

Abstract
The clinical effectiveness of systemically-administered human mesenchymal stem cells (hMSCs) depends on their capacity to engage vascular endothelium. hMSCs derived from bone marrow (BM-hMSCs) natively lack endothelial binding capacity but express a CD44 glycovariant containing N-linked sialyllactosamines that can be α(1,3)-fucosylated using fucosyltransferase-VI (FTVI) to enforce sLeX decorations, thereby creating hematopoietic cell E-/L-selectin ligand (HCELL). HCELL expression programs potent shear-resistant adhesion of circulating cells to endothelial beds expressing E-selectin. An alternative source of hMSCs is adipose tissue (A-hMSCs), and we assessed whether A-hMSCs bind E-selectin and/or possess sialyllactosamine-decorated CD44 accessible to α(1,3)-fucosylation. Similar to BM-hMSCs, we found that A-hMSCs natively lack E-selectin ligands, but FTVI-mediated cell surface α(1,3)-fucosylation induces sLeX expression and robust E-selectin binding secondary to conversion of CD44 into HCELL. Moreover, treatment with the α(1,3)-fucosyltransferase FTVII also generated expression of HCELL on both BM-hMSCs and A-hMSCs, with sLeX decorations created on N-linked glycans of the "standard" CD44 (CD44s) isoform. The finding that hMSCs from both source tissues each lack native E-selectin ligand expression prompted examination of the expression of glycosyltransferases that direct lactosaminyl glycan synthesis. These studies reveal that both types of hMSCs conspicuously lack transcripts encoding α(1,3)-fucosyltransferases but equally express glycosyltransferases critical to creation of sialyllactosamines. Collectively, these data indicate that assembly of a sialyllactosaminyl-decorated CD44s glycovariant is a conserved feature of hMSCs derived from adipose tissue and marrow, thus identifying a CD44 glycosignature of these cells and supporting the applicability of cell surface α(1,3)-fucosylation in programming migration of systemically-administered A-hMSCs to sites of tissue injury/inflammation.
Keywords: GPS, HCELL, fucosyltransferase, exofucosylation, sialyl Lewis X, E-selectin ligand, mesenchymal stem cell, sLeX
Graphical abstract
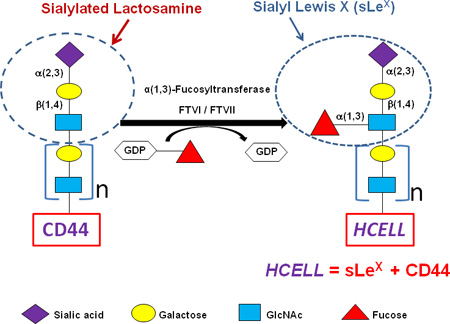
Application of Glycosyltransferase-Programmed Stereosubstitution (GPS) to enforce HCELL expression. Cell surface CD44 can be converted to the HCELL glycoform by glycan engineering via GPS. Note glycan structures (including sLeX) and component steps in the strategy to create HCELL by α(1,3)-fucosylation of “acceptor” sialylated CD44 on the hMSC membrane. The “β1-4” linkage as shown between Gal and GlcNAc defines a “Type 2” lactosamine unit. When this structure contains a fucose substitution in a 1,3-linkage to N-acetylglucosamine it is known as ‘sialylated Lewis x’ (sLeX): Treatment of hMSCs with either fucosyltransferase VI (FTVI) or fucosyltransferase VII (FTVII) drives α(1,3)-fucosylation of CD44 glycans, thereby generating sLeX and, accordingly, engendering HCELL. Data indicate that assembly of a sialyllactosaminyl-decorated CD44s glycovariant is a conserved feature of hMSCs derived from adipose tissue and marrow, thus identifying a CD44 glycosignature of these cells and supporting the applicability of cell surface α(1,3)-fucosylation in programming migration of systemically-administered A-hMSCs to sites of tissue injury/inflammation. Color key figures correspond to respective monosaccharides.
INTRODUCTION
Human mesenchymal stem cells (hMSCs) comprise a heterogeneous population of cells that support tissue growth/repair, and which can also differentiate into multiple cell types. Because hMSCs also possess potent immunomodulatory properties [1,2], these cells have received increasing attention for use in treatment of inflammatory diseases. To date, bone marrow has served as the main source of hMSCs for clinical application. However, to obtain bone marrow hMSCs (BM-hMSCs), one must perform marrow aspiration, which is a relatively invasive procedure requiring use of large-bore needles under force to penetrate the bone cortex. Alternatively, hMSCs can also be obtained from adipose tissue. These adipose-derived hMSCs (A-hMSCs) can be readily acquired through simple percutaneous fat aspiration(s) and/or from discarded tissue following liposuction/lipo-removal procedures and/or from adipose tissue procured as by-products of non-cosmetic surgeries [3–7]. Notably, comparative studies have suggested that A-hMSCs may possess more potent immunosuppressive effects than do BM-hMSCs [8–10]. The simplicity and convenience of procurement and the reported immunoregulatory profile of A-hMSCs offer significant advantages to use of these cells for treatment of inflammatory conditions.
A key prerequisite in realizing the promise of hMSC in therapy of acute and chronic inflammation is to ensure delivery of the cells to requisite sites. In all mammals, microvascular endothelial beds at all sites of tissue injury/inflammation express E-selectin. E-selectin (CD62E) is an endothelial molecule that is inducibly-expressed by inflammatory cytokines such as TNFα and IL-1 within post-capillary venules. It is a member of a family of Ca2+-dependent (“C-type”) lectins known as selectins that bind a tetrasaccharide motif consisting of a terminal sialic acid (also known as neuraminic acid; ‘NeuAc’) in α(2,3)-linkage to galactose (Gal) which is linked to an N-acetylglucosamine (GlcNAc) that is modified by fucose (Fuc). In non-epithelial cells (such as MSCs), Gal is β(1,4)-linked to GlcNAc, and this disaccharide is known as a “type 2” lactosamine unit (Galβ(1,4)-GlcNAc). The type 2 lactosamine unit can be modified with sialic acid, with fucose, or with both sialic acid and fucose: if sialic acid is linked to Gal, this trisaccharide structure is known as a “sialylated lactosamine” (or, more commonly, a “sialyllactosamine”); if fucose is α(1,3)-linked to GlcNAc, this trisaccharide structure (a “fucosylated type 2 lactosamine”) is known as “Lewis X” (LeX; also known as “CD15”). E-selectin does not bind to LeX, but does bind to the sialylated form of LeX, the tetrasaccharide structure known as ‘sialylated Lewis X’ (sLeX; also known as “CD15s”): NeuAc-α(2–3)-Gal-β(1-4)[Fuc-α(1–3)]-GlcNAc β(1-R) (see Supplemental Figures 1 and 2). By definition, sLeX is a “fucosylated sialyllactosaminyl glycan”, and it can be displayed as a protein-based glycoconjugate (i.e., a glycoprotein) and/or as a lipid-based glyconjugate (i.e., a glycolipid) [11,12,13]. This structure is recognized by mAbs such as HECA452 and CSLEX-1 [11–13] and can be operationally identified as an E-selectin ligand by using a chimeric construct comprised of E-selectin linked to the Fc region of the immunoglobulin heavy chain (E-selectin-Fc chimera (“E-Ig”)) as a probe. Importantly, the creation of sLeX within the cell’s Golgi apparatus is a conspicuously ordered process dictated by glycosyltransferases (GTs) with strict target acceptor specificities working in a defined sequence, requiring first the addition of sialic acid to a terminal type 2 lactosamine unit (mediated by GTs known as α(2–3)-sialyltransferases), followed then by the addition of fucose to GlcNAC within the sialyllactosaminyl glycan (mediated by GTs known as α(1–3)-fucosyltransferases) (see Supplemental Fig. 1). The addition of fucose to an unsialylated type 2 lactosamine prevents subsequent sialylation, thereby structurally locking the lactosamine into the trisaccharide LeX (Supplemental Fig. 1).
Prior studies from our lab have shown that BM-hMSCs express sialyllactosamines displayed on membrane CD44 that can be glycoengineered, directly on the cell surface by α(1,3)-fucosylation using fucosyltransferase VI (FTVI), to display sLeX. This sLeX-bearing CD44 glycoform is known as hematopoietic cell E-/L-selectin ligand (HCELL), a highly potent E-selectin ligand. Expression of HCELL licenses homing of blood-borne cells to tissue sites where E-selectin is expressed, initiating a cascade of events resulting in extravasation [11,12]. In the present study, we sought to identify whether A-hMSCs assemble glycoconjugates displaying sialyllactosamine decorations, and to analyze the effect(s) of α(1,3)-exofucosylation on creation of E-selectin ligands. To this end, we used two different α(1,3)-fucosyltransferases with overlapping, yet different, acceptor specificities, fucosyltransferase-VI (FTVI) and fucosyltransferase-VII (FTVII): FTVI can modify unsialylated and sialylated lactosamines to create both LeX and sLeX, respectively, but FTVII can only modify a sialylated lactosamine (thereby creating sLeX). Our studies reveal that, similar to BM-hMSCs, A-hMSCs have no native expression of E-selectin ligands, and that cell surface α(1,3)-fucosylation with either FTVI or FTVII generates the expression of HCELL equally among BM-hMSCs and A-hMSCs; in each case, sLeX decorations are created exclusively on N-linked glycans of “standard” CD44 (“CD44s”, i.e., a CD44 protein isoform that lacks peptide sequences encoded by splice exons), yielding equivalent potency in binding to E-selectin. Importantly, α(1,3)-exofucosylation with FTVI does not engender creation of CD15 in either A- or BM-MSCs, indicating that CD44 in each case is not assembled bearing non-sialylated lactosamines. Moreover, qRT-PCR analysis of GTs that direct synthesis of sialofucosylated lactosaminyl glycans reveals equivalent expression profiles among BM- and A-hMSCs. Collectively, these data provide new insights on the glycosignature of hMSC, and support the utility of cell surface glycan engineering to enforce HCELL expression as a means to direct migration of systemically-administered A-hMSCs to inflammatory sites.
MATERIALS AND METHODS
Cell lines
The human hematopoietic cell line KG1a was obtained from ATCC and cultured in RPMI 1640 media supplemented with 10% FBS and 1% penicillin-streptomycin (Gibco/Invitrogen). Human umbilical vein endothelial cells (HUVECs) were obtained from Brigham & Women’s Hospital (BHW) Pathology Core facility and cultured as described [11]. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 and 21% O2.
MSCs Isolation and culture
A-hMSCs were obtained from adult human subcutaneous adipose tissue of 20 healthy female donors, aged 32 ± 8 years, undergoing abdominal liposuction. The de-identified, discarded lipoaspirates were obtained in accordance with protocols approved by the Human Experimentation and Ethics Committees of the University of Florida, the Pennington Biomedical Research Center, and the Western Institutional Review Board (LaCell LLC). Lipoaspirates were processed according the protocol described [3,4]. All studies were performed on cells propagated within culture passages 3–5.
To isolate BM-hMSCs, bone marrow samples were obtained from harvest filters of healthy donors for patients undergoing bone marrow transplantation at Massachusetts General Hospital. Cells were obtained in accordance with protocols approved by the Human Experimentation and Ethics Committee of Partners Healthcare. Bone marrow mononuclear cells were isolated by a density gradient using Ficoll-Paque PLUS (Sigma-Aldrich). Mononuclear cells were then plated in 175 cm2 culture flasks at 160,000 cells/cm2 in cell media consisting of DMEM, 10% FBS, 1% penicillin-streptomycin and incubated at 37°C in a humidified atmosphere containing 5% CO2 and 21% O2. Cells were grown to maximal 70% confluency at each passage. All studies were performed on cells propagated within culture passages 3–5.
Immunophenotyping of hMSCs
hMSCs were incubated with a panel of primary antibodies chosen based on the known profile of BM-hMSC populations [3,4]. Flow cytometry analysis was performed using the following commercially available mAbs together with isotype-matched controls: FITC-conjugated anti-human CD13 and CD36, and PE-conjugated anti-human CD34, CD105, CD146, and CD166 (all from Beckman Coulter); FITC-conjugated mAb HECA452 (Biolegend) and anti-human CD62E (R&D Systems); PE-conjugated anti-human CD44, CD45, CD49d, CD73 and CD90, and PECy5-conjugated anti- human CD29 and CD106 (all from BD Biosciences). Data were collected using a Cytomics FC 500 MPL flow cytometer (Beckman Coulter) and analyzed with FlowJo version 10.0.6 (TreeStar).
Real-time quantitative PCR analysis of glycosyltransferase gene expression
Total RNA was purified from hMSCs samples using the RNeasy® Micro Kit, which included a DNAse I treatment step as per manufacturer's instructions (Qiagen). First-strand cDNA was synthesized from 0.5 µg total RNA using the iScriptTM cDNA synthesis kit (Bio-Rad). Primer sequences used for real-time quantitative PCR (qRT-PCR) are listed in Supplemental Table 1. Quantitation of human glycosyltransferase transcript expression was performed on the LightCycler 480 (Roche) with SYBR Green Master mix (Applied Biosystems). The relative fold differences in transcript expression were calculated by adapting the 2−ΔCt × 100 formula [14].
Glycoengineering of sLeX expression on hMSCs
For α(1,3)-exofucosylation, hMSCs were treated with 1µg of α(1,3)-linkage-specific human fucosyltransferase VII (FTVII) (obtained from R&D Systems) in HBSS without Ca2+/Mg2+ containing 10mM HEPES, 0.1% human serum albumin (HSA) and 1mM GDP-Fucose (Sigma Aldrich), at 37°C for 1h, as described previously [11]. In each experiment, we compared FTVII treatment (as described above) to controls which consisted of buffer treated cells (without enzyme in an equivalent volume of buffer under identical conditions). Recombinant human FTVI enzyme was created and used for hMSC exofucosylation as previously described [15].
Lectin Analysis of lactosaminyl glycans on hMSCs
For lectin binding analysis, hMSCs were stained with biotin-conjugated Maackia amaurensis II (MALII) (Vector laboratories), which recognizes sialic acid α(2,3)-linked to galactose, followed by staining with streptavidin-PE (Southern Biotech) for 30 min at 4°C. The binding specificity of the lectin was validated by Vibrio cholerae sialidase (Roche Molecular Biochemicals) treatment of hMSC for 30 min at 37°C in Hanks Balanced Salt Solution (HBSS) without Ca2+/Mg2+ and 1% bovine serum albumin (BSA).
Flow cytometry to assess E-Ig reactivity, HECA452 staining and CD44 expression
For flow cytometry measurement of E-selectin ligand expression, untreated and FTVII-treated hMSCs were stained in a 3-step procedure using recombinant mouse E-selectin–human Ig chimera (0.5 µg/ml; R&D Systems) in Ca2+-containing binding buffer (HBSS, 5mM HEPES, 2mM CaCl2 and 5%FBS), followed by staining with rat anti-mouse E-selectin (CD62E) mAb (R&D Systems), and then FITC-conjugated goat anti-rat IgG (Southern Biotech), each for 30 min at 4°C. Chimera buffer containing 2mM of EDTA (for Ca2+-chelation) was also used as a control for the specificity of binding of E-Selectin-Ig (EDTA group). For detection of sLeX, staining was performed using FITC-conjugated anti-human/mouse clone HECA452 (rat IgM; Southern Biotech) for 30 min at 4°C, and, also, using FITC-conjugated anti-human CD15s mAb (clone CSLEX1; IgM) (Southern Biotech) for 30 min at 4°C. CD44 staining was performed using anti-human CD44-PE mouse mAb (clone G44-26; IgG2) for 30 minutes at 4°C.
Preparation of whole cell lysates and Western blot analysis
To obtain cell lysates, hMSCs were suspended in 150mM NaCl, 50mM Tris-HCl (ph 7.4), 0.02% NaN3, 20 µg/mL PMSF and protease inhibitor cocktail (Roche), sonicated, and then solubilized in 2% Nonidet P-40 (NP-40). Protein samples were then separated on a 7.5 % SDS–PAGE electrophoresis gel (Bio-Rad). Resolved membrane proteins were transferred to Polyscreen polyvinylidene difluoride (PVDF) membranes (Bio-Rad) and blocked with 10% nonfat dry milk and 0.1% Tween20 in TBS. For assessment of E-selectin ligands, membranes were incubated with recombinant mouse E-selectin–human Ig chimera in TBS 0.1% Tween20 containing 2mM CaCl2, followed by staining with rat anti-mouse CD62E mAb (R&D Systems) and then goat anti-rat IgG-HRP (Southern Biotech). For detection of sLeX, membranes were incubated with rat HECA452 mAb, followed by staining with goat anti-rat IgM-HRP (Southern Biotech). For detection of CD44, membranes were incubated with mouse anti-human CD44 (Clone 2C5; IgG2), followed by staining with goat anti-mouse Ig-HRP (Southern Biotech). HRP conjugated antibodies were detected by chemiluminiscence using Lumi-Light Western blotting substrate (Roche).
Analysis of E-selectin ligand expression after enzymatic treatments
Sialic acid residues were removed by treatment of KG1a and hMSCs (Untreated and FTVII-treated) with 0.1U/mL Vibrio cholerae neuraminidase (Roche Molecular Biochemicals) in HBSS without Ca2+/Mg2+ and 1% BSA at 37°C for 30 min; for controls, an equivalent volume of enzyme buffer alone was added to the cells under identical conditions. The efficiency of neuraminidase treatment in each case was confirmed by absence of reactivity to HECA452 and CSLEX1 mAb (i.e., detecting loss of sLeX). The relative contribution of glycoproteins as sLeX scaffolds was determined by treatment of cells with the broad-specificity protease bromelain, followed by staining with HECA452 to assess protease-resistant sLeX expression. To remove N-glycans, protein lysates of hMSCs were treated with Peptide-N-Glycosidase (PNGase-F, New England Biolabs) according to the manufacturer’s instructions.
Immunoprecipitation of CD44
Lysates of KG1a and hMSCs were prepared as described above and precleared with protein G-agarose beads (Invitrogen). For CD44 immunoprecipitation, lysates were incubated with a cocktail of mouse anti-CD44 mAb consisting of 2C5 (R&D Systems), 515, and G44-26 (both from BD Biosciences). Immunoprecipitates were then collected with protein G-agarose beads and eluted via boiling in 1.5× reducing SDS-sample buffer. Proteins were then subjected to reducing 7.5 % SDS-PAGE and western blotting.
Assessment of hMSC differentiation capacity following exofucosylation
To analyze whether FTVII treatment had any effect on differentiation capacity, untreated and FTVII-treated hMSCs were subjected to adipogenic and osteogenic differentiation according to protocols previously described [3].
Parallel plate flow chamber assay to assess E-selectin ligand activity
To analyze the capacity of α(1,3)-fucosylated-hMSCs to engage vascular E-selectin under physiologic blood flow conditions, dynamic flow adhesion assays were performed using a parallel-plate flow chamber and binding interactions were visualized in real time using video-microscopy. All flow chamber studies were performed according to protocols previously described [11]. hMSCs were suspended in HBSS containing 10mM HEPES, 2mM CaCl2 and 5% FBS and perfused through HUVEC-containing channels at an initial shear rate of 0.5 dynes/cm2, with step-wise increments to 8 dynes/cm2, thereby mimicking hemodynamic shear conditions present in the interaction of a blood-borne cell with human vasculature [16]. The total number of interacting cells in a single 4X field of view (fixed in the middle of the flow channel) as well as velocities and shear resistance during the 5 min perfusion period were evaluated to determine the capacity of cells to interact with endothelial E-selectin [17,18]. Controls for specificity of E-selectin binding consisted of perfusion of hMSC in presence of function-blocking anti-mouse E-selectin mAb (clone 10E9.6, from BD Biosciences).
FIbronectin adhesion assay
To assess the functional impact of HCELL ligation on VLA-4 adhesiveness, we assessed binding of hMSC to fibronectin-coated wells as previously described [19]. Briefly, hMSCs were either preincubated with E-Ig immobilized on tissue culture plates or suspended in buffer alone (untreated cells), and then lifted and incubated on tissue culture plates coated with human fibronectin (Sigma), each for 1h at 37°C. Following incubation with fibronectin, plates were fixed 4% paraformaldehyde followed by staining of cells with 0.1% crystal violet. Absorbance was read at 595 nm after a wash with 10% acetic acid (BMG-Labtech microplate reader).
RESULTS
Immunophenotypic profiles of A-hMSCs and BM-hMSCs
BM-hMSCs and A-hMSCs, each isolated from more than 20 different healthy human donors, showed similar morphology when cultured. Both groups appeared homogeneous and maintained a characteristic spindle-shaped appearance through passage 5. Flow cytometry histograms were compared between A-hMSCs and BM-hMSCs based on their expression of a panel of cell surface markers including those associated with stromal, endothelial and hematopoietic cells, and ligands of E-selectin (E-Ig reactivity and sLeX antigen expression (reactivity with HECA452 mAb)) (Table 1).
Table 1.
Immunophenotypic profile of surface antigens on hMSCs derived from bone marrow (BM-hMSCs) or adipose tissue (A-hMSCs)
| Surface Markers |
BM-hMSCs | A -h MSCs | ||||
|---|---|---|---|---|---|---|
| % | MFI | % | MFI | p Value % | p Value MFI | |
| CD13 | 92 ± 2 | 19 ± 2 | 99 ± 1 | 25 ± 5 | 0.176 | 0.436 |
| CD29 | 99 ± 1 | 91 ± 11 | 99 ± 1 | 66 ± 17 | 0.830 | 0.108 |
| CD34 | 3 ± 1 | 3 ± 1 | 28 ± 6* | 9 ± 2 | 0.040 | 0.056 |
| CD36 | 10 ± 3 | 2 ± 1 | 69 ± 8* | 19 ± 3* | 0.002 | 0.052 |
| CD44 | 97 ± 2 | 219 ± 44 | 98 ± 1 | 312 ± 78 | 0.840 | 0.314 |
| CD45 | 2 ± 1 | 3 ± 1 | 0.2 ± 0.1 | 4 ± 1 | 0.034 | 0.569 |
| CD49d | 97 ± 3 | 93 ± 2 | 98 ± 1 | 176 ± 20 | 0.727 | 0.055 |
| CD62E | 2 ± 1 | 3 ± 1 | 0.5 ± 0.1 | 3 ± 1 | 0.099 | 0.670 |
| CD73 | 98 ± 3 | 88 ± 22 | 95 ± 3 | 101 ± 15 | 0.752 | 0.727 |
| CD90 | 97 ± 2 | 132 ± 9 | 94 ± 5 | 192 ± 35 | 0.709 | 0.146 |
| CD105 | 97 ± 1 | 64 ± 5 | 97 ± 1 | 77 ± 16 | 0.710 | 0.550 |
| CD106 | 87 ± 2 | 50 ± 3* | 82 ± 1 | 5 ± 1 | 0.136 | 0.001 |
| CD146 | 74 ± 7 | 22 ± 1 | 70 ± 3 | 21 ± 2 | 0.424 | 0.621 |
| CD166 | 98 ± 2 | 94 ± 2 | 97 ± 1 | 79 ± 17 | 0.685 | 0.323 |
| HECA452 | 0.9 ± 0.1 | 3 ± 1 | 2 ± 1 | 3 ± 1 | 0.283 | 0.491 |
Flow cytometry histograms were compared between BM-and A-hMSCs based on their expression of a panel of cell surface markers including those associated with stromal, endothelial, hematopoiteic cells and ligands of E-selectin. The values represent percentage surface positive and MFI (mean ± SEM). Data are representative of experiments performed on BM-and A-hMSC cultures derived from at least twenty individuals.
Statistically significant differences were calculated using paired t-test (*p<0.05).
Abbreviations: BM-hMSCs, Bone Marrow-derived human Mesenchymal Stem Cells; A-hMSCs, Adipose-derived human Mesenchymal Stem Cells; MFI, Mean Fluorescence Intensity.
Flow cytometry indicated that both BM-hMSCs and A-hMSCs met MSC surface marker criteria as established by the International Society of Cell Therapy (ISCT) and the International Federation for Adipose Therapeutics and Science (IFATS) [5]. BM-hMSC and A-hMSC populations were uniformly positive for CD73, CD90, CD105, CD44 and negative for CD45, CD62E (E-selectin) and sLeX (i.e., no HECA452-reactivity). However, significant differences were noted with respect to expression of other markers. Notably, A-hMSCs, at early passages, typically express low levels of CD34 and CD36, whereas BM-hMSCs generally lack expression of these markers. In contrast, while both BM-hMSCs and A-hMSCs characteristically display CD29, CD106 and CD166, BM-hMSCs express higher levels (higher mean fluorescence intensity (MFI) by flow cytometry) of these proteins (Table 1).
Conserved expression of sialyllactosaminyl glycans on CD44s of A-hMSCs and BM-hMSCs: FTVI and FTVII-mediated α(1,3)-exofucosylation of hMSCs converts cell surface CD44 into HCELL
Flow cytometry data indicated that A-hMSCs, similar to BM-hMSCs, natively lack expression of E-selectin ligands as assessed using HECA452 mAb and E-Ig as probes (Fig. 1A). To assess whether A-hMSCs express glycoconjugates displaying terminal sialyllactosaminyl glycans, we sought to identify whether the expression of E-selectin ligands on A-hMSCs could be enforced via exofucosylation [11]. To this end, we utilized α(1,3)-fucosyltransferases VI (FTVI) and VII (FTVII) that can each engineer sLeX glycan determinants on the cell surface via addition of fucose in α(1,3)-linkage to N-acetylglucosamine within sialylated type 2-lactosamines (Supplemental Fig. 2). Notably, the FTVI enzyme is capable of fucosylating two glycan acceptors, type 2 lactosamine or α(2,3)-sialylated type 2 lactosamine (and can thus render the fucosylated glycans LeX and sLeX, respectively), whereas FTVII specifically fucosylates only an α(2,3)-sialylated type 2 lactosamine acceptor (and can thus only create sLeX) (Supplemental Fig. 2). Treatment of BM-hMSCs and A-hMSCs with both enzymes did not yield expression of LeX (data not shown), but both cell types showed marked induction of HECA452 reactivity by both flow cytometry and western blot (Fig. 1A,B), and equivalently induced reactivity with E-Ig (Fig. 1A). We observed that A-hMSCs, like BM-hMSCs, display sLeX determinants predominantly on a glycoprotein of molecular weight ~90 kDa, a size consistent with that of standard CD44 (Figure 1C). To confirm that the observed equivalency in HECA452-reactivity among FTVII-exofucosylated A- and BM-hMSCs reflected comparable expression of sLeX determinants and not a HECA452 epitope-specific effect, we analyzed staining using another sLeX-specific mAb, CSLEX-1, and, furthermore examined for abrogation of reactivity following sialidase digestion. Similar to prior results obtained with FTVI-mediated exofucosylation [11], FTVII treatment of hMSCs from both sources engendered high CSLEX-1 (Fig. 2A) and HECA452 reactivity (Fig. 2B) and specificity for these mAb was validated by loss of reactivity following sialidase treatment (i.e., removal of the terminal sialic acid of sLeX). To further characterize the expression of sialylated lactosaminyl glycan determinants, hMSCs were incubated with nonagglutinating levels of the lectin Maackia amuriensis lectin (MALII) (which is specific for α(2,3)-sialylated type 2 lactosamines). As shown in Supplemental Fig 3, MALII bound to A- and BM-hMSCs with equivalent profiles, in each case staining dampened after sialidase treatment of the cells, indicating similar expression of cell surface sialylated type 2 lactosamines, on hMSCs derived from both sources.
Figure 1. FTVI- and FTVII-mediated α(1,3)-exofucosylation equally create sLeX determinants on hMSCs derived from either bone marrow (BM-hMSCs) or adipose tissue (A-hMSCs).

(A): Representative flow cytometry histograms of HECA452 mAb (left) and E-Ig (right) staining of hMSCs that were untreated (black line) or treated either with FTVI (dotted line) or FTVII (dashed line). Grey filled histogram represents staining with isotype control. As assessed by HECA452 and E-Ig staining, both types of hMSCs natively lack sLeX epitopes, each of which are uniformly created after FTVI or FTVII treatment. (B and C): Representative western blot analysis of whole cell lysates of untreated, FTVI- and FTVII-treated hMSCs resolved by SDS-PAGE and stained with HECA452 mAb (B) or anti-CD44 mAb (C). Lysates of KG1a cells serve as positive control for HECA452 blot results. Exofucosylation of both types of hMSCs prominently engenders a HECA452-reactive glycoprotein of mw ~90kDa (B) that has mobility profile equivalent to that of CD44 (C). For all figures, data are representative of five independent experiments. Abbreviations: FTVI, Fucosyltransferase VI; FTVII, Fucosyltransferase VII; BM-hMSCs, Bone Marrow-derived human Mesenchymal Stem Cells; A-hMSCs, Adipose-derived human Mesenchymal Stem Cells; E-Ig, E-selectin-immunoglobulin Fc chimera.
Figure 2. Sialidase treatment of FTVII–exofucosylated hMSCs from either bone marrow (BM-hMSCs) or adipose tissue (A-hMSCs) abrogates reactivity with anti-sLeX mAbs.

(A–B): Flow cytometry analysis of CSLEX-1 (A) and HECA452 mAb staining (B) of untreated, FTVII-treated and FTVII + sialidase treated A and BM-hMSCs. As shown, α(1,3)-fucosylation with FTVII generates CSLEX-1 and HECA452 reactivity equally among BM-and A-hMSCs. Abrogation of CSLEX-1 and HCEA452 reactivity, after sialidase treatment, indicate the presence of sialic acid decorations for the enforced creation of sLeX determinant. Graph values represent fold change with respect to untreated hMSCs (mean ± SEM) of at least five independent experiments. (C and D): CD44 immunoprecipitation was performed on whole cell lysates of untreated and FTVII-treated hMSCs. Representative western blot analysis of total cell lysates (T) and CD44 immunoprecipitated (IP) resolved by SDS-PAGE and stained with E-Ig (C) or anti-CD44 mAb (D). Western blot reveal a principal E-Ig reactive glycoprotein of mw ~90 kDa after α(1,3)-exofucosylation (C), which is created exclusively on the CD44 protein scaffold (~90 kDa) on both BM-and A-hMSCs (D). Western blot data are representative of experiments performed on both hMSCs culture derived from at least twenty individuals. Abbreviations: BM-hMSCs, Bone Marrow-derived human Mesenchymal Stem Cells; A-hMSCs, Adipose-derived human Mesenchymal Stem Cells; FTVII, Fucosyltransferase VII; MFI, Mean Fluorescence Intensity; E-Ig, E-selectin-immunoglobulin Fc chimera.
By both flow cytometry (Table 1) and western blot (Figs. 2C and 2D), we found that A-hMSCs, like BM-hMSCs, characteristically display high levels of CD44. To directly determine whether hMSC CD44 can be converted to HCELL by FTVII-mediated α(1,3)-exofucosylation, CD44 was immunoprecipitated from lysates of untreated and FTVII-treated A-hMSCs and BM-hMSCs and then blotted E-Ig and with anti-CD44 mAb. As shown in Figure 2C, FTVII treatment induces HCELL expression on A-hMSCs equal to that on BM-hMSCs. Of note, following FTVII treatment, the ‘standard’ (non-alternatively spliced) form of CD44 (75–100 KDa) was the only isoform on the surface of both types of hMSCs displaying sLeX (Fig. 2D).
FTVII-enforces hMSC sLeX expression predominantly on glycoproteins
To analyze the relative contributions of protein versus lipid glycoconjugate scaffolds in exofucosylation-enforced expression of sLeX on hMSCs, cells were treated with the protease bromelain before FTVII treatment. Flow cytometry and western blot data show that bromelain digestion of BM-hMSCs as well as A-hMSCs markedly reduced HECA452 reactivity and abrogated CD44 expression (Figs. 3A and 3B), indicating that pertinent sialylated lactosamine acceptors are principally presented on membrane glycoproteins, not glycolipids. Moreover, as shown in Figure 3C, digestion of FTVII-treated cells with Vibrio cholerae sialidase, markedly diminished HECA452 staining of HCELL on western blot, indicating that FTVII induces sLeX decorations on CD44. To assess whether sLeX is displayed on N-glycans or on O-glycans of A-hMSCs, cell lysates of FTVII-treated cells were digested with N-glycosidase F to release all N-Glycans. Similar to HCELL as displayed by KG1a cells and by exofucosylated BM-hMSCs [11], N-glycosidase F digestion completely eliminated HECA452-reactivity of FTVII-treated A-hMSC CD44 (Fig. 3D), indicating that the relevant sLeX decorations are presented solely on N-glycans.
Figure 3. FTVII enforces expression of sLeX predominantly on N-linked glycoproteins in both BM-hMSCs and A-hMSCs.

(A): Representative flow cytometry histograms of HECA452 mAb and anti-CD44 mAb staining in hMSCs that were untreated (grey line), FTVII-treated (bold line), treated with the protease bromelain (dotted line) or FTVII-treated followed by bromelain treatment (dashed line). Grey filled histogram represents staining with isotype control. (B): HECA452 and CD44 western blot analysis of untreated and FTVII-treated hMSCs +/− bromelain-treatment. (C): HECA452 Western blot analysis of untreated and FTVII-treated hMSCs +/− sialidase-treatment. Lysates of KG1a cells serve as a positive control. (D): HECA452 and CD44 western blot analysis of untreated and FTVII-treated A-hMSCs +/− N-glycosidase F-treatment, indicating that CD44 displays sLeX on N-glycans. Lysates of KG1a cells serve as positive control. For all figures, data are representative of experiments performed on both A-hMSCs and BM-hMSC cultures derived from at least ten individuals. Abbreviations: FTVII, Fucosyltransferase VII; BM-hMSCs, Bone Marrow-derived human Mesenchymal Stem Cells; A-hMSCs, Adipose-derived human Mesenchymal Stem Cells.
Interestingly, by western blot analysis, a HECA452- and E-Ig-reactive glycoprotein of molecular weight ~150 kDa is variably detected on lysates of both types of hMSCs, more typically in A-hMSCs, in absence of treatment with either FTVI or FTVII (Figs. 1B, 2C, 3B and 3C). Notably, as detected by flow cytometry, neither A- nor BM-hMSCs natively react with anti-sLeX mAb or with E-Ig chimera (Figs. 1A, 2A, and 2B), raising doubt that this ~150 kDa glycoprotein, if present on the membrane, is contributing to cell surface E-selectin engagement. Moreover, by western blot analysis, the HECA452 staining of this molecule is not completely abrogated by sialidase treatment of cells, which indicates that this molecule may be resistant to sialidase digestion and/or there may be intracellular stores (Fig. 3C). However, following α(1,3)-exofucosylation, western blots of lysates of both A- and BM-hMSCs have variably shown increased HECA452 and E-Ig staining of the band at ~150 kDa (Figs. 1B, 2C, 3B and 3C); these data suggest that the ~150 kDa protein is a cell surface molecule, and, contrary to HCELL, N-glycosidase F digestion does not eliminate the HECA452-reactivity of this band (Figure 3D). The identity of this glycoprotein is at present uncertain, and further studies are warranted to elucidate its structural biology.
Glycosyltransferase gene expression among hMSCs
Biosynthesis of terminal sialic acid decorations on type 2 lactosamine extensions depends on addition of sialic acid to galactose in α(2,3)-linkage which can be mediated by action of three sialyltransferases, ST3Gal-III, ST3Gal-IV, and ST3Gal-VI [20–22]. Once sialylated with α(2,3) sialic acid, these extensions can then become modified by addition of fucose in α(1,3)-linkage to the N-acetylglucosamine within the terminal sialylated lactosamine unit, a modification which can be mediated by any one of five α(1,3)-fucosyltransferases (FTIII, FTIV, FTV, FTVI, and/or FTVII), thus creating sLeX [23–25] (see supplemental Fig. 1 for schematic diagram of synthesis of the tetrasaccharide sLeX).
The expression of these glycosyltransferases relevant for lactosaminyl glycan decorations was analyzed by qRT-PCR in both A-and BM-hMSCs (Supplemental Table 1). The results indicate that there is essentially no expression of α(1,3)- fucosyltransferases FTIII, FTV, FTVI, and FTVII among A- and BM-hMSCs, yet extremely low transcript levels of the α(1,3)-fucosyltransferase FTIV are observed (Fig. 4A). Additionally, both A- and BM-hMSCs possess very low transcript levels for the α(2,3)-sialyltransferases ST3Gal-III and ST3Gal-VI (Fig. 4B), but each have noticeably higher expression of ST3Gal-IV transcripts; this pattern would predict equivalent sialylation capacity of terminal lactosamines in both A- and BM-hMSCs.
Figure 4. Analysis of Glycosyltransferase gene expression in BM-hMSCs and A-hMSCs.

qRT-PCR analysis of fucosyltransferases (A) and sialyltransferases (B) involved in sLeX creation in BM-hMSCs (black bars) and A-hMSCs (grey bars). The mRNA expression of each glycosyltransferase was normalized to GAPDH. Values are reported as mean ± SEM. Experiments were performed with a minimum of six individuals. Abbreviations: BM-hMSCs, Bone Marrow-derived human Mesenchymal Stem Cells; A-hMSCs, Adipose-derived human Mesenchymal Stem Cells.
FTVII exofucosylation increases E-Selectin ligand activity on hMSCs under physiologic blood flow conditions: Enforced expression of HCELL on A-hMSCs confers E-selectin binding activity equivalent to that of BM-hMSCs
To analyze the capacity of α(1,3)-fucosylated-hMSCs to engage vascular E-selectin under physiologic blood flow conditions, dynamic flow adhesion assays were performed on cytokine-stimulated HUVEC monolayers. Untreated hMSCs, consistent with their low reactivity with HECA452 and CSLEX-1 mAb, did not tether/roll on stimulated endothelium at physiological shear levels (1–4 dyn/cm2) and did not interact even at lower levels of shear stress. However, FTVII treatment of hMSCs resulted in a marked increase in their capacity to interact with the E-selectin expressed on HUVEC monolayers. For both BM- and A-hMSCs, these binding interactions were resistant to detachment at shear stress levels up to 8 dynes/cm2, well above post-capillary venule physiologic conditions (Figure 5A and 5B).
Figure 5. Enforced expression of HCELL on A-hMSCs confers functional E-selectin binding activity equivalent to that of BM-hMSCs.

(A–B): Parallel-plate flow chamber analysis of tethering and rolling interactions of BM-hMSCs (A) and A-hMSCs (B) on E-selectin-expressing endothelial cells. Untreated and FTVII-treated hMSCs were perfused into a parallel-plate flow chamber seeded with TNFα-stimulated HUVEC and their tethering/rolling capacity was then determined at shear stresses of 0.5, 1, 2, 4, 8 and 16 dynes/cm2. Both hMSC types exhibited similarly increased tethering/rolling adhesive interactions on HUVECs at shear stress levels of up to 16 dynes/cm2 after FTVII treatment. Values are reported as mean ± SEM. Data are representative of three independent experiments. Abbreviations: FTVII, Fucosyltransferase FTVII; HCELL, Hematopoietic Cell E/L-Selectin Ligand; BM-hMSCs, Bone Marrow-derived human Mesenchymal Stem Cells; A-hMSCs, Adipose-derived human Mesenchymal Stem Cells; HUVEC, Human Umbilical Vein Endothelial Cells.
HCELL/E-selectin interactions up-regulate adhesion of A-hMSCs to Fibronectin
To assess the impact of HCELL ligation on VLA-4 adhesiveness, A-hMSCs were exposed to E-selectin (E-Ig) and subsequently analyzed for adhesion to fibronectin. For comparison of results of E-selectin/HCELL engagement, data are shown as fold change of FTVII-treated A-hMSCs with respect to untreated A-hMSCs.
As shown in supplemental figure 4, E-Ig engagement increased binding of FTVII-treated A-hMSCs to fibronectin 2–3 fold compared to that of untreated A-hMSCs. Preincubation with function-blocking anti-mouse CD62E decreased this effect, indicating binding of FTVII-treated A-hMSCs to fibronectin was critically dependent on prior engagement of E-Ig. These results show that HCELL engagement with E-Ig activates VLA-4 adhesion to fibronectin, consistent with results of prior studies of BM-hMSCs [11,19].
FTVII exofucosylation does not affect differentiation potential of hMSCs
To assess whether exofucosylation alters differentiation capacity of hMSCs, untreated and FTVII-treated A-and BM-hMSCs were cultured in adipogenic and osteogenic differentiation cocktail [3]. Comparing untreated and FTVII-treated cells, there was no difference in the induction of lipid accumulation among BM and A-hMSCs (Fig. 6A). However, while osteoid mineralization was not different among untreated and FTVII-treated BM-hMSC, A-hMSCs showed increased mineralization in FTVII-treated cells compared to untreated cells (Fig. 6B).
Figure 6. Analysis of differentiation potential of hMSCs after FTVII treatment.

(A): Adipogenic differentiation potential of untreated versus FTVII-treated BM-hMSCs (white bars) and A-hMSCs (black bars). Intracellular lipids droplets enrichment of hMSCs under adipogenic differentiation conditions. Data presented as the ratio of Oil Red O staining under adipogenic conditions relative to untreated controls (B). Osteogenic differentiation potential of untreated versus FTVII-treated BM-hMSCs (white bars) and A-hMSCs (black bars) under osteogenic differentiation conditions. Data presented as the ratio of Alizarin Red S staining under osteogenic conditions relative to untreated controls. Values are reported as mean ± SEM. Data are representative of three independent experiments. Abbreviations: FTVII, Fucosyltransferase FTVII; BM-hMSCs, Bone Marrow-derived human Mesenchymal Stem Cells; A-hMSCs, Adipose-derived human Mesenchymal Stem Cells.
DISCUSSION
E-selectin receptor/ligand interactions are the foremost mediators of “Step 1” adhesive events that drive recruitment of circulating cells to sites of tissue injury/damage. Prior studies from our lab have shown that BM-hMSCs lack E-selectin ligands, but these cells express a glycovariant of “standard” CD44 (CD44s, i.e., CD44 lacking splice-variant peptides) that is modified by sialyllactosaminyl glycans. Importantly, these CD44 sialylated lactosamines can be specifically shaped by enzyme-mediated α(1,3)-fucosylation to become sLeX, thereby engendering the CD44 glycoform known as HCELL, a potent E-selectin ligand [11,12]. Here, we investigated whether A-hMSCs can natively bind E-selectin expressed on endothelial cells. Moreover, we analyzed whether A-hMSCs possess glycoconjugates displaying sialyllactosamine decorations and, accordingly, if enforced α(1,3)-fucosylation of A-hMSCs could program HCELL expression with resulting E-selectin binding under hemodynamic flow conditions.
Flow cytometry analysis reveals that, similar to BM-hMSCs, A-hMSCs lack expression of sLeX and are unreactive with E-selectin-Ig chimera (E-Ig), but exofucosylation of the cells using the α(1,3)-fucosyltransferases FTVI and FTVII in each case markedly increases sLeX expression and E-selectin binding (Figure 1A), and there was no expression of LeX following FTVI-mediated exofucosylation, indicating that these cells do not display unsialylated lactosaminyl glycans.
Similar to prior findings with BM-hMSCs following FTVI-mediated exofucosylation, digestion with the protease bromelain markedly reduced HECA452 staining following FTVII-mediated exofucosylation, indicating that FTVII treatment of A-hMSCs enforces sLeX determinants predominantly on cell surface glycoproteins, not glycolipids (Figures 3A and 3B). Western blot analysis under reduced SDS-PAGE conditions consistently demonstrated that, following enforced α(1,3)-exofucosylation, A-hMSCs and BM-hMSCs similarly display sLeX predominantly on CD44s (thus creating HCELL), and flow cytometry showed equivalent induction of E-Ig-reactivity between A-hMSCs and BM-hMSCs (Fig. 1A), indicating comparable HCELL expression. Though western blot studies show that HCELL is the principal glycoconjugate that displays sLeX following exofucosylation of both A-hMSCs and BM-hMSCs, we have also variably observed faint staining by both HECA-452 and E-Ig of a glycoprotein of molecular weight ~150 kDa on both untreated as well as exofucosylated cells; the identity of this glycoprotein is currently under investigation.
The extent to which sLeX impacts cell trafficking is critically dependent on the scaffold upon which it is displayed. Specifically, the core protein and/or lipid structure displaying sLeX determinants influences not only the circulating cell’s capacity to engage endothelial E-selectin, but also the efficiency of extravasation [12]. Functionally, α(1,3)-exofucosylation markedly increased the tethering/rolling capacity of A- and BM-hMSCs on TNF-stimulated endothelial cells to similarly high levels, not just at physiologic shear stress levels of 1–4 dynes/cm2 typical of post-capillary venules, but also at shear stress levels beyond 10 dynes/cm2, approaching that of arteries (Figure 5) [26]. These findings indicate that exofucosylated A- and BM-hMSCs are equally primed to migrate to endothelial beds at sites of inflammation. In particular, the finding that HCELL is the dominant glycoconjugate in presentation of sLeX on the MSC surface following exofucosylation is important for two reasons: (1) HCELL is a highly potent E-selectin ligand; and (2) Binding of this sLeX-bearing CD44 glycovariant to E-selectin critically impacts the multistep cascade of events that drive extravasation [19]. Specifically, upon cross-linking of HCELL via engagement by E-selectin, our prior studies of BM-hMSCs showed that integrin VLA-4 (also known as α4β1 or CD49d/CD29) is activated, thus allowing for VLA-4-mediated firm adherence without need for chemokine-dependent signalling [19]. To determine whether this chemokine-bypass pathway is operational in A-hMSCs, we assessed the capacity of HCELL+ A-hMSCs to bind fibronectin following HCELL cross-linking via incubation with E-Ig. We consistently observed that ligation of HCELL on A-hMSCs is sufficient to engender VLA-4 activation with resulting adhesion to fibronectin. Our present findings thus indicate that CD44 function is preserved in hMSCs derived from different host tissues, providing further support for a key role of CD44 receptor/ligand interactions in directing cell trafficking via a step 2-bypass pathway of integrin activation [11,19].
To date, the identification of hMSC populations is based on an array of biomarkers as there is no single surface marker unique to this cell population [27]. Indeed, all current hMSC biomarkers have been characterized most extensively on BM-hMSCs, and the extent to which these markers apply to non-BM-source hMSCs remains to be determined [27]. The finding that both BM- and A-hMSCs characteristically display sialylated lactosaminyl glycans and conspicuously lack surface expression of both unsialylated lactosaminyl glycans and fucosylated lactosaminyl glycans suggests that this glycosignature may serve as a distinguishing trait of hMSCs. To provide insight on the mechanistic basis of this glycobiology, we performed qRT-PCR studies of pertinent α(2,3)-sialyltransferases and α(1,3)-fucosyltransferases that modify terminal lactosaminyl glycans. The pattern of gene expression was analogous between both types of hMSCs: there was conserved expression of the sialyltransferase ST3Gal-IV and, with exception of trace levels of transcripts encoding FTIV, there was general absence of all α(1,3)-fucosyltransferases. ST3Gal-IV is the principal sialyltransferase directing synthesis of sLeX in human leukocytes [28] and thus, collectively, the observed pattern of hMSC glycosyltransferase gene expression would predict that terminal lactosaminyl glycans of both BM- and A-hMSC surfaces would be sialylated and convertible to sLeX by α(1,3)-exofucosylation. More specifically, the fact that these sialylated type 2 lactosamines are concentrated predominantly on one cell surface protein, CD44, and, moreover, exclusively on N-glycans of this protein, highlights a distinct glycoprofile of both BM- and A-hMSCs. Commensurately, using two different α(1,3)-fucosyltransferases, FTVI and FTVII, conversion of this CD44 glycovariant into HCELL occurs equally efficiently on both types of hMSCs.
Our prior studies of human hematopoietic stem cells [29] and of human neural stem cells [30] showed that, in contrast to hMSCs, these adult human stem cell populations display sialylated type 2 lactosaminyl glycans not just on CD44, but robustly on multiple membrane glycoproteins, and the content of these structures differs in a cell type-specific fashion. However, notably, α(1,3)-exofucosylation of murine MSCs also engenders HCELL expression (and no other E-selectin ligands) with relevant sLeX determinants found exclusively on N-glycans of CD44, suggesting that the assembly of sialyllactosaminyl N-glycosylations of CD44 may be a general feature of mammalian MSCs [31]. Thus, beyond implications in glycoengineering hMSCs to optimize their migration to sites of tissue injury/inflammation to facilitate relevant applications in regenerative medicine and immunotherapeutics, further studies of MSC glycan decorations are warranted because a greater understanding of the sugar-coating of MSCs may unveil novel biomarkers that will aid in identification of these cells based on this discriminating “taste”.
CONCLUSION
This study provides evidence that adipose-derived human MSCs (A-hMSCs) express surface markers similar to that of bone marrow-derived human MSCs (BM-hMSCs) and notably, like BM-hMSCs, A-hMSCs lack expression of E-selectin ligands but possess sialylated lactosamines that can be α(1,3)-fucosylated to create the canonical E-selectin binding determinant, sLeX. Our results show that α(1,3)-exofucosylation of the hMSC surface with either one of two fucosyltransferases, FTVI and FTVII, generates equivalent expression of sLeX on BM- and A-hMSCs; on both cell populations, these sLeX determinants are created predominantly on N-linked glycans of membrane CD44, thereby engendering HCELL, with resultant induction of robust E-selectin ligand activity. Gene expression studies indicate that A- and BM-hMSCs have similar patterns of mRNA encoding α(1,3)-fucosyltransferases and α(2,3)-sialyltransferases; these data, combined with biochemical evidence that A- and BM-hMSCs display similar sialylated glycoforms of CD44, suggests that patterns of conserved protein glycosylation(s) may define a common glycan signature of hMSCs. Our results thus offer insights on how elucidation of cell surface glycan decorations may yield novel biomarkers of hMSCs, and highlight the applicability of cell surface glycan engineering to enforce HCELL expression on A-hMSCs and thereby program the migration of these immunomodulatory cells to inflammatory sites.
Supplementary Material
SIGNIFICANCE STATEMENT.
Previous studies of human MSCs (hMSCs) indicate that bone marrow-derived hMSCs (BM-hMSCs) display a sialyllactosamine-decorated form of “standard” CD44 (CD44s) that can be α(1,3)-fucosylated to enforce creation of HCELL, a potent E-selectin ligand. E-selectin ligand expression programs migration of cells to all sites of inflammation. Here we sought to determine whether E-selectin ligands are natively expressed on A-hMSCs, and to analyze whether these cells express glycoconjugates that can serve as acceptors of α(1,3)-fucosylation to engender E-selectin ligands. We compared the products of exofucosylation using two different α(1,3)-fucosyltransferases, FTVI and FTVII, on both BM- and A-hMSCs, and evaluated the expression of glycosyltransferases that direct synthesis of lactosaminyl glycans in both A- and BM-hMSCs. We observed that exofucosylation equally enforces expression of the potent E-selectin ligand HCELL among A- and BM-hMSCs and that the profile of expression of lactosaminyl glycan-generating glycosyltransferases is conserved across these cells. These studies highlight conservation in the assembly of sialyllactosamine-modified CD44s among hMSCs derived from two different tissue sources, thereby identifying a characteristic “glycosignature” of these cells and supporting the utility of cell surface glycan engineering to enforce migration of A-hMSCs to sites of tissue injury/inflammation.
Acknowledgments
We thank, Nandini Mondal, Brad Dykstra and Mariana Silva for helpful discussions on the manuscript and Jeffrey Gimble MD PhD at LaCell LLC (New Orleans LA) for providing cryopreserved A-hMSC for the study. R.S. is supported by grants from the National Institutes of Health National Heart Lung Blood Institute (NHLBI grant PO1 HL107146, Program of Excellence in Glycosciences). S.F.V. acknowledges support from the “Miguel Servet” tenure track program (CP10/00438) from the Fondo de Investigación Sanitaria (FIS), co-financed by the ERDF. S.F.V. is supported by grants from the Spanish Ministry of Economy and Competitiveness-European Regional Development Fund (ERDF) (SAF2012-36186 and SAF2015-65019-R) to SF-V). J.V. is supported by grants from ERDF (PI11/0085, PI14/00228) and the Spanish Biomedical Research Center in Diabetes and Associated Metabolic Disorders (CIBERDEM) (CB07708/0012), an initiative of the Instituto de Salud Carlos III). G.P.P is supported by grant from the Ministerio de Salud Carlos III (Programa contrato de perfeccionamiento postdoctoral Sara Borrell CD10/00285).
According to the National Institutes of Health policies and procedures, the Brigham & Women’s Hospital has assigned intellectual property rights regarding HCELL and glycosyltransferase-programmed stereo-substitution (GPS) to the inventor (R.S.), who may benefit financially if the technology is licensed.
Footnotes
AUTHOR CONTRIBUTIONS
G.P.P.: study design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; C.D.: Collection of data, manuscript writing; C.R.C.: Collection of data; A.K.: sample collection; S.F.V. and J.V.: study design, manuscript writing; R.S.: Conception and design of the study, data analysis and interpretation, supervising all research, manuscript writing, final approval of manuscript.
DISCLOSURE OF POTENTIAL CONFLICT OF INTEREST
R.S.’s ownership interests were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policy. All other authors declare no competing financial interests.
REFERENCES
- 1.Di Nicola M, Carlo-Stella, Magni M, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;10:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 2.Meisel R, Zibert A, Laryea M, et al. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;12:4619–4621. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- 3.Pachón-Peña G, Yu G, Tucker A, et al. Stromal stem cells from adipose tissue and bone marrow of age-matched female donors display distinct immunophenotypic profiles. J Cell Physiol. 2011;3:843–851. doi: 10.1002/jcp.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pachón-Peña G, Serena C, Ejarque M, et al. Obesity determines the immunophenotypic profile and functional characteristics of human mesenchymal stem cells from adipose tissue. Stem Cells Transl Med. 2016;5:464–475. doi: 10.5966/sctm.2015-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourin P, Bunnell B, Casteilla L, et al. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: a joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT) Cytotherapy. 2013;15:641–648. doi: 10.1016/j.jcyt.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pires A, Mendes-Pinheiro B, Teixera F, et al. Unveiling the Differences of Secretome of Human Bone Marrow Mesenchymal Stem Cells, Adipose Tissue-Derived Stem Cells, and Human Umbilical Cord Perivascular Cells: A Proteomic Analysis. Stem Cells Dev. 2016;25:1073–1083. doi: 10.1089/scd.2016.0048. [DOI] [PubMed] [Google Scholar]
- 7.Sousa B, Parreira R, Fonseca E, et al. Human adult stem cells from diverse origins: an overview from multiparametric immunophenotyping to clinical applications. Cytometry A. 2014;1:43–77. doi: 10.1002/cyto.a.22402. [DOI] [PubMed] [Google Scholar]
- 8.Mizuno H, Tobita M, Uysal A. Concise review: Adipose-derived stem cells as a novel tool for future regenerative medicine. Stem Cells. 2012;5:804–810. doi: 10.1002/stem.1076. [DOI] [PubMed] [Google Scholar]
- 9.Melief S, Zwaginga J, Fibbe W, et al. Adipose tissue-derived multipotent stromal cells have a higher immunomodulatory capacity than their bone marrow-derived counterparts. Stem cells translational medicine. 2013;2:455–463. doi: 10.5966/sctm.2012-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purandare B, Teklemarian T, Zhao L, et al. Temporal HLA profiling and immunomodulatory effects of human adult bone marrow- and adipose-derived mesenchymal stem cells. Regen Med. 2014;1:67–79. doi: 10.2217/rme.13.82. [DOI] [PubMed] [Google Scholar]
- 11.Sackstein R, Merzaban J, Cain D, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;2:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 12.Sackstein R. Glycosyltransferase-programmed stereosubstitution (GPS) to create HCELL: engineering a roadmap for cell migration. Immunol Rev. 2009;1:51–74. doi: 10.1111/j.1600-065X.2009.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sackstein R. Directing stem cell trafficking via GPS. Methods Enzymol. 2010;479:93–105. doi: 10.1016/S0076-6879(10)79005-4. [DOI] [PubMed] [Google Scholar]
- 14.Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Dykstra B, Lee J, Mortensen L, et al. Glycoengineering of E-Selectin ligands by intrecellular versus extracellular fucosylation differentially affects osteotropism of human mesenchymal stem cells. Stem Cells. 2016 doi: 10.1002/stem.2435. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JY, Buzney C, Poznansky M, et al. Dynamic alterations in chemokine gradients induce transendothelial shuttling of human T cells under physiologic shear conditions. Journal of leukocyte biology. 2009;86:1285–1294. doi: 10.1189/jlb.0309214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyce J, Mellor E, Perkins B, et al. Human mast cell progenitors use alpha4-integrin, VCAM-1, and PSGL-1 E-selectin for adhesive interactions with human vascular endothelium under flow conditions. Blood. 2002;8:2890–2896. doi: 10.1182/blood.v99.8.2890. [DOI] [PubMed] [Google Scholar]
- 18.Burdick M, McCaffery J, kim Y, et al. Colon carcinoma cell glycolipids, integrins, and other glycoproteins mediate adhesion to HUVECs under flow. Am J Physiol Cell Physiol. 2003;4:977–987. doi: 10.1152/ajpcell.00423.2002. [DOI] [PubMed] [Google Scholar]
- 19.Thankamony S, Sackstein R. Enforced hematopoietic cell E-and L-selectin ligand (HCELL) expression primes transendotelial migration of human mesenchymal stem cells. Proc Natl Acad Sci. 2011;6:2258–2263. doi: 10.1073/pnas.1018064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okajima T, Fukumoto S, Miyazaki H, et al. Molecular cloning of a novel alpha2,3-sialyltransferase (ST3Gal-VI) that sialylates type II lactosamine structures on glycoproteins and glycolipids. J Biol Chem. 1999;17:11479–11486. doi: 10.1074/jbc.274.17.11479. [DOI] [PubMed] [Google Scholar]
- 21.Sasaki K, Watanabe E, Kawashima K, et al. Expression cloning of a novel Gal beta (1–3/1–4) GlcNAc alpha 2,3-sialyltransferase using lectin resistance selection. J Biol Chem. 1993;30:22782–22787. [PubMed] [Google Scholar]
- 22.Harduin-Lepers A, Vallejo-Ruiz V, Krzewinski-Recchi M, et al. The human sialyltransferase family. Biochimie. 2001;8:727–737. doi: 10.1016/s0300-9084(01)01301-3. [DOI] [PubMed] [Google Scholar]
- 23.Serna S, Yan S, Martin-Lomas M, et al. Fucosyltransferases as synthetic tools: glycan array based substrate selection and core fucosylation of synthetic N-glycans. J Am Chem Soc. 2011;41:16495–16502. doi: 10.1021/ja205392z. [DOI] [PubMed] [Google Scholar]
- 24.de Vries T, Knegtel R, Holmes E, et al. Fucosyltransferases: structure/function studies. Glycobiology. 2001;11:119R–128R. doi: 10.1093/glycob/11.10.119r. [DOI] [PubMed] [Google Scholar]
- 25.Escrevente C, Machado E, Brito C, et al. Different expression levels of α3/4fucosyltransferases and Lewis determinants in ovarian carcinoma tissues and cell lines. Int J Oncol. 2006;29:557–566. [PubMed] [Google Scholar]
- 26.Papaioannou T, Stefanadis C. Vascular wall shear stress: basic principles and methods. Hellenic J Cardiol. 2005;46:9–15. [PubMed] [Google Scholar]
- 27.Lv F, Tuan R, Cheung K, et al. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014;32:1408–1419. doi: 10.1002/stem.1681. [DOI] [PubMed] [Google Scholar]
- 28.Mondal N, Buffone A, Stolfa G, et al. ST3Gal-4 is the primary sialyltransferase regulating the synthesis of E-, P-, and L-selectin ligands on human myeloid leukocytes. Blood. 2015;125:687–696. doi: 10.1182/blood-2014-07-588590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merzaban J, Burdick M, Gadhoum S, et al. Analysis of glycoprotein E-selectin ligands on human and mouse marrow cells enriched for hematopoietic stem/progenitor cells. Blood. 2011;118:1774–1783. doi: 10.1182/blood-2010-11-320705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merzaban J, Imitola J, Starossom, et al. Cell surface glycan engineering of neural stem cells augments neurotropism and improves recovery in a murine model of multiple sclerosis. Glycobiology. 2015;25:1392–1409. doi: 10.1093/glycob/cwv046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdi R, Moore R, Sakai S, et al. HCELL expression on murine MSC licenses pancreatotropism and confer durable reversal of autoimmune diabetes in NOD mice. Stem cells. 2015;33:1523–1531. doi: 10.1002/stem.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.