

Abstract
Early embryonic stages of pluripotency are modeled for epigenomic studies primarily with human embryonic stem cells (ESC) or induced pluripotent stem cells (iPSCs). For analysis of DNA methylation, however, ESCs and iPSCs do not accurately reflect the DNA methylation levels found in preimplantation embryos. Whole genome bisulfite sequencing (WGBS) approaches have revealed the presence of large partially methylated domains (PMDs) covering 30–40% of the genome in oocytes, preimplantation embryos, and placenta. In contrast, ESCs and iPSCs show abnormally high levels of DNA methylation compared to inner cell mass (ICM) or placenta. Here we show that dental pulp stem cells (DPSCs), derived from baby teeth and cultured in serum-containing media, have PMDs and mimic the ICM and placental methylome more closely than iPSCs and ESCs. By principal component analysis, DPSC methylation patterns were more similar to two other neural stem cell types of human derivation (EPI-NCSC and LUHMES) and placenta than were iPSCs, ESCs or other human cell lines (SH-SY5Y, B lymphoblast, IMR90). To test the suitability of DPSCs in modeling epigenetic differences associated with disease, we compared methylation patterns of DPSCs derived from children with chromosome 15q11.2–q13.3 maternal duplication (Dup15q) to controls. Differential methylation region (DMR) analyses revealed the expected Dup15q hypermethylation at the imprinting control region, as well as hypomethylation over SNORD116, and novel DMRs over 147 genes, including several autism candidate genes. Together these data suggest that DPSCs may be useful model for epigenomic and functional studies of human neurodevelopmental disorders.
Keywords: epigenetics, epigenomics, DNA methylation, neural stem cells, teeth, dental pulp stem cells, human disease models, imprinting
Graphical abstract
The global DNA methylome profiles (right) identified from human dental pulp stem cells (DPSCs) derived from exfoliated baby teeth are more similar to those of early life tissues derived from the embryo and placenta than induced pluripotent stem cells (iPSCs) derived from skin. For investigation of human diseases with altered DNA methylation patterns, such as Dup15q syndrome, DPSCs are a relevant cell culture model.

Introduction
The epigenetic layer of DNA methylation is of increasing interest to investigate in the pathogenesis of human diseases, particularly those with potential fetal origins. The global DNA methylation landscapes of early life human tissues, such as oocytes, blastocyst, ICM, or placenta, are characterized by a distinct genome-wide hypomethylation compared to differentiated tissues post implantation [1]. Additional features of the early life DNA methylome include a bimodal distribution of DNA methylation levels and higher relative levels of gene body methylation, which makes them distinct from differentiated tissues [2–4]. However, somatic tissues and most human embryonic stem cell types only partially fulfill one of these characteristics, which is modest but significant higher methylation levels over active gene bodies [5].
The bimodal distribution of DNA methylation levels is due to the presence of partially methylated domains (PMDs) in oocyte, pre-implantation embryos, and placenta [2, 3, 6]. PMDs are characterized by repressed but inducible transcription and chromatin inaccessibility compared to highly methylated domains (HMDs) [7–10]. Following implantation, the fetal tissues developing from the inner cell mass (ICM) gain global DNA methylation and lose PMDs, while the placental cells arising from the trophectoderm retain PMDs, global hypomethylation, and relative gene body hypermethylation throughout pregnancy [2, 6]. A likely reason for the aberrant methylation patterns observed in iPSCs, ESCs, and other pluripotent stem cells is the tendency in culture for pluripotent cells to drift towards a “post-inner cell mass intermediate” stage that is characteristic of the post-implantation embryonic disk [11, 12]. Since epigenome-wide analyses are often performed on cell types derived from iPSCs and ESCs, the question arises: how well is the pre-implantation epigenome retained? Furthermore, investigations of epigenetic alterations in human disease states using iPSCs may be confounded by this in vitro modified epigenome.
Dental pulp stem cells (DPSCs) are neural crest-derived cultures derived from exfoliated baby teeth that can be differentiated into a multitude of cell types including neurons [13–16]. DPSCs also provide a practical solution to the collection of a large number of stem cells from even rare disorders since they can be easily collected from remote locations and transported to the laboratory for growth without the need for biopsy or cellular reprogramming. However, these cells have not yet been characterized by genome-wide epigenetic approaches to ensure that proper early developmental epigenetic patterns are in place prior to differentiation into disease specific cell types (i.e. neurons, chondroplasts, adiopocytes, etc.). In this study, we performed whole genome bisulfite sequencing (WGBS) of DNA methylation in DPSCs and compared methylation features and differences to the more common stem cell lines used in human genetic studies (ESCs and iPSCs), as well other lines used for human neuronal cultures (LUHMES and EPI-NSCs), to previous methylome maps of human tissues and cell lines.
Materials and Methods
Sample acquisition and DNA purification
Teeth for the generation of dental pulp stem cell lines (DPSC) were obtained from neurotypical control subjects at the Memphis Pediatric Dental Clinic. Each DPSC cell line described in Supplementary Table 1 was derived from a single donated tooth. Chromosome duplication 15q11.2–q13.3 syndrome (Dup15q) subjects were recruited through the Dup15q Alliance. All subjects gave informed consent for the storage and research use of their DPSC in accordance with the UTHSC Institutional Review Board. All teeth were processed into DPSC lines according to previously published protocols [15, 16] and frozen at early passages (2–5) for use in these studies. For these studies, cells were grown to confluence, split one time and DNA extracted using Genera Puregene Kit.
LUHMES (Lund Human Mesencephalic) cells, which were originally derived from 8-week-old female ventral mesencephalon, were a generous gift from Dr. Marcel Leist (University of Konstanz, Germany). LUHMES cells were maintained in the proliferative undifferentiated state as previously described [17]. Cells used for these experiments were at passage 14. Epidermal neural crest stem cells (EPI-NCSCs) were isolated from bulge explants of hair follicles dissected from full-thickness human skin biopsies provided by the UC Davis Body Donation Program and Department of Dermatology. EPI-NCSC sample 1 was derived from scalp cadaver tissue from an 85-year-old female, with cells at passages 7–8. EPI-NCSC sample 2 was isolated from leftover Moh’s surgery tissue from a male, age unknown, with cells at passage 6. EPI-NCSCs were cultured in the proliferative, undifferentiated state according to a previously published protocol [18]. DNA was isolated from cell pellets using the Qiagen (Valencia, CA) DNeasy Blood and Tissue Kit according to manufacturer’s instructions.
WGBS library prep
Samples were prepared as described previously. Briefly, 500 ng of DNA was bisulfite converted using Zymo’s EZ DNA Methylation-Lightning kit according to the manufacturer’s instructions. Then, 100 ng of converted DNA was used to prepare the library using the EpiGnome/TruSeq DNA Methylation. The manufacturer’s instructions were followed except we performed 14 cycles of amplification instead of 10 in the last PCR step.
Code availability
All custom scripts and code are available for download at https://github.com/kwdunaway/WGBS_Tools as well as instructions on how to use them. Brief descriptions of each script are also on the wiki of this site.
WGBS alignment
Raw FASTQ files were filtered using Illumina quality score flag, then split into two files based on adapter contamination. Reads were trimmed to remove the adapters as well as the last 10 bases before the adapters in order to remove biased hypomethylation contamination known to occur at DNA within 10 bases of the 3′ adapter [19]. Reads were then aligned to the human genome (hg38) using BS-Seeker2 [20]. Conversion efficiency was determined by analyzing methylation percentage of mitochondria DNA since it is expected to have no detectable levels of methylation [21]. All but one of the samples had over 99.5% conversion efficiency (Supplementary Table 1), which was determined by analyzing the conversion rate of the mitochondrial DNA using script ConvEff_SAM.pl.
Windowing
Average coverage of each CpG was around 3×, which is enough to confidently call 20kb windows [22]. Methylation data for CpGs found within CpG islands were masked before windowing. Windows that had less than 20 CpGs assayed for any of the 12 brain samples were also removed. Methylation was determined for each 20kb using script Window_permeth_readcentric.pl through the following formula:
Significant differential methylation between Dup15q and control was determined through an unpaired twin-tailed t-test resulting in p<0.05. Hypomethylation was called if Dup15q methylation was less than control and hypermethylation if the inverse pattern was observed.
Gene body methylation
Gene body methylation was determined using the same formula as windowing, except CpG Islands were included and a minimum of 10 CpGs for each gene was used as a cut-off. Gene body was defined as the region from the transcription start site to the transcription end site.
Principal Component Analysis
Principal component analysis (PCA) is a mathematical algorithm that reduces the dimensionality of the data down to two dimensions while retaining most of the variation in the data set [23]. Briefly, using the principal component with the highest distribution of data (PC1) as the x-axis and the second highest principal component (PC2) as the y-axis, the data is distributed as evenly across the plot as possible while maintaining distance between points as a proxy for how similar each point is to each other.
CNV determination using WGBS read depth data
CNVs were detected using a window-based approach that leveraged read depth of control samples to detect coverage differences in other samples using Line1_FASTQ.pl. In order to account for low coverage areas of the genome, a minimum coverage of 30 reads was set for all control samples [24]. A window size of 5 kb was chosen because the average coverage was around 3x. 5 kb window size was chosen because it should have 150 reads on average per window. Read depth was previously discovered to infer CNVs in Bisulfite Sequencing using ReadDepth [25].
Differentially Methylated Regions
DMRs were called using the R packages DSS and bsseq along with custom R commands [26, 27]. Example code can be found in examples/DRM_analysis.R on the github site. Briefly, a BSseq object was made for each chromosome using the methylation data for all samples used in a comparison then smoothed using BSmooth. CpGs were removed if more than one sample had zero reads covering it per condition. Significance was tested using BSmooth. tstat and DMRs were identified with dmrFinder as sets of CpGs with a t-statistic greater than the critical value for alpha = 0.05 and with a gap < 300 bases. Only DMRs with > 3 CpGs, a mean methylation difference > 10%, and an areaStat > 20 were kept. DMRs were associated with the nearest gene (max distance 5kb away).
Gene Ontology
Gene lists were determined through either DMR or PMD analysis for given conditions and uploaded as official gene symbols into DAVID Gene Ontology online tool. The following pathways were selected for analysis: SP_PIR_Keywords, UP_seq_feature, GOTERM_BP_FAT, GOTERM_CC_FAT, GOTERM_MF_FAT, KEGG_PATHWAY, PIR_tissue_specificity, UP_tissue. Pathways with a Bonferroni score of < .01 were kept and genes for each category were provided.
Results
DPSCs are a better model of global early life methylation patterns than pluripotent stem cell lines
In a global landscape view of the DNA methylome using gene body and 20 kb windowing of WGBS data, there are three major characteristics of placenta and ICM tissues: 1) bimodal distribution of 20 kb windows; 2) higher relative gene body methylation and; 3) global hypomethylation (< 70% CpG methylation). All of these characteristics are lacking in differentiated tissues (adult brain cortex and fetal liver) as well as ESC lines (H1 and H9) and iPSCs (Fig. 1a and Supplementary Table 1). These three methylome features are also in common with two transformed cell lines, including SH-SY5Y neuroblastoma and EBV-transformed B lymphoblast lines that contain PMDs [28, 29]. Two additional cell types used for neuronal differentiation, the human mesencephalic-derived LUHMES cell line [30] and neural crest stem cells isolated from human hairy skin tissue (EPI-NCSCs) [31] showed some but not all methylome features. Namely, while LUHMES and EPI-NCSCs are hypomethylated, they do not exhibit a bimodal 20 kb window pattern observed in placenta and ICM. In contrast, DPSCs grown with minimal (2–5) passages mimic the methylation pattern found in placenta and ICM tissues, although, there was some expected variability between hypomethylated patterns between individual DPSC lines (Fig. 1b and Supplementary Table 1).
Figure 1. Comparison of whole genome methylome profiles between DPSCs and other stem cells and tissues.
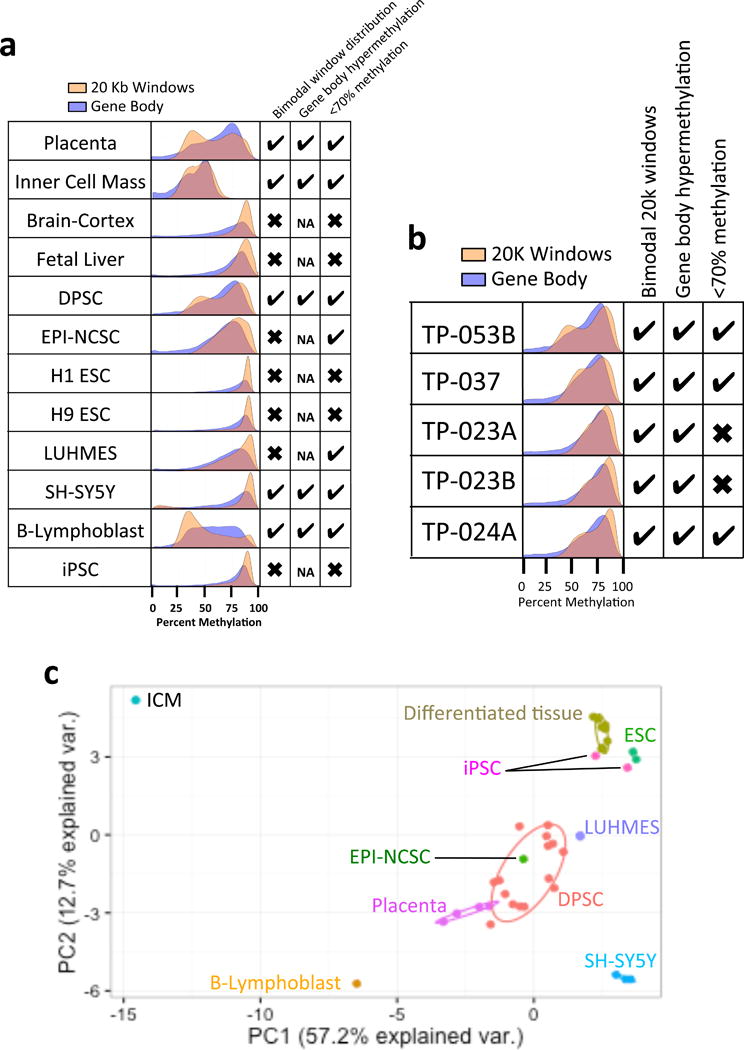
a. Histograms showing methylation patterns of gene body (blue) and 20 kb windows (gold), with CpG islands excluded. Check boxes indicate: left, whether two peaks were observed in the 20 kb window histogram; center, if gene body methylation was biased to the right (HMD) peak of the 20 kb window histogram (or NA for “not applicable” if bimodal peaks were absent); and right, hypomethylation indicated by < 70% average global mean % CpG methylation (sequencing data summaries and sources in Supplementary Table 1). b. Histograms show global mean % CpG methylation across 5 different control neurotypical DPSC cultures, revealing individual variability. c. Principal compenents analysis (PCA) of 20 kb window and gene body methylation on all samples in (a). Differentiated tissues refers to the combination of brain cortex and fetal lung samples that clustered together and closer to ESCs and iPSCs, while DPSC cluster closely with EPI-NCSC, LUHMES and placenta.
Using Principal Component Analysis (PCA) on methylation of 20 kb windows and gene body, there was a clear cluster of stem cell lines with similar methylation pattern to the four placenta samples. DPSCs, EPI-NCSCs, and LUHMES cells cluster closer to placenta than to other tissues (Fig. 1c). ESCs (H1 and H9) and iPSCs cluster closest differentiated tissues (brain cortex and fetal liver). The cell cultures SH-SY5Y and GM12878 (B-lymphoblast) all clustered independently from any other cell type. Interestingly, ICM cells were further away from any other cell type analyzed, likely due to the prominent hypomethylation of this sample. As ICM data quality could have been affected by the low cell number in this very limited tissue type [2] we used placental samples as an early life tissue for correlation analyses with stem cell lines (Figure 2 and Supplementary Figures 1–5). DPSCs showed the strongest correlation with placenta when comparing methylation levels over gene bodies (Fig. 2a,d) and 20 kb windows (Fig. 2d and S2d). A subset of genes were hypermethylated in every long-term cultured cell line compared to Placenta (Fig. 2b and S1f–k), except for B-Lyphoblast. These cells also exhibited in lower correlations (R2 < 0.65) than DPSCs (R2 ~ 0.76) and EPI-NCSCs (R2 ~ 0.74) (Fig. 2d).
Figure 2. Placenta gene body methylation correlates closest to DPSCs.
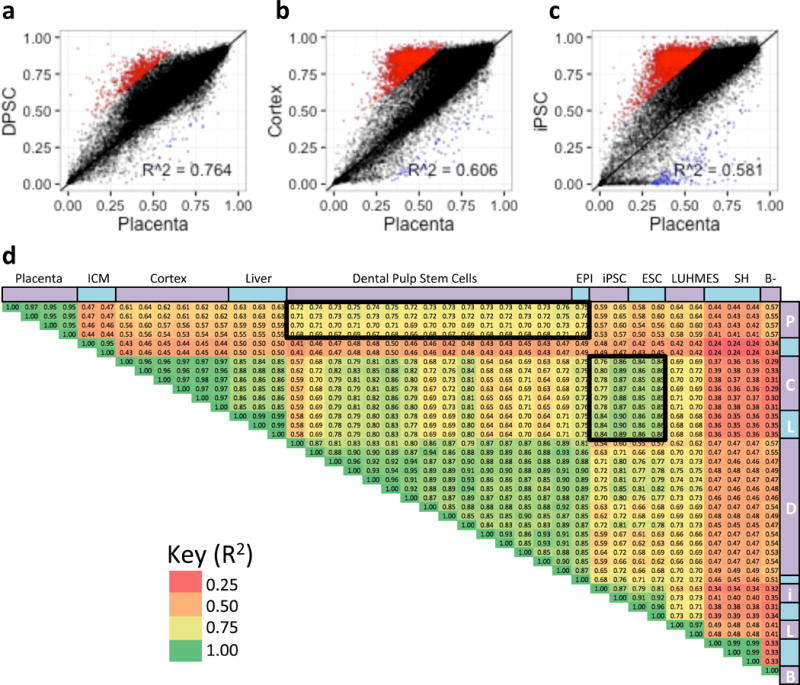
a–c. Scatterplots showing correlations of gene body mean % methylation from select samples in Figure 1a compared to placenta. Windows hypomethylated (red) or hypermethylated (blue) in placenta versus each comparison tissue were defined as greater than 30% methylation difference from Placenta. d. Pearson correlation plot of combined gene body and 20 kb window correlations (R2) of each samples compared to each other. Heat map colors correspond to R2 value.
DPSCs can be used to detect disease-relevant methylation differences in Dup15q syndrome
To determine whether DPSCs can be used effectively to detect methylation differences in a genetic neurodevelopmental disease, we investigated chromosome 15q11.2–q13.3 duplication (Dup15q) syndrome, a recurrent copy number variant (CNV) observed in autism spectrum disorders [32]. Four different short-term DPSC cultures from isodicentric Dup15q syndrome subjects were compared to five different cultures from neurotypically developing controls. To confirm that the 4× 15q11.2–q13.3 copy number was maintained in cell culture, we used read count analysis from WGBS data across the 15q region (Fig. 3a). To determine if methylation markers of 15q11.2–q13.3 duplication were maintained in DPSCs, the 2 kb Prader-Willi syndrome imprinting control region (PWS-IC) was compared between Dup15q and control DPSCs (Fig. 3b–d). Significant hypermethylation of the PWS-IC was observed in Dup15q compared to control DPSCs, as expected based on maternal origin and similar differences observed in somatic tissues [33]. A wider analysis of methylation differences over 20 kb windows across the imprinted 15q11.2–q12 locus demonstrated a hypomethylated block over the SNORD116 locus implicated in PWS, but no significant change over the entire imprinted gene cluster (Fig. 3b–c). An analysis of differentially methylated regions (DMR) in DPSC lines also detected novel DMRs associated with 73 hypomethylated and 74 hypermethylated genes in Dup15q DPSCs compared to Controls (Table 1). This gene list was compared to the SFARI gene list of autism candidates (gene.sfari.org) and several genes overlapped with genetic evidence, including RAI1, encoding retinoic acid induced 1, duplicated in the ASD Potocki-Lupski syndrome [34]. In addition, multiple genes identified from DMR analysis of Dup15q DPSCs also corresponded as those found with DMRs in Dup15q syndrome compared to control postmortem brain samples [35]. We found significant enrichment in the number of genes identified by DMRs in Dup15q brain and DPSCs over the number expected to overlap by chance for both hypomethylated and hypermethylated regions (Fig. 3e).
Figure 3. Assessment of DPSCs as a model of methylation differences in Dup15q syndrome.
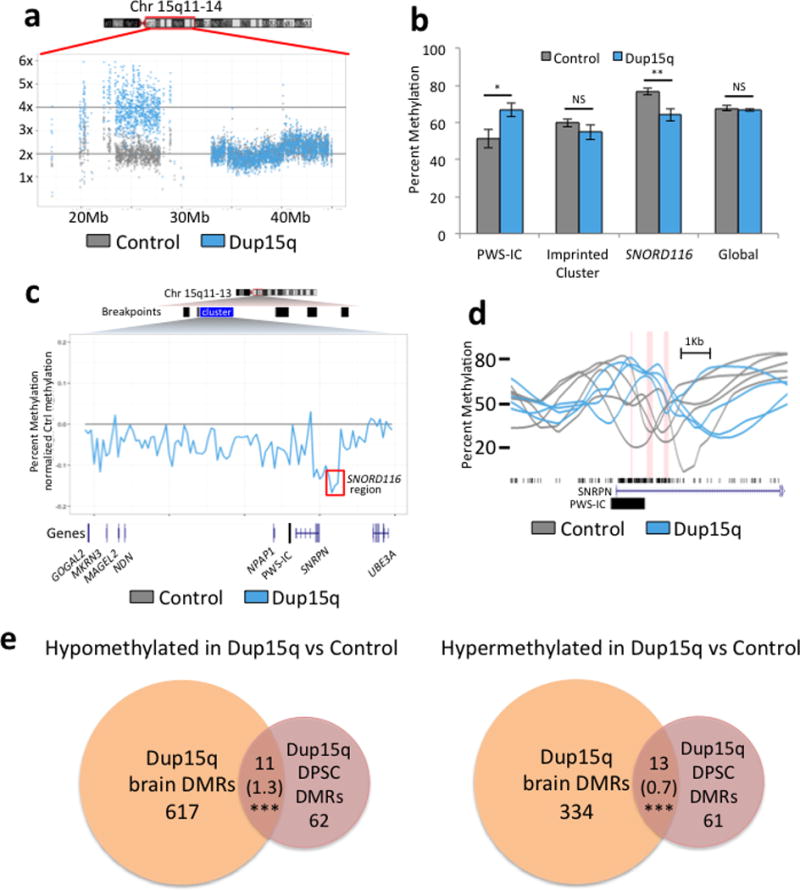
a. Normalized read coverage over the region duplicated in Dup15q syndrome showing 2× in a Control culture and 4× in a Dup15q culture, as expected for the maintenance of this large CNV in culture. b. Imprinted and global methylation patterns compared between averages from 4 Dup15q and 5 control DPSC cultures. Bar graphs demonstrate expected hypermethylation over the maternally methylated PWS-IC (chr15:24953253-24957502) but no significant change in methylation over the entire imprinted cluster or global mean % methylation. A novel DMR corresponding to the SNORD116 PWS locus (chr15:25042076-25109152) was observed to be significantly hypomethylated in Dup15q compared to control DPSCs. *P<0.05, **P<0.01, or not significant (NS) by unpaired t-test. c. Average methylation difference between an average of 5 control and 4 Dup15q DPSC cultures using 20k windows over the imprinted 15q11–12 cluster (chr15:23440000-25500000). The red box highlights the SNORD116 DMR. d. Plots of % methylation from individual control (grey) and Dup15q (blue) DPSC cultures over three significantly hypomethylated regions (red columns) around the PWS-IC. e. Venn diagrams representing overlap of genic DMRs identified from Dup15q compared to control DPSC (this study, brick red) and those identified in Dup15q compared to control postmortem brain (Dunaway et al, 2016 [35], orange). The number of genes hypomethylated in Dup15q in both systems are represented on the left and hypermethylated genes are on the right. ***P<0.001 by Fisher’s exact test for observed number of genes overlapping versus expected (expected number in parentheses). Table 1 designates specific genes that were also aberrantly methylated in Dup15q brain (either hypo- or hyper-methylated).
Table 1.
Genes overlapping with differentially methylated regions
| Genes with hypomethylated DMRs | ||||
|---|---|---|---|---|
| AATK b | FOXQ1 | LIPE b | MMP25-AS1 | RASA3 b |
| AATK-AS1 | FRAT1 | LIPE-AS1 b | NBEA* b | RRN3P1 |
| ADRA2B | GFPT2 b | LOC100049716 b | OTX2-AS1 | RTEL1-TNFRSF6B |
| AKAP10 | GJD2 | LOC100128568 | PABPC1L | SERPINB9P1 |
| AMH | GMDS-AS1 | LOC101930071 | PAPL b | SPNS2 |
| ATP8B1 | GNA15 | LOC729506 b | PAX8-AS1 | SPRNP1 |
| CAMTA1* b | GNB1L* | LSP1P3 | PAX9 | ST8SIA1 |
| CARM1 | HCN2 | MAB21L1 | PDP2 | SULT4A1 |
| CHD2# | HSBP1L1 | MEG9 b | PDXK b | TDRD9 b |
| CHST13 | IL17RD | MEI1 | PGAM2 | TMEM240 |
| COL27A1 | IRX1 | MIR3150A | PIP5KL1 | TMEM72-AS1 |
| EGFR | JSRP1 | MIR3150B | PRKCG | TNFRSF6B |
| FAM19A5 b | KCNK15 | MIR4458HG b | PRR35 | TRAF2 |
| FAM90A1 | KIAA1024 | MIR548AI b | RAI1* b | ZCCHC14 |
| FHAD1 b | LHX9 | MMP25 | ||
| Genes with hypermethylated DMRs region | ||||
| ADCY2 b | DPP9 | LHFPL2 | PRKCDBP | TEX101 b |
| ADORA2B | DUSP5 | LINC00664 | R3HDM4 b | TINCR b |
| ASB16 | EIF2AK4 b | LINC01252 b | RASL11A | TM4SF1 |
| BTN1A1 | EPHA1 | LOC100507389 | RIMS1* b | TMEM102 |
| C3 | EVA1B | LOC101926935 | RSPO3 | TMEM200C b |
| CACNA2D4 | FAM171A2 b | LOC101929124 | SHH | TNFAIP8L1 b |
| CAPN15 | FAM207A b | LOC102724927 | SLC7A5 b | TONSL |
| CDH4 | FRMD4A b | LOC339807 | SMG9 | VIPR2 |
| CHMP6 | FUK b | LOC442028 | SNRPN+ b | WNT7B b |
| CHST2 | HDAC4+ b | LOC728613 b | SNURF+ b | YPEL1 |
| CNTFR | IL21R-AS1 | LRRC14B | SPRY2 b | ZC3H4 |
| COG1 | INS-IGF2 | MVB12B b | SPTBN4 | ZFP36 |
| CRLF1 | JAK3 | NOTCH1 b | STPG2 | ZMIZ1 b |
| CYP26C1 | KCNE4 | NUDT19 | TBCD b | ZNF703 b |
| DOT1L | KLHL26 | NXN | TES | |
autism candidate, rare single gene variant
autism candidate, copy number variant
autism candidate, genetic association
DMR also found either hypermethylated or hypomethylated in Dup15q brain (Dunaway, et al, in press)
While we were successful in identifying some methylation differences associated with Dup15q samples, there were some limitations of the DPSC cultures. First, both the global (Fig 1b) and local (Fig. 3d) methylation patterns were inherently variable between individual DPSC lines for both control and Dup15q (Fig. S6, S7a–b). In addition, because of their globally hypomethylated epigenomic state and the need for expansion in culture, DPSCs appear to be susceptible to genome instability, resulting in de novo copy number variants (CNVs). In one of the Dup15q DPSC lines, a de novo CNV duplication was observed on chromosome 17q23.2–25.3 (Fig. 3e). However, the 17q duplication had minimal impacts on DNA methylation over 20 kb windows or gene bodies (Fig. S7c–d).
Discussion
Here we demonstrate a potential advantage of using DPSCs to investigate epigenetic alterations in human disorders through a comparative analysis of global DNA methylation patterns by WGBS from different tissue and stem cell types. Compared to other pluripotent or neuronal lineage human cultures, DPSCs more accurately model the early life ICM and placenta in their global methylome characteristics. However, we also found inherent difficulties of maintaining stability of both the methylome and genome in DPSC cultures. Genomic and epigenomic instabilities resulting in de novo CNVs and or methylation changes may occur in all types of human stem cell cultures, and are important to screen for with sequencing-based approaches. Our WGBS method of relatively low coverage sequencing is an approach that can be used to quantitate both whole methylome and CNV detection of stem cell cultures.
In spite of the inherent variability of methylation patterns between DPSC lines, we were able to identify both expected (PWS-IC) and novel areas of differential methylation of potential relevance to Dup15q syndrome. The SNORD116 locus is the minimal deletion region for PWS, encoding noncoding RNAs for snoRNAs as well as a long noncoding host gene 116HG involved in neuronal regulation of diurnal metabolism [36]. Interestingly, one of the Dup15q hypomethylated DMRs overlapped with the PTLS gene RAI1 which also has a role in circadian rhythms and chronobiology [34]. Furthermore, while the global hypomethylation previously observed in Dup15q syndrome postmortem brain was not detectable in the Dup15q DPSC lines compared to controls, several genes identified by the DMR analysis of DPSC cultures were in common, including RAI1 and the histone deacetylase encoding HDAC4. DPSC cultures may be useful in understanding the epigenetic alterations in early neuronal precursors to better understand epigenetic aspects of Dup15q syndrome molecular pathogenesis.
A subset of individual CpG probes from array-based methylation analyses have been significantly correlated with chronologic age and have also been used to predict epigenetic age [37]. While DNA methylation features in common to tissues containing PMDs (bimodal distribution, gene body hypermethylation, and global hypomethylation) are observed in early life preimplantation embryos and placenta, they were also observed in LUHMES cells derived from 8-week old tissues, DPSCs derived from children, and EPI-NCSCs derived from an 85-year old cadaver. Therefore, the commonalities in global DNA methylation patterns appear to be unrelated to subject chronologic age, and perhaps more reflect the maintenance of methylation patterns of stem cell types at relatively low passage numbers in culture. However, passage number of the cell lines did not appear to entirely explain the methylation differences in cell lines reported in this study (Supplementary Table 1).
Conclusions
These results demonstrate that DPSC methylation patterns were more similar to EPI-NCSC and LUHMES neural stem cell lines and placenta than were iPSCs, ESCs or other human cell lines. Furthermore, we identified novel DMRs associated with Dup15q over 147 genes, including several autism candidate genes, demonstrating the efficacy of DPSC lines in modeling epigenetic dysregulation in a human autism spectrum disorder.
Supplementary Material
Acknowledgments
This work was supported by NIH R01ES021707, NIH P01ES011269, and EPA 83543201 (JML) and NIH R21NS075709 (LTR). KD was supported by a National Institute of Environmental Health Sciences funded training program in Environmental Health Sciences (T32 ES007059). This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303 and the UC Davis MIND Institute Intellectual and Developmental Disabilities Research Center [U54 HD079125].
Footnotes
Keith Dunaway: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript
Sarita Goorha: provision of study material, collection and assembly of data, final approval of manuscript
Lauren Matelski: provision of study material, collection and assembly of data, final approval of manuscript
Nora Urraca: provision of study material and patients, final approval of manuscript
Pamela Lein: provision of study material, final approval of manuscript
Ian Korf: data analysis and interpretation, final approval of manuscript
Lawrence Reiter: conception and design, financial support, data analysis and interpretation, final approval of manuscript
Janine LaSalle: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript
Disclosures: The authors have no conflicts to disclose.
References
- 1.Nishino K, Umezawa A. DNA methylation dynamics in human induced pluripotent stem cells. Hum Cell. 2016 doi: 10.1007/s13577-016-0139-5. [DOI] [PubMed] [Google Scholar]
- 2.Guo H, Zhu P, Yan L, et al. The DNA methylation landscape of human early embryos. Nature. 2014;511:606–610. doi: 10.1038/nature13544. [DOI] [PubMed] [Google Scholar]
- 3.Schroeder DI, Blair JD, Lott P, et al. The human placenta methylome. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6037–6042. doi: 10.1073/pnas.1215145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kubo N, Toh H, Shirane K, et al. DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genomics. 2015;16:624. doi: 10.1186/s12864-015-1833-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kundaje A, Meuleman W, Ernst J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schroeder DI, Jayashankar K, Douglas KC, et al. Early Developmental and Evolutionary Origins of Gene Body DNA Methylation Patterns in Mammalian Placentas. PLoS Genet. 2015;11:e1005442. doi: 10.1371/journal.pgen.1005442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen KD, Sabunciyan S, Langmead B, et al. Large-scale hypomethylated blocks associated with Epstein-Barr virus-induced B-cell immortalization. Genome Research. 2014;24:177–184. doi: 10.1101/gr.157743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nature Genetics. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schroeder DI, LaSalle JM. How has the study of the human placenta aided our understanding of partially methylated genes? Epigenomics. 2013;5:645–654. doi: 10.2217/epi.13.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Leary T, Heindryckx B, Lierman S, et al. Tracking the progression of the human inner cell mass during embryonic stem cell derivation. Nat Biotechnol. 2012;30:278–282. doi: 10.1038/nbt.2135. [DOI] [PubMed] [Google Scholar]
- 12.Van der Jeught M, OO’Leary T, Duggal G, et al. The post-inner cell mass intermediate: implications for stem cell biology and assisted reproductive technology. Hum Reprod Update. 2015;21:616–626. doi: 10.1093/humupd/dmv028. [DOI] [PubMed] [Google Scholar]
- 13.El-Iyachi I, Goorha S, Reiter LT, et al. Effects of hTERT immortalization on osteogenic and adipogenic differentiation of dental pulp stem cells. Data in Brief. 2016 doi: 10.1016/j.dib.2016.01.009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gronthos S, Mankani M, Brahim J, et al. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A. 2000;97:13625–13630. doi: 10.1073/pnas.240309797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiraly M, Porcsalmy B, Pataki A, et al. Simultaneous PKC and cAMP activation induces differentiation of human dental pulp stem cells into functionally active neurons. Neurochem Int. 2009;55:323–332. doi: 10.1016/j.neuint.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Urraca N, Memon R, El-Iyachi I, et al. Characterization of Neurons from Immortalized Dental Pulp Stem Cells for the Study of Neurogenetic Disorders. Stem Cell Research. 2015;15:722–730. doi: 10.1016/j.scr.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scholz D, Poltl D, Genewsky A, et al. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem. 2011;119:957–971. doi: 10.1111/j.1471-4159.2011.07255.x. [DOI] [PubMed] [Google Scholar]
- 18.Clewes O, Narytnyk A, Gillinder KR, et al. Human epidermal neural crest stem cells (hEPI-NCSC)–characterization and directed differentiation into osteocytes and melanocytes. Stem cell reviews. 2011;7:799–814. doi: 10.1007/s12015-011-9255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo W, Fiziev P, Yan W, et al. BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC genomics. 2013;14:774. doi: 10.1186/1471-2164-14-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong EE, Okitsu CY, Smith AD, et al. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol. 2013;33:2683–2690. doi: 10.1128/MCB.00220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziller MJ, Hansen KD, Meissner A, et al. Coverage recommendations for methylation analysis by whole-genome bisulfite sequencing. Nat Methods. 2015;12:230–232. doi: 10.1038/nmeth.3152. 231 p following 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jolliffe I. Principal Components Analysis. New York: Springer; 2002. [Google Scholar]
- 24.Gusnanto A, Wood HM, Pawitan Y, et al. Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics. 2012;28:40–47. doi: 10.1093/bioinformatics/btr593. [DOI] [PubMed] [Google Scholar]
- 25.Miller CA, Hampton O, Coarfa C, et al. ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS One. 2011;6:e16327. doi: 10.1371/journal.pone.0016327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng H, Conneely KN, Wu H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014;42:e69. doi: 10.1093/nar/gku154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen KD, Sabunciyan S, Langmead B, et al. Large-scale hypomethylated blocks associated with Epstein-Barr virus-induced B-cell immortalization. Genome Research. 2014;24:177–184. doi: 10.1101/gr.157743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen KD, Sabunciyan S, Langmead B, et al. Large-scale hypomethylated blocks associated with Epstein-Barr virus-induced B-cell immortalization. Genome research. 2014;24:177–184. doi: 10.1101/gr.157743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroeder DI, Lott P, Korf I, et al. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome research. 2011;21:1583–1591. doi: 10.1101/gr.119131.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lotharius J, Falsig J, van Beek J, et al. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci. 2005;25:6329–6342. doi: 10.1523/JNEUROSCI.1746-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narytnyk A, Verdon B, Loughney A, et al. Differentiation of Human Epidermal Neural Crest Stem Cells (hEPI-NCSC) into Virtually Homogenous Populations of Dopaminergic Neurons. Stem cell reviews. 2014 doi: 10.1007/s12015-013-9493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finucane BM, Lusk L, Arkilo D, et al. 15q Duplication Syndrome and Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 33.Scoles HA, Urraca N, Chadwick SW, et al. Increased copy number for methylated maternal 15q duplications leads to changes in gene and protein expression in human cortical samples. Mol Autism. 2011;2:19. doi: 10.1186/2040-2392-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carmona-Mora P, Walz K. Retinoic Acid Induced 1, RAI1: A Dosage Sensitive Gene Related to Neurobehavioral Alterations Including Autistic Behavior. Curr Genomics. 2010;11:607–617. doi: 10.2174/138920210793360952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunaway KW, Islam MS, Coulson RL, et al. Cumulative Impact of Polychlorinated Biphenyl and Large Chromosomal Duplications on DNA Methylation, Chromatin, and Expression of Autism Candidate Genes. Cell Rep. 2016;17:3035–3048. doi: 10.1016/j.celrep.2016.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powell WT, Coulson RL, Crary FK, et al. A Prader-Willi locus lncRNA cloud modulates diurnal genes and energy expenditure. Human Molecular Genetics. 2013;22:4318–4328. doi: 10.1093/hmg/ddt281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.