

Abstract
Purpose
The aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, mediates a broad spectrum of biological processes, including ovarian growth and ovulation. Recently, we found that an endogenous AhR ligand (ITE) can inhibit ovarian cancer proliferation and migration via the AhR. Here, we tested whether 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, an exogenous AhR ligand) may exert similar anti-ovarian cancer activities using human ovarian cancer and non-cancerous human ovarian surface epithelial cells.
Methods
Two human ovarian cancer cell lines (SKOV-3 and OVCAR-3) and one human ovarian surface epithelial cell line (IOSE-385) were used. Cell proliferation and migration activities were determined using crystal violet and FluoroBlok insert system assays, respectively. AhR protein expression was assessed by Western blotting. Expression of cytochrome P450, family 1, member A1 (CYP1A1) and member B1 (CYP1B1) mRNA was assessed by qPCR. Small interfering RNAs (siRNAs) were used to knock down AhR expression.
Results
We found that TCDD dose-dependently suppressed OVCAR-3 cell proliferation, with a maximum effect (~70 % reduction) at 100 nM. However, TCDD did not affect SKOV-3 and IOSE-385 cell proliferation and migration. The estimated IC50 of TCDD for inhibiting OVCAR-3 cell proliferation was 4.6 nM. At 10 nM, TCDD time-dependently decreased AhR protein levels, while it significantly increased CYP1A1 and CYP1B1 mRNA levels in SKOV-3, OVCAR-3 and IOSE-385 cells, indicating activation of AhR signaling. siRNA-mediated AhR knockdown readily blocked TCDD-mediated suppression of OVCAR-3 cell proliferation.
Conclusion
Our data indicate that TCDD can suppress human ovarian cancer cell proliferation via the AhR signaling pathway and that TCDD exhibits an anti-proliferative activity in at least a subset of human ovarian cancer cells.
Keywords: TCDD, AhR, Human ovarian cancer, Proliferation, Migration
1 Introduction
Although several improvements have been made in ovarian cancer treatment over the last two decades, primarily due to the evolution of surgical techniques and chemotherapy, ovarian cancer is still the most lethal gynecological cancer with highest incidence rates in the western world [1, 2]. High degrees of heterogeneity at both the cellular and molecular level impose major challenges to the treatment of cancer, including ovarian cancer [1, 3, 4]. Thus, a better understanding of heterogeneities of ovarian cancer cells will be critical for the development of more effective therapeutic strategies [1, 4].
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that transduces extracellular signals through DNA binding-dependent and -independent mechanisms [5]. Upon binding to its ligands within the cytoplasm, AhR-ligand complexes translocate into the nucleus, in which they heterodimerize with the AhR nuclear translocator (ARNT), bind to specific enhancer sequences termed dioxin responsive elements (DRE), and activate the expression of downstream genes including those encoding cytochrome P450, family 1, member A1 (CYP1A1) and member B1 (CYP1B1) (two well-studied xenobiotic metabolizing enzymes) [5, 6]. Once activated, AhR transports back to the cytoplasm where it is degraded by the proteasome system [7]. Thus, upon binding to ligands, decreases in AhR protein and increases in CYP1A1 and/or B1 mRNA and protein levels generally indicate activation of AhR signaling. The AhR mediates a broad spectrum of biological processes, including the metabolism of dioxin and related compounds. In addition, AhR is involved in regulating processes of normal ovarian growth and function, i.e., both AhR knockout in mice and exposure of rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, a classical AhR ligand) have been found to diminish or block ovulation and to reduce the number of pre-antral and antral follicles [6, 8, 9].
It has been well-established that TCDD is a potent environmental toxicant and carcinogen [5]. Indeed, TCDD exposures have been shown to increase mortality rates from many cancers, including lung and lymphatic hematopoietic cancers [10–14]. However, epidemiological studies suggest that occupational exposure to high levels of TCDD does not increase the risk for human ovarian and endometrial cancers [15], and even may be associated with a decreased risk for endometrial and breast cancers [10, 11]. Recent evidence has further shown that TCDD can inhibit mammary and uterine tumor formation and growth in rats [16], as well as inhibit human mammary, pancreatic and esophageal cancer cell growth [17–19]. AhR is expressed in a variety of human ovarian cancer histotypes, regardless of grade or stage [20, 21]. Previously, we have reported AhR expression in human ovarian surface epithelial cells, two transformed ovarian cancer cell lines (SKOV-3 and OVCAR-3), and one immortalized human ovarian surface epithelial cell line (IOSE-385) [21]. More importantly, in the same study, we have shown that 2-(1‘H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE), an endogenous AhR ligand, can inhibit OVCAR-3 cell proliferation and SKOV-3 cell migration via the AhR. This effect was not seen in IOSE-385 cells [21]. Thus, AhR may serve as a potential therapeutic target for ovarian cancers, as well as for breast and esophageal cancers [17, 18].
To date, the functional interactions of TCDD and AhR in ovarian cancer are poorly understood. Here, we tested whether TCDD can suppress AhR-mediated proliferation and migration in two human ovarian cancer cell lines. As a control, a human ovarian surface epithelial cell line, IOSE-385 was used.
2 Materials and methods
2.1 Cell lines and culture conditions
Two human ovarian adenocarcinoma cell lines (SKOV-3 and OVCAR-3) were obtained from the American Type Culture Collection (Manassas, VA, USA), and a human immortalized ovarian surface epithelial cell line (IOSE-385) was kindly provided by Dr. Nelly Auersperg, the Canadian Ovarian Tissue Bank (University of British Columbia, Vancouver, Canada). Both cancer cell lines were isolated from ascites fluid and were classified as cisplatin-resistant and p53-deficient [22]. These two cancer cell lines, however, differ in many other aspects. For example, even though both cancer cell lines express the estrogen receptor (ER), only OVCAR-3 cells respond to estrogen due to a defective ERα expression in SKOV-3 cells [23, 24]. In addition, only OVCAR-3 cells express CA125, a major ovarian cancer biomarker [1, 25]. Thus, these two cancer cell lines may represent cisplatin-resistant ovarian cancers that differ in their response to estrogen and the expression of CA125. SKOV-3 and IOSE-385 cells were cultured in Roswell Park Memorial Institute (RPMI) 1,640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10 % fetal bovine serum (Thermo Scientific, Pittsburgh, PA, USA) and penicillin/streptomycin (Thermo Scientific) (designated here as complete medium) as described before [21, 24, 26]. OVCAR-3 cells were cultured in complete medium supplemented with 10 μg/mL insulin (Sigma-Aldrich, St. Louis, MO, USA) [21].
2.2 Cell proliferation and migration assays
Cell proliferation and migration assays were carried out as reported before [21, 27]. Previously, we found that ITE time-dependently suppresses OVCAR-3 cell proliferation, with a maximum effect on day 6 of treatment [21]. Thus, in the current study, day 6 was chosen as time point for examining the effects of TCDD. Briefly, 16 h after seeding in 96-well plates (day 0; 1,000, 5,000 and 5,000 cells/well for SKOV-3, OVCAR-3 and IOSE-385, respectively; 6 wells/dose), the cells were treated with TCDD (0.01–100 nM; Cambridge Isotope Laboratories, Andover, MA, USA) or dimethyl sulfoxide (DMSO, 0.1 %v/v, the maximum concentration used in the final TCDD solutions; vehicle control, Sigma-Aldrich) for 6 days with a daily change of the media containing TCDD and DMSO. In a preliminary study, we also treated cells with TCDD at 0.01–100 nM for 2 and 4 days, but no significant effect was observed. The cell numbers were determined using a crystal violet method, and wells containing known cell numbers were used to establish a standard growth curve for each cell line. Briefly, after treatment, cells were rinsed with phosphate buffered saline (PBS, 5 mM phosphate, 145 mM NaCl, 5 mM KCl, pH 7.5), fixed in methanol for 15 min, air-dried for 5 min and stained with 0.1 % (w/v) crystal violet for 15 min. After staining, wells were rinsed with distilled water and air dried again. Once dry, the cells were solubilized with 2 % (w/v) sodium deoxycholate solution for 30 min with gentle agitation. Finally, the absorbance was measured at 570 nm on a microplate reader. Wells containing known cell numbers (0, 2,500, 5,000, 10,000 and 20,000 cells/well; n=6/ cell density) were treated in a similar fashion to establish standard growth curves for each individual cell line. The IC50 value for TCDD-mediated inhibition of OVCAR-3 cell proliferation was estimated using an Origin data analysis and graphing software package (Version 8.1) (OriginLab Corporation, Northampton, MA, USA) as described before [21]. After cell proliferation evaluation, an optimal TCDD dose (10 nM), which significantly inhibited OVCAR-3 cell proliferation (Fig. 1), was chosen for subsequent cell migration, Western blotting and qPCR assays as described below. This TCDD dose has also been shown to effectively induce cellular responses in various other studies [28–30].
Fig. 1.
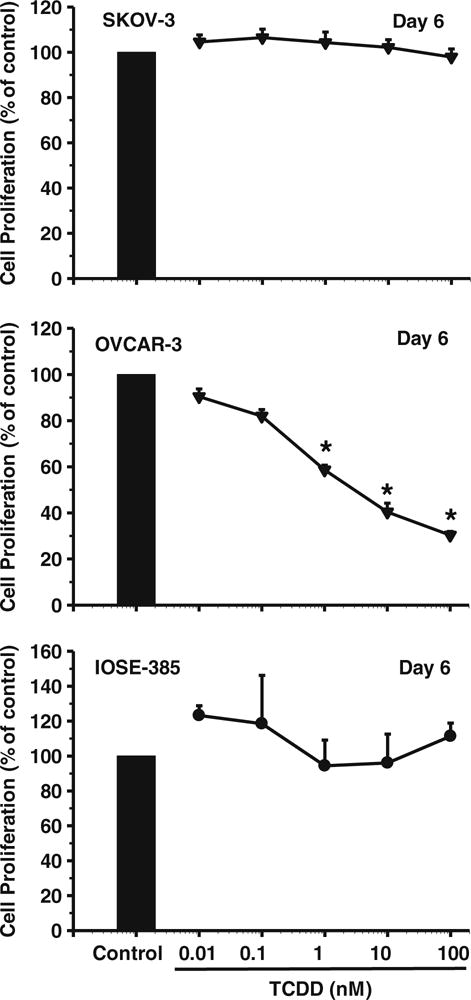
Effects of TCDD on SKOV-3, OVCAR-3 and IOSE-385 cell proliferation. Cells were treated without or with TCDD for 6 days, with a daily change of media containing TCDD or DMSO. Cell proliferation is expressed as means±SEM % of the control (n=3–4). *: Different from control at each corresponding day (p<0.05)
Cell migration was evaluated using a FluoroBlok Insert System (8.0 μm pores; BD Biosciences, San Jose, CA, USA) as described before [21, 26]. Since we previously found that OVCAR-3 cells do not exhibit any migration in this system [21, 26], here only SKOV-3 and IOSE-385 cells were assayed for their migratory responses. Briefly, after treating cells without or with TCDD (10 nM) for 6 days with a daily change of TCDD-containing medium, cells were seeded into the insert (30,000 cells/insert) in complete growth medium without or with TCDD (exactly the same media in the upper and bottom wells). After 16 h, migrated cells were stained with 0.2 μg/ml calcein AM (Life Technology, Grand Island, NY USA) and counted using the MetaMorph image analysis software package as described before [21, 26].
2.3 Western blot analysis
Western blot analyses were conducted as reported before [21, 26]. Briefly, 60–70 % confluent cell cultures were treated with a single dose of TCDD (10 nM) for 48, 24, 8, 2, 1 or 0 h. Protein samples (20 μg) were prepared using standard methods and subjected to Western blotting. The resulting membranes were probed with a rabbit anti-AhR antibody (1:2,000; Enzo Life Sciences, Farmingdale, NY, USA) [21], followed by re-probing with a mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (1:10,000; Abnova, Walnut, CA, USA). Proteins were visualized using enhanced chemiluminescence reagents (Thermo Scientific) and signals were recorded using an Epson Perfection 4,990 Photo Scanner.
2.4 RNA isolation and qPCR
RNA isolation and real-time PCR were carried out as reported before [21, 27]. Briefly, sub-confluent SKOV-3, OVCAR-3 and IOSE-385 cell cultures were treated with a single dose of TCDD (10 nM) in complete growth medium for 48, 24, 8, 2, 1 or 0 h. Total RNAwas isolated using a RNAsimple Total RNA Kit (Tiangen, Beijing, China) and converted to cDNA using a PrimeScript RT reagent kit (TaKaRa, Dalian, China). For mRNA expression analysis, qPCR was performed using SYBR Premix Ex Taq (TaKaRa, Dalian, China) on an ABI StepOnePlus System (Life Technology). The primer sequences for CYP1A1 were 5′-CACAGCACAACAAGAGACACAA-3′ (sense) and 5′-TAGCCAGGAAGAGAAAGACCTC-3′ (antisense), and the primer sequence for CYP1B1 were 5′-TTCCAAAGAAAGTTCTACAGTGTCC-3′ (sense) and 5′-CCCACCCCACACACACATAC-3′ (antisense). Relative levels of CYP1A1 and CYP1B1 transcript levels were normalized to β-actin. The primer sequences for β-actin were 5′-CCAACCGCGAGAAGATGA-3′ (sense) and 5′- CCAGAGGCGTACAGGGATAG -3′ (antisense). The real-time PCR reaction was carried out for 30 s at 95 °C for incubation, and then 15 s at 95 °C and 20 s at 56 °C for 40 cycles. To confirm the amplification specificity, the PCR products were subjected to a melting curve analysis. mRNA levels were analyzed using the 2−ΔΔCT method.
2.5 siRNA transfection
To determine whether TCDD-induced cell proliferation was AhR-dependent, siRNAs were applied to knock down AhR expression. Since TCDD inhibited the proliferation of OVCAR-3, but not SKOV-3 and IOSE-385 cells, and since TCDD had no effect on the migration of SKOV-3 and IOSE-385 cells, the AhR-specific siRNA transfection was carried out only in OVCAR-3 cells. The siRNA transfection was performed as described before [21, 31]. The AhR siRNA targeting human AhR was purchased from Dharmacon (Chicago, IL, USA) and control scrambled siRNAs (sense: 5-GAGAGGUCCCUCCCAUCUUTT-3; antisense: 5-AAGAUGGGAGGGACCUCUCTT-3) with 5′- Cy3 were synthesized by Integrated DNA Technologies (Coralville, IW, USA). After reaching 50–60 % confluence, cells were transfected with siRNA using a Lipofectamine RNAiMAX transfection reagent (Life Technology). After a 4 h transfection period, the growth medium was supplemented with serum. After the optimal dose and time were identified, additional cells were transfected for determining their proliferative responses to TCDD.
2.6 Statistical procedures
Data were analyzed using one-way ANOVA (SigmaStat; Jandel Co., San Rafael, CA, USA). When a F-test was significant, data were compared with their control using the Student-Newman-Keuls’ test or the Student t-test. p< 0.05 was considered statistically significant.
3 Results
3.1 TCDD inhibits OVCAR-3 cell proliferation
We found that, compared to the vehicle control, TCDD dose-dependently inhibited (p<0.05) OVCAR-3, but not SKOV-3 nor IOSE-385, cell proliferation (Fig. 1). At doses of 0.01 and 0.1 nM‚ TCDD did not markedly affect OVCAR-3 cell proliferation, but at doses of 1, 10, and 100 nM TCDD decreased (p<0.05) OVCAR-3 cell proliferation by 40 %, 60 %, and 70 %, respectively (Fig. 1). The estimated 50 % maximal inhibitory concentration (IC50) of TCDD for OVCAR-3 cell proliferation was estimated to be 4.63 nM. In addition, we found that the SKOV-3 and OVCAR-3 cell numbers after TCDD treatments were always higher than those initially seeded (i.e., 1,000 and 5,000 cells/well for SKOV-3 and OVCAR-3 cells, respectively). For example, the estimated OVCAR-3 cell numbers in the TCDD (10 nM) treatment groups on day 6 was 6910±1533.7. These latter data suggest that TCDD, at the doses applied here, does not have any significant cytotoxic effect on these two ovarian cancer cell lines. In addition, we found that treatment of the SKOV-3 and IOSE-385 cells with TCDD for 2, 4, and 6 days did not inhibit their migration (Fig. 2). OVCAR-3 cells were not included in this latter test (see materials and methods).
Fig. 2.
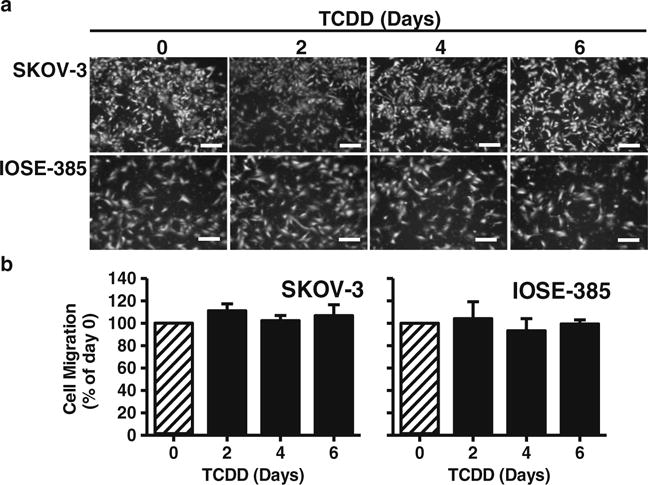
Effects of TCDD on SKOV-3 and IOSE-358 cell migration. a SKOV-3 and IOSE-358 cells were treated with TCDD (10 nM) for 0, 2, 4 or 6 days, followed by b migration assay. Migrated cells were stained and counted. Cell numbers are expressed as means±SEM % of the day 0 control (n=3). *: Different from the day 0 control (p<0.05). Bars in (a): 200 μm
3.2 TCDD down-regulates AhR protein expression
To determine whether TCDD activates the AhR, Western blot analyses were performed. After doing so, a single band was detected at ~95 kDa, corresponding to the reported human AhR molecular mass [21, 32, 33] in all three cell lines studied, i.e., SKOV-3, OVCAR-3, and IOSE-385 (Fig. 3). Subsequently, we found that a single dose of TCDD (10 nM) time-dependently decreased (p<0.05) AhR protein levels in all three cell lines (Fig. 3), indicating activation of the AhR. The protein expression patterns observed were, however, different among the cell lines tested (Fig. 3). We found that the TCDD-induced decreased in AhR protein levels started after 1, 2 and 8 h in OVCAR-3, SKOV-3 and ISOE-385 cells, respectively (Fig. 3). After 8 h, TCDD induced decreases in AhR protein levels by ~89 %, 82 % and 64 % in OVCAR-3, SKOV-3 and IOSE-385 cells, respectively (Fig. 3).
Fig. 3.
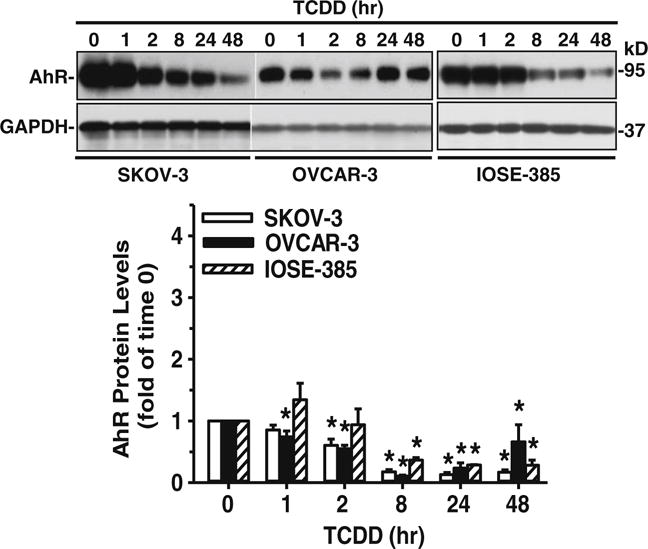
Effect of TCDD on AhR protein levels in SKOV-3, OVCAR-3 and IOSE-385 cells. Cells were treated with a single dose of TCDD (10 nM) up to 48 h and protein extracts were subjected to Western blot analysis. Data are expressed as means±SEM fold of the time 0 control (n=3–5). *: Different from the time 0 control (p<0.05)
3.3 TCDD up-regulates CYP1A1 and CYP1B1 mRNA expression
To confirm the activation of AhR downstream signaling after TCDD treatment in SKOV-3, OVCAR-3 and IOSE-385 cells, we decided to assess CYP1A1 and CYP1B1 mRNA levels by qPCR. By doing so, we found that TCDD time-dependently increased (p<0.05) CYP1A1 and CYP1B1 mRNA levels in all three cell lines tested (Fig. 4), indicating that TCDD can indeed activate AhR/CYP1A1 and AhR/CYP1B1 signaling in these cells. Again, the patterns observed among these three cell lines were different. In SKOV-3 and OVCAR-3 cells the stimulatory effect of TCDD started at 2 h and was maintained up to 48 h. In IOSE-385 cells, TCDD exposure led to an increase (p<0.05) in CYP1A1 mRNA only at 2 h, and an increase in CYP1B1 mRNA only at 2 and 8 h (Fig. 4).
Fig. 4.
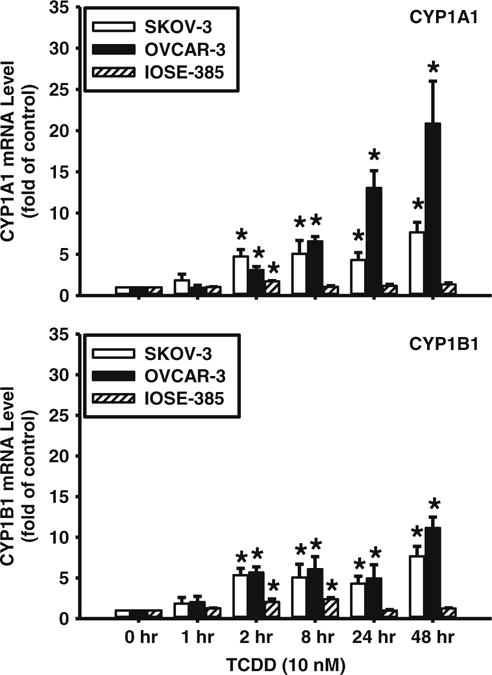
Effects of TCDD on CYP1A1 mRNA expression in SKOV-3, OVCAR-3 and IOSE-385 cells as detected by qPCR. Cells were treated with a single dose of TCDD (10 nM) up to 48 h and total mRNA extracts were subjected to qPCR. Data normalized to β-actin are expressed as means±SEM fold of the time 0 control (n=3–4). *: Different from the time 0 control (p<0.05)
3.4 AhR knockdown blocks the growth inhibitory effect of TCDD
To confirm a role of AhR in TCDD-induced inhibition of OVCAR-3 cell proliferation, AhR expression was knocked down in OVCAR-3 cells using an AhR-specific siRNA. We found that, as compared to the vehicle and scrambled siRNA controls, the siRNA at 20 nM significantly reduced (p<0.05) AhR protein levels in OVCAR-3 cells by ~97 %, 96 % and 86 % at 2, 4 and 6 days of infection, respectively (Fig. 5a). More importantly, we found that the knockdown of AhR blocked (p<0.05) the TCDD-induced inhibition of OVCAR-3 cell proliferation (Fig. 5b), thereby indicating that this TCDD-induced inhibition is AhR-dependent.
Fig. 5.
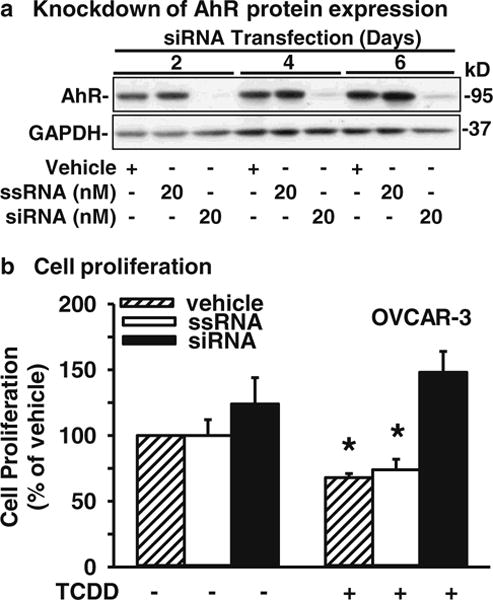
Effects of AhR knockdown on OVCAR-3 cell proliferation. a Cells were transfected with vehicle control, scrambled siRNA (ssRNA) control or AhR-specific siRNA (siRNA) for 2, 4 or 6 days. Proteins were collected and subjected to Western blot analysis. Data from 20 nM siRNA treatment are presented and expressed as means±SEM fold of the vehicle control (n=3). b After siRNA transfection for 2 days, cells were treated with TCDD (10 nM) for an additional 4 days with a daily change of TCDD, followed by cell proliferation assays. Data are expressed as means±SEM % of the vehicle control (n=5–6). *: Different from the vehicle control (p<0.05)
4 Discussion
In the current study we show that TCDD inhibits OVCAR-3 cell proliferation via the AhR, while it does not affect SKOV-3 and IOSE-358 cell proliferation and migration. These results suggest that TCDD, at the doses and treatment times studied, exhibits an anti-proliferative activity against at least a subset of ovarian cancer cells. This notion is not surprising since TCDD and other relatively non-toxic AhR ligands such as ITE, diindolylmethane (DIM) and alkyl substituted chlorinated dibenzofurans (alkyl-PCDFs) have in the past been shown to be able to suppress the growth of various cancers, including ovarian, uterine, mammary and liver cancers, both in vitro and in vivo [11–21, 34–36]. Even though TCDD, at the doses and time periods applied here, did not exhibit any significant adverse effects on non-tumorigenic IOSE-385 cells, it should be taken into consideration that its therapeutic applicability may be hampered by its potential carcinogenic effects on other cells or tissues [10–14].
The results of the current study are consistent with our recent report showing inhibitory effects of ITE on SKOV-3, OVCAR-3 and IOSE-385 cell proliferation, and on IOSE-3 cell migration [21]. However, in contrast to ITE, TCDD does not suppress SKOV-3 cell migration. These latter data suggest that, while both TCDD and ITE suppress ovarian cancer cell proliferation, they may differentially regulate ovarian cancer cell migration. In addition, this difference in suppression of TCDD on SKOV-3 and OVCAR-3 cell proliferation confirms the different phenotypes of these two ovarian cancer cell lines and emphasizes the need to recognize and understand the heterogeneity of cancer cell populations between and within patients at the cellular and the molecular levels [1, 4, 37].
To date, it is not clear what may cause the differential effect of TCDD on SKOV-3, OVCAR-3 and IOSE-385 cell proliferation. Also, the mechanisms underlying the differential effects of TCDD and ITE on SKOV-3 cell migration remain elusive. These differential effects are obviously not due to an uncoupling of TCDD from the AhR/CYP1A1 or AhR/ CYP1B1 signaling cascades, since we found that TCDD time-dependently decreases AhR protein levels and increases CYP1A1 and CYP1B1 mRNA levels in all three cell lines studied. Similarly, these differential effects are unlikely to result from a differential induction of CYP1A1 and CYP1B1 mRNA levels by TCDD, since ITE also only suppresses OVCAR-3 cell proliferation and SKOV-3 cell migration, even though ITE induces comparable increases in CYP1A1 mRNA levels in SKOV-3 and OVCAR-3 cells [21]. It appears more likely that differential regulation of signaling molecules downstream of CYP1A1 and CYP1B1 may contribute to these diverse effects. These downstream signaling molecules may include other growth-regulatory proteins such as p21 and p53 [38, 39], or protein kinases such as the mitogen-activated protein kinase (MAPK) and v-akt murine thymoma viral oncogene homolog 1 (Akt1), all of which are known to be critical for regulating normal and abnormal (cancer) cell proliferation and migration [40–42]. In addition, these different effects might at least partially be explained by differences in the expression of the carcinoma antigen 125 (CA125) and the estrogen receptor (ER) in these two ovarian cancer cell lines [23, 24]. Moreover, since TCDD was found to activate AhR/ CYP1A1 and AhR/CYP1B1 in all three cell lines studied, and since TCDD only suppressed OVCAR-3 cell proliferation but not SKOV-3 or IOSE-3 cell proliferation, these data also indicate that activation of AhR/CYP1A1 and AhR/CYP1B1 alone is not sufficient for eliciting TCDD-induced ovarian cancer cell responses.
It should be noted that TCDD is much less potent (~23 fold) than ITE, since the estimated IC50 for ITE on OVCAR-3 cell proliferation is 0.2 nM, versus 4.6 nM for TCDD [21]. Thus, given the finding that ITE does not exhibit any classic toxic effects such as induction of cleft palate and hydronephrosis, typically associated with perinatal TCDD exposure [20, 43], possibly because it is a naturally produced compound derived from two amino acids via a condensation reaction [44], ITE may serve as a candidate drug for ovarian cancer.
It has been shown that AhR alone can act as a tumor suppressor in the absence of a xenobiotic ligand in liver cancer [45]. Similarly, it has been shown that AhR knockdown can increase the invasiveness of breast cancer cells [36]. In the current study we found, however, that AhR knockdown does not alter OVCAR-3 cell proliferation in the absence of TCDD. Together with our recent observation that AhR knockdown fails to affect serum-induced SKOV-3 cell migration [21], these data suggest that AhR’s tumor suppressive effect may be cell type and/or cancer type specific. As yet, however, we cannot exclude the possibility that AhR alone can mediate other cellular processes in the absence of AhR ligands in SKOV-3 cells. In this respect, it would be interesting to explore the role of AhR in mediating chemoresistance in ovarian cancer cells, as such a role has been observed in human colorectal cancer cells [7].
In conclusion, instead of being an environmental carcinogen, particularly at high concentrations and/or upon long-term exposures [5], TCDD at the doses and time periods studied here exhibits anti-proliferative activity in at least a subset of ovarian cancer cells. These data advance our understanding of the effect of TCDD on ovarian cancer cells and normal ovarian epithelial cells, and confirm that AhR signaling plays an important role in inhibiting ovarian cancer cell growth, as suggested previously [21].
Acknowledgments
This work was supported in part by the US National Institutes of Health grant PO1 HD38843 to R.R. Magness, I.M. Bird, and J. Zheng, a Department of Obstetrics/Gynecology R & D Grant, University of Wisconsin-Madison to J. Zheng, and the National Science Foundation of China grants No. 81100429 and 81270703 to K. Wang
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
Contributor Information
Yan Li, Department of Obstetrics and Gynecology, University of Wisconsin, 202 S. Park St., Madison, WI 53715, USA.
Kai Wang, Clinical and Translational Research Center, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 200040, China.
Yi-Zhou Jiang, Department of Obstetrics and Gynecology, University of Wisconsin, 202 S. Park St., Madison, WI 53715, USA.
Xin-Wen Chang, Clinical and Translational Research Center, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 200040, China.
Cai-Feng Dai, Department of Obstetrics and Gynecology, University of Wisconsin, 202 S. Park St., Madison, WI 53715, USA; Center for Reproductive Medicine, Qilu Hospital of Shandong University, Jinan 250012, Shandong, China.
Jing Zheng, Department of Obstetrics and Gynecology, University of Wisconsin, 202 S. Park St., Madison, WI 53715, USA.
References
- 1.Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9:167–181. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- 2.Di J, Duiveman-de Boer T, Zusterzeel PL, Fig CG, Massuger LF, Torensma R. The stem cell markers Oct4A, Nanog and c-Myc are expressed in ascites cells and tumor tissue of ovarian cancer patients. Cell Oncol. 2013;36:363–374. doi: 10.1007/s13402-013-0142-8. [DOI] [PubMed] [Google Scholar]
- 3.Krijgsman O, Israeli D, van Essen HF, Eijk PP, Berens ML, Mellink CH, Nieuwint AW, Weiss MM, Steenbergen RD, Meijer GA, Ylstra B. Detection limits of DNA copy number alterations in heterogeneous cell populations. Cell Oncol. 2013;36:27–36. doi: 10.1007/s13402-012-0108-2. [DOI] [PubMed] [Google Scholar]
- 4.Bast CRB, Jr, Hennessy GBM. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Safe S, McDougal A. Mechanism of action and development of selective aryl hydrocarbon receptor modulators for treatment of hormone-dependent cancers (review) Int J Oncol. 2002;20:1123–1128. [PubMed] [Google Scholar]
- 6.Hernandez-Ochoa I, Karman BN, Flaws JA. The role of the aryl hydrocarbon receptor in the female reproductive system. Biochem Pharmacol. 2009;77:547–559. doi: 10.1016/j.bcp.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujii-Kuriyama Y, Kawajiri K. Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:40–53. doi: 10.2183/pjab.86.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clapp RW, Jacobs MM, Loechler EL. Environmental and occupational causes of cancer: new evidence 2005–2007. Rev Environ Health. 2008;23:1–37. doi: 10.1515/reveh.2008.23.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertazzi PA, Zocchetti C, Guercilena S, Consonni D, Tironi A, Landi MT, Pesatori AC. Dioxin exposure and cancer risk: a 15-year mortality study after the “Seveso accident”. Epidemiology. 1997;8:646–652. [PubMed] [Google Scholar]
- 11.Viel JF, Clement MC, Hagi M, Grandjean S, Challier B, Danzon A. Dioxin emissions from a municipal solid waste incinerator and risk of invasive breast cancer: a population-based case–control study with GIS-derived exposure. Int J Health Geogr. 2008;7:4. doi: 10.1186/1476-072X-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steenland K, Bertazzi P, Baccarelli A, Kogevinas M. Dioxin revisited: developments since the 1997 IARC classification of dioxin as a human carcinogen. Environ Health Perspect. 2004;112:1265–1268. doi: 10.1289/ehp.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin P, Chang H, Tsai WT, Wu MH, Liao YS, Chen JT, Su JM. Overexpression of aryl hydrocarbon receptor in human lung carcinomas. Toxicol Pathol. 2003;31:22–30. doi: 10.1080/01926230390173824. [DOI] [PubMed] [Google Scholar]
- 14.McGregor DB, Partensky C, Wilbourn J, Rice JM. An IARC evaluation of polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans as risk factors in human carcinogenesis. Environ Health Perspect. 1998;106(Suppl 2):755–760. doi: 10.1289/ehp.98106755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Consonni D, Pesatori AC, Zocchetti C, Sindaco R, D’Oro LC, Rubagotti M, Bertazzi PA. Mortality in a population exposed to dioxin after the Seveso, Italy, accident in 1976: 25 years of follow-up. Am J Epidemiol. 2008;167:847–858. doi: 10.1093/aje/kwm371. [DOI] [PubMed] [Google Scholar]
- 16.Kociba RJ, Keyes DG, Beyer JE, Carreon RM, Wade CE, Dittenber DA, Kalnins RP, Frauson LE, Park CN, Barnard SD, Hummel RA, Humiston CG. Results of a two-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Toxicol Appl Pharmacol. 1978;46:279–303. doi: 10.1016/0041-008x(78)90075-3. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Zong H, Li S, Zhang D, Zhang L, Xia Q. Activation of aryl hydrocarbon receptor suppresses invasion of esophageal squamous cell carcinoma cell lines. Tumori. 2012;98:152–157. doi: 10.1177/030089161209800121. [DOI] [PubMed] [Google Scholar]
- 18.Zhang S, Lei P, Liu X, Li X, Walker K, Kotha L, Rowlands C, Safe S. The aryl hydrocarbon receptor as a target for estrogen receptor-negative breast cancer chemotherapy. Endocr Relat Cancer. 2009;16:835–844. doi: 10.1677/ERC-09-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koliopanos A, Kleeff J, Xiao Y, Safe S, Zimmermann A, Buchler MW, Friess H. Increased arylhydrocarbon receptor expression offers a potential therapeutic target for pancreatic cancer. Oncogene. 2002;21:6059–6070. doi: 10.1038/sj.onc.1205633. [DOI] [PubMed] [Google Scholar]
- 20.Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:20768–20773. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Li Y, Jiang YZ, Dai CF, Patankar MS, Song JS, Zheng J. An endogenous aryl hydrocarbon receptor ligand inhibits proliferation and migration of human ovarian cancer cells. Cancer Lett. 2013;340:63–71. doi: 10.1016/j.canlet.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagopian GS, Mills GB, Khokhar AR, Bast RC,ZHS., Jr Expression of p53 in cisplatin-resistant ovarian cancer cell lines: modulation with the novel platinum analogue (1R, 2R-diaminocyclohexane) (trans-diacetato) (dichloro)-platinum (IV) Clin Cancer Res. 1999;5:655–663. [PubMed] [Google Scholar]
- 23.Lau KM, Mok SC, Ho SM. Expression of human estrogen receptor-α and -β, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci U S A. 1999;96:5722–5727. doi: 10.1073/pnas.96.10.5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li HH, Zhao YJ, Li Y, Dai DF, Jobe SO, Li XF, Yang XS, Patankar MS, Magness RR, Zheng J. Estradiol-17β and its metabolites attenuate vitamin c-suppressed human ovarian cancer cell proliferation. Reprod Sci. 2014;21:102–111. doi: 10.1177/1933719113492211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boivin M, Lane D, Piche A, Rancourt C. Ca125 (MUC16) tumor antigen selectively modulates the sensitivity of ovarian cancer cells to genotoxic drug-induced apoptosis. Gynecol Oncol. 2009;115:407–413. doi: 10.1016/j.ygyno.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Dai CF, Jiang YZ, Li Y, Wang K, Liu PS, Patankar MS, Zheng J. Expression and roles of slit/robo in human ovarian cancer. Histochem Cell Biol. 2011;135:475–485. doi: 10.1007/s00418-011-0806-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu YM, Chen X, Zhou Q, He QZ, Kang JH, Zheng J, Wang K, Duan T. ITE and TCDD differentially regulate the vascular remodeling of rat placenta via the activation of AhR. PLoS One. 2014;9:e86549. doi: 10.1371/journal.pone.0086549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bofinger DP, Feng L, Chi LH, Love J, Stephen FD, Sutter TR, Osteen KG, Costich TG, Batt RE, Koury ST, Olson JR. Effect of TCDD exposure on CYP1A1 and CYP1B1 expression in explant cultures of human endometrium. Toxicol Sci. 2001;62:299–314. doi: 10.1093/toxsci/62.2.299. [DOI] [PubMed] [Google Scholar]
- 29.Pitt JA, Feng L, Abbott BD, Schmid J, Batt RE, Costich TG, Koury ST, Bofinger DP. Expression of AhR and ARNT mRNA in cultured human endometrial explants exposed to TCDD. Toxicol Sci. 2001;62:289–298. doi: 10.1093/toxsci/62.2.289. [DOI] [PubMed] [Google Scholar]
- 30.Mueller MD, Vigne JL, Streich M, Tee MK, Raio L, Dreher E, Bersinger NA, Taylor RN. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases glycodelin gene and protein expression in human endometrium. J Clin Endocrinol Metab. 2005;90:4809–4815. doi: 10.1210/jc.2004-2064. [DOI] [PubMed] [Google Scholar]
- 31.Jiang YZ, Li Y, Wang K, Dai CF, Huang SA, Chen DB, Zheng J. Distinct roles of HIF1A in endothelial adaptations to physiological and ambient oxygen. Mol Cell Endocrinol. 2014;391:60–67. doi: 10.1016/j.mce.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang YZ, Wang K, Fang R, Zheng J. Expression of aryl hydrocarbon receptor in human placentas and fetal tissues. J Histochem Cytochem. 2010;58:679–685. doi: 10.1369/jhc.2010.955955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juan SH, Lee JL, Ho PY, Lee YH, Lee WS. Antiproliferative and antiangiogenic effects of 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, in human umbilical vascular endothelial cells. Eur J Pharmacol. 2006;530:1–8. doi: 10.1016/j.ejphar.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 34.Wormke M, Castro-Rivera E, Chen I, Safe S. Estrogen and aryl hydrocarbon receptor expression and crosstalk in human ishikawa endometrial cancer cells. J Steroid Biochem Mol Biol. 2000;72:197–207. doi: 10.1016/s0960-0760(00)00030-3. [DOI] [PubMed] [Google Scholar]
- 35.McDougal A, Gupta MS, Morrow D, Ramamoorthy K, Lee JE, Safe SH. Methyl-substituted diindolylmethanes as inhibitors of estrogen-induced growth of t47d cells and mammary tumors in rats. Breast Cancer Res Treat. 2001;66:147–157. doi: 10.1023/a:1010608000074. [DOI] [PubMed] [Google Scholar]
- 36.Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF, Thomas RS. Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Mol Endocrinol. 2010;24:359–369. doi: 10.1210/me.2009-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geurts van Kessel A. The cancer genome: from structure to function. Cell Oncol. 2014;37:155–165. doi: 10.1007/s13402-014-0177-5. [DOI] [PubMed] [Google Scholar]
- 38.Stevens EA, Mezrich JD, Bradfield CA. The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology. 2009;127:299–311. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puga A, Ma C, Marlowe JL. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem Pharmacol. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan Z, Chang X, Puga A, Xia Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons: role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochem Pharmacol. 2002;64:771–780. doi: 10.1016/s0006-2952(02)01138-3. [DOI] [PubMed] [Google Scholar]
- 41.Tan Z, Huang M, Puga A, Xia Y. A critical role for map kinases in the control of ah receptor complex activity. Toxicol Sci. 2004;82:80–87. doi: 10.1093/toxsci/kfh228. [DOI] [PubMed] [Google Scholar]
- 42.Wu R, Zhang L, Hoagland MS, Swanson HI. Lack of the aryl hydrocarbon receptor leads to impaired activation of AKT/protein kinase B and enhanced sensitivity to apoptosis induced via the intrinsic pathway. J Pharmacol Exp Ther. 2007;320:448–457. doi: 10.1124/jpet.106.111773. [DOI] [PubMed] [Google Scholar]
- 43.Henry EC, Bemis JC, Henry O, Kende AS, Gasiewicz TA. A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Arch Biochem Biophys. 2006;450:67–77. doi: 10.1016/j.abb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 44.Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, DeLuca HF. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci U S A. 2002;99:14694–14699. doi: 10.1073/pnas.232562899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan Y, Boivin GP, Knudsen ES, Nebert DW, Xia Y, Puga A. The aryl hydrocarbon receptor functions as a tumor suppressor of liver carcinogenesis. Cancer Res. 2010;70:212–220. doi: 10.1158/0008-5472.CAN-09-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]