

Abstract
Clostridium difficile has emerged as a major cause of infectious diarrhea in hospitalized patients, with increasing mortality rate and annual healthcare costs exceeding $3 billion. Since C. difficile infections are associated with the use of antibiotics, there is an urgent need to develop treatments that can inactivate the bacterium selectively without affecting commensal microflora. Lytic enzymes from bacteria and bacteriophages show promise as highly selective and effective antimicrobial agents. These enzymes often have a modular structure, consisting of a catalytic domain and a binding domain. In the current work, using consensus catalytic domain and cell-wall binding domain sequences as probes, we analyzed in silico the genome of C. difficile, as well as phages infecting C. difficile. We identified two genes encoding cell lytic enzymes with possible activity against C. difficile. We cloned the genes in a suitable expression vector, expressed and purified the protein products, and tested enzyme activity in vitro. These newly identified enzymes were found to be active against C. difficile cells in a dose-dependent manner. We achieved a more than 4-log reduction in the number of viable bacteria within 5 h of application. Moreover, we found that the enzymes were active against a wide range of C. difficile clinical isolates. We also characterized the biocatalytic mechanism by identifying the specific bonds cleaved by these enzymes within the cell wall peptidoglycan. These results suggest a new approach to combating the growing healthcare problem associated with C. difficile infections.
Keywords: enzyme, antimicrobial, lytic, bacteria, clostridium
Introduction
Clostridium difficile is a species of gram-positive, spore forming anaerobic bacteria, and the causative agent of pseudomembranous colitis (Bartlett et al., 1978). It is a part of physiological gut flora in 2–5% of the general population and in up to 20% of hospitalized patients; however, not all strains produce enterotoxin that leads to morbidity and mortality (McDonald, 2005). In the 1980s, an epidemic strain with increased toxin production— ribotype 027/NAP1/toxinotype III—was identified (McDonald et al., 2005). This hypervirulent C. difficile strain produces increased amounts of toxin (up to 16- and 23-times greater production of toxin A and B, respectively), relative to the typical clinical isolates of C. difficile (Warny et al., 2005). Due to the widespread use of broad-spectrum antibiotics, C. difficile infections (CDI) have become the most common cause of infectious diarrhea in hospitalized patients (Kelly et al., 1994). Moreover, according to the Centers for Disease Control and Prevention (CDCP), the annual incidence of CDI in the U.S. is near 0.5 million with 29,000 deaths (Lessa et al., 2015). Following extensive antibiotic therapy, C. difficile becomes opportunistic and proliferates in the microflora-depleted intestinal tract. Indeed in a recent report, 31% of patients who received antibiotics in acute medical care wards were colonized with C. difficile and 56% of these patients developed CDI (Kyne et al., 2002). As a result, extended hospital stay has been estimated to cost the U.S. healthcare system between $750 million and $3.2 billion annually (Kyne et al., 2002).
Currently, the first-line treatment for CDI in mild cases is to discontinue the use of antibiotics; oral metronidazole or vancomycin is indicated in more severe infections. Discontinuing the use of antibiotics allows re-establishment of colonic microflora; however, it can further exacerbate the underlying medical condition. When antibiotics are used to treat CDI, spore formation is often induced and/or results in recurrence (Fawley et al., 2007; Merrigan et al., 2010; Wilcox and Fawley, 2000). Spores of C. difficile remain unaffected by initial therapy, resulting in a relapse in a majority of patients when treatment is discontinued. Surgery is indicated only if recurrent infections are severe and associated with serious complications.
The recent wave of CDI in healthcare facilities highlights the urgent need to develop treatment modalities that can inactivate C. difficile selectively without affecting the commensal microflora. Enzymes from bacteria and bacteriophages show promise in the development of highly selective and effective antimicrobial agents (Fischetti, 2005; Mehta et al., 2013; Parisien et al., 2008; Schmitz et al., 2010; Schuch et al., 2002; Shockman et al., 1996). The tremendous selectivity of bacteriolytic enzymes against a narrow range of bacterial species is particularly well suited for treating C. difficile infections.
In the current work, using a consensus catalytic domain sequence and a consensus cell-wall binding domain sequence as probes, we analyzed in silico the genome of C. difficile, as well as phages infecting C. difficile. We identified two genes encoding cell lytic enzymes with possible activity against C. difficile. We cloned these two genes, expressed and purified the protein products termed CDG and CD11 (Fig. 1), and tested enzyme activity in vitro. These newly identified enzymes were found to be highly active against a wide range of C. difficile clinical isolates. We also characterized the biocatalytic mechanism by identifying the specific bonds cleaved by these enzymes within the cell wall peptidoglycan. These enzymes are promising candidates for the treatment of C. difficile infections.
Figure 1.

Two-step approach to identification of novel enzymes against C. difficile. (a) Bacillus enzyme PlyG was used as a probe to identify homologous sequences in the genome of C. difficile. Sequence alignment of PlyG and CD-G, which have homologous catalytic domains but dissimilar binding domains. (b) CD-G was then used as a probe to identify CD11. CD-G and CD11 have dissimilar catalytic domains but homologous binding domains. Identical residues are shaded black. Homologous substitutions are shaded gray. The conserved features in the catalytic domains are designated as follows, (*) amino acids forming the amidase binding site, (:) amino acids forming the substrate binding site. Identical residues are shaded in black. Homologous substitutions are shaded in gray.
Materials and Methods
Bacterial Cell Culture and Plasmids
C. difficile strain 630 was purchased from ATCC, and was stored frozen at −80°C in 20% (v/v) glycerol. The frozen glycerol stocks were used to grow cells in BHIS media. BHIS media was prepared by adding 37 g of brain-heart infusion, and 5 g yeast extract in 1-L of deionized water. This mixture was autoclaved for 20 min and then allowed to cool at room temperature. To the warm media, 10 mL of filter-sterilized 10% (w/v) L-cysteine was added. C. difficile cells were grown in 4 mL of BHIS media in a 14 mL falcon tube at 37°C in an anaerobic chamber.
In addition to the commercial strain, we also studied 13 clinical isolates of C. difficile, including strains R20291 and CD196 (Stabler et al., 2009), strains CF5, M68, M120, and BI-9 (He et al., 2010), and strains Liv024, Liv022, TL178, TL176, TL174, CD305, and UK1 (Yang et al., 2014). Strains R20291, CD196, CF5, M68, M120, BI-9, Liv024, Liv022, TL178, TL176, TL174, CD305 were gifts from Trevor Lawley, Sanger Institute, UK, and UK1 was originally provided by Joseph Sorg, Texas A&M University and now maintained in the lab as a model strain for establishing the mouse C. difficile infection (CDI) model (Steele et al., 2012; Sun et al., 2011; Wang et al., 2012). Clinical isolates of C. difficile were stored as spore stock in 20% (v/v) ethanol and were cultured overnight in BHIS media in presence of 0.1% sodium taurocholate used as germinant.
Genes for CDG and CD11 were codon optimized for E. coli expression, and were then cloned using NdeI/XhoI restriction sites in E. coli plasmid pGS-21a with an N-terminal His6 tag (GenScript). BL21 Star (DE3) chemically competent cells (Invitrogen, Thermo Fisher Scientific, Waltham, MA), optimized for reduced mRNA degradation and full-length peptide synthesis, were used as expression host.
Expression and Purification of Lytic Enzymes CDG and CD11
The lytic enzymes were expressed and purified using the Invitrogen Pro-Bond Protein Purification System (Invitrogen) under native conditions, as previously described by Mehta et al. (2013). The expression and purification conditions are described in greater detail in the supplementary materials section (S-1).
Isolation of Cell Wall Peptidoglycan of C. difficile Vegetative Cells
The cell wall isolation procedure is described in greater detail in the supplementary materials section (S-2).
Characterization of Enzyme Activity Using an LC–MS Assay
The experimental protocol is described in greater detail in the supplementary materials section (S-3).
Pull-Down Assay
The experimental protocol is described in greater detail in the supplementary materials section (S-4).
Antimicrobial Activity Assay on Vegetative Cells
Bacterial culture (1-mL) was harvested when the cell suspension reached exponential growth (absorbance of OD600 = 0.3) by spinning at 12,000 rpm for 5 min; the pellet was then washed twice and then suspended in phosphate buffered saline (PBS), pH 7.4. Cell suspension (100 μL of 107 CFU/mL) was added to each Eppendorf tube containing enzyme solution at predetermined enzyme concentrations (typically in the range of 100–250 or 0–50 μg/mL for dose dependence experiments) and the corresponding buffer control. The enzyme solution was introduced into the anaerobic chamber when the cells were harvested and ready to be treated. Final cell count was ~106 CFU/mL. Periodically, the control cell suspension (without enzyme) was diluted 10-, 100-, and 1000-fold. Fifty microliters of each of these dilutions were then plated on BHIS agar and incubated at 37°C in an anaerobic chamber for 14–16 h. The dilution, which yielded countable colonies, was used to determine the number of viable cells. Similar dilutions were used for cell suspensions treated with enzyme solution.
Antimicrobial Activity Assay Against Clinical Isolates of C. difficile
Spores of clinical isolates of C. difficile were initially prepared using methods described previously (Sorg and Dineen, 2009), and were stored as spore stock in 20% ethanol. An aliquot of spore stock was diluted in water and spun to wash off ethanol. The washing can be repeated to ensure complete removal of ethanol. Spores of individual strains were then allowed to germinate in pre-reduced BHIS containing 0.1% sodium taurocholate (used as germinant) in an anaerobic chamber and grown overnight at 37°C without shaking (Sorg and Dineen, 2009). Thirty microliters of overnight cultures were then used to inoculate into 3 mL of pre-reduced fresh BHIS medium and cells were harvested when OD600 reached 0.3 by a tabletop centrifuge at 14,000 rpm for 3 min. Cells were then washed with PBS and diluted with PBS to a final concentration of 107 CFU/mL. Hundred microliter of the suspensions were added to 900 μL of PBS or enzymes (CD11, CDG) in 1.5 mL Eppendorf tubes and incubated for 5 h inside the anaerobic chamber. Serial dilutions were made and plated on BHIS plates to determine viable cells of each after overnight incubation in the anaerobic chamber. Percentages of killing were determined by comparing the viable cell numbers from enzyme treated groups with PBS control.
Results and Discussion
In Silico Analysis to Identify Enzymes With Lytic Activity Against C. difficile Using the Bacillus anthracis Endolysin PlyG as a Probe
Most lytic enzymes have a modular structure that consists of two domains, typically an N-terminal catalytic domain and a C-terminal binding domain (Fischetti, 2005, 2008; Loessner, 2005; Loessner et al., 2002). The binding domain recognizes specific epitopes on the cell wall peptidoglycan that are unique for each bacterial species, thereby conferring lytic enzymes with exquisite selectivity towards bacterial species or even strains (Loessner et al., 2002; Mayer et al., 2011). The catalytic domain is responsible for cleaving critical bonds within the bacterial cell wall that are responsible for the highly cross-linked structure of cell wall peptidoglycan and maintaining the structural integrity of bacterial cells (Fischetti, 2005, 2008; Loessner, 2005). Based on the type of bond(s) targeted by the catalytic domains, lytic enzymes are endowed with muramidase (Porter et al., 2007), glucosaminidase (Pritchard et al., 2007), transglycosylase (Blackburn and Clarke, 2001), alanine-amidase (Schuch et al., 2002), alanoyl-glutamate endopeptidase (Korndorfer et al., 2008), glutaminyl-lysine endopeptidase (Pritchard et al., 2007), or cross-bridge endopeptidase activity (Baker et al., 2006).
Similar chemical bonds give rise to the highly cross-linked peptidoglycan structure in the genus Bacillus and Clostridium (Bourguet et al., 2012; Helgason et al., 2000; Loessner et al., 1997; Low et al., 2005; Peltier et al., 2011; Severin et al., 2004). We reasoned that the catalytic domains of enzymes that target B. anthracis and C. difficile may also be similar. To identify lytic enzymes that target C. difficile, we used the catalytic domain of the known B. anthracis bacteriophage endolysin PlyG (NCBI Acc. No YP_338200) as a probe sequence. PlyG is a N-acetylmuramoyl-L-alanine amidase that originates from the Bacillus phage Gamma (Schuch et al., 2002). We performed a sequence homology search within the open reading frames (ORF) of an in silico translated database of C. difficile genomes (GenBank). As a result, we identified, within the genome of Peptoclostridium difficile DA00211, a putative lytic enzyme sequence with a homologous catalytic domain, yet dissimilar binding domain to that of PlyG (Fig. 1a), which we termed CDG. The overall homology of CDG and PlyG is 37% identity and 48% similarity (calculated by the NCBI BLASTp algorithm), while the catalytic domain of CDG shows 37% identity and 50% similarity with that of PlyG. Based on catalytic homology to PlyG, we predicted that CDG (GenBank Acc. No EQH20562.1) would be an N-acetylmuramoyl-L-alanine amidase that hydrolyzes the amide bond between N-acetylmuramic acid and L-amino acids in the cell wall peptidoglycan. The fact that the catalytic domains of enzymes targeting bacilli and clostridia were homologous confirmed our reasoning; moreover, the lack of homology in the binding domain sequences was consistent with a role for these domains in imparting species specificity.
Next, we used the sequence of the putative binding domain of the new enzyme CDG as a probe to identify other cell lytic enzymes with activity against C. difficile 630 (P. difficile 630). We identified a new enzyme in P. difficile referred to in NCBI database as NCBI Reference Sequence No. WP_009895119.1, which we termed CD11 (Fig. 1b). It had a conserved cell-wall binding domain, but dissimilar catalytic domain, compared to CDG. The binding domains of CD11 and CDG share 56% identity and 76% similarity (calculated by the NCBI BLASTp algorithm). CD11 was also a putative N-acetylmuramoyl-L-alanine amidase (EC 3.5.1.28).
Expression and Purification of CD11 and CDG
Following identification of the purported lytic enzymes CDG and CD11 within the Clostridia genomes, we proceeded to determine whether these enzymes could kill C. difficile. The newly identified enzymes were overexpressed in E. coli and purified from the cell lysate proteins as described in the Materials and Methods section. A significant fraction of the overexpressed lytic enzymes was found in the soluble cell lysate (bands highlighted with white ovals in lanes 1 and 2 of Fig. 2a and b, respectively). The expression and purification procedure described here, consistently yielded 60–70 mg of purified CDG (shown in lane 4 of Fig. 2a) per liter of host cell culture and 90–100 mg of CD11 (shown in lane 4 of Fig. 2b) per liter of host cell culture. The molecular weights of CDG and CD11, including the N-terminal His-tag, are 29.6 and 31 kDa, respectively. Recombinant CDG and CD11 expressed in E. coli and purified from other lysate proteins showed a single band on SDS–PAGE, at a molecular weight corresponding to that of a monomeric protein (Fig. 2).
Figure 2.

SDS–PAGE analysis of soluble protein and purified protein. (a) CD-G: Lanes 1 – Soluble cell lysate, 2 – Protein marker, 3 – Flow through IMAC column, 4 – Purified CDG. (b) CD-11: Lanes 1 – Protein marker, 2 – Soluble cell lysate, 3 – Flow through IMAC column, 4 –Purified CD11.
Antimicrobial Activity of CD11 and CDG
The newly identified enzymes CD11 and CDG were tested for their antimicrobial activity against C. difficile 630 using the antimicrobial plating assay. When cells were treated with 150 μg of purified CD11 and CDG, we observed >90% killing of C. difficile cells within 2 h of treatment, and longer incubation with enzyme resulted in 99.99% killing of C. difficile cells, which corresponds to 4-log cell killing in 3.5 h (Fig. 3a). With 5.5 h incubation, the number of viable C. difficile cells was reduced to <10, thereby demonstrating near complete neutralization with CD11 and CDG (Fig. 3a). We determined the minimum amount of enzyme required for neutralization of vegetative C. difficile cells by treating cells with increasing dose of enzymes for a fixed treatment time of 3.5 h. As low as 2.5 μg/mL of CDG and CD11 treatment resulted in >90% killing of cells (Fig. 3b). Greater cell killing was observed with higher doses of enzymes, with >3-log cell killing observed using 50 μg/mL of CDG, while >4-log killing was observed using 50 μg/mL of CD11 (Fig. 3b).
Figure 3.

Effect of incubation time and enzyme concentration on bacterial killing against fresh culture of vegetative cells. (a) Kinetics of bacterial killing against vegetative cells; the enzyme concentration was 150 μg/mL. (b) Effect of enzyme concentration on bacterial killing; the treatment time was 3.5 h. PBS (
 ), CD11 (
), CD11 (
 ), and CDG (
), and CDG (
 ).
).
Selective Bactericidal Activity of CD11 and CDG
As discussed above, unlike antibiotics, lytic enzymes possess antimicrobial activity that is often exquisitely selective to bacterial species and in some cases even individual strains. CDG and CD11 were selective against C. difficile 630, while showing no bactericidal activity against B. anthracis and S. aureus cells as determined using the antimicrobial plating assay. It has been demonstrated that the selectivity of lytic enzymes is attributed to their binding domains that causes differential binding of lytic enzymes to various bacterial species. We allowed the binding of lytic enzymes to vegetative cells of C. difficile 630 and S. aureus and analyzed the fraction of bound and unbound enzymes using SDS–PAGE as described in the Materials and Methods sections above. Both CD11 and CDG bound to C. difficile cells, as indicated by the thick enzyme bands in the bound fractions in Figure 4a. Other bands not corresponding to lytic enzymes observed in the bound fraction are likely due to proteins released from the lysed cells. Trace amounts of lytic enzymes observed in the unbound fractions could be attributed to an excess amount of lytic enzymes used for cell binding, or to loosely bound enzyme released from cells during the centrifugation step. When tested on S. aureus, CD11 and CDG were primarily found in the unbound fractions, showing that both enzymes had minimal binding to S. aureus cells. The results of the binding study indicate that the interactions between C. difficile and CD11/CDG were specific.
Figure 4.

Selective binding of lytic enzymes to (a) C. difficile cells, (b) S. aureus cells. L, protein ladder; B, bound fraction; U, unbound fractions; W, wash fractions (loosely bound).
Bactericidal Activity of CD11 and CDG Against Clinical Isolates of C. difficile
The bactericidal efficacy of lytic enzymes CDG and CD11 was selective against C. difficile, and was ineffective against other species including B. anthracis and S. aureus as described in the section above. We further evaluated the efficacy of enzymes against the more relevant strains—clinical isolates—of C. difficile. The growth and cell culture conditions used were as described in the Materials and Methods section. Cells harvested from an exponential culture (OD600 ~ 0.3) were washed to remove media and then treated with 150 μg/mL of enzyme and corresponding buffer control as described in the Materials and Methods section above. For each of these strains/clinical isolates, the cell viability was reduced by 4-log as a result of 5 h treatment with enzymes (Fig. 5). While 4-log reduction in cell viability was observed for most strains, some strains were more susceptible to enzyme treatment showing more than 5-log reduction in cell viability (Fig. 5).
Figure 5.
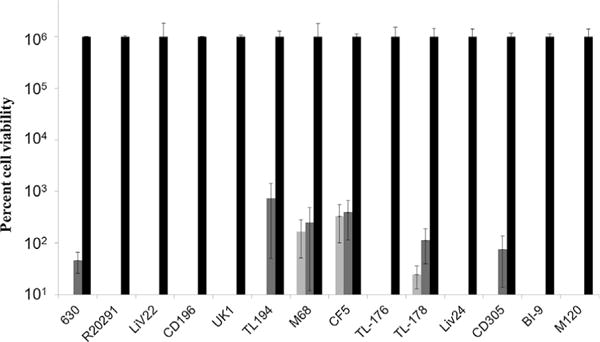
Enzyme activity against clinical isolates of C. difficile for CD11 (
 ), CDG (
), CDG (
 ), and PBS (
), and PBS (
 ). Absence of gray and/or white bars for some clinical isolates indicates no surviving bacterial cells and corresponds to a reduction in cell viability of more than 5-log.
). Absence of gray and/or white bars for some clinical isolates indicates no surviving bacterial cells and corresponds to a reduction in cell viability of more than 5-log.
Biochemical Action of CD11 and CDG Against Clinical Isolates of C. difficile
Analysis of the CD11 and CDG sequences using BLASTP revealed that while the C-terminal binding domains were highly homologous, the N-terminal catalytic domains had minimal similarity. However, BLAST search of CD11 and CDG sequences against the entire NCBI database yielded conserved domains, such that, CD11 catalytic domain belonged to N-acetylmuramoyl-L-alanine amidase (MurNAc-LAA) superfamily, and CDG catalytic domain belonged to Peptidoglycan recognition proteins (PGRP) superfamily. The MurNAc-LAA family is one of several peptidoglycan hydrolases (PGHs) found in bacterial and bacteriophage or prophage genomes that are involved in the degradation of the peptidoglycan (Marchler-Bauer and Bryant, 2004; Marchler-Bauer et al., 2009, 2011, 2013, 2015). MurNAc-LAA includes the Amidase_3 family, which is typically an autolysin that hydrolyzes the amide bond between N-acetylmuramoyl and L-amino acids in certain cell wall glycopeptides (Marchler-Bauer and Bryant, 2004; Marchler-Bauer et al., 2009, 2011, 2013, 2015). Peptidoglycan recognition proteins (PGRPs) are pattern recognition receptors that bind, and in certain cases, hydrolyze peptidoglycans (PGNs) of bacterial cell walls (Marchler-Bauer and Bryant, 2004; Marchler-Bauer et al., 2009, 2011, 2013, 2015). The PGRP superfamily includes Amidase_2 family. This family includes Zn-dependent N-Acetylmuramoyl-L-alanine Amidase (Marchler-Bauer and Bryant, 2004; Marchler-Bauer et al., 2009, 2011, 2013, 2015). This enzyme cleaves the amide bond between N-acetylmuramoyl and L-amino acids in bacterial cell walls.
Figure 6a shows a representative structure of cell wall peptidoglycan with arrows indicating putative target sites for amidase activity. Cell wall fragments that are expected to be released due to amidase activity—TP, TriP-TP, and TP-TP—are highlighted by the ovals in Figure 6a. The elution profile of muropeptide fragments released after digestion of peptidoglycan using CD11 (Fig. 6b), and CDG (Fig. 6c) consisted of peaks corresponding to TP, TriP-TP, and TP-TP based on mass spectrometry analysis, confirming that CD11 and CDG both have N-acetylmuramoyl-L-alanine amidase activity.
Figure 6.

N-acetylmuramoyl-L-alanine amidase activity of CD11 and CDG determined using LC-MS analysis. (a) Cell wall peptidoglycan structure indicating putative target site for amidase a activity. Elution profile of muropeptide fragments released after digestion of peptidogly can using (b) CD11, and (c) CDG. A – Amidase, TriP – Tripeptide consisting of L-ala – D-glu – Diaminopimelic acid, TP– Tetrapeptide consisting of L-ala – D-glu – Diaminopimelic acid – D-ala, TriP-TP– TriP cross-linked through its diaminopimelic acid to D-ala of TP, TP-TP– TP cross-linked through its diaminopimelic acid to D-ala of other TP, Ovals indicate the muropeptide fragments expected to release as a result of amidase activity. The results indicate that CDG and CD11 has N-acetylmuramoyl amidase activity.
Conclusion
Given the increasing incidence of infections with antibiotic resistant bacteria, there is a need to develop new antimicrobial agents. We have exploited the modular structure of lytic enzymes in combination with the sequence of a known lytic enzyme that targets a different bacterial species to identify putative lytic enzymes against C. difficile. We demonstrated that these enzymes were highly active against vegetative cells of C. difficile (achieving a more than 4-log reduction in the number of viable bacteria using as low as 150 μg of enzyme) and had exquisite selectivity against clinical isolates of C. difficile, while being ineffective against Bacillus or Staphylococcal species. The characterization of the biocatalytic activity of these enzymes identified the specific bonds cleaved by these enzymes within the cell wall peptidoglycan. The approach used in this work and our past work (Grover et al., 2014; Mehta et al., 2013) should enable the effective decontamination of any bacterial pathogen by sequencing its genome and using bioinformatics to identify putative lytic enzymes. In previous work, we have incorporated other lytic enzymes into antimicrobial nanocomposites (Miao et al., 2011; Pangule et al., 2010; Solanki et al., 2013). The lytic enzymes identified in this work could be encapsulated in enteric polymers (Jain et al., 2005), thereby protecting the lytic enzymes from the acidic environment in the stomach while delivering the enzymes to the colon to target C. difficile. Other approaches that could be explored in the future include the administration of probiotics engineered to express these lytic enzymes for the treatment of C. difficile associated diarrhea (CDAD) (Mayer et al., 2008). Such approaches may be very useful in combating the growing concern posed by bacterial pathogens such as C. difficile.
Supplementary Material
Acknowledgments
Contract grant sponsor: DTRA
Contract grant number: W9132T-11-C-0025
Contract grant sponsor: Korean National Research Foundation
We thank Dmitri Zagorevski for his help with LC-MS experiments and analyses, and we gratefully acknowledge discussions with Dr. K. Solanki. This work was partly supported by DTRA (W9132T-11-C-0025) and a Global Research Lab grant from the Korean National Research Foundation (2014K1A1A2043032).
Footnotes
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Baker JR, Liu C, Dong S, Pritchard DG. Endopeptidase and glycosidase activities of the bacteriophage B30 lysin. Appl Environ Microbiol. 2006;72(10):6825–6828. doi: 10.1128/AEM.00829-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett JG, Chang TW, Gurwith M, Gorbach SL, Onderdonk AB. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N Engl J Med. 1978;298(10):531–534. doi: 10.1056/NEJM197803092981003. [DOI] [PubMed] [Google Scholar]
- Blackburn NT, Clarke AJ. Identification of four families of peptidoglycan lytic transglycosylases. J Mol Evol. 2001;52(1):78–84. doi: 10.1007/s002390010136. [DOI] [PubMed] [Google Scholar]
- Bourguet FA, Souza BE, Hinz AK, Coleman MA, Jackson PJ. Characterization of a novel lytic protein encoded by the Bacillus cereus E33L gene ampD as a Bacillus anthracis antimicrobial protein. Appl Environ Microbiol. 2012;78(8):3025–3027. doi: 10.1128/AEM.06906-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawley WN, Underwood S, Freeman J, Baines SD, Saxton K, Stephenson K, Owens RC, Jr, Wilcox MH. Efficacy of hospital cleaning agents and germicides against epidemic Clostridium difficile strains. Infect Control Hosp Epidemiol. 2007;28(8):920–925. doi: 10.1086/519201. [DOI] [PubMed] [Google Scholar]
- Fischetti VA. Bacteriophage lytic enzymes: Novel anti-infectives. Trends Microbiol. 2005;13(10):491–496. doi: 10.1016/j.tim.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Fischetti VA. Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol. 2008;11(5):393–400. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover N, Paskaleva EE, Mehta KK, Dordick JS, Kane RS. Growth inhibition of Mycobacterium smegmatis by mycobacteriophage-derived enzymes. Enzyme Microb Technol. 2014;63:1–6. doi: 10.1016/j.enzmictec.2014.04.018. [DOI] [PubMed] [Google Scholar]
- He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HM, Quail MA, Rance R. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci. 2010;107(16):7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgason E, Økstad OA, Caugant DA, Johansen HA, Fouet A, Mock M, Hegna I, Kolstø A-B. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensisone species on the basis of genetic evidence. Appl Environ Microbiol. 2000;66(6):2627–2630. doi: 10.1128/aem.66.6.2627-2630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D, Panda AK, Majumdar DK. Eudragit S100 entrapped insulin microspheres for oral delivery. AAPS PharmSciTech. 2005;6(1):E100–E107. doi: 10.1208/pt060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med. 1994;330(4):257–262. doi: 10.1056/NEJM199401273300406. [DOI] [PubMed] [Google Scholar]
- Korndorfer IP, Kanitz A, Danzer J, Zimmer M, Loessner MJ, Skerra A. Structural analysis of the L-alanoyl-D-glutamate endopeptidase domain of Listeria bacteriophage endolysin Ply500 reveals a new member of the LAS peptidase family. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt 6):644–650. doi: 10.1107/S0907444908007890. [DOI] [PubMed] [Google Scholar]
- Kyne L, Hamel MB, Polavaram R, Kelly CP. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin Infect Dis. 2002;34(3):346–353. doi: 10.1086/338260. [DOI] [PubMed] [Google Scholar]
- Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessner MJ. Bacteriophage endolysins—Current state of research and applications. Curr Opin Microbiol. 2005;8(4):480–487. doi: 10.1016/j.mib.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Loessner MJ, Kramer K, Ebel F, Scherer S. C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol Microbiol. 2002;44(2):335–349. doi: 10.1046/j.1365-2958.2002.02889.x. [DOI] [PubMed] [Google Scholar]
- Loessner MJ, Maier SK, Daubek-Puza H, Wendlinger G, Scherer S. Three Bacillus cereus bacteriophage endolysins are unrelated but reveal high homology to cell wall hydrolases from different bacilli. J Bacteriol. 1997;179(9):2845–2851. doi: 10.1128/jb.179.9.2845-2851.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low LY, Yang C, Perego M, Osterman A, Liddington RC. Structure and lytic activity of a Bacillus anthracis prophage endolysin. J Biol Chem. 2005;280(42):35433–35439. doi: 10.1074/jbc.M502723200. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Bryant SH. CD-search: Protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M. CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37(suppl 1):D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39(suppl 1):D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. CDD: Conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41(D1):D348–D352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43(D1):D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MJ, Garefalaki V, Spoerl R, Narbad A, Meijers R. Structure-based modification of a Clostridium difficile-targeting endolysin affects activity and host range. J Bacteriol. 2011;193(19):5477–5486. doi: 10.1128/JB.00439-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MJ, Narbad A, Gasson MJ. Molecular characterization of a Clostridium difficile bacteriophage and its cloned biologically active endolysin. J Bacteriol. 2008;190(20):6734–6740. doi: 10.1128/JB.00686-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald LC. Clostridium difficile: Responding to a new threat from an old enemy. Infect Control Hosp Epidemiol. 2005;26(8):672–675. doi: 10.1086/502600. [DOI] [PubMed] [Google Scholar]
- McDonald LC, Killgore GE, Thompson A, Owens RC, Jr, Kazakova SV, Sambol SP, Johnson S, Gerding DN. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353(23):2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- Mehta KK, Paskaleva EE, Azizi-Ghannad S, Ley DJ, Page MA, Dordick JS, Kane RS. Characterization of AmiBA2446, a novel bacteriolytic enzyme active against Bacillus species. Appl Environ Microbiol. 2013;79(19):5899–5906. doi: 10.1128/AEM.02235-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrigan M, Venugopal A, Mallozzi M, Roxas B, Viswanathan V, Johnson S, Gerding DN, Vedantam G. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol. 2010;192(19):4904–4911. doi: 10.1128/JB.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Pangule RC, Paskaleva EE, Hwang EE, Kane RS, Linhardt RJ, Dordick JS. Lysostaphin-functionalized cellulose fibers with antistaphylococcal activity for wound healing applications. Biomaterials. 2011;32(36):9557–9567. doi: 10.1016/j.biomaterials.2011.08.080. [DOI] [PubMed] [Google Scholar]
- Pangule RC, Brooks SJ, Dinu CZ, Bale SS, Salmon SL, Zhu G, Metzger DW, Kane RS, Dordick JS. Antistaphylococcal nanocomposite films based on enzyme-nanotube conjugates. ACS Nano. 2010;4(7):3993–4000. doi: 10.1021/nn100932t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien A, Allain B, Zhang J, Mandeville R, Lan CQ. Novel alternatives to antibiotics: Bacteriophages, bacterial cell wall hydrolases, and antimicrobial peptides. J Appl Microbiol. 2008;104(1):1–13. doi: 10.1111/j.1365-2672.2007.03498.x. [DOI] [PubMed] [Google Scholar]
- Peltier J, Courtin P, El Meouche I, Lem ee L, Chapot-Chartier M-P, Pons J-L. Clostridium difficile has an original peptidoglycan structure with a high level of N-acetylglucosamine deacetylation and mainly 3-3 cross-links. J Biol Chem. 2011;286(33):29053–29062. doi: 10.1074/jbc.M111.259150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter CJ, Schuch R, Pelzek AJ, Buckle AM, McGowan S, Wilce MC, Rossjohn J, Russell R, Nelson D, Fischetti VA, Whisstock JC. The 1.6A crystal structure of the catalytic domain of PlyB, a bacteriophage lysin active against Bacillus anthracis. J Mol Biol. 2007;366(2):540–550. doi: 10.1016/j.jmb.2006.11.056. [DOI] [PubMed] [Google Scholar]
- Pritchard DG, Dong S, Kirk MC, Cartee RT, Baker JR. LambdaSa1 and LambdaSa2 prophage lysins of Streptococcus agalactiae. Appl Environ Microbiol. 2007;73(22):7150–7154. doi: 10.1128/AEM.01783-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz JE, Schuch R, Fischetti VA. Identifying active phage lysins through functional viral metagenomics. Appl Environ Microbiol. 2010;76(21):7181–7187. doi: 10.1128/AEM.00732-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch R, Nelson D, Fischetti VA. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature. 2002;418(6900):884–889. doi: 10.1038/nature01026. [DOI] [PubMed] [Google Scholar]
- Severin A, Tabei K, Tomasz A. The structure of the cell wall peptidoglycan of Bacillus cereus RSVF1, a strain closely related to Bacillus anthracis. Microb Drug Resist. 2004;10(2):77–82. doi: 10.1089/1076629041310082. [DOI] [PubMed] [Google Scholar]
- Shockman GD, Daneo-Moore L, Kariyama R, Massidda O. Bacterial walls, peptidoglycan hydrolases, autolysins, and autolysis. Microb Drug Resist. 1996;2(1):95–98. doi: 10.1089/mdr.1996.2.95. [DOI] [PubMed] [Google Scholar]
- Solanki K, Grover N, Downs P, Paskaleva EE, Mehta KK, Lee L, Schadler LS, Kane RS, Dordick JS. Enzyme-based listericidal nanocomposites. Sci Rep. 2013;3:1584. doi: 10.1038/srep01584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg JA, Dineen SS. Laboratory maintenance of Clostridium difficile. Curr Protoc Microbiol. 2009;12:A:9A.1:9A.1.1–9A.1.10. doi: 10.1002/9780471729259.mc09a01s12. [DOI] [PubMed] [Google Scholar]
- Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10(9):R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele J, Chen K, Sun X, Zhang Y, Wang H, Tzipori S, Feng H. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis. 2012;205(3):384–391. doi: 10.1093/infdis/jir748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wang H, Zhang Y, Chen K, Davis B, Feng H. Mouse relapse model of Clostridium difficile infection. Infect Immun. 2011;79(7):2856–2864. doi: 10.1128/IAI.01336-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun X, Zhang Y, Li S, Chen K, Shi L, Nie W, Kumar R, Tzipori S, Wang J. A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect Immun. 2012;80(8):2678–2688. doi: 10.1128/IAI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366(9491):1079–1084. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- Wilcox MH, Fawley WN. Hospital disinfectants and spore formation by Clostridium difficile. Lancet. 2000;356(9238):1324. doi: 10.1016/S0140-6736(00)02819-1. [DOI] [PubMed] [Google Scholar]
- Yang Z, Liu W, Schmidt D, Shi L, Li S, Chen K, Sheng J, Tremblay JM, Yu H. A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J Infect Dis. 2014;210(6):964–972. doi: 10.1093/infdis/jiu196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.