

Abstract
In plants, imprinted gene expression occurs in endosperm seed tissue and is sometimes associated with differential DNA methylation between maternal and paternal alleles1. Imprinting is theorized to have been selected for because of conflict between parental genomes in offspring2, but most studies of imprinting have been conducted in Arabidopsis thaliana, an inbred primarily self-fertilizing species that should have limited parental conflict. We examined embryo and endosperm allele-specific expression and DNA methylation genome-wide in the wild outcrossing species Arabidopsis lyrata. Here we show that the majority of A. lyrata imprinted genes also exhibit parentally-biased expression in A. thaliana, suggesting that there is evolutionary conservation in gene imprinting. Surprisingly, we discovered substantial interspecies differences in methylation features associated with paternally expressed imprinted genes (PEGs). Unlike in A. thaliana, the maternal allele of many A. lyrata PEGs was hypermethylated in the CHG context. Increased maternal allele CHG methylation was associated with increased expression bias in favor of the paternal allele. We propose that CHG methylation maintains or reinforces repression of maternal alleles of PEGs. These data suggest that while the genes subject to imprinting are largely conserved, there is flexibility in the epigenetic mechanisms employed between closely related species to maintain monoallelic expression. This supports the idea that imprinting of specific genes is a functional phenomenon, and not simply a byproduct of seed epigenomic reprogramming.
Keywords: genomic imprinting, DNA methylation, endosperm, Arabidopsis, CHG gene body hypermethylation, parental conflict
Genomic imprinting is a form of epigenetic gene regulation in flowering plants and mammals in which alleles of genes are expressed in a parent-of-origin dependent manner. Allele-specific gene expression profiling has identified hundreds of imprinted genes in A. thaliana, maize, and rice endosperm, the functions of which are largely unknown3–10. Allelic differences in DNA methylation and chromatin modification between maternal and paternal alleles are important for establishing and maintaining imprinted expression1. The emerging picture from multiple species is that the paternal allele of PEGs is associated with DNA methylation, while the silent maternal allele is hypomethylated and bears the Polycomb Repressive Complex 2 (PRC2) mark H3K27me3 11,12.
Several evolutionary theories have been proposed to describe processes that would select for fixation of this unusual pattern of gene expression13. The kinship or parental conflict theory posits that imprinting is selected for because of asymmetric relatedness among kin2,13. In species where the maternal parent directly provisions growing progeny and has offspring by multiple males, maternally and paternally inherited genomes are predicted to have conflicting interests with regard to the extent of maternal investment. Paternally inherited alleles are expected to favor maternal investment at the expense of half-siblings.
Low conservation of imprinting between A. thaliana and monocots14, limited conservation between rice and maize14, evidence for intraspecific variation in imprinting6, and lack of strong phenotypes for some imprinted gene mutants has cast doubt on whether imprinting of particular genes is functionally important. Additionally, although some imprinted genes are associated with differential methylation, it has been suggested that imprinted expression is simply a byproduct of endosperm DNA methylation changes – changes that could have a primary function outside of imprinting regulation15,16. We were motivated by these considerations and by predictions of the parental conflict theory to compare imprinting and seed DNA methylation between two closely related species that differ in breeding strategy. A. lyrata and A. thaliana diverged approximately 13 million years ago17. Although A. thaliana outcrosses to some extent in the wild, as an obligate outcrosser A. lyrata should be subject to a higher degree of parental conflict than A. thaliana and should therefore be under greater pressure to maintain imprinting.
To identify A. lyrata imprinted genes, we performed mRNA-seq on parental strains and F1 hybrid embryo and endosperm tissue derived from crosses between the sequenced A. lyrata strain MN47 (MN) and a strain from Karhumäki (Kar) (Supplementary Figure 1, Supplementary Figure 2, Supplementary Tables 1 and 2). After reannotating A. lyrata genes based on our extensive RNA-seq data (see Supplementary Methods), sequence polymorphisms between MN and Kar were used to quantify the contributions of each parental genome to gene expression. All possible pairwise comparisons (n=12) of parent-of-origin bias among three MN × Kar and four Kar × MN reciprocal cross replicates were performed to identify imprinted genes using the same criteria we previously applied to A. thaliana6. Only genes that were defined as imprinted in at least 40% of comparisons were included in the final set (Figure 1, Supplementary Tables 3 and 4, see Supplementary Methods for details of imprinting criteria). This analysis yielded 49 paternally expressed imprinted genes (PEGs) and 35 maternally expressed imprinted genes (MEGs) in endosperm (Figure 1A). Allele-assignment calls for thirteen genes, including both imprinted and non-imprinted genes, were validated by pyrosequencing (Supplementary Figure 3). As expected3,5, there was little evidence for imprinting in embryos (Figure 1A).
Figure 1. Identification of imprinted genes in A. lyrata and comparison to A. thaliana.
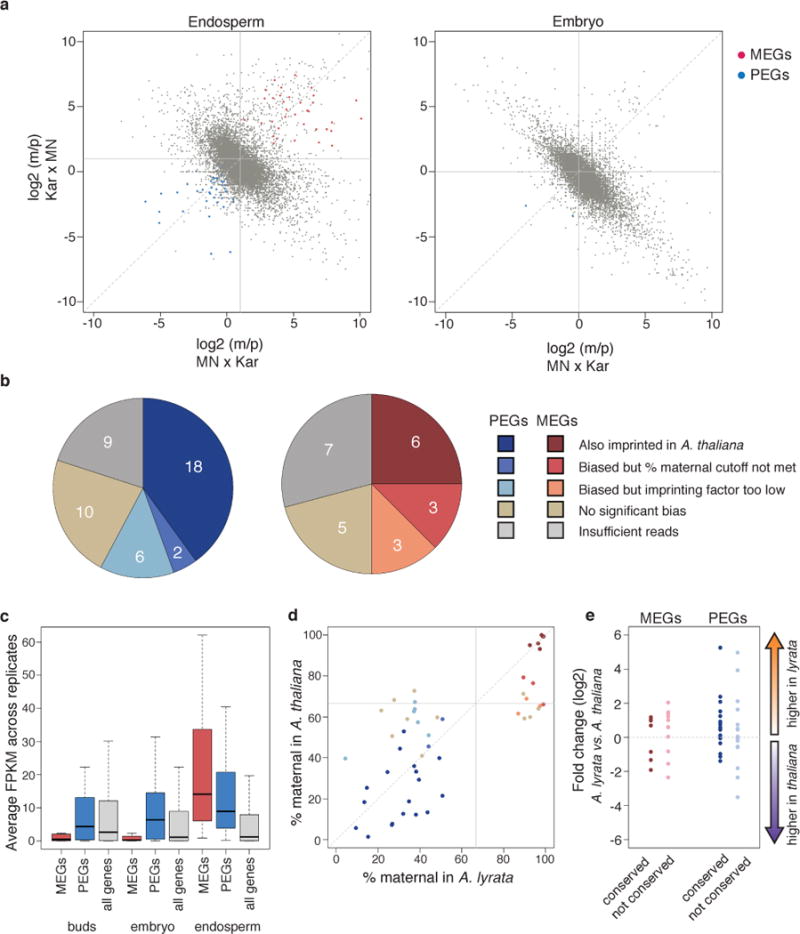
a) Comparison of maternal to paternal transcript ratio (m/p) from reciprocal crosses for all genes. Counts from biological replicates were pooled for plotting. b) Imprinting conservation between A. lyrata and A. thaliana. Reason for lack of imprinting in A. thaliana indicated. c) Average gene expression in Kar × Kar tissues. Outliers not shown. d) % maternal reads for imprinted genes. Colors as in (b). e) Relative gene expression levels in A. lyrata vs. A. thaliana endosperm for imprinted genes. Log2 ratios were calculated using DESeq2.
We compared A. lyrata and A. thaliana endosperm imprinted genes (Figure 1, Supplementary Figure 4, Supplementary Table 4). Of the A. lyrata PEGs for which there were sufficient data available in A. thaliana, 72% (26/36) were also paternally biased in A. thaliana with 50% (18/36) meeting all stringent criteria for being designated as a PEG in both species (Figure 1B). Conserved PEGs encoded DNA binding proteins and genes related to chromatin modification, among others (Supplementary Table 4). Of the A. lyrata MEGs for which there were sufficient data in A. thaliana, 70% (12/17) were also significantly maternally biased in A. thaliana, with 35% (6/17) meeting all criteria for being called as a MEG in both datasets (Figure 1B). The conserved MEGs included the Polycomb group gene FIS2, the F-box gene SDC, another F-box gene, and three genes encoding DNA binding proteins. While previous research has identified somewhat more imprinted genes in A. thaliana than what we describe in A. lyrata, these studies involved multiple accessions and assessed imprinting for a greater total number of genes6. The majority of genes that were imprinted in A. thaliana but not in A. lyrata lacked sufficient data to make an imprinting designation in A. lyrata (Supplementary Figure 4). Thus, it is presently unclear whether the number of imprinted genes differs significantly between the species. All of the genes that are commonly imprinted among A. thaliana and cereals14 were also imprinted in A. lyrata.
Many mammalian imprinted genes are clearly involved in growth regulation, including genes for nutrient uptake and feeding behavior18. By contrast, we find that proteins encoded by conserved plant imprinted genes are predicted to regulate or effect the expression of many other genes (chromatin proteins and transcription factors) or protein abundance (F-boxes). We also found that some pathways, rather than orthologous genes, were imprinted in both species, as has been previously noted for imprinting of different subunits of the PRC2 complex among Arabidopsis and cereals19. In A. thaliana, the large subunit of RNA Polymerase IV, NRPD1, which functions in RNA-directed DNA methylation (RdDM)20, is a PEG5,6. Although we did not find evidence for imprinting of the NRPD1 gene in A. lyrata, homologues of two other genes involved in RdDM were PEGs (Supplementary Table 4): NRPD4/NRPE4/RDM2 (AL946699), which encodes a common subunit of Pol IV and Pol V, and RRP6L1 (AL337734), which encodes an exosomal protein that impacts RdDM. Thus, in both species the function of RdDM in the endosperm is under paternal influence, but this is achieved via different genes.
The kinship theory is essentially an argument about optimal total gene expression levels in offspring13. We therefore evaluated the expression levels and patterns of imprinted genes. MEGs appear to be primarily endosperm-specific genes; they have much lower than average expression in embryos and flower buds, and much higher than average expression in the endosperm (Figure 1C). Conversely, PEGs were more highly expressed in all tissues than genes on average, and showed more modest expression increases in endosperm, suggesting that the expression of MEGs and PEGs is regulated differently. We also compared the percent maternal transcripts for homologous imprinted A. lyrata and A. thaliana genes (Figure 1D). Conserved MEGs and PEGs exhibited similar degrees of parental bias in the two species (Figure 1D). However, comparison of the A. thaliana and A. lyrata gene expression level for individual imprinted genes indicated that the overall expression level of PEGs was higher in A. lyrata than in A. thaliana (Figure 1E). These findings are consistent with stronger selection for higher expression of PEGs in species with greater parental conflict, such as obligate outcrossers13.
In A. thaliana, active DNA demethylation by the 5-methylcytosine DNA glycosylase DME in the central cell (the female gamete that is the progenitor of the endosperm) before fertilization is essential for establishing gene imprinting at many loci1. Imprinting of many A. thaliana genes, particularly PEGs, is correlated with maternal allele demethylation of proximal sequences corresponding to fragments of transposable elements6,21. A. lyrata PEGs were somewhat enriched for the presence of TEs in 5′ regions compared to all genes, with 30 out of 49 PEGs (61%) associated with at least one TE within 2 kb 5′, compared to 51% of all genes (Supplementary Table 4). To test if the relationship between methylation and imprinting was conserved in A. lyrata, we profiled methylation genome-wide in MN × MN flower bud, embryo, and endosperm tissue by whole genome bisulfite sequencing. Shared and novel endosperm methylation features were observed compared to A. thaliana (Figure 2, Figure 3, Supplementary Figure 5, Supplementary Table 5). In plants, DNA methylation is found in CG, CHG, and CHH sequence contexts. CG methylation was strongly decreased in TEs and in the 5′ and 3′ regions of genes in endosperm relative to other tissues (Figure 2A, Supplementary Figure 5). By profiling allele-specific DNA methylation in F1 embryo and endosperm from Kar females crossed to MN males, we determined that maternally inherited DNA was primarily responsible for endosperm CG hypomethylation (Figure 2B). These data suggest that, like in A. thaliana, A. lyrata maternally-inherited genomes are actively demethylated before fertilization6,21,22.
Figure 2. A. lyrata endosperm exhibits an unusual methylation profile.
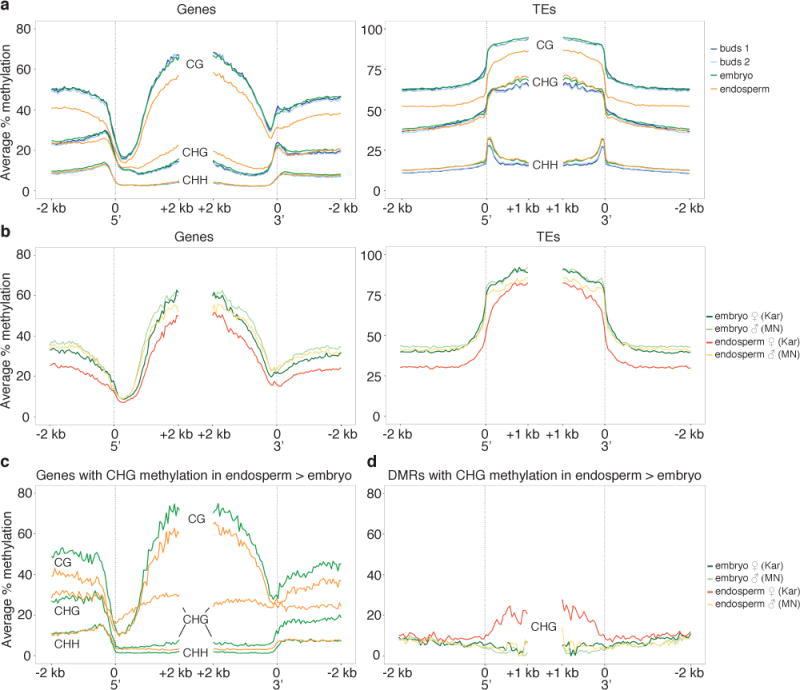
a) Average % methylation for genes (left) and TEs (right) in MN tissues. b) Average allelic % CG methylation in Kar × MN embryo and endosperm. c) Average % methylation for 1606 genes containing a MN × MN endosperm CHG hypermethylated DMR. Green, embryo; orange, endosperm. d) Average % CHG methylation in Kar × MN embryo and endosperm, with separate profiles for the maternal (Kar) and paternal (MN) alleles, over regions CHG hypermethylated in MN × MN endosperm.
Figure 3. A. lyrata PEGs are associated with maternal allele CHG gene body hypermethylation.
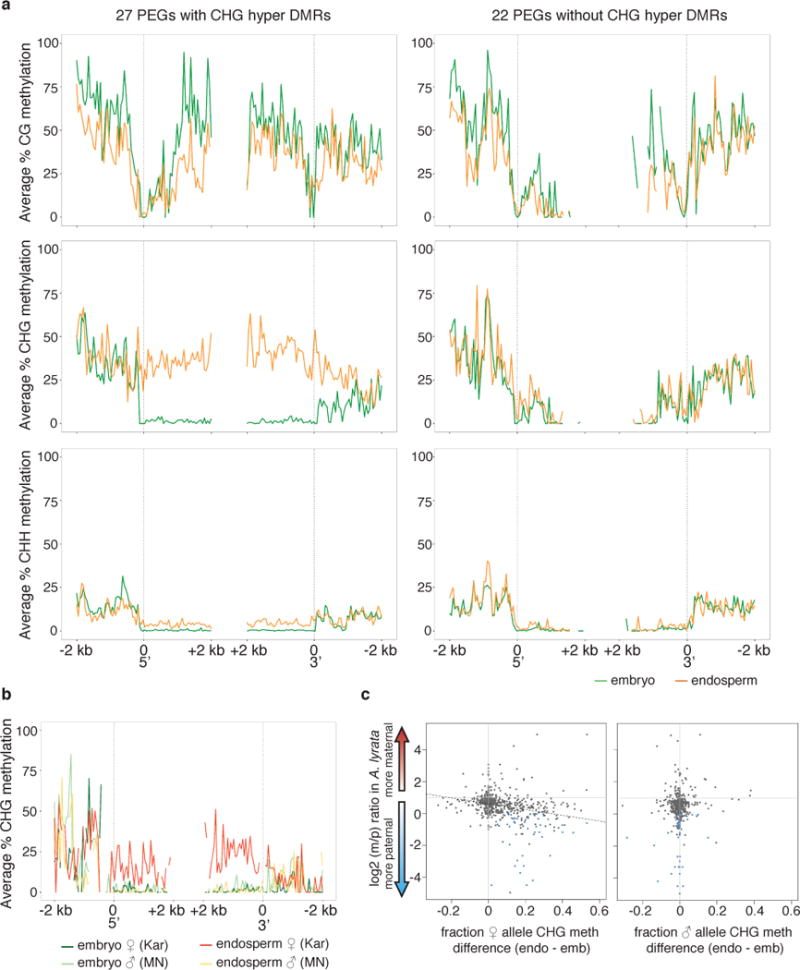
a) Average MN methylation profiles for 27 PEGs containing an endosperm CHG hypermethylated DMR (left) and the 22 PEGs without a CHG DMR (right). b) Maternal and paternal allele CHG methylation for 27 CHG DMR PEGs c) Average maternal to paternal transcript ratio (m/p) for all genes containing a CHG endosperm hypermethylated DMR plotted as a function of difference in average CHG methylation on the maternal allele (left) or paternal allele (right) between endosperm and embryo. CHG methylation difference calculated within DMRs. Blue dots, PEGs.
By contrast, we were surprised to discover that A. lyrata endosperm had a non-CG DNA methylation profile distinct from A. thaliana. This was unexpected because DNA methylation patterns in A. lyrata vegetative tissues display similar features to A. thaliana, although overall methylation levels are higher (Supplementary Figure 5). We found that average CHG methylation in gene bodies was increased in endosperm compared to embryo (Figure 2A), a phenotype not observed in wild type A. thaliana endosperm profiled at similar developmental stages6,22 (Supplementary Figure 5). To determine whether differences in aggregate methylation profiles represented small changes in many regions or larger changes in specific regions of the genome, we compared embryo and endosperm methylation profiles to identify differentially methylated regions (DMRs)6. Like in A. thaliana, the most abundant class of DMRs were less CG methylated in the endosperm compared to the embryo, with 38% of these falling within 2 kb 5′ of genes and 34% within 2 kb 3′ of genes (Supplementary Table 6). Regions that gained CHG methylation in MN × MN endosperm displayed markedly different characteristics; 84% fell within gene bodies, corresponding to 1606 genes (Figure 2C, Supplementary Table 6). CHG endosperm hypermethylated DMRs were also longer than all other DMR types (mean length = 564 bp with 400 bp standard deviation) (Supplementary Table 6). CHG gene body hypermethylation was also observed in Kar × MN endosperm, although on fewer genes (n=194). Allele-specific analysis of methylation indicated that endosperm CHG hypermethylation was specific to maternally inherited alleles (Figure 2D).
Methylation within gene bodies is usually restricted to the CG context, which is maintained after DNA replication by the maintenance methyltransferase MET1. CHG methylation, normally not found in genes, is maintained by the DNA methyltransferase CMT3, which directly binds to the repressive histone modification H3K9me223. When accompanied by H3K9me2, CHG gene body methylation is associated with transcriptional repression24. We found that gain of gene body CHG methylation in A. lyrata endosperm was associated with reduced gene expression (Supplementary Figure 6). Of the CHG hypermethylated genes with enough coverage to evaluate differential expression (n=1225), 338 were significantly less expressed in endosperm than in embryo, compared to 159 significantly more highly expressed in endosperm. This represents a significant enrichment of CHG hypermethylated genes among genes less expressed in endosperm than embryo (P(x ≥ 338) = 1.766 × 10−21, hypergeometric test) and a significant depletion among genes upregulated in endosperm (P(× ≤ 159) = 2.04 × 10−10, see Supplementary Methods). The mechanism responsible for CHG gene body hypermethylation in A. lyrata endosperm remains unclear. We found significant overlap between A. thaliana genes that gain CHG or H3K9me2 in ibm1 mutants and CHG hypermethylation of orthologous genes in A. lyrata endosperm (Supplementary Figure 7). IBM1 encodes a histone lysine demethylase that prevents accumulation of H3K9me2, and thus accumulation of CHG methylation, in genes24. IBM1 transcript abundance was lower in the endosperm than embryo (Supplementary Figure 7). In A. thaliana, methylation in the long intron of IBM1 is required for proper transcript splicing and production of an enzymatically active protein25. We found that A. lyrata IBM1 exhibited decreased CG and non-CG methylation and increased accumulation of RNA-seq reads in the long intron in endosperm relative to embryo (Supplementary Figure 7). However, A. thaliana endosperm also had reduced methylation in the long intron and decreased IBM1 transcript abundance compared to the embryo (Supplementary Figure 7). Thus, differences in IBM1 expression alone are not sufficient to explain CHG hypermethylation in A. lyrata endosperm compared to A. thaliana, although reduced IBM1 activity is likely part of the mechanism.
Several of the observed endosperm methylation features were correlated with gene imprinting. More than half of the A. lyrata MEGs and approximately one third of PEGs were associated with endosperm CG hypomethylated DMRs in the 2 kb region upstream of the transcriptional start site, whereas only 11% of non-imprinted genes were similarly associated with these DMRs (Figure 3, Supplementary Table 4, Supplementary Figure 8, Supplementary Figure 9). CG hypomethylation occurred specifically on the maternally inherited allele (Supplementary Figure 8). Thus, reduction of CG methylation by active demethylation is likely also an important component of the A. lyrata imprinting mechanism. We found a striking and non-mutually exclusive association between PEGs and endosperm CHG hypermethylation. Almost 60% of PEG gene bodies (n=27) were CHG hypermethylated, and about one-third were also associated with a 5′ or 3′ CG hypomethylated DMR (Supplementary Table 4). The average methylation profile of PEGs containing a CHG endosperm hypermethylated DMR indicated a very strong increase in CHG methylation across the entire gene body, which was specific to the maternally inherited allele (Figure 3, Figure 4). Results were validated for two PEGs, homologues of AT5G10950 and AT5G26210, by locus-specific BS-PCR (Figure 4, Supplementary Figure 10). In both A. thaliana and A. lyrata these genes were associated with CG or CHH endosperm hypomethylated DMRs in 5′ regions, but were additionally associated with gene body CHG hypermethylated DMRs in A. lyrata. Interestingly, gain of CHG methylation on the maternal allele was often accompanied by loss of CG gene body methylation, while paternally inherited alleles retained CG gene body methylation and had a similar methylation profile to embryo alleles (Figure 4, Supplementary Table 4). For the 22 PEGs lacking a gene body CHG hypermethylated DMR, half had a CG hypomethylated DMR in the flanking regions 2 kb 5′ or 3′, more like typical A. thaliana PEGs (Supplementary Table 4). Interestingly, these genes largely lacked CG gene body methylation in all tissues (Figure 3A). Thus, there appear to be at least two classes of PEGs in terms of methylation features (Figure 3A), which may correspond to different modes of epigenetic regulation. PEGs conserved with A. thaliana are found in both classes, although the majority (12/18) are CHG hypermethylated (Supplementary Table 4).
Figure 4. Conserved PEGs exhibit distinct methylation profiles between species.
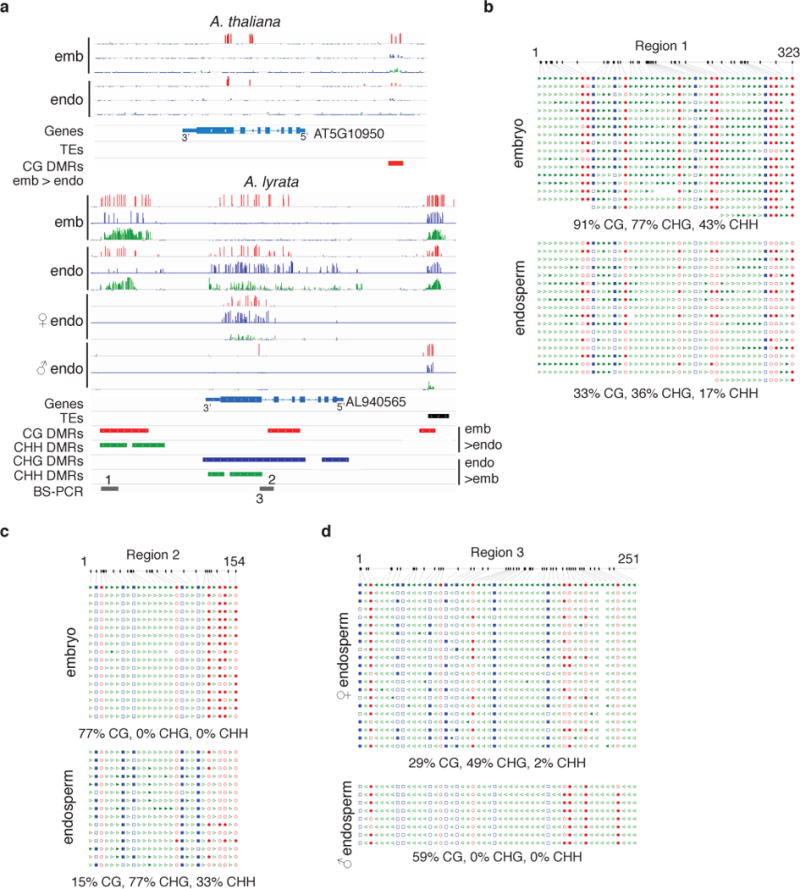
a) Bisulfite-seq methylation profile and DMRs in A. thaliana (Ler)6 and A. lyrata (MN × MN and allele-specific Kar × MN profiles shown) embryo and endosperm around a conserved PEG. Red tracks, CG methylation; blue, CHG methylation; green, CHH methylation. Tick marks below the line indicate Cs with sufficient coverage but no methylation; all tracks shown from 0–100%. b–c) Bisulfite-PCR validation of MN × MN DMRs indicated in (a). The first line is the reference sequence. d) Allele-specific methylation profiles in region 3 from Kar × MN endosperm.
To determine if there was a quantitative relationship between gain of CHG methylation and allelic expression bias, we plotted the difference in CHG methylation between maternal alleles in the embryo and endosperm relative to the ratio of maternal to paternal allele transcripts (Figure 3C). The degree to which CHG methylation was gained on the maternal allele in endosperm relative to embryo was positively correlated with the extent of paternal allele expression bias in endosperm. In addition, PEGs were clearly distinct from other genes that gained CHG gene body methylation; they tended to exhibit greater gain of CHG methylation (Figure 3C) and were also hypermethylated along more of their length than all CHG hypermethylated genes (56% vs. 29%). Thus, a greater extent and amount of maternal allele CHG hypermethylation is correlated with more paternally biased transcription. These data suggest that CHG methylation, perhaps accompanied by gain of H3K9me2, represses the maternal alleles of PEGs. It is unknown whether gene body CHG methylation is established on maternal alleles before or after fertilization. Demethylation of the IBM1 regulatory intron (Supplementary Figure 7) could be initiated before fertilization in the central cell, leading to its downregulation and an increase in CHG methylation specifically on maternal alleles, which would then be maintained after fertilization. Alternatively, if maternal allele CHG methylation occurs post-fertilization, then CMT3 must be able to distinguish maternally and paternally inherited alleles. Retention of CG gene body methylation on the paternal alleles of PEGs (Figure 4) could possibly protect them from gain of CHG methylation. Interestingly, gain of gene body CHG methylation was also recently shown to occur in both A. thaliana endosperm and embryos when wild type plants were pollinated by diploid hypomethylated pollen26. Diploid pollen creates triploid seeds with tetraploid endosperm that usually abort, but seed abortion is suppressed when the pollen is hypomethylated due to mutations in met1. Many of the genes that gain CHG methylation and have reduced expression in triploid rescued seeds are PEGs26. However, this phenotype appears to be distinct from what we observed; the CHG methylation gain is much more modest than what we have described in wild type A. lyrata endosperm, and only one conserved PEG was affected26. Our data further suggest that gene body CHG hypermethylation is not a state restricted to mutant tissues, but can occur in a developmentally regulated manner that could be important for maintaining gene expression programs.
This is the first study to compare imprinting between two closely related plant species that differ in breeding strategy. A. lyrata and A. thaliana homologous imprinted genes are epigenetically modified in a distinct manner despite the close relatedness of the species (Figure 4). Allele-specific maintenance of gene repression by the PRC2 complex is an important component of the imprinting mechanism in A. thaliana and other species11,12. The PRC2 complex silences the hypomethylated maternal allele of PEGs, while the methylated paternal allele is expressed. Several studies have suggested that H3K9me2 and H3K27me3 are repressive marks that can substitute for one another in mutant contexts26,27. We suggest that this substitution can also occur in wild type tissues, and favor the hypothesis that in A. lyrata endosperm the maternal allele of at least a subset of PEGs is repressed by CHG methylation/H3K9me2. Overall, our results point to high conservation of imprinting accompanied by a distinct epigenetic signature, at least for PEGs. If the mechanism of imprinting is different but the genes that are imprinted are the same, this argues that imprinting is not simply a byproduct of endosperm methylation dynamics, but that imprinted expression of specific genes is under selection. Thus, the means by which monoallelic expression can be achieved are plastic, but the genes subject to this regulation are conserved.
METHODS
Plant material
Arabidopsis lyrata MN47 (MN) seeds were obtained from the Arabidopsis Biological Resource Center (CS22696); seeds from the Karhumäki (Kar) strain were a gift from Dr. Outi Savolainen, University of Oulu, Finland. Plants were grown in a greenhouse with 16 hr of light at 21°C or in a growth chamber with 16 hr of light (120 μMol), 20°C and 50% humidity, vernalized at 4°C for a month after rosettes had formed, and then returned to the greenhouse/growth chamber. With the exception of MN × Kar crosses (female parent in cross is listed first), flowers were not emasculated prior to pollination. MN plants, which are able to self-pollinate at low frequency, were emasculated and pollinated 2 days later. Seeds were dissected into endosperm, embryo, and seed coat portions at seed development stages ranging from torpedo to bent cotyledon, around 14–19 days post pollination, depending on growth temperature, genotype, and age of the maternal parent. In addition, flower bud tissue was collected from MN plants and from Kar × MN F1 hybrid plants.
mRNA-Seq
RNA was isolated from endosperm, embryo and seed coat samples using the RNAqueous Micro Kit (Ambion). Input for mRNA-seq library construction varied from 120 to 800 ng DNase I-treated RNA. Strand-specific libraries were prepared by the Whitehead Institute Genome Technology Core using the Integenex PolyA prep protocol (Wafergen Biosystems) or using the Illumina TruSeq Stranded mRNA Kit. Libraries were multiplexed and sequenced on an Illumina HiSeq 2000 machine by the Whitehead Institute Genome Technology Core using a paired end protocol. See Supplementary Table 1 for details of library prep for specific samples. Reads were aligned to the MN47 reference genome28 using Tophat v2.0.1329. See Supplementary Methods for details on mRNA-seq analysis parameters, SNP discovery, and updated annotation of the A. lyrata genome.
Imprinting analysis
Imprinted genes were identified using our previously described method (Supplementary Methods)5,6. To assess whether A. lyrata imprinted genes were also imprinted in A. thaliana, we examined data from Pignatta et al. 20146. If the A. thaliana homologue of the A. lyrata imprinted gene was not called as an imprinted gene, we took the A. thaliana reciprocal cross comparison that showed the strongest degree of parental bias and determined why the gene was not called imprinted (see categories in Figure 1B). If any A. lyrata imprinted genes had the same A. thaliana homologue it was only counted once. To perform the reverse analysis, in which we assessed whether all genes considered imprinted in Pignatta et al. 2014 (the union set of MEGs and PEGs) were also imprinted in A. lyrata, we determined for each of the 12 comparisons in our A. lyrata data whether the gene was called imprinted, and if not, why (Supplementary Figure 4). If more than 7 comparisons lacked data, a gene was considered to have insufficient reads. If >40% of comparisons with data called the gene imprinted, it was considered imprinted. If <40% of comparisons with data called the gene imprinted or it did not meet the % maternal cutoff (but met all other imprinting criteria), the gene was considered parentally biased but not meeting the % maternal cutoff. Similarly, if <40% of comparisons called the gene imprinted and it failed the % maternal cutoff and failed the imprinting factor (IF) cutoff but met initial parental bias p-value cutoffs, it was considered parentally biased but with imprinting factor too low. All other genes were considered to have no significant parental bias. For A. thaliana imprinted genes with multiple A. lyrata homologues that differed in imprinting status, the imprinting status of the most parentally biased homologue was used to obtain counts in Supplementary Figure 4.
Whole genome bisulfite sequencing
Genomic DNA was extracted in duplicate from MN flower buds and from seeds dissected into embryo and endosperm from MN × MN and Kar × MN crosses. DNA was isolated from fresh tissue using the DNeasy Plant Mini Kit (Qiagen) (buds) or a CTAB method (embryo and endosperm) and RNase treated. 250–500 ng of DNA was used for bisulfite treatment with the MethylCode Bisulfite Conversion Kit (Invitrogen). Libraries were constructed using the EpiGnome Methyl-seq Kit (Epicentre Biotechnologies) and were sequenced on an Illumina HiSeq 2000 using a 40×40 or 100×100 paired end protocol at the Whitehead Institute Genome Technology Core (see Supplementary Table 5). Reads were aligned to the genome using Bismark v.0.13.030. To identify differentially methylated regions, the genome was divided into consecutive 300 bp windows that overlapped by 200 bp. Each window was assessed as a potential DMR between two samples (e.g. embryo and endosperm) using the method described in Pignatta et al.6. See Supplementary Methods for additional details.
Supplementary Material
Acknowledgments
M.G. thanks members of the NESCent working group on Testing Theories of Genomic Imprinting for many stimulating discussions. We thank Outi Savolainen for kindly providing A. lyrata Karhumäki seeds, the Whitehead Institute Bioinformatics and Research Computing group for assistance, and P.R. Satyaki and Ben Williams for comments on the manuscript. This research was funded by NSF grants MCB 1121952 and 1453459 to M.G. C.L.P. is supported by an NSF graduate research fellowship.
Footnotes
Data access
High throughput sequencing data has been deposited in NCBI GEO under accession GSE76076.
AUTHOR CONTRIBUTIONS
M.G. conceived the project, M.K. performed experiments, C.L.P. developed and implemented computational analyses, M.K., C.L.P., and M.G. analyzed data and wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Gehring M. Genomic Imprinting: Insights From Plants. Annu Rev Genet. 2013;47:187–208. doi: 10.1146/annurev-genet-110711-155527. [DOI] [PubMed] [Google Scholar]
- 2.Haig D, Westoby M. Parent-specific gene expression and the triploid endosperm. Am Nat. 1989;134:147–155. [Google Scholar]
- 3.Hsieh TF, et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc Natl Acad Sci USA. 2011;108:1755–1762. doi: 10.1073/pnas.1019273108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolff P, et al. High-resolution analysis of parent-of-origin allelic expression in the Arabidopsis Endosperm. PLoS Genet. 2011;7:e1002126. doi: 10.1371/journal.pgen.1002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gehring M, Missirian V, Henikoff S. Genomic Analysis of Parent-of-Origin Allelic Expression in Arabidopsis thaliana Seeds. PLoS ONE. 2011;6:e23687. doi: 10.1371/journal.pone.0023687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pignatta D, et al. Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. Elife. 2014:e03198. doi: 10.7554/eLife.03198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo M, et al. A genome-wide survey of imprinted genes in rice seeds reveals imprinting primarily occurs in the endosperm. PLoS Genet. 2011;7:e1002125. doi: 10.1371/journal.pgen.1002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waters AJ, et al. Parent-of-Origin Effects on Gene Expression and DNA Methylation in the Maize Endosperm. Plant Cell. 2012;23:4221–4233. doi: 10.1105/tpc.111.092668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xin M, et al. Dynamic expression of imprinted genes associates with maternally controlled nutrient allocation during maize endosperm development. Plant Cell. 2013;25:3212–3227. doi: 10.1105/tpc.113.115592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang M, et al. Extensive, clustered parental imprinting of protein-coding and noncoding RNAs in developing maize endosperm. Proc Natl Acad Sci USA. 2011;108:20042–20047. doi: 10.1073/pnas.1112186108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang M, et al. Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize. Genome Res. 2014;24:167–176. doi: 10.1101/gr.155879.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreno-Romero J, Jiang H, Santos-González J, Köhler C. Parental epigenetic asymmetry of PRC2-mediated histone modifications in the Arabidopsis endosperm. EMBO J. 2016;35:1298–1311. doi: 10.15252/embj.201593534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patten MM, et al. The evolution of genomic imprinting: theories, predictions and empirical tests. Heredity. 113:119–128. doi: 10.1038/hdy.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waters AJ, et al. Comprehensive analysis of imprinted genes in maize reveals allelic variation for imprinting and limited conservation with other species. Proc Natl Acad Sci USA. 2013;110:19639–19644. doi: 10.1073/pnas.1309182110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger F, Vu TM, Li J, Chen B. Hypothesis: Selection of Imprinted Genes is Driven by Silencing Deleterious Gene Activity in Somatic Tissues. Cold Spring Harb Symp Quant Biol. 2012 doi: 10.1101/sqb.2012.77.014514. [DOI] [PubMed] [Google Scholar]
- 16.Kawashima T, Berger F. Epigenetic reprogramming in plant sexual reproduction. Nat Rev Genet. 2014;15:613–624. doi: 10.1038/nrg3685. [DOI] [PubMed] [Google Scholar]
- 17.Beilstein MA, Nagalingum NS, Clements MD, Manchester SR, Mathews S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc Natl Acad Sci USA. 2010;107:18724–18728. doi: 10.1073/pnas.0909766107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morison IM, Reeve AE. A catalogue of imprinted genes and parent-of-origin effects in humans and animals. Hum Mol Genet. 1998;7:1599–1609. doi: 10.1093/hmg/7.10.1599. [DOI] [PubMed] [Google Scholar]
- 19.Danilevskaya ON, et al. Duplicated fie genes in maize: expression pattern and imprinting suggest distinct functions. Plant Cell. 2003;15:425–438. doi: 10.1105/tpc.006759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matzke MA, Mosher RA. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet. 2014;15:394–408. doi: 10.1038/nrg3683. [DOI] [PubMed] [Google Scholar]
- 21.Gehring M, Bubb KL, Henikoff S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science. 2009;324:1447–1451. doi: 10.1126/science.1171609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibarra CA, et al. Active DNA Demethylation in Plant Companion Cells Reinforces Transposon Methylation in Gametes. Science. 2012;337:1360–1364. doi: 10.1126/science.1224839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du J, et al. Dual Binding of Chromomethylase Domains to H3K9me2-Containing Nucleosomes Directs DNA Methylation in Plants. Cell. 2012;151:167–180. doi: 10.1016/j.cell.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saze H, Shiraishi A, Miura A, Kakutani T. Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science. 2008;319:462–465. doi: 10.1126/science.1150987. [DOI] [PubMed] [Google Scholar]
- 25.Rigal M, Kevei Z, Pélissier T, Mathieu O. DNA methylation in an intron of the IBM1 histone demethylase gene stabilizes chromatin modification patterns. EMBO J. 2012;31:2981–2993. doi: 10.1038/emboj.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schatlowski N, et al. Hypomethylated Pollen Bypasses the Interploidy Hybridization Barrier in Arabidopsis. Plant Cell. 2014;26:3556–3568. doi: 10.1105/tpc.114.130120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deleris A, et al. Loss of the DNA Methyltransferase MET1 Induces H3K9 Hypermethylation at PcG Target Genes and Redistribution of H3K27 Trimethylation to Transposons in Arabidopsis thaliana. PLoS Genet. 2012;8:e1003062. doi: 10.1371/journal.pgen.1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu TT, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 2011;43:476–481. doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.