

Abstract
Infections with Shiga toxin (STx)–producing bacteria cause more than a million deaths each year and have no definitive treatment. To exert its cytotoxic effect, STx invades cells through retrograde membrane trafficking, escaping the lysosomal degradative pathway. We found that the widely available metal manganese (Mn2+) blocked endosome-to-Golgi trafficking of STx and caused its degradation in lysosomes. Mn2+ targeted the cycling Golgi protein GPP130, which STx bound in control cells during sorting into Golgi-directed endosomal tubules that bypass lysosomes. In tissue culture cells, treatment with Mn2+ yielded a protection factor of 3800 against STx-induced cell death. Furthermore, mice injected with nontoxic doses of Mn2+ were completely resistant to a lethal STx challenge. Thus, Mn2+ may represent a low-cost therapeutic agent for the treatment of STx infections.
Shiga toxin (STx)–producing bacteria of the Shigella genus and enterohemorrhagic Escherichia coli (EHEC) species infect more than 150 million individuals each year and cause more than a million deaths (1). There is no definitive medical treatment. Indeed, treatment with antibiotics is contraindicated because it increases the risk of STx release and life-threatening disease (1–3). STx consists of a catalytically toxic A subunit bound to a B subunit that mediates its membrane trafficking from the cell surface through endosomes, the Golgi apparatus, and the endoplasmic reticulum (ER), where the toxin translocates to the cytosol and inactivates ribosomes (4). Direct trafficking of STx from early endosomes to the Golgi, bypassing late endosomes and lysosomes, is a crucial step that allows STx to avoid degradation. Small-molecule inhibitors targeting this step hold therapeutic promise, but the STx interactions responsible for its endosomal sorting remain unclear.
Exposure of cells to 50 to 500 μM manganese (Mn2+) induces degradation of GPP130 (5, 6), a membrane protein that cycles between the Golgi and endosomes (7, 8). Because GPP130 plays a role, albeit undefined, in endosome-to-Golgi trafficking of STx (9), we asked whether Mn2+ acts as a small-molecule inhibitor of STx. To investigate the effect of Mn2+ on STx trafficking, we used a fluorescently tagged version of the B subunit of STx (STxB) that exhibits transport kinetics identical to the holotoxin (10, 11). Whereas STxB efficiently trafficked from the cell surface to the Golgi in control cells, cells exposed to 500 μM Mn2+ lacked GPP130, and STxB accumulated in peripheral punctate structures resembling endosomes (Fig. 1, A and B). We confirmed that STxB did not reach the ER (fig. S1) by using a STxB version with a KDEL tag, which increases its retention in the ER (12).
Fig. 1.
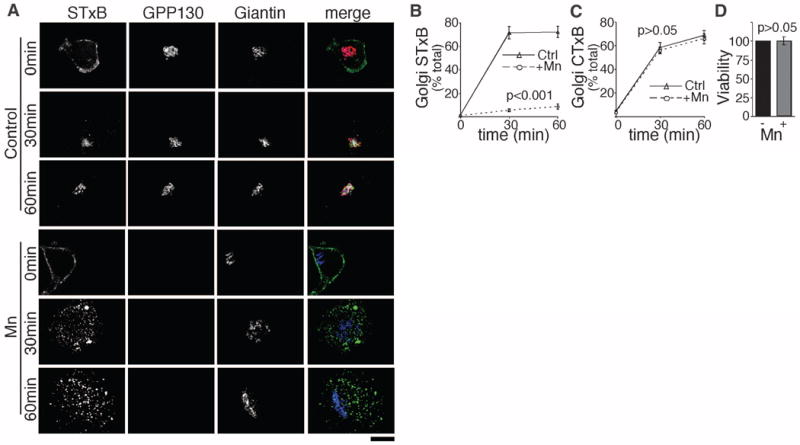
Manganese specifically blocks STxB trafficking. (A) STxB transport for the indicated times in HeLa cells untreated or pretreated with 500 μM Mn2+ for 4 hours. Scale bar, 10 μm. (B and C) Percentage of cellular STxB or CTxB in the Golgi at the indicated times postinternalization (mean ± SE; 20 cells per point). See fig. S2 for CTxB images. (D) Normalized cell viability by methylthiazolylphenyl-tetrazolium bromide (MTT) assay. Mn2+ was 500 μM for 12 hours (mean ± SE; n = 3 experiments).
The Mn-induced block in STxB trafficking was specific. There was no difference between Mn2+-treated and control cells in cholera toxin B subunit (CTxB) trafficking (Fig. 1C and fig. S2), which follows the same route as STx to the Golgi (13). Moreover, Mn2+ did not affect epidermal growth factor (EGF) internalization and degradation (fig. S3) or ER-to-cell-surface trafficking of vesicular stomatitis virus G protein (fig. S4). Further, Mn2+ did not alter the localization or trafficking of GP73 and TGN46, endogenous proteins that constitutively cycle between the Golgi and endosomes, or Lamp2, which traffics from the Golgi to lysosomes (fig. S5). Finally, 500 mM Mn2+ did not affect cell viability (Fig. 1D).
The accumulation of STxB in intracellular punctae implies that Mn2+ blocks STxB after internalization. Indeed, STxB moved to Rab5-positive early endosomes and, instead of exiting to the Golgi, trafficked to Rab7-positive late endosomes (Fig. 2A). Mn2+ did not alter Rab5 and Rab7 distribution (fig. S6). Exit to the Golgi involves tubules that extend from early endosomes (14). To determine whether Mn2+ blocks this step, we compared the early endosome localization of internalized STxB to EGF, which is excluded from tubular extensions and traffics to late endosomes. As expected, STxB was detected in endosomal tubules in control cells while EGF was excluded (Fig. 2, B and C, and fig. S7). STxB tubules contained the retromer component SNX-1 (fig. S8), confirming that they were bona fide Golgi-directed transport intermediates (14). GPP130 was also detected on STxB tubules and endosomes when trafficking of both proteins was synchronized (fig. S9). The sorting of STxB into endosomal tubules was abolished in Mn2+-treated cells (Fig. 2, B and C). The block in tubulation appeared specific because the sorting of CTxB into endosomal tubules was not affected (fig. S10). Sorting of STxB into endosomal tubules was also blocked after small interfering RNA–induced depletion of GPP130 (fig. S11), suggesting that GPP130 is the target of Mn.
Fig. 2.
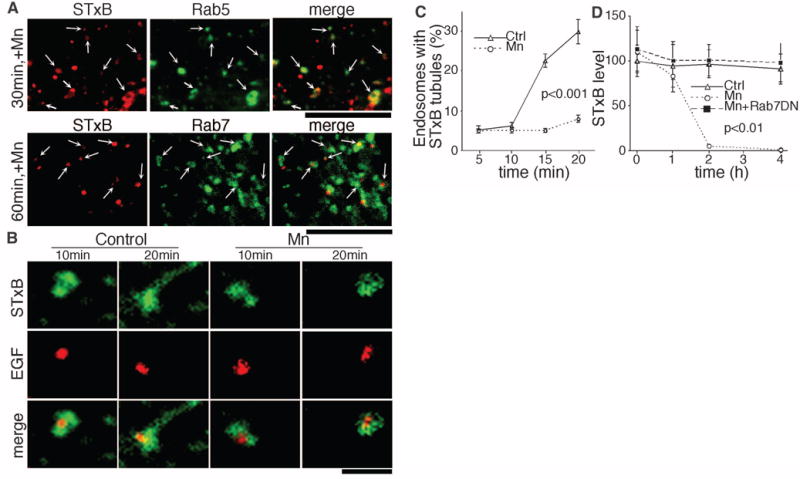
Manganese alters sorting at endosome, causing STxB degradation. (A) STxB localization compared with Rab5-GFP or Rab7-GFP at the indicated times postinternalization in Mn2+-treated cells. Scale bar, 4 μm. Arrows indicate overlap. (B) Endosomal tubulation of STxB in comparison with EGF at the indicated times postinternalization. See fig. S7A for full images. Scale bar, 1 μm. (C) Percentage of endosomes per cell with STxB tubules at the indicated times (mean ± SE; 10 cells per point; 100 to 150 endosomes per cell). See fig. S7B for EGF tubules. (D) Cellular STxB at the indicated times normalized to the control at 0 hours postinternalization (mean ± SE; 15 cells per point). See fig. S12 for images.
The presence of STxB in Rab7-positive endosomes suggests that Mn2+ may induce toxin degradation. Indeed, Mn2+ caused a dramatic loss of STxB (Fig. 2D and fig. S12). Degradation occurred after a lag and was blocked by dominant-negative Rab7 (Rab7-T22N), implying that STxB trafficked to, and was degraded in, lysosomes (Fig. 2D). Rab7-T22N did not affect STxB trafficking in control cells (fig. S13) because STxB normally bypasses late endosomes (15). Thus, Mn2+ diverts STxB to late endosomes and lysosomes, where it is degraded. This is important from a therapeutic perspective because Mn-treated cells will not contain residual toxin that could escape to the cytosol over time.
To determine whether GPP130 is the Mn2+ target, we used a rescue approach with the goal of restoring endosome-to-Golgi trafficking of STxB by expression of an Mn2+-insensitive GPP130 construct. GPP130 contains a short cytosolic domain, a single transmembrane domain, a coiled-coil lumenal stem domain, and an acidic C terminus (fig. S14). The stem contains three targeting determinants: residues 36 to 87 and 176 to 245 confer Golgi localization, and residues 88 to 175 mediate endosome-to-Golgi cycling (16). Deletion of any of these makes GPP130 Mn2+ insensitive (5). Based on this, we generated GPP1301-175-GFP (green fluorescent protein) and verified that it was Mn2+ insensitive but retained its ability to traffic between the Golgi and endosomes (fig. S15). Expression of this construct restored the ability of STxB to traffic to the Golgi after Mn2+ (Fig. 3, A and B), indicating that GPP130 was the target of Mn2+. As a control, we generated GPP130Δ88–175-GFP, an Mn2+-insensitive construct that lacked residues 88 to 175 required for endosome-to-Golgi cycling. This construct failed to traffic out of the Golgi (fig. S15) and also failed to restore STxB trafficking (Fig. 3, A and B).
Fig. 3.
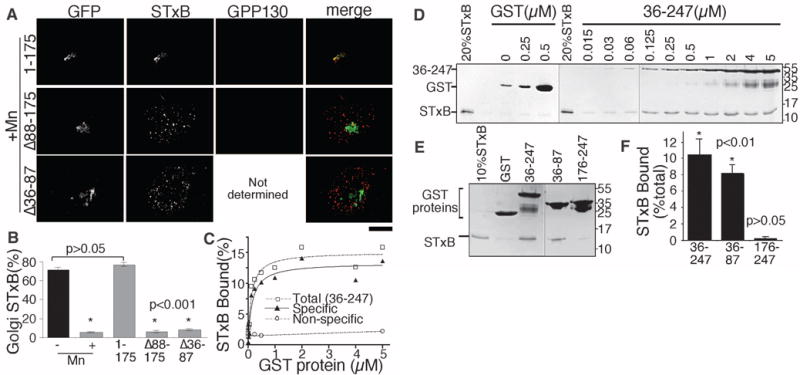
GPP130 is the cellular Mn2+ target and directly binds STxB to mediate its endosome-to-Golgi trafficking. (A) STxB localization 30 min postinternalization in Mn-treated cells expressing the indicated constructs. Endogenous GPP130 was detected using an antibody to the acidic domain. Scale bar, 10 μm. (B) Percentage of cellular STxB in Golgi from (A). Data for control and Mn2+ groups without GPP130 transfection are replotted from Fig. 1B (mean ± SE; 15 cells each). (C and D) Quantitation of His-STxB recovery after incubation with the indicated concentrations of either glutathione S-transferase (GST) or GST-GPP13036–247 and the corresponding Coomassie-stained gels. (E and F) Coomassie-stained gels and quantitation of His-STxB recovery after incubation with 5 μM of the indicated GST constructs (mean ± SE; n = 3 experiments).
Of the known cellular factors involved in STxB trafficking, all but GPP130 are cytosolic, raising the possibility that STx evolved to avoid the degradative pathway by binding GPP130 in the lumen of early endosomes. Indeed, we observed robust, direct, and specific binding of STxB to the GPP130 stem domain exhibiting a dissociation constant (Kd) of 150 nM (Fig. 3, C and D). Binding mapped to residues 36 to 87 (Fig. 3, E and F). Because deleting these residues yielded a construct, GPP130Δ36–87-GFP, that cycled yet was Mn2+ insensitive (fig. S15), we could use the rescue assay to test the functional importance of STx binding to GPP130. GPP130Δ36–87-GFP failed to rescue STxB trafficking to the Golgi (Fig. 3, A and B), indicating that the GPP130 lumenal stem domain directly interacts with STxB to mediate STxB sorting into endosomal tubules (schematized in fig. S16). It is possible that other toxins similarly coopt proteins in the GPP130 pathway to escape degradation.
To determine whether Mn2+ protects cells against Shiga toxicity, we first performed a dose-response analysis in control cells using STx1. STx1, secreted by EHEC, has a B subunit identical to STx secreted by Shigella, and the A subunit differs in only one position (17). As expected (18), the lethal dose (LD50) of STx1 was 0.05 ng/ml (Fig. 4A). Mn2+ protected STx1-treated HeLa cells (Fig. 4, A and B, and fig. S17A), and 50% cell death was not evident, even at a concentration 2000 times as high as the LD50. The estimated protection factor was 3800 (Fig. 4B). Mn2+ by itself did not compromise viability (fig. S17).
Fig. 4.
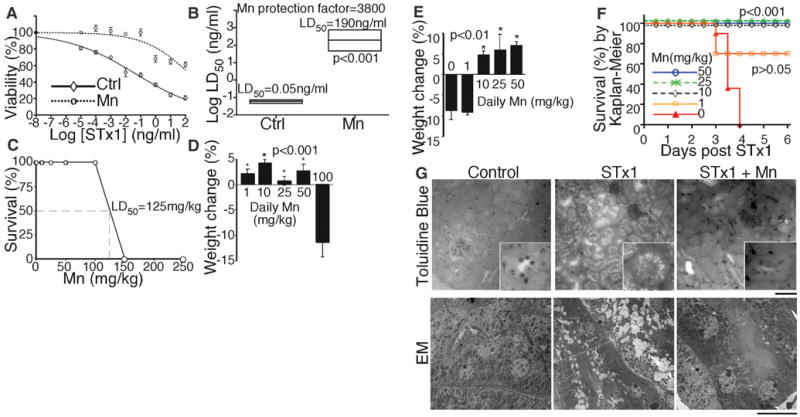
Treatment with Mn2+ protects against STx1-induced death. (A) Cell viability by MTT assay after 24 hours exposure to STx1 at the indicated concentrations. Mn2+ sample was pretreated with 500 μM for 4 hours and then Mn2+ was reduced to 125 μM during STx1 exposure (mean ± SE; n = 3 experiments). (B) LD50 with boxed 95% confidence interval from (A). (C) Fraction of mice surviving injection of Mn2+ at the indicated concentration (n = 4 mice per dose). (D) Body weight change after 96 hours of daily Mn2+ injections at the indicated concentrations (n = 4 mice per dose, except n = 2 for 100 mg/kg). Only animals with depressed body weight exhibited locomotive or any other abnormalities. (E) Body weight change of mice injected with STx1 only (n = 6 mice) or STx1 and Mn2+ at the indicated concentration (n = 6 mice for 50 mg/kg Mn2+ and 4 for other doses). Final weight recorded on day of death or on day 8 for survivors. (F) Survival curves from (E). (G) Histology of kidneys by toluidine blue staining (scale bar, 100 μm; inset, 4×) and electron microscopy (scale bar, 10 μm).
Finally, we investigated whether Mn2+ protects mice during lethal Shiga toxicity. Intra-peritoneal injection was used because this model has a definitive end point (death in 3 to 4 days) and recapitulates features of STx-induced renal damage evident in humans (19–21). To identify a test dose for Mn2+, a concentration series was injected that yielded an apparent LD50 of 125 mg per kg of weight (mg/kg) (Fig. 4C). Because Mn2+ is cleared from the system within hours (22), we also tested daily injections, and doses up to 50 mg/kg were not toxic and did not change body weight (Fig. 4D). Thus, as a proof of protection dose, we used 50 mg/kg Mn2+ once daily beginning 5 days before STx1 exposure, followed by 25 mg/kg Mn2+ once daily after toxin exposure. Life-threatening complications of STx infections in humans develop days after onset of enteric symptoms, providing an opportunity for treatment after diagnosis. Each mouse received a single injection of 25 μg/kg STx1. Mice with no Mn2+ treatment became agitated and restless within 24 hours, lost 5 to 10% of body weight at 48 to 72 hours (Fig. 4E), and died at 72 to 96 hours (Fig. 4F). In contrast, all Mn-treated animals remained healthy and survived for the duration of the study (Fig. 4, E and F). Complete protection was also evident with daily Mn2+ injections of 25 mg/kg and 10 mg/kg, but not 1 mg/kg (Fig. 4, E and F). As expected (21), histologic examination of the kidneys of STx1-treated mice revealed extensive damage in the cortical convoluted tubules, whereas animals protected by Mn2+ showed no STx1-induced renal damage (Fig. 4G). Thus, Mn2+ effectively protects against STx-induced toxicity and death in vivo even during fulminant systemic toxicosis.
In conclusion, Mn2+ may be effective in the management of STx infections. In contrast to other experimental strategies (20, 23–25), Mn2+ is an essential nutrient, its toxicology is well studied (26, 27), and it is already approved for oral and intravenous use. The low cost and wide availability of Mn2+ make it amenable for use in developing countries, where >95% of STx infections occur. Further, it may be possible to combine Mn2+ with antibiotic therapy because Mn2+ may block the toxic effects of STx released from dying bacteria.
Supplementary Material
Acknowledgments
We thank D. R. Smith, T. H. Lee, J. S. Minden, J. L. Brodsky, and M. A. Puthenveedu for advice; N. N. Urban and J. Dry-Henich for help with animal work; Y. Creeger for fluorescence-activated cell sorting; S. Truschel and B. Ballou for protein preparation; and J. Suhan for electron microscopy. Supported by NIH grant R01GM084111 to A.D.L. and an American Heart Association fellowship to S.M. A patent on the use of Mn2+ to treat Shiga toxicosis is pending. The data reported in this paper are tabulated in the main text and in the supporting online material.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/335/6066/332/DC1
Materials and Methods
References (28, 29)
References and Notes
- 1.Ochoa T, Cleary TG. In: Oski’s Pediatrics: Principles and Practice. McMillan JA, et al., editors. Lippincott Williams and Wilkins; Philadelphia: 2006. pp. 1116–1121. [Google Scholar]
- 2.Mohawk KL, Melton-Celsa AR, Zangari T, Carroll EE, O’Brien AD. Microb Pathog. 2010;48:131. doi: 10.1016/j.micpath.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. N Engl J Med. 2000;342:1930. doi: 10.1056/NEJM200006293422601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraser ME, Chernaia MM, Kozlov YV, James MN. Nat Struct Biol. 1994;1:59. doi: 10.1038/nsb0194-59. [DOI] [PubMed] [Google Scholar]
- 5.Mukhopadhyay S, Bachert C, Smith DR, Linstedt AD. Mol Biol Cell. 2010;21:1282. doi: 10.1091/mbc.E09-11-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukhopadhyay S, Linstedt AD. Proc Natl Acad Sci USA. 2011;108:858. doi: 10.1073/pnas.1013642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linstedt AD, Mehta A, Suhan J, Reggio H, Hauri HP. Mol Biol Cell. 1997;8:1073. doi: 10.1091/mbc.8.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puri S, Bachert C, Fimmel CJ, Linstedt AD. Traffic. 2002;3:641. doi: 10.1034/j.1600-0854.2002.30906.x. [DOI] [PubMed] [Google Scholar]
- 9.Natarajan R, Linstedt AD. Mol Biol Cell. 2004;15:4798. doi: 10.1091/mbc.E04-05-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mallard F, Johannes L. Methods Mol Med. 2003;73:209. doi: 10.1385/1-59259-316-x:209. [DOI] [PubMed] [Google Scholar]
- 11.Materials and methods are available as supporting material on Science Online.
- 12.Johannes L, Tenza D, Antony C, Goud B. J Biol Chem. 1997;272:19554. doi: 10.1074/jbc.272.31.19554. [DOI] [PubMed] [Google Scholar]
- 13.Sandvig K, van Deurs B. Gene Ther. 2005;12:865. doi: 10.1038/sj.gt.3302525. [DOI] [PubMed] [Google Scholar]
- 14.Popoff V, et al. J Cell Sci. 2007;120:2022. doi: 10.1242/jcs.003111. [DOI] [PubMed] [Google Scholar]
- 15.Mallard F, et al. J Cell Biol. 1998;143:973. doi: 10.1083/jcb.143.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachert C, Lee TH, Linstedt AD. Mol Biol Cell. 2001;12:3152. doi: 10.1091/mbc.12.10.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraser ME, et al. J Biol Chem. 2004;279:27511. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 18.Shin IS, et al. BMB Rep. 2009;42:310. doi: 10.5483/bmbrep.2009.42.5.310. [DOI] [PubMed] [Google Scholar]
- 19.Ishikawa S, et al. Infect Immun. 2003;71:3235. doi: 10.1128/IAI.71.6.3235-3239.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohawk KL, O’Brien AD. J Biomed Biotechnol. 2011;2011:258185. doi: 10.1155/2011/258185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tesh VL, et al. Infect Immun. 1993;61:3392. doi: 10.1128/iai.61.8.3392-3402.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki H, Wada O. Environ Res. 1981;26:521. doi: 10.1016/0013-9351(81)90227-9. [DOI] [PubMed] [Google Scholar]
- 23.Saenz JB, Doggett TA, Haslam DB. Infect Immun. 2007;75:4552. doi: 10.1128/IAI.00442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stechmann B, et al. Cell. 2010;141:231. doi: 10.1016/j.cell.2010.01.043. [DOI] [PubMed] [Google Scholar]
- 25.Fukuda S, et al. Nature. 2011;469:543. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- 26.Moreno JA, et al. Toxicol Sci. 2009;112:394. doi: 10.1093/toxsci/kfp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aschner M, Erikson KM, Herrero Hernández E, Tjalkens R. Neuromol Med. 2009;11:252. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.