

Abstract
Neutrophil migration and its role during inflammation has been the focus of increased interest in the past decade. Advances in live imaging and the use of new model systems have helped to uncover the behaviour of neutrophils in injured and infected tissues. Although neutrophils were considered to be short-lived effector cells that undergo apoptosis in damaged tissues, recent evidence suggests that neutrophil behaviour is more complex and, in some settings, neutrophils might leave sites of tissue injury and migrate back into the vasculature. The role of reverse migration and its contribution to resolution of inflammation remains unclear. Here, we discuss the different cues within tissues that mediate neutrophil forward and reverse migration in response to injury or infection, and the implications of these mechanisms to human disease.
TOC blurb
Neutrophils follow a multitude of signals to reach sites of injury or infection. Understanding how this occurs and what the fate of these neutrophils is provides insight into how immune responses are controlled and chronic inflammation avoided. Here, the authors describe the movement of neutrophils during inflammation.
Neutrophils are the most abundant leukocytes in the blood and they lead the first wave of host defense to infection or tissue damage. Neutrophils are powerful effector cells that destroy infectious threats through phagocytosis, degranulation, reactive oxygen species (ROS) and neutrophil extracellular traps1–3. Neutrophil loss by disease or therapy-induced side effects has devastating consequences that are characterized by recurrent severe infections. However, excess infiltration and activation of neutrophils at a site of tissue damage can cause chronic inflammation, limit injury repair and lead to loss of organ function1,4. Neutrophils mediate tissue damage through the release of cytokines, proteases and other factors contained in their cytoplasmic granules and also by regulating the activity of the adaptive immune response, including both T and B cell activation. Therefore, the migration and activation of neutrophils must be finely controlled. Recent advances in in vivo imaging techniques and new in vitro systems incorporating microfluidics and three-dimensional models have enabled researchers to directly visualize and quantify neutrophil behaviour (Box 1). These developments have spawned more complex studies regarding the role of neutrophils in the context of tissue homeostasis and disease, including wound healing, chronic inflammation, infection and cancer. As the role of neutrophils as key modulators of the immune response becomes clearer, it is becoming more important to understand neutrophil migration in the context of both acute and chronic inflammation and how migration is altered in disease.
Box 1. Imaging neutrophil migration.
Substantial progress has been made in understanding the mechanisms that regulate neutrophil migration using live imaging. Most of this work has involved the use of primary human or mouse neutrophils140 or human-derived neutrophil-like cell lines (HL-60141 or PLB-985142). Microfluidic devices are one of the most powerful recent tools used to study the movement of neutrophils in stable chemokine gradients94,143–145. Recent studies using microfluidics have highlighted mechanisms that differentially regulate neutrophil migration in two-dimensional compared with three-dimensional environments146. These in vitro studies are complemented by more recent work imaging neutrophil migration within the tissue microenvironment using zebrafish and mice. Intravital imaging in mouse models has been developed to directly visualize and measure neutrophil migration and function in specific organs after acute tissue damage. These models mainly focus on peripheral tissues in mice that are amenable to imaging, including the cornea147, ear dermis7,68 and cremaster muscle110,148–150. Other methods create “windows” through which immune cell infiltration can be visualized in the brain151 and lungs152. Neutrophil recruitment to the liver has been studied for many years by externalizing it to allow for high-resolution intravital imaging33,153,154. Nevertheless, these models are invasive, often requiring animal sacrifice shortly after the experiment and are generally not amenable to long-term studies. In addition, these models do not provide high temporal and spatial resolution, making it difficult to analyse behaviours of early recruited neutrophils.
Zebrafish have been used increasingly for the study of innate immune cell function, including neutrophil migration155–157. Transparent larvae allow in vivo imaging of neutrophil migration in the context of a whole living organism on the scale of days. Zebrafish neutrophils have the same morphology, behaviour and function as mammalian neutrophils158,159 and zebrafish have been used successfully to model human neutrophil disorders155,156. With fluorescent tags, specific cells and subcellular structures can be visualized, and the use of tracking software enables researchers to easily measure multiple parameters of neutrophil motility, including speed and directionality, early after tissue damage. In fact, neutrophil reverse migration was first visualized in zebrafish101 and zebrafish are currently being used to elucidate the mechanisms governing reverse migration108. Zebrafish larvae are ideal for screening chemical compounds — with such screens a fungal-derived compound that inhibits neutrophil chemotaxis160 and a compound that promotes neutrophil inflammation resolution107 were recently discovered.
This Review highlights the signals that regulate neutrophil migration in acute tissue damage, on the basis of three main phases: an early neutrophil recruitment phase induced by short-term signals, an amplification phase of neutrophil infiltration in response to more persistent signals and the resolution of inflammation, which may include neutrophil reverse migration. We also consider the differences and similarities between ‘sterile’ wound-induced and infection-induced neutrophil infiltration. Finally, the potential therapeutic relevance of modulating either neutrophil forward or reverse migration is discussed.
Neutrophil forward migration
Acute tissue damage generates a wide variety of signals produced by complex networks that establish chemoattractant gradients throughout tissues. As ‘leader’ cells in host defense responses, neutrophils must sense, prioritize and integrate all of these chemotactic cues into a migration response towards the damage5,6. In this section, we focus on the different signals that mediate neutrophil forward migration to a site of damage. This migration is thought to occur in phases (Figure 1): early neutrophil recruitment (sometimes referred to as “scouting”) followed by amplification of the response that is mediated by both tissue-resident and early-recruited cells and results in robust neutrophil infiltration from the bloodstream7,8. The process of neutrophil extravasation from the vasculature into the tissue has recently been reviewed in detail9,10. Here, we highlight some of the signals in these phases, including damage-associated molecular patterns (DAMPs), hydrogen peroxide (H2O2), lipid mediators and chemokines.
Figure 1. The phases of neutrophil recruitment.

The recruitment of neutrophils to a site of damage occurs in several phases. Damage-associated molecular patterns (DAMPs) are released at a tissue injury site and promote the release of hydrogen peroxide (H2O2) and direct the recruitment of early-arriving neutrophils through the SRC family kinase LYN (a). DAMPs also induce the production of CXC-chemokine ligand 8 (CXCL8) family chemokines and leukotrienes from surrounding tissue cells to further recruit neutrophils (b). Early-arriving neutrophils are then themselves activated to both directly and indirectly promote further secretion of CXCL8 family chemokines and leukotriene B4 (LTB4) to induce neutrophil recruitment from the circulation and amplification of the response (c). In an infection, extra layers of signalling exist to prolong and amplify neutrophil infiltration, including the release of pathogen-associated molecular patterns (PAMPs) and the involvement of other recruited immune cells, such as macrophages, dendritic cells (DCs) and T cells (d). IL, interleukin; PI3K, phosphoinositide 3-kinase; TNF, tumour necrosis factor.
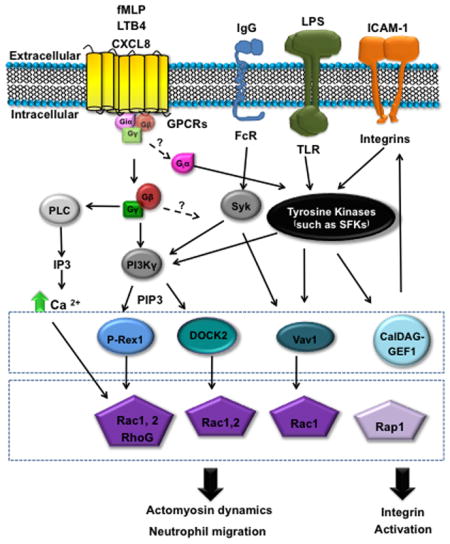
Neutrophils express more than 30 different receptors that can sense pro-inflammatory mediators and modulate neutrophil migration, function and behaviour including G protein-coupled receptors (GPCRs), Fc receptors, adhesion receptors, cytokine receptors and pattern recognition receptors (PRRs)11. Many of these receptors can directly modulate neutrophil polarization and migration (Box 2). In addition to activating migration, the signalling pathways downstream of these receptors can also affect neutrophil transcriptional activity, phagocytosis, apoptosis, degranulation and ROS production12. These cues often converge on common intracellular signalling pathways through phosphoinositide 3-kinase-γ (PI3Kγ), phospholipase C-β2 (PLCβ2) or PLCβ3 and/or extracellular signal-regulated kinase (ERK) signalling. These signalling pathways are discussed in more detail in Box 2 and have recently been reviewed6,11,13. Importantly, the pool of exogenous signals that neutrophils respond to after a wound is complex and it is likely that no single cue is absolutely required for migration.
Box 2. Neutrophil signalling in motility.
There is an extensive literature on downstream signalling mechanisms that regulate neutrophil directed migration to specific chemokines and other signals6,11,161–164 and only general principles are discussed here. Directed migration is mediated by chemoattractant signalling through G protein-coupled receptors (GPCRs) and other receptors that induce the asymmetric localization and activation of key signalling molecules, including phosphoinositide 3-kinase (PI3K), phospholipase C (PLC) and members of the RHO GTPase family, such as RAC165. The asymmetric distribution of the PI3K product phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3), at the front of polarized neutrophils is a hallmark of gradient sensing induced by chemoattractants165–167. The localized recruitment of PtdIns(3,4,5)P3 mediates the recruitment of specific adaptor proteins and guanine nucleotide-exchange factors, such as DOCK2 and P-REX1 respectively, that lead to the activation of RAC signalling and polarized actin polymerization during directed migration168,169. Although many attractant cues signal through PI3K and RAC, there is evidence that signalling hierarchies exist in which some chemoattractants are more dependent on p38 mitogen-activated protein kinase or PI3K signalling in vitro170. Specificity can be provided by the interaction of chemokine receptors with different ligands or the binding of receptors to specific downstream binding partners as has been described with binding of CXC-chemokine receptor 2 (CXCR2) to vasodilator-stimulated phosphoprotein (VASP)38. In addition, different GPCR kinases have been reported to bind to CXCR1 and CXCR2, providing specificity in the regulation of receptor internalization and therefore an additional layer of regulation171.
Less is known about the key signals that mediate neutrophil migration in vivo. Zebrafish are an important tool for the analysis of signalling in vivo and recent studies have demonstrated that both RAC signalling172 and calcium flux173 at the leading edge are sufficient to guide cell movement in live animals. However, despite recent progress, a challenge for future investigation remains in determining how neutrophils prioritize competing cues in vivo through the modulation of specific downstream intracellular signalling pathways that mediate responses to tissue damage.
Detection of early signals by neutrophils
The first signals that are responsible for early neutrophil recruitment released from damaged and necrotic cells after tissue injury are likely to be DAMPs14. Neutrophils recognize DAMPs by specific PRRs, including Toll-like receptors (TLRs) and NOD-like receptors (NLRs). The wide variety of DAMP molecules includes DNA, proteins (such as high-mobility group box 1 (HMGB1)), N-formyl peptides, extracellular matrix components, ATP and uric acid 15–17). The exact signals that govern this early stage in mice are largely unknown but are probably sensed by GPCRs as treatment of neutrophils with pertussis toxin, which inhibits GPCR-Gαi signalling, limits this early chemotaxis towards the damage site7. The signals promoting the early recruitment of neutrophils have been investigated in more detail in larval zebrafish, as this model is amenable to live imaging of early neutrophil recruitment after tissue damage (Box 1).
In larval zebrafish, one of the major early signals known to be directly sensed by neutrophils after wounding and to promote early forward migration is H2O2. After a tail wound in zebrafish up to a 200 μm radial gradient of H2O2 is formed, beginning only three minutes after wounding, with levels peaking at 20 minutes post-wounding18, and this H2O2 gradient is also observed in wounded Drosophila embryos19. This early H2O2 gradient is primarily produced by the enzyme dual oxidase 1 (DUOX1) in epithelial cells, and depletion of this enzyme in either zebrafish or Drosophila inhibits neutrophil migration to a wound18,19. Importantly, H2O2 also promotes chemotaxis of human20 and mouse21 neutrophils in vitro. SRC family kinases (SFKs) were found to directly sense this H2O2 gradient and are required in zebrafish for early neutrophil recruitment to wounds and in human neutrophils for chemotaxis to H2O2 in vitro20. Upon H2O2 exposure, a specific cysteine residue in the zebrafish SFK LYN is oxidized, promoting the autophosphorylation and activation of LYN and this activates multiple downstream pathways including ERK signalling20. The exact mechanism by which LYN directs the migration of neutrophils towards the source of H2O2 is unknown but it is likely that the signalling converges on the activation of PI3K, RAC and ERK to mediate directed migration (Box 2). This H2O2-LYN signalling seems to be important only for early neutrophil recruitment in zebrafish20. Moreover, neutrophils from mice deficient in three SFKs transplanted into a wild-type animal were still able to infiltrate to a site of inflammation, although mice deficient in these SFKs are protected from autoantibody-induced arthritis because SFK were necessary for the inflammatory microenvironment22.
The many DAMPs that are released from damaged and necrotic cells also serve to directly modulate neutrophils. For example, ATP can be sensed by two different classes of receptors, P2Ys (which are GPCRs) or P2Xs (ligand-gated ion channels)23. Zebrafish treated with apyrase or inhibitors of P2Y and P2X signalling had fewer neutrophils recruited to a wound24. Although ATP can directly induce monocyte and macrophage migration in vitro through the activation of P2Y2 receptors25, it is unclear if ATP directly activates chemotaxis of neutrophils. It has been shown that neutrophil-released ATP can act as an autocrine signal at the leading edge of the cell to amplify chemotactic signals through P2Y2 receptors and increase the efficiency of chemotaxis26. The autocrine effects of ATP and its metabolite adenosine at the cell front and rear of neutrophils, through the P2Y2 receptor and A2a receptor, respectively, promote efficient chemotaxis through differential effects on mechanistic target of rapamycin (mTOR) and mitochondrial activity27. There is also evidence that the presence of ATP can enhance the migration of neutrophils towards N-formyl peptides or CXC-chemokine ligand 8 (CXCL8) by activating RHOA28,29.
Early neutrophil chemotaxis and activation is also triggered by N-formyl peptides, such as fMet-Leu-Phe (fMLP). Although these peptides can be derived from bacterial proteins, they are also released from mitochondria after tissue damage and activate human neutrophils by binding to the GPCRs FPR1, FPR2 and FPR330–32. In vitro, FPR1-specific antibodies block neutrophil migration to disrupted mitochondrial products31 or necrotic cells33. FPR1 blockade or deficiency also prevented the guidance of recruited neutrophils to necrotic cells in a localized hepatic injury in mice,33 highlighting the importance of this signal as a short-range cue for proper neutrophil localization to a site of injury.
Other early signals produced by tissue and tissue-resident cells
The initial signals can be short-lived; studies in zebrafish suggest that the peak of H2O2 production is around 30 minutes after injury and the signal is completely cleared one hour later by the myeloperoxidase activity of wound-associated neutrophils34. For more sustained neutrophil recruitment, long-term and long-range signals are also activated, largely composed of chemokines and lipid mediators.
The CXCL8 family comprises some of the main chemokines that are involved in neutrophil migration, including CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7 and CXCL8. These chemokines are sensed by neutrophils through the GPCRs CXCR1 and CXCR235,36, and activate downstream signalling to vasodilator-stimulated phosphoprotein (VASP), PI3K and SFKs to mediate neutrophil directed migration37,38 (Box 2). These chemokines can be produced by both immune cells (including neutrophils, macrophages and T cells) and non-leukocytes (including epithelial and endothelial cells) in response to both injury and infection. Human CXCL8 has been known to be a neutrophil chemoattractant for over 25 years39 and the role of CXCL8-family–CXCR signalling in neutrophil migration has been confirmed in vivo in both mice and zebrafish40–44. To attract more distant neutrophils, CXCL8 family chemokines, among other chemokines, can bind to glycosaminoglycans on cell walls and in the extracellular matrix to create chemokine gradients along the tissues and structures through which neutrophils migrate33,42,45. When HEK293T cells expressing fluorescently tagged CXCL8 were transplanted into zebrafish larvae, the CXCL8 accumulated locally around the transplanted cells but then spread outwards into the vasculature, forming immobilized gradients42. Such intravascular extracellular matrix-bound chemokine gradients have also been observed in mouse models of acute hepatic injury, reaching as far away as 650 μm33, and when CXCL8 was injected intradermally in rabbits or ex vivo in human skin it appeared bound to the lumen of venular endothelial cells46.
Lipid mediators are also strong inducers of neutrophil chemotaxis. These mediators are derived from arachidonic acid. After arachidonic acid release by activated cytosolic phospholipase A2 (cPLA2), arachidonic acid is metabolized through multiple enzymatic pathways into a wide variety of molecules each with specific functions. The lipid mediator that is known to have the largest effect on neutrophil migration is leukotriene B4 (LTB4), which is produced from arachidonic acid by 5-lipoxygenase and sensed by the GPCR BLT1 to induce neutrophil polarization and migration (Box 2). Although some LTB4 is produced relatively early after wounding to recruit neutrophils, it also functions in the amplification stage of neutrophil recruitment and is discussed in detail in the next section. Other lipid mediators can also induce neutrophil chemotaxis, including 5-oxo-ETE and 5-KETE47.
In most cases, the production of these chemokines and lipid mediators is activated by DAMPs that are released after cell damage. Pre-made stores of chemokines can be rapidly released via exocytosis from endothelial cells that are activated by DAMPs48,49. DAMPs also activate core enzymatic and transcriptional pathways, such as nuclear factor-κB (NF-κB) and AP-1, that result in the production of chemokines and lipid mediators. Recently, in two different mouse models, DAMP-mediated activation of the NLRP3 inflammasome by ATP was linked to persistent neutrophil recruitment33,50. Tissue injury also results in the release of intracellular ion stores, creating alterations in neighbouring cell osmolarity. In vivo in zebrafish larvae, cells surrounding tissue wounds can sense changes in osmolarity through the activation of cPLA2 and release of arachidonic acid, which is metabolized into 5-KETE51. Knockdown of expression of cPLA2 or OXE-R (the GPCR for 5-KETE) inhibited leukocyte recruitment to a wound by decreasing both neutrophil velocity and directionality51.
Amplification of neutrophil recruitment
Neutrophil recruitment is exponentially amplified in a phase sometimes termed “neutrophil swarming”. To promote this phase, more signals must arise from the early recruited cells — including neutrophils and macrophages — to recruit more neutrophils both directly and indirectly through the further activation of tissue and tissue-resident cells8,52. This is the next step in the cascade of signals that promote neutrophil forward migration and involves many of the same signals that are involved in earlier phases, including LTB4 and CXCL8 family chemokines7,52.
LTB4 can amplify neutrophil responses to primary chemoattractants in multiple ways. In vitro, in response to fMLP, human neutrophils generate local microgradients of LTB4 at their leading edge, acting as an autocrine signal and stabilizing neutrophil polarization53. Longer-range gradients of LTB4 are also created to recruit new neutrophils53. In a mouse model of laser-induced dermal tissue injury, LTB4 receptor-deficient neutrophils close to a wound were able to undergo chemotaxis to the wound but more distant neutrophils required the LTB4 receptor to be efficiently recruited, suggesting that LTB4 is specifically required for persistent and long-term neutrophil infiltration54. This defect in recruitment is similar to that observed when all GPCR-Gαi2 signalling is inhibited54, providing evidence that LTB4 is a major signal in this phase. In addition to the requirement for sensing LTB4 through its receptor, neutrophils are also a source of LTB4 and neutrophils genetically lacking 5-lipoxygenase also could not swarm to the wound54. LTB4 production is also important for neutrophil recruitment in mouse models of inflammatory arthritis55,56.
The amplification step is also likely to be initiated through the recognition of DAMPs by the early-arriving cells. In addition to acting as direct neutrophil chemoattractants, N-formyl peptides also activate ERK1, ERK2 and p38 mitogen-activated protein kinase signalling pathways57 and stimulate LTB4 production by neutrophils53. Cyclic ADP ribose (cADPR) (a metabolite of NAD+) has also been shown to have a role in this amplification stage in a mouse model of laser-induced dermal tissue injury7. Treatment of neutrophils with a competitive inhibitor of cADPR signalling did not affect early neutrophil recruitment but prevented the amplification phase of the neutrophil response7. In a mouse model, neutrophil swarming correlated with cell death of some of the initially recruited cells and this probably produces more DAMPs54. The exact signals that these dying cells release to induce swarming is unclear because neutrophils deficient in single GPCRs including Fpr1, Fpr2, Cxcr2 and P2rx7 could migrate short distances normally when injected into the damaged tissue in this model54, and it is likely that a combination of signals are released.
Other secondary signals are also produced by these early-arriving neutrophils. In a model of inflammatory arthritis, LTB4-activated neutrophils produce chemokines, such as CXCL2, and the cytokine interleukin-1β (IL-1β), which activates tissue endothelial cells, fibroblast-like cells and macrophages to produce more ligands for CXCR1, CXCR2 and CCR1, further amplifying neutrophil recruitment58. Activated neutrophils also express matrix metalloproteinases (MMPs), which can cleave CXCL8 family chemokines to increase their chemotactic effect59,60. Collagen in the extracellular matrix can also be cleaved by neutrophil MMP activity to create collagen-derived chemotactic peptides61.
In addition to early recruited neutrophils, recruited monocytes and macrophages amplify the neutrophil response. Pro-inflammatory monocytes and macrophages can be recruited by initial DAMP signals but neutrophils also recruit these cells through production of macrophage chemokines and through further activation of surrounding non-immune tissue cells, setting off a feed-forward loop of neutrophil recruitment62.
Additional levels of signalling in infection
The tight control of neutrophil recruitment after infection is arguably even more important than after tissue injury. While inflammation must be maintained until the infection is cleared, neutrophils can also provide niches within which pathogens can replicate63. Neutrophil swarms have been observed after infection with multiple types of pathogen, including bacteria64 and protozoa65. Neutrophil infiltration in response to early recruited neutrophil cell death54 is likely to be even more pronounced in infections, as many pathogens can cause necrosis of host cells66. Neutrophil recruitment can also be altered by pathogens that carry virulence factors that directly interfere with or promote neutrophil recruitment67–69. Probably one of the most important differences in infections is the presence of PAMPs (pathogen-associated molecular patterns) in addition to tissue DAMPs, creating an extra level of danger signals (reviewed in REF.70). Another major variable in infection is the duration of damage, especially if the pathogen is not cleared. These factors lead to a much wider variety of immune cells participating in and modulating neutrophil recruitment including macrophages68,71, dendritic cells72, mast cells73 and T cells74.
As in a sterile tissue injury, tissue and tissue-resident cells can sense the presence of infection-induced damage and transmit the signal to neutrophils through the production of chemokines. Perivascular macrophages produce CXCL1 and CXCL2 to promote neutrophil extravasation at sites of bacterial infection in mouse ear skin68, whereas dendritic cells produce CXCL1 and CXCL2 after a Leishmania infection72. Mast cells can indirectly recruit neutrophils to a site of infection through the production of tumour necrosis factor (TNF)75 or a tryptase76, both of which promote chemokine production from tissue cells. Mast cells also directly recruit neutrophils through the production of leukotrienes such as LTB4 and LTC477.
The same amplification of neutrophil recruitment through feed-forward signalling and macrophage-neutrophil-tissue cell crosstalk is also observed after infection. In a mouse model of uropathogenic Escherichia coli infection, tissue-resident macrophages (LY6Clow) first sense the infection and produce a variety of chemokines, of which CXCL1 is the most important, to recruit both neutrophils and non-resident inflammatory macrophages (LY6Chi)71. However, these inflammatory macrophages then produce TNF to further activate the tissue-resident macrophages to release preformed stores of CXCL2 that further recruit neutrophils to the infection site71. In Staphylococcus aureus infection, early-arriving immune cells phagocytose the bacteria, leading to inflammasome activation by the bacterial cell wall component peptidoglycan and IL-1β secretion78,79. IL-1β then activates non-immune tissue cells to produce more pro-inflammatory cytokines and neutrophil chemokines80.
The marked involvement of the adaptive immune system has a key role in the persistent neutrophil inflammation that can be induced by infection. In several models, T cells and T cell-produced cytokines are important for neutrophil recruitment to infection, including Streptococcus pneumonia81 and Helicobacter pylori82 infections. Many factors contribute to the crosstalk between T cells and neutrophils in the context of infection83, but the involvement of the cytokine IL-17 seems to be particularly important84,85. In a cutaneous model of S. aureus infection, resistance to infection depends on the presence of neutrophils86, and both αβ T cells and IL-17-producing T cells have been shown to be important for the neutrophil response74. IL-17 stimulates both tissue cells and leukocytes to produce a variety of cytokines, including CXCL8 family chemokines and granulocyte-colony stimulating factor (G-CSF), which promotes the production and maturation of neutrophils from the bone marrow87,88. After infection with S. aureus, mice with lowered IL-17 levels due to γδ T cell deficiency had lowered levels of CXCL1 and CXCL2 production and consequently impaired neutrophil recruitment89. In this case, decreased neutrophil recruitment resulted in poor infection outcome which could be rescued by administration of recombinant IL-1789.
Distinguishing an infection from a wound
It is crucial for the host to distinguish whether or not a tissue injury has an associated infection, as this determines both the strength and length of the appropriate response. Responses to infection, such as an S. aureus skin infection, can create especially large neutrophil swarms90. Exactly how the host integrates information on the persistence of damage and diversity of signals after wounding or infection into differing responses is not clear and this question is relevant for the development of anti-inflammatory therapies that prevent chronic inflammation but retain a sufficient response to infection. Although many DAMPs and PAMPs signal through similar receptors and converge on similar signalling pathways, there is evidence that cells can interpret and respond to these signals differently91.
A few studies have directly compared the requirements for neutrophil recruitment to a wound versus an infection. In zebrafish larvae, H2O2 was found to be required for the early neutrophil response to a wound but not to P. aeruginosa or S. iniae infections92. Moreover, IL-1β and MYD88-mediated signalling were required for neutrophil responses to a wound but not to E. coli infection93. There is also evidence that neutrophils migrate differently to host-produced versus pathogen-produced signals. In vitro, human neutrophils retrotaxed after responding to a gradient of LTB4, whereas the neutrophils were “trapped” after they arrived at a source of the fungal cell wall component zymosan94.
One mechanism for the distinction between a wound and an infection involves CD24 and its ability to specifically trap DAMPs (including HMGB1, HSP70, and HSP90) and prevent their binding to receptors95,96. CD24 interacts with Siglec G, which associates with SH2 domain-containing protein tyrosine phosphatase 1 (SHP1), a negative regulator of NF-κB activation97. In this manner, CD24 decreases NF-κB-mediated activation of tissue-resident cells such as DCs, and thereby reduces the amplification stage of neutrophil recruitment97.
It has been proposed that neutrophils have phenotypic heterogeneity and functional plasticity (Box 3); several markers of different neutrophil phenotypes have been identified, although disease associations and functional characteristics of these phenotypes need to be established98. Differing signals at a wound or infection probably define the phenotype of the responding neutrophils (either by altering their phenotype after they arrive at the site or by specifically recruiting certain subsets that pre-exist in the circulation). A future challenge will be to determine the distinct neutrophil subsets that exist in different types of damage including ‘sterile’ versus infected tissue and to probe the complex signalling networks that integrate diverse inputs into a specific response.
Box 3. Neutrophil heterogeneity and plasticity.
During the past decade, evidence has been accumulating for the existence of neutrophil subsets with phenotypic heterogeneity and functional plasticity in different models106,174,175. Perhaps the most well-characterized example is in the tumour microenvironment, where neutrophils develop polarity that is similar to the M1/M2 polarization described for macrophages. Tumour-associated neutrophils can be designated also as N1 (anti-tumorigenic and pro-inflammatory) and N2 (pro-tumorigenic and immunosuppressive)176. Tumour-associated N2 neutrophils are characterized by high expression of CXC-chemokine receptor 4 (CXCR4), vascular endothelial growth factor and matrix metalloproteinase 9 and are induced in the presence of high levels of transforming growth factor-β (TGFβ). By contrast, N1 neutrophils express pro-inflammatory cytokines and chemokines, are able to kill cancer cells and are induced by inhibition of TGFβ signalling. Analogous neutrophil subsets may exist in damaged or infected tissues, although these phenotypes have not been well characterized. Subsets of neutrophils have also been defined by surface receptor expression and density (low-density neutrophils versus high- or normal-density neutrophils). For example, CXCR4 expression is increased in aged or senescent neutrophils177 and is associated with neutrophil trafficking to the bone marrow. Other changes in the expression of surface proteins on neutrophils have been described, including CD63 expression in the airways of patients with cystic fibrosis178, intercellular adhesion molecule 1 expression associated with systemic inflammation and reverse migration120 or CD177 expression associated with autoimmune diseases174,179. Low-density neutrophils have been associated mostly with autoimmune disorders180, sepsis181, HIV infection182,183 and cancer176. In addition to heterogeneity, neutrophil phenotypes can be plastic. For example, neutrophils under specific inflammatory settings have been reported to differentiate into other myeloid cells types83, including dendritic cells (DCs), and purified bone marrow-derived neutrophils can differentiate into a hybrid population characterized by the expression of DC and neutrophil markers after in vitro treatment with granulocyte/macrophage colony-stimulating factor, tumour necrosis factor and IL-4, and this has been verified both in vitro and in vivo184. It is likely that the environment (pro- or anti-inflammatory as well as the tissue or organ microenvironment) helps to dictate neutrophil differentiation and modulate the specialization and function of these cells.
Reverse migration of neutrophils
In a successful response to an acute injury, it is crucial to prevent tissue damage by promoting the local resolution of inflammation through the removal of neutrophils from the site of injury62. This clearance of neutrophils can occur through apoptosis or necrosis and subsequent phagocytosis by macrophages99. However, some early evidence suggested that neutrophils at inflamed sites do not necessarily undergo apoptosis. Hughes et al. used a rat model of glomerular capillary injury to track the fate of radiolabelled neutrophils and found that ≥70% of neutrophils that entered inflamed glomerular capillaries were able to return to the main circulation and did not undergo apoptosis at the site of inflammation100. More recent studies have demonstrated that neutrophils can leave sites of tissue damage in a process termed neutrophil reverse migration which describes the interstitial migration of neutrophils away from inflamed sites. Neutrophils have also been reported to re-enter the vasculature in a distinct process referred to as neutrophil reverse transendothelial migration (rTEM).
Reverse neutrophil migration within tissues away from the site of injury was first directly visualized in vivo in zebrafish larvae, in which it was demonstrated that not all recruited neutrophils die at the site of injury and most leave the site101. In subsequent studies using zebrafish, it was shown that neutrophils that leave a wound can reverse transmigrate into the vasculature (rTEM) and traffic to distal sites post-injury102,103. Moreover, Buckley and colleagues described the ability of human neutrophils to reverse transmigrate through an endothelial monolayer in vitro, identifying markers characteristic of these reverse-transmigrated neutrophils (ICAM1hiCXCR1low) and found this neutrophil phenotype in the peripheral blood of patients with systemic inflammation104. Since these early studies, neutrophil reverse migration and/or rTEM has been visualized in multiple models including zebrafish105–108, mice109,110 and in vitro human neutrophils94,111 (Figure 2). In vitro studies using microfluidics demonstrated that more than 90% of human neutrophils can reverse their direction away from a chemoattractant and migrate away continually for distances greater than 1,000 μm111. Together, these studies have suggested that reverse migration is a possible mechanism to locally resolve inflammation and a potential novel target for drug therapy in diseases characterized by excessive neutrophil infiltration. However, a caveat is that neutrophil reverse migration may lead to activated neutrophils being redistributed to other locations in the body, contributing to inflammation elsewhere. It is also important to note that many aspects of neutrophil reverse migration remain controversial including: the exact mechanism(s), the fate of reverse-migrated neutrophils and the occurrence of reverse migration in human disease.
Figure 2. Mechanisms that may be involved in neutrophil reverse migration.

Several models for reverse migration have been developed: in vitro microfluidic assays or transwell assays (a), larval zebrafish wounding (b), and mouse ischemia-reperfusion injury to model reverse transendothelial migration (rTEM) (c). These studies have reported mechanisms that regulate both neutrophil reverse migration in interstitial tissues and rTEM. a. Chemoattractants, such as CXC-chemokine ligand 8 (CXCL8), can act as chemorepellents at high concentrations in vitro, referred to as fugetaxis (i). Reverse neutrophil transmigration through endothelial cell monolayers has been reported; reverse transmigrated neutrophils have been characterised by high expression of intercellular adhesion molecule 1 (ICAM1) and low expression of CXC-chemokine receptor 1 (CXCR1) (ii). In vitro analysis in microfluidics has identified factors that regulate neutrophil forward and reverse migration (iii). The pro-resolving lipid mediator lipoxin A4 (LX4A) induces neutrophil reverse migration whereas zymosan induces neutrophil trapping. b. The reverse migration and rTEM of neutrophils has been visualized in larval zebrafish tail wounds using photoconversion (i). The activation of hypoxia-inducible factor 1α (HIF1α) in zebrafish neutrophils inhibits neutrophil reverse migration (ii), whereas the migration of macrophages to the wound via reactive oxygen species (ROS)-SRC family kinase (SFK) signalling and their direct interaction with neutrophils promotes neutrophil reverse migration (iii). c. A mouse ischemia-reperfusion injury model is used to model neutrophil rTEM or “hesitant” TEM. In this model, junctional adhesion molecule C (JAM-C) on endothelial cells modulates “complete” TEM and leukotriene B4 (LTB4) induces neutrophil elastase expression, which cleaves JAM-C, leading to an increase in rTEM (i, adapted from Colom et al, Immunity, 2015). Increased rTEM in this model leads to higher numbers of neutrophils with rTEM markers (ICAM1hi CXCR1low) at secondary sites such as the lungs (ii).
Potential mechanisms of reverse migration
The cues that mediate neutrophil reverse migration from sites of tissue damage remain largely unknown. Reverse migration can be confused with the terms fugetaxis or retrotaxis112, mechanisms that may have only a partial role in neutrophil reverse migration in vivo. Several mechanisms have been proposed to explain the reverse migration of neutrophils from inflamed tissues, including a competition between chemoattractant sources at the wound and vasculature. A neutrophil may prioritize competing cues through the downregulation of specific chemokine receptors and/or receptor desensitization. It has also been postulated that new chemorepellent cues may be released at the wound to mediate neutrophil reverse movement. Finally, other factors may influence the potential to leave damaged tissues, including decreased levels of wound chemoattractants and/or neutrophil-intrinsic transcriptional changes113. Neutrophils constantly sense a variety of stimuli and must process and prioritize all of these inputs to determine their behaviour and it is likely that a process as complex as reverse migration occurs through a combination of these mechanisms.
Relatively few studies have shown a direct link between neutrophil reverse migration and specific pathways (Figure 2). One pathway that has been specifically implicated in neutrophil migration out of wounded tissue in larval zebrafish is the hypoxia-inducible factor 1α (HIF1α) pathway105. In addition to reducing neutrophil apoptosis, HIF1α activation in zebrafish neutrophils increases the retention of neutrophils at the site of tissue injury105. It is known that in vitro chemoattractants such as CXCL8 can act as chemorepellents in higher concentrations114 and therefore neutrophil behaviour might change as the cell migrates up a chemokine gradient. Data indicating that reverse-migrated human neutrophils have lowered cell-surface expression of CXCR1104 provides some evidence for the idea that neutrophils might stop responding to chemoattractants at the wound because of receptor internalization or downregulation. Other tissue-resident cells are also implicated in the control of neutrophil reverse migration. In zebrafish, direct contact of neutrophils with macrophages at a wound induces neutrophils to migrate away, and autocrine redox-SFK signalling in macrophages is required for the resolution of neutrophil inflammation108. This is an especially intriguing observation in the context of patients with chronic granulomatous disease (CGD) and deficient ROS signalling who develop chronic neutrophil inflammation in tissues115.
The relative levels of different lipid mediators can also promote the resolution of inflammation and may be involved in neutrophil reverse migration. As discussed, LTB4 is a neutrophil chemoattractant, but after lipid-mediator class switching pro-inflammatory lipid mediator production pathways are altered to instead produce pro-resolution mediators such as lipoxin A4 (LXA4)116,117. Other pro-resolution lipid mediators include resolvins and protectins and stop the influx of new neutrophils118. They are also implicated in promoting the resolution of existing neutrophil inflammation: LXA4 can enhance the reverse migration of human neutrophils in vitro in a microfluidic device111.
Besides reverse migrating through tissue away from an injury, neutrophils may also re-enter the circulation through rTEM. Neutrophils that were recruited to a tissue wound can be observed back in the circulation in zebrafish102,103. In mice, in which there are greater challenges to imaging than in zebrafish, the Nourshargh laboratory has used “disturbed” neutrophil TEM after ischemia-reperfusion injury as a model for rTEM110,119. In this model, junctional adhesion molecule C (JAM-C) at endothelial cell junctions modulates “complete” TEM and a decrease in the levels of JAM-C expression after injury increases the occurrence of rTEM110. Moreover, LTB4 stimulates the production of neutrophil elastase, which cleaves JAM-C, increasing rates of rTEM119, suggesting that LTB4 may be involved in both forward and reverse transmigration to damaged tissue.
What determines whether neutrophils reverse migrate or die at the site of damage? Different neutrophil subsets might be predisposed to reverse migrate from the time they leave the circulation or due to transcriptional changes induced at the injury site98,120 (see Box 3). In vitro, neutrophils that encounter the fungal cell wall component zymosan have decreased reverse migration94,111, and it is tempting to speculate that neutrophils are less likely to reverse migrate away from sites of infection, at least in part to prevent the dissemination of intracellular pathogens.
It has been proposed that neutrophils do not actively undergo directed reverse migration, but they are simply randomly redistributed away from the wound. By simulating neutrophil behaviour with different mathematical models based on neutrophil migration measurements made in zebrafish, several studies suggested that the migration of neutrophils away from a wound could be best described by a diffusion process rather than a fugetaxis process107,121,122. Therefore reverse migration of neutrophils might be a result of a loss of sensitivity towards recruitment and/or retention signals rather than active repulsion induced by the release of chemorepellent from the wound or active attraction due to chemoattractants in the blood or vascular endothelial cells. Overall, the mechanisms controlling reverse migration are still not clear. It is likely that multiple processes contribute to the migration of neutrophils away from a site of inflammation and back into the circulation, possibly combined in a reverse migration cascade.
What is the phenotype and fate of reverse-migrated neutrophils?
Do reverse-migrated neutrophils have a different phenotype compared to “naïve” neutrophils? Neutrophils in peripheral tissues are known to be more active transcriptionally and translationally than their counterparts in the circulation123. Neutrophils that have undergone rTEM in vitro were found to express specific markers: ICAM1hi and CXCR1low104,110, and these markers have been used to identify neutrophils that had presumably undergone rTEM in peripheral human blood104 and in secondary sites of inflammation in mice110. In addition, human neutrophils that have undergone rTEM in vitro are less susceptible to apoptosis and produce more ROS104, and mouse neutrophils expressing rTEM markers in vivo also produce higher levels of ROS110. By contrast, although zebrafish reverse-migrated neutrophils (not necessarily neutrophils that underwent rTEM) were found to have an activated morphology, they did not have altered ROS production or response to a secondary insult, whether a tissue wound or an infection106. There is no clear evidence, therefore, that all reverse-migrated neutrophils display a pro- or anti-inflammatory phenotype. These neutrophils may in fact have varying fates or phenotypes according to where and how they are recruited out of the blood into tissues in the first place and what they encounter at the injury site.
Where do neutrophils go after reverse migrating out of the tissue? Zebrafish neutrophils can be found in tissues or the circulation for at least two days after leaving a wound102. The fate of these neutrophils is important for patients, as systemic inflammation after severe trauma can lead to multiple organ failure124. Recently, human patients with acute pancreatitis that developed acute lung injury were found to have a higher level of neutrophils carrying rTEM markers in their circulation125. In a mouse model of acute pancreatitis, increasing the level of neutrophils carrying rTEM markers in the circulation and pulmonary vasculature through genetic deletion of JAM-C increased the severity of lung injury and systemic inflammation125. Similarly, after ischemia-reperfusion injury in mice, neutrophils with rTEM markers were found re-localized in the lungs110 and experimentally increasing the rates of rTEM resulted in increased neutrophil re-localization and organ damage in the lungs, liver and heart119.
Neutrophils have also been shown to leave inflamed or infected sites, travel through the lymphatics and re-localize to the lymph nodes126–128 or bone marrow109 to affect host defense. These neutrophils can shuttle live pathogens to lymph nodes127 and modulate lymphocyte proliferation109,126. Neutrophil migration from inflamed skin to lymph nodes depends on CD11b and CXCR4, and CD11b deficiency in neutrophils decreases T cell proliferation in draining lymph nodes126. The transport of antigen from intradermally injected modified vaccinia Ankara virus to the bone marrow by neutrophils can also initiate the expansion of viral-targeting memory CD8+ T cells109. Much more work needs to be done to further clarify the phenotype and fate of reverse migrated neutrophils and their implications for human disease.
Neutrophil migration: a drug target
The failure to appropriately resolve inflammation can have disastrous effects. Excessive and persistent infiltration of neutrophils into tissues has a role in multiple inflammatory diseases, including rheumatoid arthritis129, pulmonary fibrosis130 and multiple organ failure124, making neutrophil behaviour a potential target for drug therapies. One concern with anti-inflammatory therapies is achieving the resolution of inflammation at a specific site without causing systemic immunosuppression. Another major goal is the identification of molecular targets that distinguish infection from wound-induced inflammation so as to not impair the body’s ability to fight invading pathogens. Drugs that induce neutrophil apoptosis or necrosis at an injury site would reduce neutrophil numbers but the clearance of dead cells might increase overall local inflammation. Therefore, both neutrophil forward and reverse migration are attractive targets for anti-inflammatory therapies. We discuss exciting new targets in these pathways below and highlight drugs that are in the clinical trial pipeline in Table 1.
Table 1.
Drugs in current clinical trials that specifically target neutrophil migration signals
| Drug name | Target | Disease or model | Stage | Reference |
|---|---|---|---|---|
| SCH 527123 | CXCR2 | Severe asthma, allergen-induced asthma, COPD, psoriasis and colon cancer | Phase I–II | 185 |
| Reparixin | CXCR1 and CXCR2 | Ischemia-reperfusion injury; lung, pancreatic islet and kidney transplantation, and breast cancer | Phase II–III | 186 |
| DF 2156A | CXCR1 and CXCR2 | Active bullous pemphigoid | Phase II | - |
| AZD-8309 | CXCR1 and CXCR2 | LPS-induced airway inflammation | Phase I | 187 |
| SB-656933 | CXCR2 | Ulcerative colitis, cystic fibrosis and ozone airway inflammation | Phase I–II | 132 |
| GSK1325756 | CXCR2 | Influenza virus infection and healthy volunteers | Phase II | 188 |
| AZD5069 | CXCR2 | Healthy volunteers and LPS-induced airway inflammation | Phase I |
189 190 |
| QBM076 | Not known | COPD | Phase II | - |
COPD, chronic obstructive pulmonary disease; CXCR, CXC-chemokine receptor; LPS, lipopolysaccharide. More details at clinicaltrials.gov.
Targeting forward migration
To inhibit neutrophil migration to a site of damage, neutrophil motility machinery, primary signals that recruit neutrophils or the amplification stage of inflammation could be targeted. Colchicine, a microtubule inhibitor, is a known inhibitor of neutrophil chemotaxis and impairs neutrophil recruitment to sites of inflammation in vivo. It is used to treat human conditions including gout, chronic pericarditis and familial Mediterranean fever131. To target neutrophil recruitment signals, the early phase of signalling may be more promising as the amplification phase involves the activation of multiple complex pathways. Conversely, inhibiting specific pathways in the amplification phase could fine-tune the neutrophil response without completely eliminating it. Several clinical trials are currently assessing the effect of CXCR1 and CXCR2 antagonists and inhibitors (Table 1) on inflammatory conditions ranging from cancer to autoimmune diseases (clinicaltrials.gov), and some have reported promising results132,133. However, chemokine receptor antagonist therapy has failed in the past due to differences between animal models and human patients and redundancy of the targets134. Other clinical trials are targeting lipid mediator-induced inflammation with LTB4 receptor antagonists. Resolvins are an exciting class of molecules as they can inhibit neutrophil transmigration in vitro135, and two different resolvin molecules, including chemically synthesized molecules, can significantly decrease the number of neutrophils at a site of damage at least partially through decreased levels of neutrophil chemokines including CXCL1 and LTB4135,136.
Targeting reverse migration
An alternative approach to decrease chronic inflammation is to promote neutrophil reverse migration from local sites of damage. Resolvins and protectins may also resolve inflammation by promoting the removal of leukocytes from injured tissue sites137. A drug screen in zebrafish yielded a novel drug known as tanshinone IIA, which is derived from a Chinese medicinal herb and seems to accelerate apoptosis of neutrophils and promote their reverse migration107. However, targeting this mechanism raises several concerns, including the fate and activation state of reverse migrated neutrophils. Disseminated neutrophils exhibiting a strong pro-inflammatory or anti-inflammatory phenotype could have deleterious effects such as promoting distal organ damage or systemic immunosuppression, respectively. A better characterization of reverse migrated neutrophils in vivo is necessary before further advances in the development of therapies that target this mechanism.
Future perspectives
Remarkable progress has been made in the past decade in understanding the mechanisms that regulate neutrophil migration, including the cells and signals that attract neutrophils to sites of either tissue injury or infection. However, neutrophil reverse migration remains an understudied phenomenon and future work should focus on several key questions to drive the development of new therapies for neutrophil inflammatory disorders. First, the question of whether neutrophil reverse migration is “good” or “bad” for inflammation outcome is still largely unanswered. Although excessive and prolonged neutrophil infiltration can lead to the development of chronic tissue inflammation, the migration of neutrophils back into the circulation might cause systemic inflammation and tissue damage at distal organs. It is likely that the answer to this question depends on the nature, magnitude and location of the inflammation. In the case of infection, premature neutrophil reverse migration could lead to failure to clear the infection and dissemination of intracellular pathogens to other sites. In the case of tissue injury, reverse migration might have positive effects at the local site of inflammation, as the depletion of neutrophils can improve wound healing138,139, but may have negative effects systemically such as multiple organ failure. Another issue is the diversity of neutrophil phenotypes (Box 3). In cancer models, it has been proposed that, similarly to macrophages, neutrophil phenotypes can exist on a spectrum ranging from pro-inflammatory, anti-tumour “N1” neutrophils to anti-inflammatory, pro-tumour “N2” neutrophils. Does a similar plasticity exist at sites of tissue damage or infection? Does the phenotype of the neutrophil have any role in its ability to stay at the injury site or reverse migrate away? In cases of chronic inflammation, is it possible to specifically promote the reverse migration of N1 neutrophils while leaving pro-resolving N2 neutrophils at the wound? Or is it the reverse migration specifically of these N1 neutrophils that can cause systemic inflammation and distal organ damage? Does the outcome of an injury re-programme reverse migrating neutrophils to relay specific messages to different locations in the body? Although many unanswered questions remain, it is clear that neutrophil migration has a crucial role in inflammatory pathology. Fortunately, recent models and technologies have been developed that will enable the further elucidation of forward migration signals and the role of reverse migration in the pathology of inflammatory diseases.
Key Points.
Complementary models have been developed to study neutrophil migration, including microfluidics and live imaging using mice and zebrafish.
Neutrophil migration in response to injury or infection occurs in phases: early recruitment, amplification and resolution.
Early-recruited neutrophils modulate the amplification phase both directly and indirectly, through the activation of tissue and tissue-resident cells, producing sustained signals such as the CXC-chemokine ligand 8 family chemokines.
Activated neutrophils at a site of inflammation do not necessarily undergo apoptosis but in some circumstances might undergo reverse migration away from the site of damage (reverse neutrophil migration) and/or re-enter the circulation (reverse transendothelial migration (rTEM)).
Neutrophil forward and reverse migration may be attractive targets for anti-inflammatory therapies.
Acknowledgments
This work was supported by National Institutes of Health Grant GM074827 to A.H. E.E.R. is supported by an individual fellowship from NIAAD of the National Institutes of Health under award number F32AI113956 and SDO is supported by a EMBO fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
- Reactive oxygen species (ROS)
Chemically reactive molecules containing oxygen that, when produced in large amounts, have pro-inflammatory and antimicrobial effects. Physiological levels of ROS have been shown to regulate cellular signalling pathways.
- Neutrophil extracellular traps (NETs)
Fibrous networks that are released into the extracellular environment by neutrophils. They are composed mainly of DNA, but also contain chromatin and proteins from neutrophil granules. NETs act as a mesh that traps microorganisms and exposes them to neutrophil-derived effector molecules.
- DAMPs
Cues that are derived from stressed, damaged or dead cells. These factors are highly diffusible through tissues and can be either protein-derived or non-protein-derived.
- PAMPs
Molecules that are derived from pathogens such as bacteria, fungi or viruses; they are highly diffusible through tissues and can be either protein-derived or non-protein-derived.
- Reverse neutrophil migration
The movement of neutrophils away from injured tissues within the interstitium.
- Reverse transendothelial migration
The movement of neutrophils back into the vasculature after being in tissues.
- Fugetaxis
Also known as retrotaxis. The movement of cells away from a source of chemoattractant in vitro.
Biographies
Sofia de Oliveira obtained her Ph.D. from Lisbon Medical School, University of Lisbon, Portugal, focusing on neutrophil recruitment to tissues using zebrafish. She is currently a postdoctoral fellow in Anna Huttenlocher’s laboratory at the University of Wisconsin, Madison, Wisconsin, USA, with interests in inflammation and cancer.
Emily Rosowski obtained her Ph.D. in biology from Massachusetts Institute of Technology, Cambridge, Massachusetts, USA, and is currently a postdoctoral fellow in Anna Huttenlocher’s laboratory at the University of Wisconsin. She is interested in how innate immune cells orchestrate responses to pathogens and how pathogens can subvert these responses.
Anna Huttenlocher is the Vilas Distinguished Research Professor in the Departments of Medical Microbiology and Pediatrics at the University of Wisconsin. She is a physician scientist with interests in cell migration and innate immunity in the context of wound healing, infection and cancer.
References
- 1.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature reviews. Immunology. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C. Neutrophils and immunity: challenges and opportunities. Nature reviews. Immunology. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 3.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 4.Caielli S, Banchereau J, Pascual V. Neutrophils come of age in chronic inflammation. Curr Opin Immunol. 2012;24:671–677. doi: 10.1016/j.coi.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley K. Integration of inflammatory signals by rolling neutrophils. Immunol Rev. 2002;186:8–18. doi: 10.1034/j.1600-065x.2002.18602.x. [DOI] [PubMed] [Google Scholar]
- 6.Gambardella L, Vermeren S. Molecular players in neutrophil chemotaxis--focus on PI3K and small GTPases. J Leukocyte Biol. 2013;94:603–612. doi: 10.1189/jlb.1112564. [DOI] [PubMed] [Google Scholar]
- 7.Ng LG, et al. Visualizing the neutrophil response to sterile tissue injury in mouse dermis reveals a three-phase cascade of events. The Journal of investigative dermatology. 2011;131:2058–2068. doi: 10.1038/jid.2011.179. [DOI] [PubMed] [Google Scholar]
- 8.Lämmermann T. In the eye of the neutrophil swarm-navigation signals that bring neutrophils together in inflamed and infected tissues. J Leukocyte Biol. 2015 doi: 10.1189/jlb.1MR0915-403. [DOI] [PubMed] [Google Scholar]
- 9.Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41:694–707. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nature Reviews Immunology. 2014;14:232–246. doi: 10.1038/nri3641. [DOI] [PubMed] [Google Scholar]
- 11.Futosi K, Fodor S, Mócsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. 2013;17:638–650. doi: 10.1016/j.intimp.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, Ye RD. Role of G protein-coupled receptors in inflammation. Acta Pharmacologica Sinica. 2012;33:342–350. doi: 10.1038/aps.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- 14.Pittman K, Kubes P. Damage-associated molecular patterns control neutrophil recruitment. Journal of innate immunity. 2013;5:315–323. doi: 10.1159/000347132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broggi A, Granucci F. Microbe- and danger-induced inflammation. Mol Immunol. 2015;63:127–133. doi: 10.1016/j.molimm.2014.06.037. [DOI] [PubMed] [Google Scholar]
- 16.Vénéreau E, Ceriotti C, Bianchi ME. DAMPs from Cell Death to New Life. Frontiers in immunology. 2015;6:422. doi: 10.3389/fimmu.2015.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cordeiro JVV, Jacinto A. The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nature reviews. Molecular cell biology. 2013;14:249–262. doi: 10.1038/nrm3541. [DOI] [PubMed] [Google Scholar]
- 18.Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreira S, Stramer B, Evans I, Wood W, Martin P. Prioritization of Competing Damage and Developmental Signals by Migrating Macrophages in the Drosophila Embryo. Curr Biol. 2010;20:464–470. doi: 10.1016/j.cub.2010.01.047. [DOI] [PubMed] [Google Scholar]
- 20.Yoo SK, Starnes TW, Deng Q, Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature. 2011;480:109–112. doi: 10.1038/nature10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klyubin IV, Kirpichnikova KM, Gamaley IA. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur J Cell Biol. 1996;70:347–351. [PubMed] [Google Scholar]
- 22.Kovács M, et al. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. The Journal of experimental medicine. 2014;211:1993–2011. doi: 10.1084/jem.20132496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baroja-Mazo A, Barberà-Cremades M, Pelegrín P. The participation of plasma membrane hemichannels to purinergic signaling. Biochimica et biophysica acta. 2013;1828:79–93. doi: 10.1016/j.bbamem.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 24.de Oliveira S, et al. ATP modulates acute inflammation in vivo through dual oxidase 1-derived H2O2 production and NF-κB activation. Journal of immunology (Baltimore, Md : 1950) 2014;192:5710–5719. doi: 10.4049/jimmunol.1302902. [DOI] [PubMed] [Google Scholar]
- 25.Elliott MR, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science (New York, NY ) 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 27.Bao Y, et al. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. The Journal of cell biology. 2015;210:1153–1164. doi: 10.1083/jcb.201503066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kukulski F, et al. Extracellular ATP and P2 receptors are required for IL-8 to induce neutrophil migration. Cytokine. 2009;46:166–170. doi: 10.1016/j.cyto.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lecut C, et al. P2X1 ion channels promote neutrophil chemotaxis through Rho kinase activation. Journal of immunology (Baltimore, Md : 1950) 2009;183:2801–2809. doi: 10.4049/jimmunol.0804007. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raoof M, Zhang Q, Itagaki K, Hauser CJ. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. The Journal of trauma. 2010;68:1328. doi: 10.1097/TA.0b013e3181dcd28d. [DOI] [PubMed] [Google Scholar]
- 32.Li L, et al. New development in studies of formyl-peptide receptors: critical roles in host defense. J Leukocyte Biol. 2015 doi: 10.1189/jlb.2RI0815-354RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (New York, NY ) 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 34.Pase L, et al. Neutrophil-delivered myeloperoxidase dampens the hydrogen peroxide burst after tissue wounding in zebrafish. Current biology : CB. 2012;22:1818–1824. doi: 10.1016/j.cub.2012.07.060. [DOI] [PubMed] [Google Scholar]
- 35.Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert review of clinical immunology. 2014;10:593–619. doi: 10.1586/1744666X.2014.894886. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi Y. The role of chemokines in neutrophil biology. Frontiers in bioscience : a journal and virtual library. 2008;13:2400–2407. doi: 10.2741/2853. [DOI] [PubMed] [Google Scholar]
- 37.Sai J, Raman D, Liu Y, Wikswo J, Richmond A. Parallel phosphatidylinositol 3-kinase (PI3K)-dependent and Src-dependent pathways lead to CXCL8-mediated Rac2 activation and chemotaxis. The Journal of biological chemistry. 2008;283:26538–26547. doi: 10.1074/jbc.M805611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neel NF, et al. VASP is a CXCR2-interacting protein that regulates CXCR2-mediated polarization and chemotaxis. J Cell Sci. 2009;122:1882–1894. doi: 10.1242/jcs.039057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindley I, et al. Synthesis and expression in Escherichia coli of the gene encoding monocyte-derived neutrophil-activating factor: biological equivalence between natural and recombinant neutrophil-activating factor. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:9199–9203. doi: 10.1073/pnas.85.23.9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Oliveira S, et al. Cxcl8 (IL-8) mediates neutrophil recruitment and behavior in the zebrafish inflammatory response. Journal of immunology (Baltimore, Md : 1950) 2013;190:4349–4359. doi: 10.4049/jimmunol.1203266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng Q, et al. Localized bacterial infection induces systemic activation of neutrophils through Cxcr2 signaling in zebrafish. J Leukocyte Biol. 2013;93:761–769. doi: 10.1189/jlb.1012534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarris M, et al. Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Current biology : CB. 2012;22:2375–2382. doi: 10.1016/j.cub.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 43.Cacalano G, et al. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science (New York, NY ) 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- 44.Devalaraja RM, et al. Delayed wound healing in CXCR2 knockout mice. The Journal of investigative dermatology. 2000;115:234–244. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monneau Y, Arenzana-Seisdedos F, Lortat-Jacob H. The sweet spot: how GAGs help chemokines guide migrating cells. J Leukocyte Biol. 2015 doi: 10.1189/jlb.3MR0915-440R. [DOI] [PubMed] [Google Scholar]
- 46.Middleton J, et al. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–395. doi: 10.1016/s0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 47.Powell WS, Gravel S, MacLeod RJ, Mills E, Hashefi M. Stimulation of human neutrophils by 5-oxo-6,8,11,14-eicosatetraenoic acid by a mechanism independent of the leukotriene B4 receptor. The Journal of biological chemistry. 1993;268:9280–9286. [PubMed] [Google Scholar]
- 48.Øynebråten I, et al. Characterization of a novel chemokine-containing storage granule in endothelial cells: evidence for preferential exocytosis mediated by protein kinase A and diacylglycerol. Journal of immunology (Baltimore, Md : 1950) 2005;175:5358–5369. doi: 10.4049/jimmunol.175.8.5358. [DOI] [PubMed] [Google Scholar]
- 49.Hol J, Wilhelmsen L, Haraldsen G. The murine IL-8 homologues KC, MIP-2, and LIX are found in endothelial cytoplasmic granules but not in Weibel-Palade bodies. J Leukocyte Biol. 2010;87:501–508. doi: 10.1189/jlb.0809532. [DOI] [PubMed] [Google Scholar]
- 50.Iyer SS, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Enyedi B, Kala S, Nikolich-Zugich T, Niethammer P. Tissue damage detection by osmotic surveillance. Nat Cell Biol. 2013;15:1123–1130. doi: 10.1038/ncb2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sadik CD, Luster AD. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukocyte Biol. 2012;91:207–215. doi: 10.1189/jlb.0811402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Afonso PV, et al. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev Cell. 2012;22:1079–1091. doi: 10.1016/j.devcel.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lämmermann T, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371–375. doi: 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. Journal of immunology (Baltimore, Md : 1950) 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 56.Chen M, et al. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. The Journal of experimental medicine. 2006;203:837–842. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM. N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury. 2015;46:975–984. doi: 10.1016/j.injury.2015.03.028. [DOI] [PubMed] [Google Scholar]
- 58.Chou RC, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33:266–278. doi: 10.1016/j.immuni.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681. [PubMed] [Google Scholar]
- 60.Tester AM, et al. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PloS one. 2007;2 doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Afonso PV, McCann CP, Kapnick SM, Parent CA. Discoidin domain receptor 2 regulates neutrophil chemotaxis in 3D collagen matrices. Blood. 2013;121:1644–1650. doi: 10.1182/blood-2012-08-451575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nature reviews. Immunology. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 63.Peters NC, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science (New York, NY ) 2008;321:970–974. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kreisel D, et al. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chtanova T, et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity. 2008;29:487–496. doi: 10.1016/j.immuni.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silva MT. Bacteria-induced phagocyte secondary necrosis as a pathogenicity mechanism. J Leukocyte Biol. 2010;88:885–896. doi: 10.1189/jlb.0410205. [DOI] [PubMed] [Google Scholar]
- 67.Gonzalez CD, Ledo C, Giai C, Garófalo A, Gómez MI. The Sbi Protein Contributes to Staphylococcus aureus Inflammatory Response during Systemic Infection. PloS one. 2015;10 doi: 10.1371/journal.pone.0131879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abtin A, et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol. 2014;15:45–53. doi: 10.1038/ni.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spinner JL, Hasenkrug AM, Shannon JG, Kobayashi SD, Hinnebusch BJ. Role of the Yersinia YopJ protein in suppressing interleukin-8 secretion by human polymorphonuclear leukocytes. Microbes Infect. 2016;18:21–29. doi: 10.1016/j.micinf.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- 71.Schiwon M, et al. Crosstalk between sentinel and helper macrophages permits neutrophil migration into infected uroepithelium. Cell. 2014;156:456–468. doi: 10.1016/j.cell.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sacramento L, et al. TLR9 signaling on dendritic cells regulates neutrophil recruitment to inflammatory foci following Leishmania infantum infection. Infect Immun. 2015 doi: 10.1128/IAI.00975-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abraham SN, John AL. Mast cell-orchestrated immunity to pathogens. Nature Reviews Immunology. 2010 doi: 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krishna S, Miller LS. Innate and adaptive immune responses against Staphylococcus aureus skin infections. Seminars in immunopathology. 2012;34:261–280. doi: 10.1007/s00281-011-0292-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- 76.Huang C, et al. Induction of a selective and persistent extravasation of neutrophils into the peritoneal cavity by tryptase mouse mast cell protease 6. Journal of immunology (Baltimore, Md : 1950) 1998;160:1910–1919. [PubMed] [Google Scholar]
- 77.Malaviya R, Abraham SN. Role of mast cell leukotrienes in neutrophil recruitment and bacterial clearance in infectious peritonitis. J Leukocyte Biol. 2000;67:841–846. doi: 10.1002/jlb.67.6.841. [DOI] [PubMed] [Google Scholar]
- 78.Miller LS, et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. Journal of immunology (Baltimore, Md : 1950) 2007;179:6933–6942. doi: 10.4049/jimmunol.179.10.6933. [DOI] [PubMed] [Google Scholar]
- 79.Shimada T, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell host & microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miller LS, et al. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity. 2006;24:79–91. doi: 10.1016/j.immuni.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 81.Sun K, Salmon SL, Lotz SA, Metzger DW. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect Immun. 2007;75:1196–1202. doi: 10.1128/IAI.01403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kabir S. The role of interleukin-17 in the Helicobacter pylori induced infection and immunity. Helicobacter. 2011;16:1–8. doi: 10.1111/j.1523-5378.2010.00812.x. [DOI] [PubMed] [Google Scholar]
- 83.Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood. 2014;124:710–719. doi: 10.1182/blood-2014-03-453217. [DOI] [PubMed] [Google Scholar]
- 84.Isailovic N, Daigo K, Mantovani A, Selmi C. Interleukin-17 and innate immunity in infections and chronic inflammation. J Autoimmun. 2015;60:1–11. doi: 10.1016/j.jaut.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 85.Rendon JL, Choudhry MA. Th17 cells: critical mediators of host responses to burn injury and sepsis. J Leukocyte Biol. 2012;92:529–538. doi: 10.1189/jlb.0212083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mölne L, Verdrengh M, Tarkowski A. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect Immun. 2000;68:6162–6167. doi: 10.1128/iai.68.11.6162-6167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ley K, Smith E, Stark MA. IL-17A-producing neutrophil-regulatory Tn lymphocytes. Immunol Res. 2006;34:229–242. doi: 10.1385/IR:34:3:229. [DOI] [PubMed] [Google Scholar]
- 88.Xu S, Cao X. Interleukin-17 and its expanding biological functions. Cellular & Molecular Immunology. 2010;7:164–174. doi: 10.1038/cmi.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cho JS, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. The Journal of clinical investigation. 2010;120:1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liese J, Rooijakkers SH, van Strijp JA, Novick RP, Dustin ML. Intravital two-photon microscopy of host-pathogen interactions in a mouse model of Staphylococcus aureus skin abscess formation. Cell Microbiol. 2013;15:891–909. doi: 10.1111/cmi.12085. [DOI] [PubMed] [Google Scholar]
- 91.Tan RS, Ho B, Leung BP, Ding JL. TLR cross-talk confers specificity to innate immunity. International reviews of immunology. 2014;33:443–453. doi: 10.3109/08830185.2014.921164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deng Q, Harvie EA, Huttenlocher A. Distinct signalling mechanisms mediate neutrophil attraction to bacterial infection and tissue injury. Cell Microbiol. 2012;14:517–528. doi: 10.1111/j.1462-5822.2011.01738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yan B, et al. IL-1β and reactive oxygen species differentially regulate neutrophil directional migration and Basal random motility in a zebrafish injury-induced inflammation model. Journal of immunology (Baltimore, Md : 1950) 2014;192:5998–6008. doi: 10.4049/jimmunol.1301645. [DOI] [PubMed] [Google Scholar]
- 94.Hamza B, Irimia D. Whole blood human neutrophil trafficking in a microfluidic model of infection and inflammation. Lab on a chip. 2015;15:2625–2633. doi: 10.1039/c5lc00245a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Y, Chen GYY, Zheng P. CD24-Siglec G/10 discriminates danger- from pathogen-associated molecular patterns. Trends Immunol. 2009;30:557–561. doi: 10.1016/j.it.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen GYY, Brown NK, Zheng P, Liu Y. Siglec-G/10 in self-nonself discrimination of innate and adaptive immunity. Glycobiology. 2014;24:800–806. doi: 10.1093/glycob/cwu068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen GYY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science (New York, NY ) 2009;323:1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kruger P, et al. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Path. 2015;11 doi: 10.1371/journal.ppat.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nature reviews. Immunology. 2013;13:59–66. doi: 10.1038/nri3362. [DOI] [PubMed] [Google Scholar]
- 100.Hughes J, et al. Neutrophil fate in experimental glomerular capillary injury in the rat. Emigration exceeds in situ clearance by apoptosis. The American journal of pathology. 1997;150:223–234. [PMC free article] [PubMed] [Google Scholar]
- 101.Mathias JR, et al. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukocyte Biol. 2006;80:1281–1288. doi: 10.1189/jlb.0506346. [DOI] [PubMed] [Google Scholar]
- 102.Yoo SK, Huttenlocher A. Spatiotemporal photolabeling of neutrophil trafficking during inflammation in live zebrafish. J Leukocyte Biol. 2011;89:661–667. doi: 10.1189/jlb.1010567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hall C, et al. Transgenic zebrafish reporter lines reveal conserved Toll-like receptor signaling potential in embryonic myeloid leukocytes and adult immune cell lineages. J Leukocyte Biol. 2009;85:751–765. doi: 10.1189/jlb.0708405. [DOI] [PubMed] [Google Scholar]
- 104.Buckley CD, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukocyte Biol. 2006;79:303–311. doi: 10.1189/jlb.0905496. [DOI] [PubMed] [Google Scholar]
- 105.Elks PM, et al. Activation of hypoxia-inducible factor-1α (Hif-1α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood. 2011;118:712–722. doi: 10.1182/blood-2010-12-324186. [DOI] [PubMed] [Google Scholar]
- 106.Ellett F, Elks PM, Robertson AL, Ogryzko NV, Renshaw SA. Defining the phenotype of neutrophils following reverse migration in zebrafish. J Leukocyte Biol. 2015 doi: 10.1189/jlb.3MA0315-105R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Robertson AL, et al. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Science translational medicine. 2014;6 doi: 10.1126/scitranslmed.3007672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tauzin S, Starnes TW, Becker FB, Lam PYY, Huttenlocher A. Redox and Src family kinase signaling control leukocyte wound attraction and neutrophil reverse migration. The Journal of cell biology. 2014;207:589–598. doi: 10.1083/jcb.201408090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Duffy D, et al. Neutrophils transport antigen from the dermis to the bone marrow, initiating a source of memory CD8+ T cells. Immunity. 2012;37:917–929. doi: 10.1016/j.immuni.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 110.Woodfin A, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12:761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hamza B, et al. Retrotaxis of human neutrophils during mechanical confinement inside microfluidic channels. Integrative biology : quantitative biosciences from nano to macro. 2014;6:175–183. doi: 10.1039/c3ib40175h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vianello F, Olszak IT, Poznansky MC. Fugetaxis: active movement of leukocytes away from a chemokinetic agent. Journal of molecular medicine (Berlin, Germany) 2005;83:752–763. doi: 10.1007/s00109-005-0675-z. [DOI] [PubMed] [Google Scholar]
- 113.Starnes TW, Huttenlocher A. Neutrophil reverse migration becomes transparent with zebrafish. Advances in hematology. 2012;2012:398640. doi: 10.1155/2012/398640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tharp WG, et al. Neutrophil chemorepulsion in defined interleukin-8 gradients in vitro and in vivo. J Leukocyte Biol. 2006;79:539–554. doi: 10.1189/jlb.0905516. [DOI] [PubMed] [Google Scholar]
- 115.Kuijpers T, Lutter R. Inflammation and repeated infections in CGD: two sides of a coin. Cellular and molecular life sciences : CMLS. 2012;69:7–15. doi: 10.1007/s00018-011-0834-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harbor perspectives in biology. 2015;7 doi: 10.1101/cshperspect.a016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40:315–327. doi: 10.1016/j.immuni.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Serhan CN, Chiang N. Resolution phase lipid mediators of inflammation: agonists of resolution. Curr Opin Pharm. 2013;13:632–640. doi: 10.1016/j.coph.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Colom B, et al. Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity. 2015;42:1075–1086. doi: 10.1016/j.immuni.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Beyrau M, Bodkin JV, Nourshargh S. Neutrophil heterogeneity in health and disease: a revitalized avenue in inflammation and immunity. Open biology. 2012;2:120134. doi: 10.1098/rsob.120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Holmes GR, et al. Drift-Diffusion Analysis of Neutrophil Migration during Inflammation Resolution in a Zebrafish Model. Advances in hematology. 2012;2012:792163. doi: 10.1155/2012/792163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Holmes GR, et al. Repelled from the wound, or randomly dispersed? Reverse migration behaviour of neutrophils characterized by dynamic modelling. Journal of the Royal Society, Interface / the Royal Society. 2012;9:3229–3239. doi: 10.1098/rsif.2012.0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. 2011;32:452–460. doi: 10.1016/j.it.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tsukamoto T, Chanthaphavong RS, Pape HCC. Current theories on the pathophysiology of multiple organ failure after trauma. Injury. 2010;41:21–26. doi: 10.1016/j.injury.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 125.Wu D, et al. Reverse-migrated neutrophils regulated by JAM-C are involved in acute pancreatitis-associated lung injury. Scientific reports. 2016;6:20545. doi: 10.1038/srep20545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nature communications. 2015;6:7139. doi: 10.1038/ncomms8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abadie V, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843–1850. doi: 10.1182/blood-2005-03-1281. [DOI] [PubMed] [Google Scholar]
- 128.Maletto BA, et al. Presence of neutrophil-bearing antigen in lymphoid organs of immune mice. Blood. 2006;108:3094–3102. doi: 10.1182/blood-2006-04-016659. [DOI] [PubMed] [Google Scholar]
- 129.Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nature reviews. Rheumatology. 2014;10:593–601. doi: 10.1038/nrrheum.2014.80. [DOI] [PubMed] [Google Scholar]
- 130.Cantin AMM, Hartl D, Konstan MW, Chmiel JF. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2015;14:419–430. doi: 10.1016/j.jcf.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 131.Cocco G, Chu DCC, Pandolfi S. Colchicine in clinical medicine. A guide for internists. European journal of internal medicine. 2010;21:503–508. doi: 10.1016/j.ejim.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 132.Lazaar AL, et al. SB-656933, a novel CXCR2 selective antagonist, inhibits ex vivo neutrophil activation and ozone-induced airway inflammation in humans. Br J Clin Pharmacol. 2011;72:282–293. doi: 10.1111/j.1365-2125.2011.03968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Moss RB, et al. Safety and early treatment effects of the CXCR2 antagonist SB-656933 in patients with cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2013;12:241–248. doi: 10.1016/j.jcf.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 134.Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nature reviews. Drug discovery. 2009;8:23–33. doi: 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- 135.Dalli J, et al. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem Biol. 2013;20:188–201. doi: 10.1016/j.chembiol.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Eickmeier O, et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal immunology. 2013;6:256–266. doi: 10.1038/mi.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dovi JV, He LKK, DiPietro LA. Accelerated wound closure in neutrophil-depleted mice. J Leukocyte Biol. 2003;73:448–455. doi: 10.1189/jlb.0802406. [DOI] [PubMed] [Google Scholar]
- 139.Li L, Yan B, Shi YQQ, Zhang WQQ, Wen ZLL. Live imaging reveals differing roles of macrophages and neutrophils during zebrafish tail fin regeneration. The Journal of biological chemistry. 2012;287:25353–25360. doi: 10.1074/jbc.M112.349126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Paoliello-Paschoalato AB, et al. Isolation of healthy individuals’ and rheumatoid arthritis patients’ peripheral blood neutrophils by the gelatin and Ficoll-Hypaque methods: comparative efficiency and impact on the neutrophil oxidative metabolism and Fcγ receptor expression. J Immunol Methods. 2014;412:70–77. doi: 10.1016/j.jim.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 141.Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:2458–2462. doi: 10.1073/pnas.75.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tucker KA, Lilly MB, Heck L, Rado TA. Characterization of a new human diploid myeloid leukemia cell line (PLB-985) with granulocytic and monocytic differentiating capacity. Blood. 1987;70:372–378. [PubMed] [Google Scholar]
- 143.Berthier E, Surfus J, Verbsky J, Huttenlocher A, Beebe D. An arrayed high-content chemotaxis assay for patient diagnosis. Integrative biology : quantitative biosciences from nano to macro. 2010;2:630–638. doi: 10.1039/c0ib00030b. [DOI] [PubMed] [Google Scholar]
- 144.Montanez-Sauri SI, Beebe DJ, Sung KE. Microscale screening systems for 3D cellular microenvironments: platforms, advances, and challenges. Cellular and molecular life sciences : CMLS. 2015;72:237–249. doi: 10.1007/s00018-014-1738-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Boneschansker L, Yan J, Wong E, Briscoe DM, Irimia D. Microfluidic platform for the quantitative analysis of leukocyte migration signatures. Nature communications. 2014;5:4787. doi: 10.1038/ncomms5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yamahashi Y, et al. Integrin associated proteins differentially regulate neutrophil polarity and directed migration in 2D and 3D. Biomed Microdevices. 2015;17:100. doi: 10.1007/s10544-015-9998-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mantopoulos D, Cruzat A, Hamrah P. In vivo imaging of corneal inflammation: new tools for clinical practice and research. Seminars in ophthalmology. 2010;25:178–185. doi: 10.3109/08820538.2010.518542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ley K, et al. Sequential contribution of L- and P-selectin to leukocyte rolling in vivo. The Journal of experimental medicine. 1995;181:669–675. doi: 10.1084/jem.181.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity. 2007;26:773–783. doi: 10.1016/j.immuni.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Emre Y, Jemelin S, Imhof BA. Imaging Neutrophils and Monocytes in Mesenteric Veins by Intravital Microscopy on Anaesthetized Mice in Real Time. Journal of Visualized Experiments. 2015 doi: 10.3791/53314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jenne CN, Wong CH, Petri B, Kubes P. The use of spinning-disk confocal microscopy for the intravital analysis of platelet dynamics in response to systemic and local inflammation. PloS one. 2011;6 doi: 10.1371/journal.pone.0025109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Looney MR, et al. Stabilized imaging of immune surveillance in the mouse lung. Nat Methods. 2011;8:91–96. doi: 10.1038/nmeth.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wong J, et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. The Journal of clinical investigation. 1997;99:2782–2790. doi: 10.1172/JCI119468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Slaba I, et al. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology (Baltimore, Md ) 2015;62:1593–1605. doi: 10.1002/hep.28003. [DOI] [PubMed] [Google Scholar]
- 155.Walters KB, Green JM, Surfus JC, Yoo SK, Huttenlocher A. Live imaging of neutrophil motility in a zebrafish model of WHIM syndrome. Blood. 2010;116:2803–2811. doi: 10.1182/blood-2010-03-276972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Deng Q, Yoo SK, Cavnar PJ, Green JM, Huttenlocher A. Dual roles for Rac2 in neutrophil motility and active retention in zebrafish hematopoietic tissue. Dev Cell. 2011;21:735–745. doi: 10.1016/j.devcel.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Henry KM, Loynes CA, Whyte MK, Renshaw SA. Zebrafish as a model for the study of neutrophil biology. J Leukocyte Biol. 2013;94:633–642. doi: 10.1189/jlb.1112594. [DOI] [PubMed] [Google Scholar]
- 158.Lieschke GJ, Oates AC, Crowhurst MO, Ward AC, Layton JE. Morphologic and functional characterization of granulocytes and macrophages in embryonic and adult zebrafish. Blood. 2001;98:3087–3096. doi: 10.1182/blood.v98.10.3087. [DOI] [PubMed] [Google Scholar]
- 159.Harvie EA, Huttenlocher A. Neutrophils in host defense: new insights from zebrafish. J Leukocyte Biol. 2015;98:523–537. doi: 10.1189/jlb.4MR1114-524R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Berthier E, et al. Low-volume toolbox for the discovery of immunosuppressive fungal secondary metabolites. PLoS Path. 2013;9 doi: 10.1371/journal.ppat.1003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Graziano BR, Weiner OD. Self-organization of protrusions and polarity during eukaryotic chemotaxis. Curr Opin Cell Biol. 2014;30:60–67. doi: 10.1016/j.ceb.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Deng Q, Huttenlocher A. Leukocyte migration from a fish eye’s view. J Cell Sci. 2012;125:3949–3956. doi: 10.1242/jcs.093633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Baker MJ, Pan D, Welch HC. Small GTPases and their guanine-nucleotide exchange factors and GTPase-activating proteins in neutrophil recruitment. Current opinion in hematology. 2016;23:44–54. doi: 10.1097/MOH.0000000000000199. [DOI] [PubMed] [Google Scholar]
- 164.Mócsai A, Walzog B, Lowell CA. Intracellular signalling during neutrophil recruitment. Cardiovascular research. 2015;107:373–385. doi: 10.1093/cvr/cvv159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Weiner OD. Regulation of cell polarity during eukaryotic chemotaxis: the chemotactic compass. Curr Opin Cell Biol. 2002;14:196–202. doi: 10.1016/s0955-0674(02)00310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kölsch V, Charest PG, Firtel RA. The regulation of cell motility and chemotaxis by phospholipid signaling. J Cell Sci. 2008;121:551–559. doi: 10.1242/jcs.023333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Afonso PV, Parent CA. PI3K and chemotaxis: a priming issue? Science signaling. 2011;4 doi: 10.1126/scisignal.2002019. [DOI] [PubMed] [Google Scholar]
- 168.Kunisaki Y, et al. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. The Journal of cell biology. 2006;174:647–652. doi: 10.1083/jcb.200602142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Welch HC, et al. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- 170.Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. The Journal of cell biology. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Raghuwanshi SK, et al. The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. Journal of immunology (Baltimore, Md : 1950) 2012;189:2824–2832. doi: 10.4049/jimmunol.1201114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yoo SK, et al. Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Dev Cell. 2010;18:226–236. doi: 10.1016/j.devcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Beerman RW, et al. Direct In Vivo Manipulation and Imaging of Calcium Transients in Neutrophils Identify a Critical Role for Leading-Edge Calcium Flux. Cell reports. 2015;13:2107–2117. doi: 10.1016/j.celrep.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hu N, et al. Differential expression of granulopoiesis related genes in neutrophil subsets distinguished by membrane expression of CD177. PloS one. 2014;9 doi: 10.1371/journal.pone.0099671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Welin A, et al. The human neutrophil subsets defined by the presence or absence of OLFM4 both transmigrate into tissue in vivo and give rise to distinct NETs in vitro. PloS one. 2013;8 doi: 10.1371/journal.pone.0069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fridlender ZG, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hartl D, et al. Infiltrated neutrophils acquire novel chemokine receptor expression and chemokine responsiveness in chronic inflammatory lung diseases. Journal of immunology (Baltimore, Md : 1950) 2008;181:8053–8067. doi: 10.4049/jimmunol.181.11.8053. [DOI] [PubMed] [Google Scholar]
- 178.Tirouvanziam R, et al. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4335–4339. doi: 10.1073/pnas.0712386105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bauer S, et al. Proteinase 3 and CD177 are expressed on the plasma membrane of the same subset of neutrophils. J Leukocyte Biol. 2007;81:458–464. doi: 10.1189/jlb.0806514. [DOI] [PubMed] [Google Scholar]
- 180.Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Seminars in immunopathology. 2013;35:455–463. doi: 10.1007/s00281-013-0375-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Drifte G, Dunn-Siegrist I, Tissières P, Pugin J. Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Critical care medicine. 2013;41:820–832. doi: 10.1097/CCM.0b013e318274647d. [DOI] [PubMed] [Google Scholar]
- 182.Bowers NL, et al. Immune suppression by neutrophils in HIV-1 infection: role of PD-L1/PD-1 pathway. PLoS Path. 2014;10 doi: 10.1371/journal.ppat.1003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cloke T, et al. Phenotypic alteration of neutrophils in the blood of HIV seropositive patients. PloS one. 2013;8 doi: 10.1371/journal.pone.0072034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Matsushima H, et al. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood. 2013;121:1677–1689. doi: 10.1182/blood-2012-07-445189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nair P, et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2012;42:1097–1103. doi: 10.1111/j.1365-2222.2012.04014.x. [DOI] [PubMed] [Google Scholar]
- 186.Opfermann P, et al. A pilot study on reparixin, a CXCR1/2 antagonist, to assess safety and efficacy in attenuating ischaemia-reperfusion injury and inflammation after on-pump coronary artery bypass graft surgery. Clin Exp Immunol. 2015;180:131–142. doi: 10.1111/cei.12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Leaker BR, Barnes PJ, O’Connor B. Inhibition of LPS-induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respiratory research. 2013;14:137. doi: 10.1186/1465-9921-14-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Miller BE, et al. The pharmacokinetics and pharmacodynamics of danirixin (GSK1325756)--a selective CXCR2 antagonist--in healthy adult subjects. BMC pharmacology & toxicology. 2015;16:18. doi: 10.1186/s40360-015-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jurcevic S, et al. The effect of a selective CXCR2 antagonist (AZD5069) on human blood neutrophil count and innate immune functions. Br J Clin Pharmacol. 2015;80:1324–1336. doi: 10.1111/bcp.12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nicholls DJ, et al. Pharmacological characterization of AZD5069, a slowly reversible CXC chemokine receptor 2 antagonist. The Journal of pharmacology and experimental therapeutics. 2015;353:340–350. doi: 10.1124/jpet.114.221358. [DOI] [PubMed] [Google Scholar]