

Supplemental Digital Content is available in the text
Keywords: Dyspareunia, Estradiol, Estrogen therapy, Menopause, Vaginal atrophy
Abstract
Objective:
To evaluate the safety and efficacy of TX-004HR vaginal estradiol soft-gel capsules for moderate-to-severe dyspareunia associated with postmenopausal vulvar and vaginal atrophy.
Methods:
In this randomized, double-blind, placebo-controlled, phase 3 study, postmenopausal women with a self-identified most bothersome symptom of dyspareunia received 4, 10, or 25 μg TX-004HR or placebo for 12 weeks. Four co-primary efficacy endpoints were change from baseline to week 12 in percentages of superficial and parabasal cells, vaginal pH, and severity of dyspareunia. Secondary endpoints included severity of vaginal dryness and vulvar and/or vaginal itching or irritation. Endometrial histology and adverse events (AEs) were included in the safety endpoints.
Results:
In all, 764 women were randomized (modified intent-to-treat population, n = 747; mean age 59 y). Compared with placebo, all three doses of TX-004HR significantly improved the four co-primary endpoints (P < 0.0001 for all, except dyspareunia with 4 μg, P = 0.0149). Changes in cytology, pH, and dyspareunia were also significant at weeks 2, 6, and 8. Vaginal dryness and vaginal itching/irritation improved. Sex hormone binding globulin concentrations did not change with treatment. TX-004HR was well-tolerated, with no clinically meaningful differences in treatment-emergent AEs versus placebo, and no treatment-related serious AEs or deaths.
Conclusions:
TX-004HR (4, 10, and 25 μg) was safe, well-tolerated, and effective for treating moderate-to-severe dyspareunia within 2 weeks with minimal systemic estrogen exposure. This novel product may be a potential new treatment option for women experiencing postmenopausal vulvar and vaginal atrophy.
Postmenopausal vulvar and vaginal atrophy (VVA) is the thinning, drying, and loss of elasticity of the vaginal epithelium associated with the reduction in serum estrogen after menopause.1 Up to approximately 70% of postmenopausal women show clinical evidence of VVA,2 and roughly half report symptoms associated with VVA, including vaginal dryness, irritation, itching, dysuria, and pain or bleeding with sexual activity.3,4 These clinical signs and symptoms of VVA are a component of the more encompassing term of genitourinary syndrome of menopause (GSM).5 VVA is progressive without treatment,6 and can negatively affect quality of life.7 A recent survey of 3,046 postmenopausal women with symptoms of VVA reported that only 7% of the women currently used prescription-only therapy to treat their VVA,8 and in the United States, 32 million postmenopausal women are thought to have symptomatic VVA.8,9 Thus, it is believed that the majority of postmenopausal women with symptomatic VVA are not being treated.
Low-dose vaginal estrogens are recognized as safe and effective treatment options for women with moderate-to-severe symptoms of VVA.10,11 Local low-dose vaginal estrogens result in lower levels of circulating estrogen than oral or transdermal therapies. The Working Group on Women's Health and Well-Being in Menopause, whose participants have affiliations with a number of medical societies and professional organizations, has proposed to the US Food and Drug Administration (FDA) the removal of the bold-font boxed warning from the product label of vaginal estrogens to help facilitate the prescription of these products, since the current boxed warning may be only relevant to higher doses of systemic estrogen, but not low-dose vaginal estrogens.12
Despite the positive risk-benefit profile of vaginal estrogen treatments, many women are dissatisfied with currently available products.8 Creams require a reusable applicator (which may be unsanitary) and can be messy, inconvenient, and difficult to properly administer.13,14 Currently available vaginal tablets also have the inconveniences of an applicator which may be difficult to use and may cause vaginal abrasions.15 Vaginal discharge lasting for days and perceived lack of efficacy have also been associated with vaginal tablets.16 An analysis of prescription renewals in the United States showed that most women discontinue use of vaginal creams and vaginal tablets after an average use of only 45 and 103 days, respectively.17 Vaginal rings are not widely used, but can require dexterity for insertion and can dislodge,1,10 and many women do not favor a long-term vaginal device. Women also have concerns regarding long-term safety and the risks of systemic absorption related to some of the available vaginal products.16 Further, many women prefer to use estrogen identical to their endogenous estrogen (ie, 17β-estradiol) as opposed to conjugated equine estrogens, which are contained in some vaginal products.18
TX-004HR (TherapeuticsMD, Inc, Boca Raton, FL) is a vaginal 17β-estradiol product in development, designed to rapidly and effectively treat symptoms of VVA without increasing serum estradiol levels, and to provide easy insertion with complete and rapid dissolution to minimize vaginal discharge. The muco-adhesive soft-gel capsule (VagiCap) contains solubilized estradiol and is administered without an applicator for ease of use and improved user experience. Estradiol is released after dissolution of the soft-gel capsule within the vagina, and does not require vaginal secretions to activate the process.
Phase 1 studies have shown that women have two to three times lower systemic estrogen concentrations 24 hours after vaginal insertion of 10 and 25 μg TX-004HR than with a regulatory approved and commercially available low-dose vaginal estradiol tablet at identical doses.19 The primary objective of this study was to evaluate the safety and efficacy of three doses of the investigational drug, TX-004HR (4, 10, and 25 μg), compared with placebo, in postmenopausal women at 12 weeks for the treatment of moderate-to-severe dyspareunia and VVA, components of GSM. Efficacy outcomes included changes from baseline in vaginal superficial and parabasal cells, vaginal pH, and the severity of dyspareunia associated with VVA as the most bothersome symptom (MBS).
METHODS
Study design
This 12-week, multicenter, double-blind, randomized, placebo-controlled, phase 3 trial was conducted at 89 sites in the United States and Canada (NCT02253173) from October 2014 to October 2015. Eligible women were randomized (1:1:1:1) to either TX-004HR 4, 10, or 25 μg, or matching placebo in soft-gel capsules specifically designed for vaginal insertion. Randomization was achieved using a computer-generated randomization schedule prepared by a statistician before the start of the study. Women were instructed to digitally self-administer one soft-gel capsule into the vagina at approximately the same time of day, daily for 2 weeks, and then twice weekly (approximately 3-4 d apart) for 10 weeks. All study staff, sponsor representatives, and study participants were blinded throughout the study until database lock, with the blind to be broken only to protect participant safety in emergency situations.
The study was conducted in accordance with applicable laws and regulations, including, but not limited to, the International Conference on Harmonization Guideline for Good Clinical Practice and the ethical principles that have their origins in the Declaration of Helsinki. The study protocol and the informed consent form were approved by an independent institutional review board (IRB).
Study participants
Participants signed written informed consent before any study-related activities. Postmenopausal women 40 to 75 years of age were enrolled if they had ≤5% superficial cells on vaginal cytological smear; vaginal pH > 5.0; MBS of moderate-to-severe vaginal pain associated with sexual activity (dyspareunia); onset of moderate-to-severe dyspareunia after menopause; body mass index ≤38 kg/m2; and were sexually active (with vaginal penetration) and anticipated having sexual activity during the trial period. Participants with an intact uterus must have had a screening endometrial biopsy sample sufficient for analysis.
Women were excluded from the study if they had a history or active presence of clinically important medical disease that might confound the study or be detrimental to their health. Key exclusion criteria included endometrial hyperplasia or cancer; undiagnosed vaginal bleeding; liver or kidney disorder; thromboembolic disorders; cerebrovascular accident, stroke, or transient ischemic attack; myocardial infarction or ischemic heart disease; malignancy; or endocrine disease; or any clinically important abnormalities on screening physical examination, assessments, mammogram, electrocardiogram (ECG), or laboratory tests. Women with a recent history of alcohol or drug abuse, or a history of sexual or spousal abuse were excluded, as were current heavy smokers or users of e-cigarettes or marijuana.
Women could not have used oral products containing estrogens, progestins, androgens, or selective estrogen receptor modulators (SERMs) within 8 weeks; transdermal hormone products within 4 weeks; vaginal hormone products (rings, creams, gels) within 4 weeks; intrauterine progestins within 8 weeks; progestin implants/injectables or estrogen pellets/injectables within 6 months; vaginal lubricants or moisturizers within 7 days before vaginal pH assessment during screening; investigational drugs within 60 days; or an intrauterine device within 12 weeks before screening. Concomitant medications were allowed during the study (and were to be recorded in diaries), with the exception of investigational drugs; estrogen, progestin, androgen, or SERM-containing medications other than TX-004HR, and also prescription and nonprescription medications/remedies known to treat VVA, including vaginal lubricants and moisturizers.
Efficacy parameters
The four co-primary efficacy endpoints analyzed for each dose were change from baseline to week 12 compared with placebo in (1) percentage of vaginal superficial cells, (2) percentage of vaginal parabasal cells, (3) vaginal pH, and (4) severity of the MBS of dyspareunia associated with VVA. Secondary efficacy endpoints included change from baseline to weeks 2, 6, and 8 compared with placebo for the four above endpoints. Additional secondary endpoints were change from baseline to weeks 2, 6, 8, and 12 in the severity of vaginal dryness and vulvar and/or vaginal itching or irritation associated with VVA.
Vaginal cytology was assessed by a vaginal smear, and vaginal pH was measured by comparing an indicator strip applied to the lateral vaginal wall with a colorimetric scale at screening, and at weeks 2, 6, 8, and 12. Participants were asked to identify their MBS and self-assess their symptoms of VVA including vaginal pain associated with sexual activity, vaginal dryness, and vulvar and/or vaginal itching or irritation using the VVA Symptom Self-Assessment Questionnaire (scoring: 0 = none, 1 = mild, 2 = moderate, 3 = severe) at screening. Women were re-assessed by this questionnaire at weeks 2, 6, 8, and 12.
Safety
Safety endpoints included vital signs, clinical laboratory tests (blood chemistry, hematology, hormone levels, urine analysis), ECG, physical and gynecological examination findings, pap smears, endometrial biopsies, and adverse events (AEs). AEs included undesirable medical conditions occurring at any time during all study phases including the washout period, whether or not a study treatment had been administered. An AE was considered treatment-emergent if it occurred after study drug administration, or if it was pre-existing and worsened during 120 days postdose follow-up. Participants were given a diary with instructions to record product use, sexual activity, symptoms/complaints, and other medications. AEs, concomitant medications, and vital signs were recorded and assessed at each study visit from screening to week 12.
Endometrial biopsies were collected at baseline and at week 12, or end of treatment. A diagnosis of endometrial hyperplasia from the biopsy was based on agreement of two of three reads by independent pathologists blinded to treatment.
Statistical analyses
A sample size of 140 participants per treatment arm was calculated using data from the product literature to achieve 50% to >99% power to detect a 30% to 71% change in the severity of dyspareunia (0.005 one-tailed significance level) in the modified intent-to-treat (MITT) population.
The intent-to-treat (ITT) and safety populations were the same and included all women who were randomized and received at least one dose of TX-004HR. Those in the ITT population with baseline values for each co-primary endpoint and at least one follow-up value for any of the co-primary endpoints formed the MITT population, which was the primary efficacy population.
Statistical analyses were performed using SAS Release 9.2. Baseline and demographic variables were descriptively summarized by treatment group. A stepwise procedure was used to account for the multiple doses and endpoints. Primary and secondary endpoints were compared with placebo using analyses of covariance (ANCOVAs) based on mixed-effects model repeated measures (MMRM) methods with participant as the random effect and treatment group and visit (2, 6, 8, and 12 wks) as the fixed effects, with an interaction term for visit and treatment. Baseline measures and age were covariates. Pair-wise comparisons were performed using ANCOVA for each dose of TX-004HR versus placebo. Data comparisons were considered statistically significant at an alpha level of 0.05. No last observation carried forward methods for efficacy were used for missing or invalid (when multiple laboratory data provided contradictory or ambiguous results) data, as per MMRM methods.
The number and proportion of women reporting each AE and serious AE in each treatment group were summarized. An AE was counted once for a participant if they had the same AE more than once. Actual values and change from baseline in blood chemistry parameters were summarized for each treatment group.
RESULTS
Participant disposition and baseline characteristics
Of 2,183 women screened, 764 were eligible and randomized to TX-004HR 4 μg (n = 191), 10 μg (n = 191), 25 μg (n = 190), or placebo (n = 192; Fig. 1), and were included in the safety population. Ninety-two percent (704/764) of the participants completed the study, and 94% (703/747) of women in the MITT population completed the study (Fig. 1).
FIG. 1.
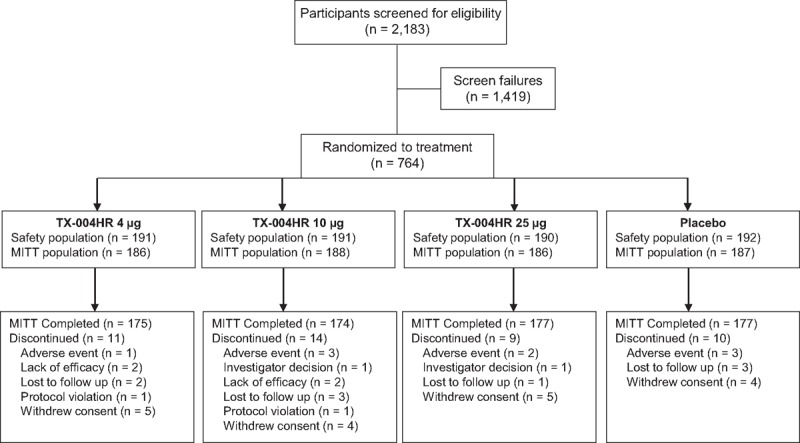
Participant disposition through the study. MITT, modified intent-to-treat.
Demographic and baseline characteristics of the MITT population were similar among groups (Table 1). Women had a mean age of 59 years, ranging from 40 to 75 years, and a mean BMI of 27 kg/m2; the majority were white. No significant differences between each treatment group versus placebo were found in baseline superficial cells, parabasal cells, vaginal pH, and severity score for dyspareunia (Table 1).
TABLE 1.
Participant demographics and baseline characteristics (MITT population)
| Characteristic | TX-004HR 4 μg (n = 186) | TX-004HR 10 μg (n = 188) | TX-004HR 25 μg (n = 186) | Placebo (n = 187) |
| Age, mean (SD), y | 59.8 (6.0) | 58.6 (6.3) | 58.8 (6.2) | 59.4 (6.0) |
| Race, n (%) | ||||
| White | 162 (87.1) | 165 (87.8) | 161 (86.6) | 160 (85.6) |
| Black or African American | 20 (10.8) | 21 (11.2) | 24 (12.9) | 21 (11.2) |
| Asian | 3 (1.6) | 2 (1.1) | 1 (0.5) | 1 (0.5) |
| BMI, mean (SD), kg/m2 | 26.6 (4.9) | 26.8 (4.7) | 26.8 (4.8) | 26.6 (4.6) |
| Hysterectomized, n (%) | 87 (46.8) | 86 (45.7) | 85 (45.7) | 73 (39.0) |
| Superficial cells, mean (SD), % | 1.3 (1.2) | 1.2 (1.2) | 1.3 (1.2) | 1.3 (1.3) |
| Parabasal cells, mean (SD), % | 52.3 (39.2) | 51.3 (38.0) | 53.5 (38.3) | 52.0 (39.2) |
| Vaginal pH, mean (SD) | 6.3 (0.9) | 6.3 (0.8) | 6.3 (0.9) | 6.3 (1.0) |
| Dyspareunia, severity score, mean (SD) | 2.7 (0.5) | 2.6 (0.5) | 2.7 (0.4) | 2.7 (0.5) |
BMI, body mass index; MITT, modified intent-to-treat.
In the MITT population, the mean severity score for vaginal dryness was 2.4 at baseline, with approximately 93% of women having moderate-to-severe vaginal dryness. The mean severity score for vulvar and/or vaginal itching or irritation was 1.2, with approximately 42% of women having moderate-to-severe itching or irritation. No significant differences between each treatment group versus placebo were found in these baseline parameters.
Vaginal cytology, vaginal pH, and dyspareunia (co-primary endpoints)
Compared with placebo, 4, 10, and 25 μg TX-004HR significantly improved the four co-primary endpoints from baseline to week 12 in the percentage of superficial cells, the percentage of parabasal cells, vaginal pH, and the severity score for dyspareunia (P < 0.0001 for all, except P = 0.0149 for dyspareunia with 4 μg; Table 2; Fig. 2A-D). The increase in the percentage of superficial cells ranged from 17% to 23% for TX-004HR doses, compared with 6% for placebo, whereas the decrease in parabasal cells ranged from 41% to 46% for TX-004HR doses versus 7% for placebo from baseline to week 12. The percentages of superficial and parabasal cells and vaginal pH also all significantly improved from baseline to weeks 2, 6, and 8 for all three doses of TX-004HR compared with placebo (P < 0.0001 for all; Table 2).
TABLE 2.
Change from baseline (LS means ± SE) at weeks 2, 6, 8, and 12 for the 4 co-primary endpoints (MITT population)
| % Superficial cells | % Parabasal cells | Vaginal pH | Dyspareunia (score) | ||||||||||
| TX-004HR | Week | n | LS mean ± SE | Pa | n | LS mean ± SE | Pa | n | LS mean ± SE | Pa | n | LS mean ± SE | Pa |
| 4 μg | 2 | 186 | 31.4 ± 1.50 | <0.0001 | 186 | −40.2 ± 1.72 | <0.0001 | 186 | −1.23 ± 0.06 | <0.0001 | 145 | −0.99 ± 0.07 | 0.0260 |
| 6 | 172 | 18.4 ± 1.54 | <0.0001 | 172 | −39.4 ± 1.75 | <0.0001 | 172 | −1.32 ± 0.07 | <0.0001 | 148 | −1.30 ± 0.07 | 0.0069 | |
| 8 | 164 | 19.0 ± 1.56 | <0.0001 | 164 | −41.9 ± 1.77 | <0.0001 | 164 | −1.35 ± 0.07 | <0.0001 | 140 | −1.52 ± 0.07 | 0.0003 | |
| 12 | 170 | 17.5 ± 1.54 | <0.0001 | 170 | −40.6 ± 1.76 | <0.0001 | 170 | −1.32 ± 0.07 | <0.0001 | 151 | −1.52 ± 0.07 | 0.0149 | |
| 10 μg | 2 | 188 | 31.9 ± 1.50 | <0.0001 | 188 | −44.4 ± 1.71 | <0.0001 | 188 | −1.37 ± 0.06 | <0.0001 | 147 | −1.08 ± 0.07 | 0.0019 |
| 6 | 170 | 16.9 ± 1.54 | <0.0001 | 170 | −43.6 ± 1.75 | <0.0001 | 170 | −1.40 ± 0.07 | <0.0001 | 150 | −1.37 ± 0.07 | 0.0009 | |
| 8 | 165 | 17.4 ± 1.56 | <0.0001 | 165 | −43.8 ± 1.76 | <0.0001 | 165 | −1.46 ± 0.07 | <0.0001 | 136 | −1.64 ± 0.07 | <0.0001 | |
| 12 | 171 | 16.7 ± 1.54 | <0.0001 | 171 | −44.1 ± 1.75 | <0.0001 | 171 | −1.42 ± 0.07 | <0.0001 | 154 | −1.69 ± 0.07 | <0.0001 | |
| 25 μg | 2 | 184 | 38.9 ± 1.50 | <0.0001 | 184 | −45.6 ± 1.72 | <0.0001 | 184 | −1.30 ± 0.07 | <0.0001 | 140 | −1.02 ± 0.07 | 0.0105 |
| 6 | 173 | 22.7 ± 1.53 | <0.0001 | 173 | −45.6 ± 1.75 | <0.0001 | 173 | −1.48 ± 0.07 | <0.0001 | 150 | −1.48 ± 0.07 | <0.0001 | |
| 8 | 166 | 23.9 ± 1.55 | <0.0001 | 166 | −45.1 ± 1.76 | <0.0001 | 166 | −1.45 ± 0.07 | <0.0001 | 129 | −1.62 ± 0.08 | <0.0001 | |
| 12 | 174 | 23.2 ± 1.53 | <0.0001 | 174 | −45.6 ± 1.75 | <0.0001 | 174 | −1.34 ± 0.07 | <0.0001 | 159 | −1.69 ± 0.07 | <0.0001 | |
| Placebo | 2 | 185 | 6.1 ± 1.50 | — | 185 | −7.0 ± 1.72 | — | 186 | −0.28 ± 0.06 | — | 141 | −0.76 ± 0.07 | — |
| 6 | 176 | 5.4 ± 1.53 | — | 176 | −9.2 ± 1.74 | — | 176 | −0.30 ± 0.07 | — | 159 | −1.03 ± 0.07 | — | |
| 8 | 167 | 6.0 ± 1.55 | — | 167 | −7.9 ± 1.76 | — | 167 | −0.38 ± 0.07 | — | 143 | −1.15 ± 0.07 | — | |
| 12 | 172 | 5.6 ± 1.54 | — | 172 | −6.7 ± 1.75 | — | 174 | −0.28 ± 0.07 | — | 163 | −1.28 ± 0.07 | — | |
LS, least square; MITT, modified intent-to-treat.
aTX-004HR versus placebo, based on mixed-effects model repeated measures.
FIG. 2.
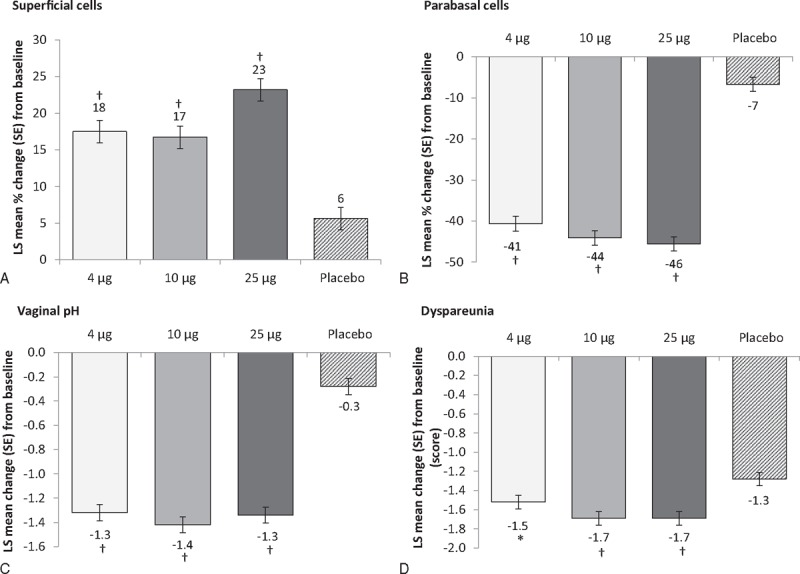
Co-primary endpoints: change from baseline to week 12 in (A) percentage of superficial cells; (B) percentage of parabasal cells; (C) vaginal pH units, and (D) dyspareunia severity scores with three doses of TX-004HR compared with placebo. ∗P < 0.05; †P < 0.0001 versus placebo (MITT population, n = 747). LS, least square; MITT, modified intent-to-treat.
At week 12, mean dyspareunia severity scores for women assigned to TX-004HR were 1.1, 0.9, and 1.0 for 4, 10, and 25 μg, respectively, whereas the mean score for women assigned to placebo was 1.4. The MBS of dyspareunia also significantly improved for women taking TX-004HR at all doses compared with placebo at weeks 2, 6, and 8 (Table 2).
Vaginal dryness and vulvar and/or vaginal itching or irritation
Vaginal dryness significantly improved with all three doses of TX-004HR at week 12 from baseline compared with placebo (P < 0.01 for 4 μg, P < 0.0001 for 10 μg and 25 μg; Fig. 3A) in the MITT population. Statistically significant differences in vaginal dryness scores were noted between TX-004HR 10 and 25 μg versus placebo at week 2 (P < 0.01 for both), and with all doses of TX-004HR versus placebo at week 6 (P < 0.01 for 4 μg, P < 0.001 for 10 μg and 25 μg) and week 8 (P < 0.05 for 4 μg, P < 0.0001 for 10 μg, and P < 0.001 for 25 μg).
FIG. 3.
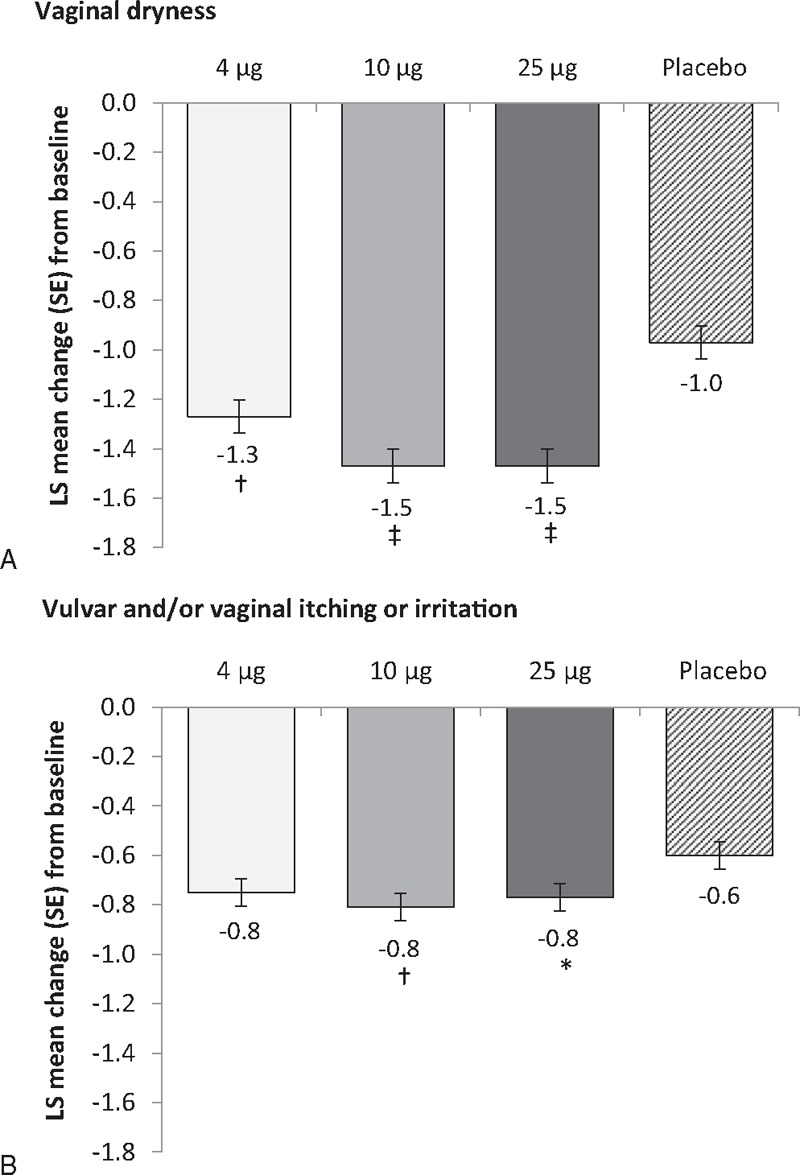
Effect of each of three doses of TX-004HR at 12 weeks compared with placebo on the secondary endpoints of (A) severity of vaginal dryness and (B) severity of vulvar and/or vaginal itching or irritation (MITT population, n = 747). ∗P < 0.05; †P < 0.01; ‡P < 0.0001 versus placebo. LS, least square; MITT, modified intent-to-treat.
The mean severity score for vulvar and/or vaginal itching or irritation decreased to 0.4 at week 12 for women in both 10 and 25-μg groups, which was significantly greater than the change observed from baseline with placebo (P = 0.0055 and P = 0.0263, respectively; Fig. 3B). The mean severity score at week 12 was 0.5 (P = 0.0503 vs placebo) in the 4-μg group.
Safety
TX-004HR had a favorable safety profile and was well-tolerated. No clinically significant differences in AEs were observed between treatment and placebo groups (Table 3). Headache was the most commonly reported TEAE, followed by vaginal discharge, nasopharyngitis, and vulvovaginal pruritus (Table 3). Headache was the only treatment-related TEAE that was numerically more frequent in women receiving TX-004HR than those receiving placebo (3.7% for 4-μg dose vs 3.1% for placebo). Vaginal discharge was reported by numerically fewer women in any of the TX-004HR groups than by women in the placebo group. Most TEAEs were mild to moderate in severity. Few participants (1.8%) discontinued the study due to AEs.
TABLE 3.
Number (%) of TEAEs reported for ≥3% in any treatment arm of the safety population
| Preferred term | TX-004HR 4 μg (n = 191) | TX-004HR 10 μg (n = 191) | TX-004HR 25 μg (n = 190) | Placebo (n = 192) |
| Any participant with reported TEAE | 97 (50.8) | 94 (49.2) | 93 (48.9) | 111 (57.8) |
| Headache | 12 (6.3) | 14 (7.3) | 6 (3.2) | 15 (7.8) |
| Vaginal discharge | 5 (2.6) | 6 (3.1) | 4 (2.1) | 13 (6.8) |
| Nasopharyngitis | 5 (2.6) | 6 (3.1) | 7 (3.7) | 10 (5.2) |
| Vulvovaginal pruritus | 4 (2.1) | 3 (1.6) | 7 (3.7) | 10 (5.2) |
| Back pain | 9 (4.7) | 1 (0.5) | 4 (2.1) | 8 (4.2) |
| Urinary tract infection | 5 (2.6) | 5 (2.6) | 8 (4.2) | 4 (2.1) |
| Upper respiratory tract infection | 5 (2.6) | 6 (3.1) | 3 (1.6) | 5 (2.6) |
| Oropharyngeal pain | 1 (0.5) | 0 (0) | 6 (3.2) | 1 (0.5) |
TEAE, treatment-emergent adverse event.
Nine serious TEAEs were reported in eight women; however, none was considered related to treatment. Complete heart block, appendicitis, endophthalmitis, and chronic obstructive pulmonary disease were each reported by a different participant in the 25-μg group. Sinus node dysfunction and ankle fracture were both reported for one women, and arthralgia and malignant melanoma were each reported for one women in the 10-μg group. None of the women in the 4-μg group had reports of serious TEAEs. One woman in the placebo group was reported to have a cervical myelopathy. No deaths occurred during the study.
No diagnoses of endometrial hyperplasia or malignancy from endometrial biopsies were observed at week 12. Total cholesterol numerically decreased from baseline to week 12 by a mean of 0.024 to 0.07 mmol/L in the treatment groups, and by 0.008 mmol/L in the placebo group. No clinically meaningful increases in triglycerides were observed in any active treatment groups compared with placebo. Sex hormone binding globulin (SHBG) concentrations (measured in a subset of 72 women) did not increase with treatment relative to placebo or baseline at week 12. No clinically significant changes in any laboratory parameters were found.
DISCUSSION
This phase 3 clinical trial demonstrated that TX-004HR at 4, 10, and 25-μg doses is safe and effective for treating vaginal changes and self-reported symptoms of VVA in postmenopausal women. Statistically significant and clinically meaningful improvements in all of the four prespecified co-primary endpoints (increase in the percentage of vaginal superficial cells, decrease in the percentage of vaginal parabasal cells and vaginal pH, and decrease in severity of the MBS of dyspareunia) were observed as early as 2 weeks with all three doses of TX-004HR as compared with placebo, and were sustained throughout the 12-week trial. Additionally, statistically significant and potentially clinically meaningful improvements were found for the secondary endpoints of vaginal dryness and vulvar and/or vaginal irritation or itching. These improvements were achieved without increasing systemic estrogen concentrations (4 and 10 μg) or with negligible (25 μg) systemic estrogen exposure, as reported in pharmacokinetic studies.20 TX-004HR was also well-tolerated with no clinically significant differences found between treatment and placebo groups in any AEs or treatment-related AEs, and no treatment-related serious AEs.
The results reported here extend the findings of a phase 2 study in which TX-004HR demonstrated early onset of action in the clinical signs of VVA with statistically significantly improved changes compared with placebo.19 Caution comparing across studies must be utilized; however, when comparing the magnitude of effect with TX-004HR in our study with those reported for other products with similar study designs and for a similar indication, the changes with TX-004HR were numerically greater. For example, significant improvements in co-primary endpoints from a randomized controlled trial of a 10-μg vaginal estradiol tablet were numerically smaller with the 10-μg estradiol tablet (change of 13% in superficial cells, −37% in parabasal cells, and −1.3 in vaginal pH)21 than with what was observed in our study with the 10-μg TX-004HR dose (change of 17% in superficial cells, −44% in parabasal cells, and −1.4 in vaginal pH) at 12 weeks. In addition, whereas improvements in some objective (cell and pH) endpoints were seen with the estradiol tablet within 2 weeks of treatment, the participant-reported improvements in a composite score of subjective symptoms were not observed until 8 weeks of therapy,21 which may be perceived as a disadvantage for many users. However, that clinical trial did not assess individual symptoms.21 A second separate, randomized controlled trial of 10 and 25-μg estradiol tablets similarly did not find significant improvements over placebo in the composite score of vaginal symptoms with either dose until 7 weeks of treatment (wk 2, NS).22 Likewise, the SERM, ospemifene, was evaluated in another separate clinical trial for the treatment of dyspareunia, and statistically significant improvements were not observed until week 12.23
Importantly, the results reported here showed significant improvement in dyspareunia within 2 weeks with all three doses of TX-004HR, with reductions in severity scores from 1.5 to 1.7 points at week 12, which were comparable or superior to reductions of 1.2 to 1.6 points reported for other currently approved dyspareunia treatments.15,24,25 Additionally, vaginal dryness improved by week 2 with 10 and 25 μg TX-004HR and by week 6 with 4 μg TX-004HR; such improvements have been associated with high user satisfaction. Furthermore, TX-004HR 10 and 25 μg showed significant improvement in vaginal irritation and/or itching at week 12, whereas none of the currently available marketed products reported improvements in these symptoms.26,27
Of note in this study was a large placebo response in the subjective but not objective measures. This may be explained, in part, by the potential lubricating properties of the excipient, Miglyol, found in the placebo vaginal capsules, and also in the active capsules. Nonetheless, the effect of TX-004HR on subjective endpoints was superior to the placebo.
Based on a large survey of postmenopausal women in the United States, only a small proportion (7%) of women are thought to receive prescription vaginal estrogen therapy alone for their VVA,4,9 probably due to lack of information about available treatments, avoidance of discussion of the topic with healthcare practitioners, or dissatisfaction with currently available products.4,7,8 Eliminating the need for an applicator or individually measuring doses is intended to give women a more positive user experience and thus potentially better compliance, resulting in overall better efficacy of treatment.
The results with TX-004HR in this study exemplify the purpose of local vaginal estrogen therapies: rapid symptom resolution without increasing systemic estrogen concentrations. A pharmacokinetic study of TX-004HR found that the mean area under the concentration-time curve (AUC) and average concentration (Cavg) for estradiol were not significantly different from placebo with 4 and 10 μg TX-004HR.20 Although statistically higher AUC for estradiol was observed with the 25-μg dose, estradiol levels remained within the postmenopausal range, with no evidence of accumulation by day 84. Although there was negligible systemic absorption, rapid efficacy was observed within 2 weeks of dosing with all doses of TX-004HR.
The low doses of this novel vaginal estradiol soft-gel capsule, TX-004HR, were well-tolerated.28 Indeed, the four most commonly reported TEAEs, including vaginal discharge and vulvovaginal pruritus, were experienced by fewer women in any TX-004HR group than in the placebo group, and were mostly mild to moderate in severity. By comparison, in a 12-week study of the efficacy of ospemifene, vaginal discharge was reported more than six times more frequently in the ospemifene group than in the placebo group.23 Genital pruritus was also reported four times more frequently in women treated with Vagifem 10-μg tablets than with placebo in a 12-month randomized study.15 Importantly, endometrial findings after TX-004HR were benign as no hyperplasia or malignancies were reported in biopsies at 12 weeks.
One of the limitations of this study was the narrowly defined inclusion criteria. Additionally, women were required to have identified moderate-to-severe dyspareunia as their MBS; however, it is well-known that most women with moderate-to-severe dyspareunia associated with postmenopausal VVA also suffer from multiple other vulvar and vaginal symptoms. The concept of the MBS has been referred to as a false construct that may not provide a full clinical evaluation of a product's efficacy. Another limitation of this study is that women were mostly white, in good health, and had an average BMI of 26.7 kg/m2, and thus, may not be representative of the US population of postmenopausal women with VVA.
CONCLUSIONS
TX-004HR at doses of 4, 10, and 25 μg was safe and effective for treating postmenopausal women with VVA and moderate-to-severe dyspareunia, and also vaginal dryness and vaginal itching or irritation. Onset of effect was seen as early as 2 weeks and was maintained throughout the study. TX-004HR was well-tolerated as reported here and systemic estrogen exposure was negligible to very low as demonstrated by the pharmacokinetic study. TX-004HR is a potential novel product that may provide a promising new treatment option for women experiencing symptomatic menopausal VVA.
Supplementary Material
Acknowledgments
The authors would like to thank the investigators of the REJOICE Study Group (see Supplemental Digital Content 1 for an Appendix listing the investigators). The authors would also like to acknowledge the medical writing assistance of Jolene Mason, PhD and Dominique Verlaan, PhD of Precise Publications, LLC, which was supported by TherapeuticsMD.
Footnotes
Funding/support: TherapeuticsMD sponsored the study and funded the medical writing support provided by Jolene Mason, PhD and Dominique Verlaan, PhD of Precise Publications, LLC.
Financial disclosure/conflicts of interest: Dr Constantine consults to multiple pharmaceutical companies, including, but not limited to, TherapeuticsMD and has stock options with TherapeuticsMD. Dr Kushner consults to pharmaceutical companies including but not limited to TherapeuticsMD. Dr Simon has served (within the past year) or is currently serving as a consultant to or on the advisory boards of: AbbVie, Inc, AMAG Pharmaceuticals, Inc, Amgen Inc, Apotex, Inc, Ascend Therapeutics, JDS Therapeutics, LLC, Merck & Co, Inc, Noven Pharmaceuticals, Inc, Novo Nordisk, Nuelle, Inc, Perrigo Company, PLC, Radius Health, Inc, Regeneron Pharmaceuticals, Inc, Roivant Sciences, Inc, Sanofi S.A., Sermonix Pharmaceuticals, Inc, Shionogi Inc, Sprout Pharmaceuticals, Symbiotec Pharmalab, TherapeuticsMD; and has also served (within the past year) or is currently serving on the speaker's bureaus of: Amgen Inc, Eisai, Inc, Merck, Noven Pharmaceuticals, Inc (New York, NY), Novo Nordisk, Shionogi Inc; and in the last year has received or is currently receiving grant/research support from: AbbVie, Inc, Actavis, PLC, Agile Therapeutics, Bayer Healthcare LLC, New England Research Institute, Inc, Novo Nordisk, Palatin Technologies, Symbio Research, Inc, TherapeuticsMD; and is a stockholder (direct purchase) in Sermonix Pharmaceuticals. Dr Pickar was formerly an employee of Wyeth Research; has received consultant fees from Wyeth/Pfizer, Besins Healthcare, Shionogi Inc, Metagenics, Radius Health, Inc, and TherapeuticsMD; and has stock options with TherapeuticsMD. Dr Archer (within the past 3 years) has received research support from Actavis (previously Allergan, Watson Pharmaceuticals, Warner Chilcott), Bayer Healthcare, Endoceutics, Glenmark, Merck (previously Schering Plough, Organon), Radius Health, Shionogi Inc, and TherapeuticsMD; has served as consultant to Abbvie (previously Abbott Laboratories), Actavis (previously Allergan, Watson Pharmaceuticals, Warner Chilcott), Agile Therapeutics, Bayer Healthcare, Endoceutics, Exeltis (previously CHEMO), Ferring Pharmaceuticals, InnovaGyn, Merck (previously Schering Plough, Organon), Pfizer, Radius Health, Sermonix Pharmaceuticals, Shionogi, Inc, Teva Women's Healthcare, and TherapeuticsMD; and has received Speakers’ Bureau honorarium for Ascend Therapeutics, Merck (previously Schering Plough, Organon), Noven, and Pfizer. Dr Bernick is a board member and an employee of TherapeuticsMD with stock/stock options. Ms Gasper, Dr Graham, and Dr Mirkin are employees of TherapeuticsMD with stock/stock options.
REFERENCES
- 1.Mac Bride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc 2010; 85:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gass ML, Cochrane BB, Larson JC, et al. Patterns and predictors of sexual activity among women in the hormone therapy trials of the Women's Health Initiative. Menopause 2011; 18:1160–1171. [DOI] [PubMed] [Google Scholar]
- 3.Santoro N, Komi J. Prevalence and impact of vaginal symptoms among postmenopausal women. J Sex Med 2009; 6:2133–2142. [DOI] [PubMed] [Google Scholar]
- 4.Simon JA, Kokot-Kierepa M, Goldstein J, Nappi RE. Vaginal health in the United States: results from the Vaginal Health: Insights, Views & Attitudes survey. Menopause 2013; 20:1043–1048. [DOI] [PubMed] [Google Scholar]
- 5.Portman DJ, Gass ML. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women's Sexual Health and the North American Menopause Society. Menopause 2014; 21:1063–1068. [DOI] [PubMed] [Google Scholar]
- 6.Dennerstein L, Dudley EC, Hopper JL, Guthrie JR, Burger HG. A prospective population-based study of menopausal symptoms. Obstet Gynecol 2000; 96:351–358. [DOI] [PubMed] [Google Scholar]
- 7.Nappi RE, Kokot-Kierepa M. Women's voices in the menopause: results from an international survey on vaginal atrophy. Maturitas 2010; 67:233–238. [DOI] [PubMed] [Google Scholar]
- 8.Kingsberg SA, Wysocki S, Magnus L, Krychman ML. Vulvar and vaginal atrophy in postmenopausal women: findings from the REVIVE (Real Women's Views of Treatment Options for Menopausal Vaginal Changes) survey. J Sex Med 2013; 10:1790–1799. [DOI] [PubMed] [Google Scholar]
- 9.US Census Bureau. Age and sex composition: 2010 Census Briefs. Issued May 2011. Available at: http://www.census.gov/prod/cen2010/briefs/c2010br-03.pdf Accessed April 14, 2015. [Google Scholar]
- 10.North American Menopause Society. Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20:888–902. [DOI] [PubMed] [Google Scholar]
- 11.De Villiers TJ, Pines A, Panay N, et al. Updated 2013 International Menopause Society recommendations on menopausal hormone therapy and preventive strategies for midlife health. Climacteric 2013; 16:316–337. [DOI] [PubMed] [Google Scholar]
- 12.Manson JE, Goldstein SR, Kagan R, et al. Why the product labeling for low-dose vaginal estrogen should be changed. Menopause 2014; 21:911–916. [DOI] [PubMed] [Google Scholar]
- 13.Freedman MA. Perceptions of dyspareunia in postmenopausal women with vulvar and vaginal atrophy: findings from the REVIVE survey. Womens Health (Lond Engl) 2014; 10:445–454. [DOI] [PubMed] [Google Scholar]
- 14.Minkin MJ, Maamari R, Reiter S. Postmenopausal vaginal atrophy: evaluation of treatment with local estrogen therapy. Int J Womens Health 2014; 6:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vagifem (estradiol vaginal tablets). Prescribing Information. Bagsvaerd, Denmark: Novo Nordisk Pharmaceuticals Inc; 2012. [Google Scholar]
- 16.Wysocki S, Kingsberg S, Krychman M. Management of vaginal atrophy: implications from the REVIVE study. Clin Med Insights Reprod Health 2014; 8:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Portman D, Shulman L, Yeaw J, et al. One-year treatment persistence with local estrogen therapy in postmenopausal women diagnosed as having vaginal atrophy. Menopause 2015; 22:1197–1203. [DOI] [PubMed] [Google Scholar]
- 18.Fishman JR, Flatt MA, Settersten RA., Jr Bioidentical hormones, menopausal women, and the lure of the “natural” in U.S. anti-aging medicine. Soc Sci Med 2015; 132:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pickar JH, Amadio JM, Hill JM, Bernick BA, Mirkin S. A randomized, double-blind, placebo-controlled phase 2 pilot trial evaluating a novel, vaginal softgel capsule containing solubilized estradiol. Menopause 2016; 23:506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archer DF, Constantine G, Kushner H, et al. TX-004HR Vaginal estradiol effectively treats vulvar and vaginal atrophy (VVA) with negligible to low systemic absorption of estradiol. Poster presented at: the Endocrine Society's 98th Annual Meeting & Expo, Boston, MA, 2016. [Google Scholar]
- 21.Simon J, Nachtigall L, Gut R, Lang E, Archer DF, Utian W. Effective treatment of vaginal atrophy with an ultra-low-dose estradiol vaginal tablet. Obstet Gynecol 2008; 112:1053–1060. [DOI] [PubMed] [Google Scholar]
- 22.Bachmann G, Lobo RA, Gut R, Nachtigall L, Notelovitz M. Efficacy of low-dose estradiol vaginal tablets in the treatment of atrophic vaginitis: a randomized controlled trial. Obstet Gynecol 2008; 111:67–76. [DOI] [PubMed] [Google Scholar]
- 23.Portman DJ, Bachmann GA, Simon JA. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause 2013; 20:623–630. [DOI] [PubMed] [Google Scholar]
- 24.Premarin (conjugated estrogens tablets, USP). Prescribing Information. Philadelphia, PA: Wyeth Pharmaceuticals Inc; 2010. [Google Scholar]
- 25.Shionogi, Inc, Osphena (ospemifene) tablets, for oral use. Prescribing Information. 2013. [Google Scholar]
- 26.Portman D, Palacios S, Nappi RE, Mueck AO. Ospemifene, a non-oestrogen selective oestrogen receptor modulator for the treatment of vaginal dryness associated with postmenopausal vulvar and vaginal atrophy: a randomised, placebo-controlled, phase III trial. Maturitas 2014; 78:91–98. [DOI] [PubMed] [Google Scholar]
- 27.Eriksen PS, Rasmussen H. Low-dose 17 beta-estradiol vaginal tablets in the treatment of atrophic vaginitis: a double-blind placebo controlled study. Eur J Obstet Gynecol Reprod Biol 1992; 44:137–144. [DOI] [PubMed] [Google Scholar]
- 28.Ulrich LS, Naessen T, Elia D, Goldstein JA, Eugster-Hausmann M. Endometrial safety of ultra-low-dose Vagifem 10 microg in postmenopausal women with vaginal atrophy. Climacteric 2010; 13:228–237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.