5831–E7834, R6231–D7534), missing from PRV and HCMV NEC. Interface 2 has two similarly located salt bridges in α-herpesviruses (HSV-1: D10431–R16734 and D23231–R15834; PRV: D7131–R15334 and D19531–R14434) but not in HCMV. The extensive interdigitation of side chains along interface 1 suggests that it may be rigid. By contrast, interface 2 is relatively smooth and may permit some motion between UL31 and UL34. Indeed, UL31 and UL34 within HSV-1, PRV, and HCMV structures have distinct relative orientations (Fig. 3). Although some of the UL31/UL34 interactions are conserved among α-herpesviruses and even β-herpesviruses, the majority of contacts between UL31 (UL53) and UL34 (UL50), especially at interface 2, appear species specific.
Although UL31 and UL34 from PRV as well as UL53 and UL50 from HCMV could be purified individually (Leigh et al., 2015; Lorenz et al., 2015), this was not possible with the HSV-1 UL31 or UL34 due to their tendency to precipitate after solubility tag removal (Bigalke et al., 2014). The structures suggest that in the absence of their respective binding partners, both UL31 and UL34 would expose hydrophobic patches, normally buried at the interface, which could lead to aggregation. Additionally, the “hook” in UL31 is likely misfolded in the absence of UL34.
4. The Hexagonal Lattice of the Nec: Structure and Function
4.1 The NEC Assembles into a Hexagonal Lattice In Vitro
One mechanism of membrane deformation used by proteins is forming an ordered array or coat (Zimmerberg and Kozlov, 2006). Consistent with its ability to bud membranes, NEC assembles into honeycomb-like hexagonal lattices on membranes both in vitro (Bigalke et al., 2014) and in vivo (Hagen et al., 2015). The ability of NEC to oligomerize was first observed in vitro with the purified recombinant HSV-1 NEC lacking the UL34 TM region (aka soluble NEC) (Bigalke et al., 2014). Soluble NEC is a heterodimer in solution, but upon incubation with synthetic lipid vesicles, it oligomerized, as detected by crosslinking and cryoelectron microscopy. The latter technique revealed a coat-like NEC array on the inner surface of lipid vesicles that appeared as a honeycomb lattice in top views (Fig. 5) and spikes emanating from the membrane toward the interior of the vesicle in side views. Given that the NEC cannot cross membranes, the vesicles with inner coats represent the product of budding and scission (Bigalke et al., 2014). The hexagonal lattice forming prior to or during budding could not be visualized (Bigalke et al., 2014) likely because budding in vitro occurs very fast. These observations suggest that NEC drives budding by a rapid and likely cooperative formation of this coat-like hexagonal lattice that efficiently scaffolds the membrane from the inside.
Fig. 5.
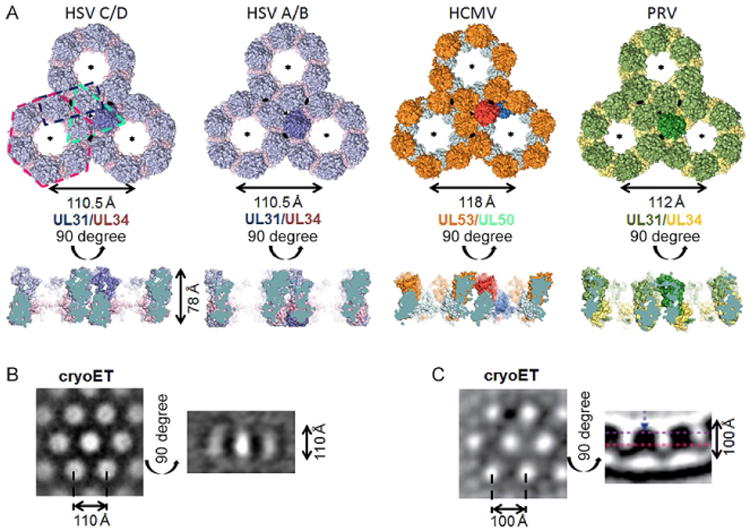
Comparison of hexagonal lattice in HSV-1 (two crystal lattices), HCMV (crystal lattice), and PRV (model derived from cryoET data). (A) For each lattice, three connected hexameric rings are shown side by side in a top view, perpendicular to the sixfold symmetry axis, and a side view. One NEC heterodimer is highlighted in every lattice. The two-, three-, and sixfold axes in each lattice are indicated by lense, triangle, and star symbols, respectively. Representative dimer, trimer, and hexamer are indicated by dashed lines in the HSV-1 C/D lattice. The hexameric rings are very similar in both HSV-1 lattices and the HCMV lattice but differ in the PRV lattice model. HSV-1 A/B is rings are turned toward each other in a 10.5 degree angle compared to the C/D lattice. The PRV lattice model is the only curved lattice in the side view while the rest of the lattices are planar. All crystal lattices are ∼78 Å thick. For HSV-1 NEC (B) and PRV NEC (C), cryoET lattices are shown for comparison. The lattices are thicker than in the crystal structures due to the presence of the membrane-proximal regions, absent from all crystallized NEC constructs. The diameter of the hexameric ring in the PRV cryoET lattice is slightly smaller than other ring diameters due to the curvature of the lattice and the positioning of the slice (purple dashed line in side view). HSV-1 NEC cryoET image in (B) is reprinted from Bigalke, J.M., Heldwein, E.E., 2015b. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J., 34, 2921–2936. PRV NEC cryoET image in (C) is reprinted from Hagen, C., Dent, K.C., Zeev-Ben-Mordehai, T, Grange, M., Bosse, J.B., Whittle, C., Klupp, B.G., Siebert, C.A., Vasishtan, D., Bauerlein, F.J., Cheleski, J., Werner, S., Guttmann, P., Rehbein, S., Henzler, K., Demmerle, J., Adler, B., Koszinowski, U., Schermelleh, L., Schneider, G., Enquist, L.W., Plitzko, J.M., Mettenleiter, T.C., Grunewald, K., 2015. Structural basis of vesicle formation at the inner nuclear membrane. Cell, 163, 1692–1701. http://dx.doi.org/10.1016/j.cell.2015.11.029, under Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/.
4.2 Hexagonal Lattice in NEC Crystals Resembles NEC Coats
The HSV-1 NEC crystallized in space group P6 with two NEC heterodimers in the asymmetric unit, NECAB and NECCD. Close inspection of the crystal packing revealed two hexagonal lattices stacked on top of each other, one formed by NECAB and the other by NECCD (Fig. 5). The two NEC heterodimers are nearly identical and each forms a hexagonal lattice with the periodicity of 110.5 Å and the thickness of 78.0 Å. Nonetheless, the two lattices are distinct as described in Section 4.3.
The geometry and the dimensions of the NEC crystal lattice in the crystals resemble both the NEC coats visualized by cryoEM on the inner surface of the budded vesicles obtained in vitro (Bigalke et al., 2014) and the coats on the inner surface of the perinuclear vesicles formed in cells transfected with PRV UL31 and UL34 or infected with PRV (Hagen et al., 2015). Both the crystal lattice and the membrane coat have hexagonal symmetry and the periodicity of ∼110 Å (Fig. 5). The main difference between the two honeycomb lattices, the crystal and the membrane coat, is their thickness. While the crystal lattice is 78 Å thick, the thickness of the NEC coat in budded vesicles, excluding the lipid bilayer, is ∼110 Å (Bigalke et al., 2014), leaving ∼30-Å thick density in the vicinity of the membrane unaccounted for by the crystal structure (Fig. 5). This additional density seen at the MP end of the spikes forming the membrane coats can be attributed to the 50 additional residues at the N-terminus of UL31, 14 additional residues at the N-terminus of UL34, and 35 additional residues at the C-terminus of UL34, missing from the crystallized NEC construct. All three missing regions are expected to co-localize at the MP end of the NEC and extend the NEC spike by ∼30 Å toward the membrane producing a characteristic fence-like pattern in side view cryoEM projections of the HSV-1 NEC membrane coats (Bigalke et al., 2014) and an “archway” motif in the PRV perinuclear vesicles (Hagen et al., 2015). The similarity in the architecture of the two NEC crystal lattices and the membrane coats strongly suggests that the crystal lattice recapitulates the membrane coats in the budded vesicles and that the NEC/NEC interactions observed in the crystals are relevant to the NEC-mediated budding.
Interestingly, one of the HCMV NEC crystal forms also took space group P6 (Walzer et al., 2015), although this is a rather unusual space group for protein crystals. The periodicity of the hexagonal lattice observed in the HCMV NEC crystals is ∼118 Å and the thickness is ∼74 Å (Fig. 4). Slightly different dimensions of the HSV-1 and CMV crystal lattices are likely due to the differences in the structures of the NEC heterodimers themselves, especially, the positioning of helix α4 in UL34 vs UL50 that results in HCMV NEC having a broader pedestal relative to HSV-1 and PRV NEC (Fig. 3). Despite these differences, the ability of the NEC from both α- and β-herpesviruses to form hexagonal lattices implies a similar strategy for NEC coat formation and similar budding mechanism. We anticipate that the yet uncharacterized NEC from γ-herpesviruses will share this property.
4.3 Analysis of NEC/NEC Interactions Within the Crystal Lattices
To give rise to the hexagonal lattice, the NEC molecules interact at two-, three-, and sixfold symmetry axes or, in other words, form dimers, trimers, and hexamers (Fig. 5). Two observations support the idea that the hexamers (rather than the dimers or the trimers) are the building blocks of the NEC lattice. First, the interactions that form the hexamers are more extensive and more conserved (16%) than interactions at the dimeric (13%) and trimeric (2%) interfaces. Second, the two HSV-1 NEC molecules in the asymmetric unit, NECAB and NECCD, form nearly identical hexamers but two distinct lattices, which we refer to here as A/B and C/D, by forming different sets of interactions at the two- and threefold symmetry axes (Figs. 5 and 6). It is helpful to think of the hexagonal lattice in terms of two sets of interactions: those involved in the formation of the hexameric rings (hexameric interfaces) and those that link individual hexameric rings to each other (interhexameric interfaces). The latter include interactions at the dimeric and trimeric interfaces. Comparisons of the three hexagonal crystal lattices: the two HSV-1 NEC lattices, A/B and C/D, and the HCMV NEC lattice follow.
Fig. 6.
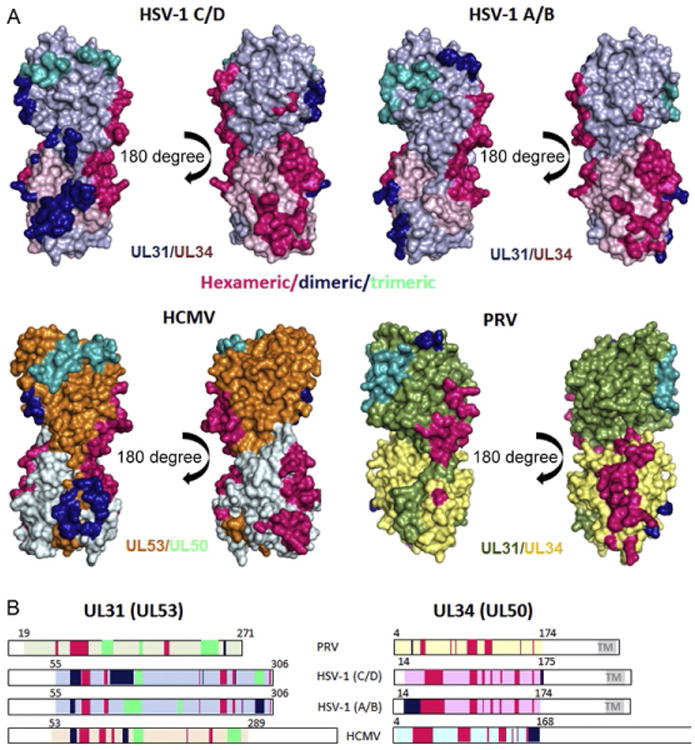
Detailed comparison of hexameric and interhexameric interfaces in HSV-1 A/B, C/D, and HCMV crystal lattices, and the PRV lattice model. (A) Interfaces are colored hotpink (hexameric), dark blue (dimeric), and teal (trimeric) to match the representative hexamer, dimer, and trimer in Fig. 5A. Bar diagrams (B) are colored according to the same color scheme. Hexameric interfaces are very similar in both HSV-1 and the HCMV lattices. In PRV, this interface is shifted toward the center of the molecule in this orientation. These differences are also apparent in the bar diagram. HSV-1 C/D and HCMV lattices also have overlapping dimeric and trimeric interfaces, whereas in the HSV-1 A/B lattice, the trimeric and dimeric interactions are almost completely reversed. Interfaces in the PRV lattice differ from those in the other three lattices.
In all three lattices, the hexameric rings are formed by UL34/UL34 and UL34/UL31 interactions and are mainly mediated by loops (Fig. 6). In UL34 (and HCMV UL50), these are the loops just prior to, between or after β1–β2, β4–β5, β6, β8, and β9; for UL31 (and HCMV UL53), these are β1–β2, β3–β4, and P8. Although few residues at the hexameric interface are conserved among α- and β-herpesviruses (∼16%), the hexamers observed in the HCMV NEC crystal lattice are very similar.
Although the hexamers in all three lattices are essentially the same in HSV-1 and are very similar in HCMV, they are arranged in two distinct ways: one that is similar in the HSV-1 C/D and HCMV lattices and the other, a different one, in the HSV-1 A/B lattice. Nevertheless, all interhexameric interactions primarily involve helices rather than loops (Fig. 6). In the HSV-1 C/D and HCMV lattices, these are helices α2, α3, α4, η2, η3, α5, α10 in UL31 (or UL53) and helix α4 plus the loop before in UL34 (or UL50). Of these, only η2, η3, α5, and α10 in UL31 (or UL53) are involved in interactions at the trimeric interface, whereas all others mediate the interactions at the dimeric interface. Helix α3 in UL31 (UL53), located at the dimeric interface, contributes one residue to the Zn-binding site and appears to be held in place by Zn coordination. In this way, Zn binding could potentially contribute to the formation of the hexagonal lattice.
The interhexameric interactions in the HSV-1 A/B lattice are slightly different from the above described. Although nearly the same set of residues is involved in interhexameric interactions (Fig. 6B) in A/B and C/D lattices, residues that lie at the dimeric interface in the C/D lattice map to the trimeric interface in the A/B lattice and vice versa. The dimeric interactions in the A/B lattice include residues in UL31 α2, which contact α1, β1, and the loop in between of UL34. There is a second dimeric interaction with another NEC molecule, which is mediated by residues in UL31 α10. The trimeric interactions are partly identical (η2—3), but instead of α10, the loop prior to β5 is involved in addition to α4. A close comparison of NECAB and NECCD reveals a small shift of helices α4 and α2 in UL31. These helices participate in threefold symmetry contacts within the NECAB lattice but two- and threefold symmetry contacts within the NECCD lattice. Ajuxtaposition of the A/B and C/D lattices reveals that the hexameric rings adopt somewhat different orientations, reminiscent of a cogwheel that had been turned 10.5 degree counterclockwise from C/D to A/B (Fig. 5). As the result, even though the residues involved in interhexameric interactions are the same, residues located at the dimeric interface in the A/B lattice mediate interactions at the trimeric interface in the C/D lattice.
Whether both lattices observed in the crystals of HSV-1 NEC are biologically relevant is currently unclear. The similarities between the HCMV and HSV-1 C/D (but not A/B) lattices favor the HSV-1 C/D as the biologically relevant lattice. Nevertheless, the ability of the NEC hexamers to interact in more than one way suggests plasticity in the hexameric arrangement.
Helix α4 of UL34 was not resolved in the HSV-1 NEC structure possibly because the last few residues of this helix were absent from the crystallization construct. In the HSV-1 C/D lattice, this helix would interfere with the hexamer formation due to steric hindrance at the dimeric interface. But, it would not affect the hexagonal lattice formed by the HSV-1 A/B lattice. Although this helix has been postulated to play a regulatory role in oligomerization (Bigalke and Heldwein, 2015b), its presence in the HCMV NEC lattice suggests that the absence of α434 in HSV-1 NEC crystal lattice could instead be a crystallographic artifact, meaning that the flat crystal lattice requires that α434 becomes disordered. A slightly curved lattice, such as that observed in NEC vesicle coats, could potentially accommodate this helix. Interestingly, HSV-1 NEC crystals were thin, fragile plates that tended to warp, and so HSV-1 NEC may have a tendency to form a slightly curved lattice.
As noted earlier, the hexameric interactions are mediated by residues located in loops and in β hairpins, whereas the interhexameric interactions are mainly mediated by α-helices. It is tempting to speculate that interactions mediated by flexible loops are stronger because the residues have more conformational freedom and can therefore achieve a better fit, similarly to interlacing fingers. This idea is consistent with the notion that the hexameric rings are the building blocks of the hexagonal lattice. In contrast, interactions mediated by helices may be weaker, which would make interhexameric interactions more pliant. Such plasticity may be important in transitioning from flat, strictly hexagonal packing to a curved coat, discussed in more detail in the following sections.
4.4 The NEC Assembles into a Hexagonal Lattice at the INM
Direct evidence that the NEC forms honeycomb coats in cells was recently obtained by cryoelectron microscopy and tomography of the perinuclear vesicles formed in uninfected cells stably expressing PRV UL31 and UL34 (Hagen et al., 2015). The NEC formed 10 nm thick coats that lined the inner surface of perinuclear vesicles and were, on average, 107 nm in diameter. Subtomogram averaging revealed a curved hexagonal lattice composed of interconnected yet distinct layers: MP and MD. Both MP and MD lattices have the periodicity of ∼11 nm, which is consistent with the honeycomb lattice observed in vitro and in crystals. The MD layer is ∼7 nm thick, which resembles the NEC lattice observed in crystals and is thus formed by the crystallized UL31/UL34 core complex. The MP layer is ∼3 nm thick and is likely formed by the MP regions of UL31 and UL34, absent from the crystal structure and has a distinct appearance. Notably, “holes” in the MD lattice line up with dome-shaped protrusions in the MP lattice. These “domes,” being located at the sixfold symmetry axes, are likely composed of MP regions of six NEC heterodimers forming each hexamer (Fig. 7).
Fig. 7.
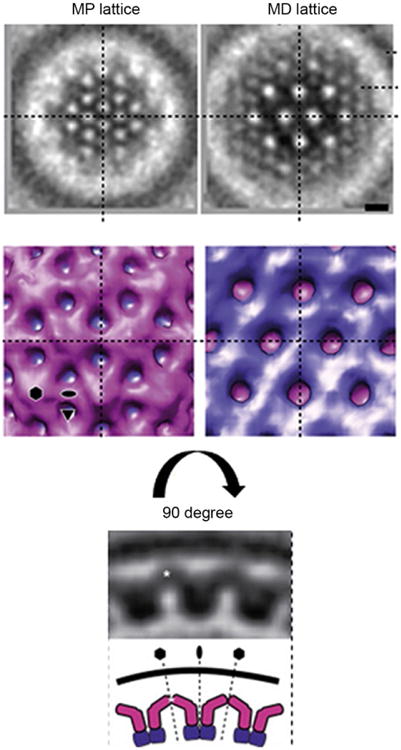
Subtomogram averaging of PRV NEC coat. Slices through the membrane-proximal (MP) and membrane-distal (MD) regions are shown to highlight two layers of the NEC hexagonal lattice. The MD slice corresponds to the lattices seen in crystal structures of HSV-1 and HCMV NEC as well the hexagonal lattice seen previously by cryoET in HSV-1 NEC coats formed in vitro. The MP slice shows a distinct lattice likely formed by MP regions, absent from all crystallized NEC constructs. It links the NEC core structure to the UL34 transmembrane anchor and to the membrane. A 90 degree-related side view below shows the MP region (pink) forming an archway in the vicinity of the membrane whereas the MD region (purple) is mainly involved in forming the hexagonal NEC lattice. Reprinted from Hagen, C., Dent, K.C., Zeev-Ben-Mordehai, T., Grange, M., Bosse, J.B., Whittle, C., Klupp, B.G., Siebert, C.A., Vasishtan, D., Bauerlein, F.J., Cheleski, J., Werner, S., Guttmann, P., Rehbein, S., Henzler, K., Demmerle, J., Adler, B., Koszinowski, U., Schermelleh, L., Schneider, G., Enquist, L.W., Plitzko, J.M., Mettenleiter, T.C., Grunewald, K., 2015. Structural basis of vesicle formation at the inner nuclear membrane. Cell, 163, 1692–1701. http://dx.doi.org/10.1016/j.cell.2015.11.029, under Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/.
The curvature of the MD lattice follows that of the vesicle membrane. Locally, the curvature is achieved by hexamers being tilted relative to one another at twofold interfaces, which is consistent with the hexamer being the building block of the honeycomb lattice. The wide range of coat curvatures that the NEC can achieve is likely permitted by a high degree of flexibility within the MP layer of the NEC lattice. This is consistent with the lack of defined secondary structure predicted for the MP regions of UL31 and UL34.
To obtain an atomic model of the NEC coat, the crystal structure of PRV NEC heterodimer was fitted into the curved PRV NEC coat model obtained by 3D subtomogram averaging (Hagen et al., 2015), yielding an atomic model of the curved PRV NEC coat (PDB ID 5FKI) (Zeev-Ben-Mordehai et al., 2015). Comparison of the interfaces mediating hexameric and interhexameric interactions in the crystal lattices in HSV-1 and HCMV NEC vs the PRV NEC lattice model shows large differences between the crystal lattices and the model (Fig. 6). First, the hexamers look different (Figs. 5 and 6). A close analysis of the interactions forming the hexamer reveals large differences at the interfaces. The hexameric interface in the PRV lattice is significantly smaller than the hexameric interface in the HSV-1 and HCMV lattices (43 vs ∼60 residues). The interhexameric interacting regions are likewise completely different (Fig. 6).
The differences between the crystal lattices and the model suggest that the packing of the individual NEC heterodimers changes greatly during transition from the flat to the curved array. Yet, residues that have been shown to be important for NEC function in HSV-1, do not map to the interfaces in the model PRV lattice (see Section 4.5). Besides, as the building blocks of the lattice, hexamers would be expected to shift in their position to each other to form a curved lattice rather than change their assembly completely. Therefore, an alternative explanation is that the discrepancy between the crystal lattices and the model could be due to an imprecise fit of the NEC heterodimer into the low-resolution tomogram. Fitting hexameric rings instead of single NEC molecules, based on the HSV-1 crystal lattice, could potentially yield a more accurate atomic model of the NEC coat. Regardless, further studies are necessary to determine how individual NEC heterodimers form the honeycomb coat and what conformational changes the NEC lattice undergoes during transition from the flat array to the spherical coat.
The NEC coats were also observed in HSV-1-infected cells by cryoelectron microscopy and tomography (Hagen et al., 2015). The NEC formed coats that lined the inner surface of the perinuclear viral particles, formed during egress. Moreover, cryoelectron tomography of HSV-1-infected nuclei provided snapshots of the entire budding process (Hagen et al., 2015). The NEC formed ∼100 nm flat ordered patches at the nucleoplasmic side of the INM when nucleocapsids were in close contact to the INM. The NEC patches did not extend beyond the capsid budding sites and were not observed in the absence of capsids, which implies that contact with the capsid is important for coat nucleation. The coat curved and expanded during budding of the INM into the perinuclear space. As the capsid budding progressed, the NEC formed a tight coat around the capsid. During subsequent deenvelopment, the NEC coat was left behind at the cytoplasmic face of the ONM.
Although ∼70% of perinuclear vesicles contained capsids, perinuclear vesicles lacking capsids were also apparent, raising the possibility that capsids are not the sole trigger of NEC coat nucleation. These capsid-less coats had a smaller diameter, ∼115nm, relative to the diameter of the coats in the perinuclear viral particles. The finding that in the absence of capsids, the NEC coat assembly produces vesicles of a size somewhat smaller than capsids implies that the capsid, rather than the NEC, determines the minimum diameter of the enveloped perinuclear viral particles.
Although the ordered nature of the NEC coats was evident, due to technical limitations, their precise geometry could not be investigated. However, the appearance and the diameter of capsid-less perinuclear coats in infected and transfected cells are similar, and one may conclude that the NEC forms honeycomb coats during nuclear egress in infected cells.
4.5 NEC Oligomerization Is Required for Budding
The hexagonal coats observed on the inner surface of budded vesicles raise the idea that the formation of the hexagonal lattice is a prerequisite for NEC-mediated budding (Bigalke et al., 2014; Hagen et al., 2015). The crystal structure of the hexagonal NEC lattice provided detailed information on the positions of individual residues to test this idea by mutational analysis.
4.5.1 Structural Basis for the Nonbudding Phenotype of Several NEC Mutants
Several UL31 and UL34 mutants in HSV-1 and PRV defective in viral replication yet capable of forming NEC and localizing to the INM have been reported (Bjerke et al., 2003; Passvogel et al., 2013, 2014; Roller et al., 2010). In the crystal structure of HSV-1 NEC, some of these mutations map to the hexameric interface and likely disrupt NEC function by perturbing its oligomerization (Bjerke et al., 2003; Passvogel et al., 2014; Roller et al., 2010). One such mutant, containing a double point mutation D35A/E37A within UL34, had a strong defect in replication due to a block in capsid nuclear budding (Roller et al., 2010). Although capsids could be seen juxtaposed with the INM and the INM curving slightly around them, the budding was blocked. The dominant-negative phenotype of this mutant pointed to a possible defect in oligomerization. Subsequent in vitro experiments confirmed that the double mutant was defective in budding and in forming the hexagonal coats on budded membranes (Bigalke et al., 2014).
In the crystal structure of HSV-1 NEC, residues D3534 and E3734 are located within a flexible loop but only E3734 is at the hexameric interface (Fig. 8). Indeed, the E37A34 mutation alone was sufficient to convey the nonbudding phenotype in vitro (Bigalke and Heldwein, 2015b). This mutation likely destabilizes the hexameric interfaces thereby hindering lattice assembly. The dominant-negative effect of the mutation suggests that, when present in sufficient amounts, it can “poison” the formation of the NEC lattice even in the presence of the WT UL34. PRV E2434, which corresponds to HSV-1 E3734, is unresolved in the PRV NEC crystal structure but does not map to the hexameric interface in the PRV lattice model.
Fig. 8.
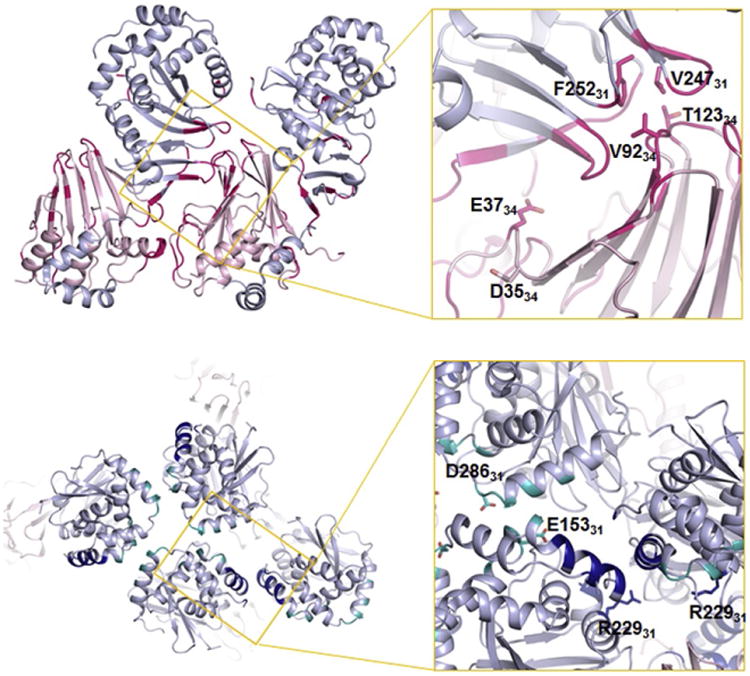
Position of mutant residues that disturb HSV-1 NEC activity within the hexagonal lattice. Two protomers within the hexameric ring are shown, with the interface colored in hotpink. The hexameric interface is critical for NEC function because mutations of residues at this interface result in a nonbudding phenotype. Mutations at the interhexameric interfaces (lower panel: dimeric interface) in dark blue, trimeric interface in teal also reduce budding efficiency but to a lower extent. The suppressor mutation that showed an enhanced budding activity (R229L) is also shown at the dimeric interface. Adapted from Bigalke, J.M., Heldwein, E.E., 2015b. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J., 34, 2921–2936.
4.5.2 Mutations That Disrupt Oligomerization Interfaces Hinder Budding
To test the importance of the hexagonal lattice in budding, a panel of residues at hexameric and interhexameric (dimeric or trimeric) interfaces were mutated to either disrupt electrostatic interactions or introduce bulky side chains that could not be accommodated at the interface. The goal was to destabilize the hexagonal lattice by perturbing either contacts within the hexamers or between the hexamers. Several mutations at the hexameric interface (V92F34, T123Q34, F252Y31, and V247F31) (Fig. 8) strongly reduced the NEC budding in vitro, which demonstrated that the disruption of the hexameric interface through steric hindrance lead to reduced budding. Therefore, formation of the hexagonal lattice is necessary for budding. Unexpectedly, mutations at the interhexameric interface (D286R31 and E153R31) (Fig. 8) had a more modest effect on budding (Bigalke and Heldwein, 2015b). These observations pinpoint the hexameric interface as being more important for budding, but a firm conclusion awaits a comprehensive mutational analysis of the interfaces. Although mutations that block budding in vitro would be expected to block capsid budding in infected cells, this has not yet been confirmed.
4.5.3 Mutation That Restores Budding of Nonbudding NEC Mutants May Reinforce the Hexagonal Lattice
A spontaneous suppressor mutation that restored budding to the non-budding mutant virus containing a dominant-negative mutation in UL34, described in Section 4.5.1, has been reported (Roller et al., 2010). A single mutated residue in UL31, R229L, was responsible for the suppressor phenotype. This mutation also restored the defect in in vitro budding (Bigalke and Heldwein, 2015b). Surprisingly, in the crystal structure of the HSV-1 NEC, residue R22931 does not map to the hexameric interface. Instead, residue R22931 is located at the dimeric interface in the C/D lattice (Fig. 8). In the A/B lattice, the R22931 side chain does not participate in any interactions, although curvature could potentially bring it closer to other residues at the trimeric interface.
Interestingly, the compensatory effect of the mutation R229L31 is not limited to the dominant-negative mutant of UL34. In vitro, it also rescued budding of other UL34 mutants that disrupted the hexameric interface, e.g., V92F34 (Bigalke and Heldwein, 2015b). While V9234 is at the hexameric interface, it is not near E3734. Therefore, the finding that the same mutation could rescue nonbudding phenotypes of either UL34 mutant was remarkable. Whether the mutation could rescue budding of mutants that disrupted interhexameric interactions is yet unknown. How R229L31 mutation can restore WT budding efficiency of nonbudding UL34 mutants without being in the vicinity of the mutated residues is unclear. However, in vitro experiments have shown that the suppressor mutation alone enhanced budding efficiency ∼1.4-fold (Bigalke and Heldwein, 2015b). These findings support the hypothesis that rather than acting to restoring disrupted contacts at the hexameric interface, the suppressor mutation R229L31 has a generally beneficial effect on budding. Given its location in the vicinity of the interhexameric interface, one possible mechanism could be to stabilize the NEC lattice by reinforcing contacts between the hexamers.
5. Nec Lattice Curvature
The hexagonal crystal lattice formed by the NEC is flat, whereas the honeycomb coats are spherical. While flat arrays permit strictly symmetrical hexagonal packing works, formation of a curved array, such as a coat, requires distortions, or defects, in hexagonal packing. While models of purely hexagonal NEC coats have been proposed (Bigalke et al., 2014; Hagen et al., 2015), their veracity is yet unclear. A spherical particle characterized by hexagonal symmetry is typically achieved through a regular inclusion of pentagons, which generates a so-called polyhedron, e.g., an icosahedral viral capsid. But, the NEC coats observed in vitro and in cells lack obvious icosahedral symmetry (Bigalke et al., 2014; Hagen et al., 2015). In some cases, however, the hexagonal lattice can be closed by incorporation of irregular defects, such as observed in the immature HIV capsids formed by Gag (Briggs et al., 2009; Schur et al., 2015) and the early poxvirus envelope (Heuser, 2005) formed by D13 (Hyun et al., 2011). It is tempting to speculate that the NEC coat could arise from incorporation of such irregular defects into a hexagonal lattice. The two distinct interhexamer arrangements observed in the HSV-1 NEC crystals may indicate that the lateral packing of hexamers has some flexibility, which could provide a way for introducing occasional lattice disruptions so that the spherical NEC coats are formed (Figs. 5 and 9). For example, hexamers may have to rotate slightly while the curved lattice is being formed. Perfectly hexagonal patches of NEC lattice, which have been observed in NEC coats (Bigalke et al., 2014; Hagen et al., 2015), would be interrupted by irregularities resulting in a curved array (Fig. 9).
Fig. 9.
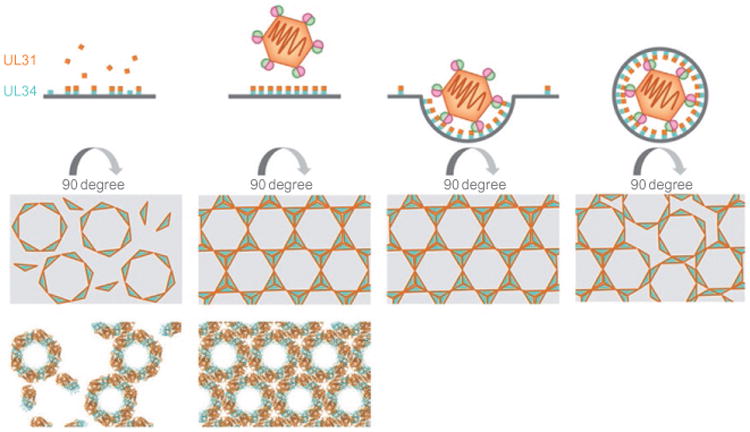
Model of NEC-mediated budding during nuclear egress. UL34 localizes to the INM by means of its C-terminal transmembrane helix. Soluble UL31 is present in the nucleus and binds to UL34 at the INM. Hexameric rings assemble and eventually connect to form a lattice. Upon capsid binding, possibly through UL17/UL25 or the major capsid protein, the NEC forms a coat that deforms the membrane around the capsid. To form a spherical object, disruptions in the hexagonal lattice are likely required. NEC coats lack obvious icosahedral symmetry, and these disruptions may be of irregular nature. Reprinted from Bigalke, J.M., Heldwein, E.E., 2016. Nuclear exodus: herpesviruses lead the way. Annu. Rev. Virol., 3.
Curiously, known viral coats composed of spherical hexagonal lattices with irregular defects are temporary and are not retained in mature viral particles. The immature HIV capsid is transformed into a mature capsid characterized by “broken” polyhedral symmetry (Briggs et al., 2009). The early poxvirus envelope formed by D13, which drives the formation of membrane crescents and their coalescence into a spherical particle, is disassembled shortly after the immature viral particle is formed (Condit et al., 2006). Given that the NEC coat is disassembled during deenvelopment (Hagen et al., 2015; Skepper et al., 2001) and is not a part of the mature virion, it is tempting to speculate that hexagonal coats containing irregularities possess characteristics that make them susceptible to modifications that may ease their disassembly.
6. Regulation of Nec Lattice Assembly
The NEC has a robust membrane vesiculation activity both in vitro (Bigalke et al., 2014; Lorenz et al., 2015) and in cells stably expressing the UL31 and UL34 (Desai et al., 2012; Klupp et al., 2007; Luitweiler et al., 2013). But during infection, empty perinuclear vesicles are rarely observed (Hagen et al., 2015; Klupp et al., 2011). Therefore, in infected cells, the intrinsic budding ability of the NEC is likely regulated to avoid nonproductive budding. Given that NEC oligomerization is the driving force for the vesiculation, formation of the NEC lattice would need to be inhibited until the mature capsid comes along. Indeed, it was recently found that the flat NEC coat only assembled on the INM once the capsid approached the membrane (Hagen et al., 2015).
6.1 Regulation of NEC Lattice Assembly by Membrane-Interacting Regions of NEC
The presence of the honeycomb lattice in the crystals of HSV-1 and HCMV NEC demonstrates the capacity of the NEC to self-assemble in solution, at least, at the high protein concentration achieved in crystal setups. Of note, NEC constructs that formed crystal lattice lacked membrane-interacting regions. By contrast, longer HSV-1 NEC constructs containing intact membrane-interacting regions, such as those used in the in vitro budding assay (Fig. 2), oligomerized only in the presence of membranes (Bigalke et al., 2014). These observations suggest that the membrane-interacting regions within the NEC could inhibit their ability to oligomerize correctly in the absence of membranes and that their displacement (in the longer NEC construct upon membrane binding) or removal (in the NEC used for crystallization) is required for the NEC coat self-assembly. Membrane-interacting regions could, therefore, be a part of the regulatory mechanism that controls NEC-mediated budding. However, in infected cells, UL34 is already localized to the INM by means of its C-terminal TM region. Therefore, additional regulatory factors could be needed to keep the MP region in its inhibitory conformation to prevent premature coat formation. This can potentially be achieved either by posttranslational modifications or by protein binding, discussed in the following sections.
6.2 Regulation of NEC Lattice Assembly by Phosphorylation
The budding activity of the HSV-1 NEC could be regulated by posttranslational modification, specifically, phosphorylation by the viral kinase US3 (Mou et al., 2009). Several serines within the N-terminus of UL31 are phosphorylated by the US3 kinase during replication (Mou et al., 2009). Substitution of these serines with glutamates, which mimic phosphorylation, blocked capsid budding. By contrast, their replacement with alanines, to mimic unphosphorylated state, led to both the apparent NEC aggregation at the INM, presumably, due to uncontrolled NEC-mediated vesiculation, and aberrant accumulation of perinuclear virions in herniations, implying a block in deenvelopment (Mou et al., 2009). Phosphorylation could thus function to prevent the NEC from oligomerizing prematurely, that is, until the arrival of a capsid. Dephosphorylation would then be a prerequisite for NEC oligomerization. Additionally, phosphorylation may also play a role during the deenvelopment step.
6.3 Regulation of NEC Lattice Assembly by Capsid
Another regulatory input could be provided by the capsid itself. Given that primarily mature, genome-containing capsids bud into the INM (Klupp et al., 2011), it is possible that the trigger for the NEC oligomerization is provided by a surface component of a mature capsid. The NEC recruits viral capsids to the INM (Yang and Baines, 2011), and it has been proposed that the auxiliary capsid protein UL25 or the major capsid protein VP5 or both directly bind UL31 (Yang and Baines, 2011; Yang et al., 2014). A mature capsid, with multiple binding sites for the NEC, would create avidity effects that could help drive the formation of extended NEC patches and ultimately, the entire coat. Transmitting the signal from putative capsid-binding site within UL31 at the MD end of the NEC to the MP region would require large conformational changes within the NEC.
How the budding activity of NEC is inhibited in infected cells and how this inhibition is relieved in the presence of the capsid are an area ripe for future investigations.
7. Nec and Capsid Deenvelopment at the Onm
Capsid budding at the INM, which leads to the formation of the perinuclear viral particles, is an essential step in herpesvirus nuclear egress. But, nuclear egress is not completed until the membrane of the perinuclear viral particle fuses with the ONM, and the capsids are released into the cytosol. This final stage of nuclear egress is termed deenvelopment. At its core, deenvelopment is membrane fusion, but relatively little is known about the mechanism of this process, and even the nature of the fusogen is still uncertain. Viral glycoproteins that are essential for initial entry into host cells, i.e., gB, gD, and gH/gL, are present in perinuclear virions (Stannard et al., 1996), which make gB, the conserved herpesvirus fusogen acting during viral entry an obvious candidate for a fusogen enabling deenvelopment. Yet, while gB appears to be required for deenvelopment in HSV-1 and EBV infected cells (Farnsworth et al., 2007; Lee and Longnecker, 1997; Wright et al., 2009), this is not the case for other herpesviruses, such as PRV or KSHV (Klupp et al., 2008; Krishnan et al., 2005). Herpesvirus genomes do not appear to encode any other fusogens and could instead hijack host fusogens. Thus, the identity of the fusogen mediating fusion during deenvelopment remains a mystery.
Although deenvelopment is primarily thought of as the fusion of the membrane of the perinuclear viral particle with the ONM, capsid release into the cytosol would also require the disassembly of the stable NEC coat. How the hexagonal NEC coat gets disassembled is yet unclear, but phosphorylation of UL31 by the US3 kinase appears important. US3 is present in perinuclear viral particles, and in its absence, these particles accumulate in the perinuclear space (Reynolds et al., 2002), similarly to what is observed with serine-to-alanine UL31 mutant (Mou et al., 2009). Phosphorylation of the NEC after primary envelopment may lead to structural rearrangements that disrupt the hexagonal lattice, thereby enabling deenvelopment. By interfering with oligomerization, phosphorylation of the NEC would both inhibit budding in the absence of the capsid and promote disassembly of the NEC coat during deenvelopment. It is tempting to speculate that the disassembly of the NEC lattice and fusion is coordinated, with one event possibly triggering the other thereby increasing the efficiency of the deenvelopment process.
8. Summary
Herpesviral capsids are translocated from the nucleus into the cytoplasm by an unusual mechanism—termed nuclear egress—whereby capsids bud at the INM (primary envelopment), and the resulting primary virions fuse with the ONM (deenvelopment). The conserved NEC, located at the INM, is essential for nuclear egress. Recent studies have established the NEC as a virally encoded budding machine and illuminated the mechanism of NEC-mediated membrane budding that is driven by the formation of a honeycomb coat. The current model suggest that the NEC forms hexagonal patches in the vicinity of the nucleocapsid and that these patches expand as the budding process progresses until a complete hexagonal coat is formed around the capsid. Crystal structures of the NEC from several herpesviruses have revealed the architecture of the NEC heterodimer and highlighted conserved features. In particular, the hexagonal lattices observed in NEC crystals provided an atomic-level view of interactions forming the NEC coat. These findings have galvanized the field of herpesvirus egress and enable further exploration of the mechanism of NEC-regulated membrane budding. The NEC is the first viral membrane-budding machinery that is capable of both membrane scaffolding and scission, which makes it a fascinating system for further studies. The budding activity of the NEC is likely subject to both positive and negative control by viral proteins, in addition to posttranslational modifications, but the regulatory mechanism has not yet been elucidated. Future studies need to investigate the mechanisms by which the NEC coat assembly is inhibited in the absence of capsid, how capsids trigger the NEC activity, and how the NEC coat is disassembled during deenvelopment. Ultimately, the detailed understanding of the NEC-mediated nuclear budding will aid in the development of therapeutics to control herpesvirus infections.
Acknowledgments
This work on nuclear egress in the Heldwein lab was supported by the NIH grants 1R21AI097573, 1R01GM111795, and the Burroughs Wellcome Fund. J.M.B. is a recipient of a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft GZ: BI 1658/1-1.
References
- Adell MA, Migliano SM, Teis D. ESCRT-III and Vps4: a dynamic multipurpose tool for membrane budding and scission. FEBS J. 2016 doi: 10.1111/febs.13688. [Epub ahead of print] http://dx.doi.org/10.1111/febs.13688. [DOI] [PubMed]
- Bergerat A, De Massy B, Gadelle D, Varoutas PC, Nicolas A, Forterre P. An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature. 1997;386:414–417. doi: 10.1038/386414a0. [DOI] [PubMed] [Google Scholar]
- Bigalke JM, Heldwein EE. The great (nuclear) escape: new insights into the role of the nuclear egress complex of herpesviruses. J Virol. 2015a;89:9150–9153. doi: 10.1128/JVI.02530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigalke JM, Heldwein EE. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015b;34:2921–2936. doi: 10.15252/embj.201592359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigalke JM, Heuser T, Nicastro D, Heldwein EE. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun. 2014;5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerke SL, Cowan JM, Kerr JK, Reynolds AE, Baines JD, Roller RJ. Effects of charged cluster mutations on the function of herpes simplex virus type 1 UL34 protein. J Virol. 2003;77:7601–7610. doi: 10.1128/JVI.77.13.7601-7610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. Structure and assembly of immature HIV. Proc Natl Acad Sci USA. 2009;106:11090–11095. doi: 10.1073/pnas.0903535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck A, Wagner M, Ruzsics Z, Lotzerich M, Iglesias M, Singh IR, Koszinowski UH. Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J Virol. 2004;78:8026–8035. doi: 10.1128/JVI.78.15.8026-8035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson LA, Hurley JH. In vitro reconstitution of the ordered assembly of the endosomal sorting complex required for transport at membrane-bound HIV-1 Gag clusters. Proc Natl Acad Sci USA. 2012;109:16928–16933. doi: 10.1073/pnas.1211759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpp LN, Galler R, Bonaldo MC. Interaction between the yellow fever virus nonstructural protein NS3 and the host protein Alix contributes to the release of infectious particles. Microbes Infect. 2011;13:85–95. doi: 10.1016/j.micinf.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Chang YE, Roizman B. The product of the UL31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J Virol. 1993;67:6348–6356. doi: 10.1128/jvi.67.11.6348-6356.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BJ, Lamb RA. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology. 2008;372:221–232. doi: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condit RC, Moussatche N, Traktman P. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res. 2006;66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- Corless L, Crump CM, Griffin SD, Harris M. Vps4 and the ESCRT-III complex are required for the release of infectious hepatitis C virus particles. J Gen Virol. 2010;91:362–372. doi: 10.1099/vir.0.017285-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump CM, Yates C, Minson T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J Virol. 2007;81:7380–7387. doi: 10.1128/JVI.00222-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison AJ, Eberle R, Ehlers B, Hayward GS, Mcgeoch DJ, Minson AC, Pellett PE, Roizman B, Studdert MJ, Thiry E. The order Herpesvirales. Arch Virol. 2009;154:171–177. doi: 10.1007/s00705-008-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J. Reconstitution of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J Virol. 2012;86:594–598. doi: 10.1128/JVI.05988-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Bio-chem Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- Effantin G, Dordor A, Sandrin V, Martinelli N, Sundquist WI, Schoehn G, Weissenhorn W. ESCRT-III CHMP2A and CHMP3 form variable helical polymers in vitro and act synergistically during HIV-1 budding. Cell Microbiol. 2013;15:213–226. doi: 10.1111/cmi.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina A, Feederle R, Raffa S, Gonnella R, Santarelli R, Frati L, Angeloni A, Torrisi MR, Faggioni A, Delecluse HJ. BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J Virol. 2005;79:3703–3712. doi: 10.1128/JVI.79.6.3703-3712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci USA. 2007;104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J Virol. 2002;76:364–378. doi: 10.1128/JVI.76.1.364-378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C, Ott M, Raschbichler V, Nagel CH, Binz A, Sodeik B, Bauerfeind R, Bailer SM. The herpes simplex virus protein pUL31 escorts nucleocapsids to sites of nuclear egress, a process coordinated by its N-terminal domain. PLoS Pathog. 2015;11:e1004957. doi: 10.1371/journal.ppat.1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonnella R, Farina A, Santarelli R, Raffa S, Feederle R, Bei R, Granato M, Modesti A, Frati L, Delecluse HJ, Torrisi MR, Angeloni A, Faggioni A. Characterization and intracellular localization of the Epstein-Barr virus protein BFLF2: interactions with BFRF1 and with the nuclear lamina. J Virol. 2005;79:3713–3727. doi: 10.1128/JVI.79.6.3713-3727.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bauerlein FJ, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grunewald K. Structural basis of vesicle formation at the inner nuclear membrane. Cell. 2015;163:1692–1701. doi: 10.1016/j.cell.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J Cell Biol. 2005;169:269–283. doi: 10.1083/jcb.200412169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogue IB, Bosse JB, Hu JR, Thiberge SY, Enquist LW. Cellular mechanisms of alpha herpesvirus egress: live cell fluorescence microscopy of pseudorabies virus exocytosis. PLoS Pathog. 2014;10:e1004535. doi: 10.1371/journal.ppat.1004535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 2012;31:4204–4220. doi: 10.1038/emboj.2012.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH. ESCRTs are everywhere. EMBO J. 2015;34:2398–2407. doi: 10.15252/embj.201592484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun JK, Accurso C, Hijnen M, Schult P, Pettikiriarachchi A, Mitra AK, Coulibaly F. Membrane remodeling by the double-barrel scaffolding protein of poxvirus. PLoS Pathog. 2011;7:e1002239. doi: 10.1371/journal.ppat.1002239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol. 2011;9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- Jose J, Snyder JE, Kuhn RJ. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009;4:837–856. doi: 10.2217/fmb.09.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Mettenleiter TC. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J Virol. 2000;74:10063–10073. doi: 10.1128/jvi.74.21.10063-10073.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci USA. 2007;104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. Glycopro-teins required for entry are not necessary for egress of pseudorabies virus. J Virol. 2008;82:6299–6309. doi: 10.1128/JVI.00386-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Mettenleiter TC. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J Virol. 2011;85:8285–8292. doi: 10.1128/JVI.00741-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan HH, Sharma-Walia N, Zeng L, Gao SJ, Chandran B. Envelope glycoprotein gB of Kaposi's sarcoma-associated herpesvirus is essential for egress from infected cells. J Virol. 2005;79:10952–10967. doi: 10.1128/JVI.79.17.10952-10967.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Longnecker R. The Epstein-Barr virus glycoprotein 110 carboxyterminal tail domain is essential for lytic virus replication. J Virol. 1997;71:4092–4097. doi: 10.1128/jvi.71.5.4092-4097.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh KE, Sharma M, Mansueto MS, Boeszoermenyi A, Filman DJ, Hogle JM, Wagner G, Coen DM, Arthanari H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc Natl Acad Sci USA. 2015;112:9010–9015. doi: 10.1073/pnas.1511140112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Jiang S, Wang J, Mo C, Zeng Z, Yang Y, Chen C, Li X, Cui W, Huang J, Peng T, Cai M. Characterization of the nuclear import and export signals of pseudorabies virus UL31. Arch Virol. 2015;160:2591–2594. doi: 10.1007/s00705-015-2527-7. [DOI] [PubMed] [Google Scholar]
- Liang L, Baines JD. Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J Virol. 2005;79:3797–3806. doi: 10.1128/JVI.79.6.3797-3806.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, Mettenleiter TC, Antonin W. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem. 2015;290:6962–6974. doi: 10.1074/jbc.M114.627521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotzerich M, Ruzsics Z, Koszinowski UH. Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J Virol. 2006;80:73–84. doi: 10.1128/JVI.80.1.73-84.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luitweiler EM, Henson BW, Pryce EN, Patel V, Coombs G, Mccaffery JM, Desai PJ. Interactions of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex: ORF69 is a potent factor for remodeling cellular membranes. J Virol. 2013;87:3915–3929. doi: 10.1128/JVI.03418-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, Coen DM. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015;34:2937–2952. doi: 10.15252/embj.201592651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter TC, Klupp BG, Granzow H. Herpesvirus assembly: an update. Virus Res. 2009;143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Mou F, Wills E, Baines JD. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol. 2009;83:5181–5191. doi: 10.1128/JVI.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Kuhn RJ, Rossmann MG. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science. 2002;297:854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- Olmos Y, Hodgson L, Mantell J, Verkade P, Carlton JG. ESCRT-III controls nuclear envelope reformation. Nature. 2015;522:236–239. doi: 10.1038/nature14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DJ, Crump CM, Graham SC. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses. 2015;7:5084–5114. doi: 10.3390/v7092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Trube P, Schuster F, Klupp BG, Mettenleiter TC. Mapping of sequences in pseudorabies virus pUL34 that are required for formation and function of the nuclear egress complex. J Virol. 2013;87:4475–4485. doi: 10.1128/JVI.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Janke U, Klupp BG, Granzow H, Mettenleiter TC. Identification of conserved amino acids in pUL34 which are critical for function of the pseudorabies virus nuclear egress complex. J Virol. 2014;88:6224–6231. doi: 10.1128/JVI.00595-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC. Functional characterization of nuclear trafficking signals in pseudorabies virus pUL31. J Virol. 2015;89:2002–2012. doi: 10.1128/JVI.03143-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. U(L) 31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol. 2001;75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol. 2002;76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller RJ, Zhou Y, Schnetzer R, Ferguson J, Desalvo D. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol. 2000;74:117–129. doi: 10.1128/jvi.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller RJ, Bjerke SL, Haugo AC, Hanson S. Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J Virol. 2010;84:3921–3934. doi: 10.1128/JVI.01638-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology. 2011;411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Lamb RA. Viral membrane scission. Annu Rev Cell Dev Biol. 2013;29:551–569. doi: 10.1146/annurev-cellbio-101011-155838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Jing X, Leser GP, Lamb RA. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell. 2010;142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam MD, Evans BT, Coen DM, Hogle JM. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J Virol. 2009;83:2996–3006. doi: 10.1128/JVI.02441-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnee M, Wagner FM, Koszinowski UH, Ruzsics Z. A cell free protein fragment complementation assay for monitoring the core interaction of the human cytomegalovirus nuclear egress complex. Antiviral Res. 2012;95:12–18. doi: 10.1016/j.antiviral.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Schur FK, Hagen WJ, Rumlova M, Ruml T, Muller B, Krausslich HG, Briggs JA. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature. 2015;517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- Shen H, Chen K. BM61 of Bombyx mori nucleopolyhedrovirus: its involvement in the egress of nucleocapsids from the nucleus. FEBS Lett. 2012;586:990–995. doi: 10.1016/j.febslet.2011.12.040. [DOI] [PubMed] [Google Scholar]
- Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. 2014;1079:105–116. doi: 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- Skepper JN, Whiteley A, Browne H, Minson A. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment→deenvelopment→reenvelopment pathway. J Virol. 2001;75:5697–5702. doi: 10.1128/JVI.75.12.5697-5702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stannard LM, Himmelhoch S, Wynchank S. Intra-nuclear localization of two envelope proteins, gB and gD, of herpes simplex virus. Arch Virol. 1996;141:505–524. doi: 10.1007/BF01718314. [DOI] [PubMed] [Google Scholar]
- Sundquist WI, Krausslich HG. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med. 2012;2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GM, Hanson PI, Kielian M. Ubiquitin depletion and dominant-negative VPS4 inhibit rhabdovirus budding without affecting alphavirus budding. J Virol. 2007;81:13631–13639. doi: 10.1128/JVI.01688-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungricht R, Kutay U. Establishment of NE asymmetry-targeting of membrane proteins to the inner nuclear membrane. Curr Opin Cell Biol. 2015;34:135–141. doi: 10.1016/j.ceb.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Ungricht R, Klann M, Horvath P, Kutay U. Diffusion and retention are major determinants of protein targeting to the inner nuclear membrane. J Cell Biol. 2015;209:687–703. doi: 10.1083/jcb.201409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votteler J, Sundquist WI. Virus budding and the ESCRT pathway. Cell Host Microbe. 2013;14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzer SA, Egerer-Sieber C, Sticht H, Sevvana M, Hohl K, Milbradt J, Muller YA, Marschall M. Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J Biol Chem. 2015;290:27452–27458. doi: 10.1074/jbc.C115.686527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert T, Wunder C, Lippincott-Schwartz J, Hurley JH. Membrane scission by the ESCRT-III complex. Nature. 2009;458:172–177. doi: 10.1038/nature07836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CC, Wisner TW, Hannah BP, Eisenberg RJ, Cohen GH, Johnson DC. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J Virol. 2009;83:11847–11856. doi: 10.1128/JVI.01397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Baines JD. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci USA. 2011;108:14276–14281. doi: 10.1073/pnas.1108564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Wills E, Lim HY, Zhou ZH, Baines JD. Association of herpes simplex virus pUL31 with capsid vertices and components of the capsid vertex-specific complex. J Virol. 2014;88:3815–3825. doi: 10.1128/JVI.03175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Huang Z, Wei D, Hu Z, Yang K, Pang Y. Identification of Autographa californica nucleopolyhedrovirus ac93 as a core gene and its requirement for intranuclear microvesicle formation and nuclear egress of nucleocapsids. J Virol. 2011;85:11664–11674. doi: 10.1128/JVI.05275-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeev-Ben-Mordehai T, Weberruss M, Lorenz M, Cheleski J, Hellberg T, Whittle C, El Omari K, Vasishtan D, Dent KC, Harlos K, Franzke K, Hagen C, Klupp BG, Antonin W, Mettenleiter TC, Grunewald K. Crystal structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep. 2015;13:2645–2652. doi: 10.1016/j.celrep.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HY, Yamada H, Jiang YM, Yamada M, Nishiyama Y. Intracellular localization of the UL31 protein of herpes simplex virus type 2. Arch Virol. 1999;144:1923–1935. doi: 10.1007/s007050050715. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]