

Abstract
The molecular pathways regulating lymphoid priming, fate, and development of multipotent bone marrow hematopoietic stem and progenitor cells (HSPCs) that continuously feed thymic progenitors remain largely unknown. While Notch signal is indispensable for T cell specification and differentiation, the downstream effectors are not well understood. PRL2, a protein tyrosine phosphatase that regulates hematopoietic stem cell proliferation and self-renewal, is highly expressed in murine thymocyte progenitors. Here we demonstrate that protein tyrosine phosphatase PRL2 and receptor tyrosine kinase c-Kit are critical downstream targets and effectors of the canonical Notch/RBPJ pathway in early T cell progenitors. While PRL2 deficiency resulted in moderate defects of thymopoiesis in the steady state, de novo generation of T cells from Prl2 null hematopoietic stem cells (HSCs) was significantly reduced following transplantation. Prl2 null HSPCs also showed impaired T cell differentiation in vitro. We found that Notch/RBPJ signaling upregulated PRL2 as well as c-Kit expression in T cell progenitors. Further, PRL2 sustains Notch-mediated c-Kit expression and enhances SCF/c-Kit signaling in T cell progenitors, promoting effective DN1-DN2 transition. Thus, we have identified a critical role for PRL2 phosphatase in mediating Notch and c-Kit signals in early T cell progenitors.
Keywords: PRL2, T cell progenitors, ETPs, Notch, c-Kit, and thymopoiesis
Graphical abstract
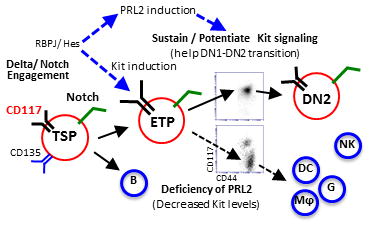
Cooperation of PRL2 phosphatase with Notch and c-Kit signals is critical for early T cell differentiation. Delta/Notch engagement induces the expression of both c-Kit and PRL2 in ETPs. PRL2 then sustains and/or potentiates Kit signaling to facilitate DN1-DN2 transition. In the absence of PRL2, c-Kit expression is decreased and decreased c-Kit expression promotes ETP differentiation toward myeloid lineages. Thus, sustaining high level of c-Kit expression by PRL2 is crucial for ETP expansion, survival, and commitment during early stage T cell development.
INTRODUCTION
The thymic microenvironment supports the differentiation and proliferation of the bone marrow-derived progenitors as they enter and migrate through the thymus [1–2]. The lymphoid-primed multipotent progenitors (LMPPs) are thought to be the thymus-seeding progenitors (TSPs) that differentiate into early T lineage progenitors (ETPs) [1–2]. ETPs share fundamental characteristics with bone marrow progenitors, such as high expression of the cell-surface markers c-Kit and CD44, the absence of markers of mature cells and multilineage developmental potential. After arriving in the thymus, ETPs lose the potential to generate B cells, whereas their potential to develop into myeloid cells, natural killer cells and dendritic cells is retained at the CD4−CD8− double-negative 2a (DN2a) stage [1–2]. ETPs express high levels of receptor tyrosine kinase c-Kit that persists in DN2a cells but is gradually downregulated in DN2b/DN3 cells, where final T cell commitment is accomplished by the expression of a number of transcription factors, including GATA3 and Bcl11b [3–6].
C-Kit plays a critical role in maintaining postnatal T lymphopoiesis in thymus [3–7]. Mice lacking c-Kit (c-Kitw/w) displayed age-dependent accumulation of DN1 cells and had massive reduction of DN2 to CD4+CD8+ double-positive (DP) cells in the thymus [5]. Mice with partial loss-of-function (LOF) c-Kit mutations also showed similar T cell phenotypes. In vivo and in vitro studies that utilized c-Kit inhibitors demonstrated that c-Kit was critical for early thymocyte development [7–10]. Although c-Kit is critically important for post-transplant T cell reconstitution, it is dispensable for post-transplant B cell and myeloid cell formation [11–12], indicating that T cell generation is more dependent on c-Kit activity than other lineages. While it has been postulated that the severe hematopoietic defects seen in Kit mutant mice may be due to cumulative effects from HSCs to progenitors [11–13], the role of c-Kit signaling in ETPs is largely unknown.
Delta/Notch association is one of the most important signals provided by the thymic environment to initiate T cell differentiation [1–2]. Although molecular mechanisms of early T cell differentiation have been extensively investigated, the downstream effectors of Notch signaling require further clarification. Given that Notch activation is essential for T-lineage specification of lymphomyeloid progenitors seeding the thymus [1] and that progression along T cell lineage further requires cooperative signaling provided by SCF and receptor tyrosine kinase c-Kit, it is important to delineate how Notch activation links to SCF/c-Kit signaling in T cell progenitors [14].
The phosphatase of regenerating liver (PRL) family of phosphatases, consisting of PRL1, PRL2, and PRL3, represents an intriguing group of proteins being validated as biomarkers and therapeutic targets in human cancer [15–17]. We have been investigating the role of PRL2 in development and cancer [18–21]. We generated Prl2 deficient mice and found that PRL2 is required for extra-embryonic development and associates the oncogenic properties of PRL2 with its ability to negatively regulate PTEN, thereby activating the PI3K-Akt pathway [18]. To determine the role of PRL2 in hematopoiesis, we analyzed HSC behavior in Prl2 deficient mice. We found that Prl2 deficiency impairs HSC self-renewal as revealed by serial bone marrow transplantation assays [19–20]. Moreover, we observed that Prl2 null hematopoietic stem and progenitor cells (HSPCs) are more quiescent and show reduced activation of the AKT and ERK signaling. While stem cell factor (SCF) is an early acting cytokine that activates the receptor tyrosine kinase KIT and promotes HSC maintenance, how SCF/KIT signaling is regulated in hematopoietic stem and progenitor cells is poorly understood. We found that PRL2 is important for SCF-mediated HSPC proliferation and loss of PRL2 decreased the ability of oncogenic KIT/D814V mutant in promoting hematopoietic progenitor cell proliferation. Thus, PRL2 plays critical roles in regulating HSC self-renewal, at least in part, through mediating SCF/KIT signaling [19–20]. We also found that PRL2 deficiency impairs Kit signaling and spermatogenesis [21]. Thus, the defects seen in PRL2-deficient hematopoietic and testis spermatogonia cells recapitulate some phenotypes of c-Kit mutant mice [3–7], suggesting that PRL2 may regulate SCF/c-Kit signaling during development [19–21].
Here we report a functional requirement for PRL2 in T cell development. We observed that PRL2 is highly expressed in early stage thymic progenitors. While PRL2 deficiency resulted in moderate defects of thymopoiesis in the steady state, de novo generation of T cells from Prl2 null HSCs was significantly reduced following transplantation. Prl2 null HSPCs also showed impaired T cell differentiation in vitro. We found that Notch/RBPJ signaling upregulated PRL2 as well as c-Kit expression in T cell progenitors. We further discovered that PRL2 enhanced SCF/c-Kit signaling in T cell progenitors, promoting efficient DN1 to DN2 transition. Thus, we have identified a critical role for the PRL2 phosphatase in mediating Notch and c-Kit signals during T cell development.
MATERIALS AND METHODS
Mice
Wild type C57BL/6 (CD45.2+), B6.SJL (CD45.1+), and F1 mice (CD45.2+ CD45.1+) mice were purchased from the Jackson Laboratories. Prl2−/−, RBPJflox/flox-Mx1-cre+, and wild type mice were maintained in the Indiana University Animal Facility according to IACUC-approved protocols, and kept in Thorensten units with filtered germ-free air. All mice were 7–12 weeks of age at the time of analysis.
Flow cytometry
Flow cytometry analysis of hematopoietic stem and progenitor cells was performed as described previously [18]. Briefly, hematopoietic cells isolated from bone marrow, thymus, or spleen were stained on ice for 30 min with the Biotin-conjugated lineage markers and then were stained with fluorochrome-conjugated antibodies. Definition of each stem and progenitor population is shown in Supplementary Table 1. Flow antibodies were purchased from Biolegend, eBioscience or BD Bioscience (listed in Supplementary Table 2). Experiments were performed on FACSAria and FACSLSR II cytometers (BD Biosciences) and analyzed by using the FlowJo Version 9.3.3 software (TreeStar).
Real-time PCR
For quantitative reverse-transcription polymerase chain reaction (RT-PCR), total RNA was isolated using the RNeasy Plus Micro Kit (QIAGEN), and then subjected to reverse transcription with random hexamers (SuperScript III kit; Invitrogen). Quantitative real-time PCR was performed with SYBR Green (Qiagen) on an ABI PRISM 7500 system. Gene specific primers were designed to flank introns so that products from cDNA could be distinguished from possible genomic DNA contamination. Primer sequences are listed in Supplementary Table 3.
In vitro methylcellulose colony-forming cell assay
Clonogenic progenitors were determined in methylcellulose medium (MethoCult GF M3234, StemCell Technologies) with cytokines (SCF, TPO, EPO, IL-3, and GM-CSF) using 2 x 104 BMMCs per well (6-well plate). Colonies were scored after 7 days of the culture.
Apoptosis assays
Thymocytes were harvested and cultured in the presence of IL-7 (5ng/ml) for 48hrs with or without 2Gy irradiation. Cell viability was evaluated by PI/Annexin V staining in combination with lineage markers, including CD25, CD44, and CD117.
In vitro T-cell differentiation assays
OP9-DL1 cells were maintained and cultured as previously described [22]. Sorted 200 of KSLs or 1x105 of DN3 cells from Prl2+/+ and Prl2−/− mice were cultured with OP9-DL1 cells in the presence of IL-7 (10 ng/ml). The fusion proteins Delta-like1/Fc or Jagged1/Fc (R&D) were coated onto 96 well plate at the concentration of 5 ng/ml or 20 ng/ml respectively [23]. Sorted LMPPs, ETPs, or DN2 cells were also applied onto coated plate in the presence of Flt3 ligand (10ng/ml), IL-7 (10ng/ml) and graded dose of SCF (0.2 to 25 ng/ml). Cells were harvested for measuring cell proliferation and differentiation.
Transplantation assays
Bone marrow mononuclear cell and HSC transplantation assays were performed as described previously [18]. Briefly, we injected 200 CD48−CD150+KSL cells from wild type or Prl2 null mice (CD45.2+) plus 3 × 105 competitor bone marrow cells (CD45.1+) into lethally irradiated F1 mice (CD45.1+CD45.2+).
Production of Retrovirus
Retroviral particles were produced by transfection of Phoenix E cells with the MSCV-IRES-GFP or MSCV-Notch-ICN1-IRES-GFP plasmids, according to standard protocols. Mouse hematopoietic progenitor cells were infected with high-titer retroviral suspensions in the presence of retronectin. Twenty-four hours after infection, the GFP positive cells were sorted by FACS.
Luciferase assay
293 cells were transfected with human PRL2 promoter driven luciferase reporter plasmids containing either RBPJ binding sites or mutant RBPJ binding sites. Luciferase activity was assayed 24 hours after transfection according to manufacturer’s instructions (Promega).
Statistical Analysis
We used either Student’s t test or two-way analysis of variance to determine statistical significance. *, P<0.05; **, P<0.01; ***, P<0.001; ns, not significant.
RESULTS
PRL2 deficiency alters postnatal thymopoiesis
To determine the role of PRL2 in T cell development, we examined the spleen and thymus of Prl2+/+ and Prl2−/− mice. PRL2 deficiency resulted in marked reduction of splenocyte and thymocyte counts compared to that of the Prl2+/+ mice (Fig. 1A). Although both T cell and B cell generation were normal in Prl2−/− spleen (Supplementing information Fig. S1A), T cell production is altered in Prl2−/− thymus. While the frequency of DN cells in Prl2−/− thymus was increased, the frequency of DP cells was decreased (Fig. 1B). The absolute number of DN4 and DP cells was lower in Prl2−/− thymus than that of the Prl2+/+ thymus (Fig. 1C). Within DN cells, the frequency of DN2a, DN2b, and DN3 was higher in Prl2−/− thymus than that of the Prl2+/+ thymus, whereas the frequency of DN1 and ETP was comparable (Supplementing information Fig. S1B). Prl2−/− thymus showed higher frequency of Gr1+ myeloid cells and B220+ cells compared to Prl2+/+ thymus (Supplementing information Fig. S1C). To determine the impact of PRL2 deficiency on thymic microenvironment, we examined SCF and DLL expression in stromal cells from Prl2+/+ and Prl2−/− thymus. We found that the expression of SCF and DLL were comparable in Prl2+/+ and Prl2−/− cells (Supplementing information Fig. S1D), suggesting that Prl2−/− thymic microenvironment is normal. We also found comparable numbers of lymphoid-primed multipotent progenitors (LMPPs) and common lymphoid progenitors (CLPs) in both Prl2+/+ and Prl2−/− bone marrow (Supplementing information Fig. S1E). Given that PRL2 is important for HSPC proliferation [18], we measured the cell cycle status of T cell progenitors and found that Prl2−/− DN2a and DN2b cells were more quiescent than Prl2+/+ cells (Fig. 1D). Prl2−/− T cell progenitors did not show increased apoptosis in the presence or the absence of irradiation in vitro (Fig. 1E). Thus, PRL2 deficiency specifically alters postnatal thymopoiesis.
Figure 1. Prl2−/− mice have moderate T cell defects in steady state.

(A) Splenocytes and thymocytes from 8-week-old Prl2+/+ and Prl2−/− mice were isolated and counted. Absolute splenocyte and thymocyte numbers from Prl2−/− mice are reduced to 50% of Prl2+/+ mice. ***P<0.001, n=10. (B) Analysis of CD4+, CD8+, DP, and DN thymocytes in Prl2+/+ and Prl2−/− mice by flow cytometry. The frequency of DN cells is increased in Prl2−/− thymus compared to Prl2+/+ thymus. **P<0.01, n=7. (C) Analysis of DN1, ETP, DN2, DN3, DN4 and DP thymocytes in Prl2+/+ and Prl2−/− mice by flow cytometry. Absolute number of DN4 and DP cells is decreased in Prl2−/− thymus compared to Prl2+/+ thymus. **P<0.01, n=7. (D) Steady state cell cycle status of thymocytes was determined by Ki67 and DAPI staining. The percentage of cells at the G0 phase of the cell cycle was shown. Percentage of quiescent (G0) Prl2−/− DN2a and DN2b thymocytes is slightly increased compared to Prl2+/+ cells. *P<0.05, n=6. (E) Staining for Annexin V and propidium iodide (PI) in thymocytes isolated from Prl2+/+ and Prl2−/− mice after 48 h culture in serum-free medium, in the presence or the absence of irradiation (2Gy).
PRL2 deficiency impairs T cell development following HSC transplantation
Given that c-Kit loss-of-function (LOF) bone marrow cells show decreased T cell reconstitution following transplantation [11–13], we evaluated T cell development in recipient mice repopulated with Prl2+/+ or Prl2−/− BM cells. In competitive transplantation assays, Prl2−/− BM cells showed significantly reduced T cell production in recipient thymus compared to Prl2+/+ cells at 18 weeks following transplantation (Fig. 2A). Next, we performed HSC transplantation and found that Prl2−/− HSCs showed markedly reduced T cell production in the peripheral blood (PB) of recipient mice compared to that of the Prl2+/+ cells (Fig. 2B), indicating that the de novo T cell generation from HSCs was impaired. The number of donor-derived CD3+ cells in the PB of recipient mice repopulated with Prl2−/− HSCs was 30-fold less than that of the Prl2+/+ cells (Fig. 2C). While the absolute number of Prl2−/− HSCs in the femurs of the recipient mice was 6-fold lower than that of the Prl2+/+ HSCs, the T cell defect in the peripheral blood of recipient mice repopulated with Prl2−/− HSCs was more dramatic than B cell or myeloid cell defect (Fig. 2C). Donor cell engraftment in the recipient thymus was similar to our observation in PB (Prl2+/+: 91±6.7 vs. Prl2−/−: 4.6±0.3 %, ***p<0.0001); however, Prl2−/− HSCs showed robust ETP chimerism (60%), which was much higher than overall thymocyte chimerism and close to that of the Prl2+/+ cells (91.8±10.1 vs. 59.6±13.5%) (Fig. 2D and S2A). Subsequently, Prl2−/− HSCs showed decreased DN2 production, which was less than 10% (Supplementing information Fig. S3A). Consistent with our observations in the thymus, T cell production in the recipient spleen repopulated with Prl2−/− HSCs was lower than that of the Prl2+/+ cells (Supplementing information Fig. S3B). We also calculated the frequency of donor-derived T cell progenitor populations for each developmental stage. The frequency of Prl2+/+ ETPs was approximately 0.01%, and the frequency of other T cell progenitor cell population was increased during T cell differentiation. However, the frequency of ETP derived from Prl2−/− HSCs was 30-fold higher than that of the Prl2+/+ cells (Fig. 2E and 2F). The frequency of DN2 and DN3 cells derived from Prl2−/− HSCs was moderately increased (Fig. 2E and 2F), indicating that the DN1 to DN2 transition requires PRL2.
Figure 2. PRL2 deficiency impairs T cell development following hematopoietic stem cell transplantation.

(A) The frequency of donor-derived cells in spleen, thymus, and bone marrow of recipient mice was determined 18 weeks following bone marrow transplantation. Prl2−/− cells show decreased engraftment in thymus compared to Prl2+/+ cells. (B) The frequency of donor-derived cells in the peripheral blood (PB) of recipient mice was determined every 4 weeks following transplant Prl2+/+ or Prl2−/− HSCs into lethally-irradiated recipient mice. CD3+ T cell recovery was significantly lower in recipients repopulated with Prl2−/− HSCs than that of the Prl2+/+ HSCs. (C) Absolute number of donor derived LT-HSCs in 2 femurs (left panel). Absolute number of PB T cells generated from Prl2−/− HSCs was remarkably reduced compared with that of the Prl2+/+ HSCs. (D) Achievement of donor-derived chimerism in each differentiation stage at 16 weeks after transplantation is shown. (E) Representative flow cytometry plot of donor-derived ETPs. Number indicated the percentage of ETPs in total CD45.2 positive cells. (F) The frequency of different T cell population in donor-derived cells is shown (***P<0.001, **P<0.01, n=8).
PRL2 deficiency impairs T cell development in vitro
Next, we investigated the effect of PRL2 deficiency on in vitro T cell generation utilizing both the Delta-Like1 (DLL1) expressing OP9 (OP9-DL1) stromal cells [22] and the feeder-free T cell culture system with Delta-like1-Fc fusion protein (DLL1-Fc) [23]. When sorted KSL cells were cultured on OP9-DL1 cells, the number of cumulative produced cells from 500 Prl2−/− KSLs was reduced up to 100 fold compared to that of the Prl2+/+ cells (Fig. 3A). The total double positive (DP) cell count was also significantly decreased in the Prl2−/− group compared to the Prl2+/+ group (Fig. 3B), demonstrating that PRL2 is critical for T cell expansion in vitro. To determine the stage specificity, we cultured KSLs or DN3 cells on OP9-DL1 cells and then harvested all cells for flow cytometry analysis. After 14 days of culture, Prl2−/− KSLs showed reduced ETP production compared to Prl2+/+ KSLs (Fold increase: 184±14 vs. 12.5±4.3, ****p<0.0001; Fig. 3C). Prl2−/− KSLs showed reduced DN1 production as well (data not shown). While DN1 contains heterogeneous cells with varied T cell differentiation capacities [24], we found that KSLs from either genotype generated comparable number of non-ETP DN1 cells (17.1±6.1 vs. 7.1±1.2, p=0.05; Fig. 3C). When Prl2−/− DN3 cells were plated onto OP9-DL1 cultures, we observed moderate decrease in cell expansion compared to Prl2+/+ cells (Fold increase: 4.8±0.2 vs. 3.2±0.3, **p<0.01; Fig. 3D), suggesting that PRL2 specifically regulates early T cell lineage development.
Figure 3. PRL2 deficiency impairs T cell development in vitro.

(A and B) Purified Prl2+/+ and Prl2−/− KSL cells were cultured on DL-OP9 cells in the presence of IL-7. Cells were then harvested at different time point and stained with various markers to measure T cell differentiation. Prl2−/− KSLs show marked reduction of cumulative total cell count (A) and double positive T cell count (B) compared with Prl2+/+ cells. (C and D) To determine stage specificity, sorted KSLs and DN3 were cultured on OP-DL1 cells for 8 days or 15 days, respectively. Production of ETPs from KSLs and production of total nucleated cells (TNCs) from DN3 cells were calculated. Prl2−/− cells show profound expansion defects at early stage (C) than that of the later stage (D). (E) Sorted LMPPs, ETPs, or DN2 cells were cultured on DLL1-Fc coated plate in the presence of graded dose of SCF. Total cell count was determined at day 12. Prl2−/− cells show defective expansion compared with Prl2+/+ cells, even in the presence of high dose of SCF.
Although early stage T cell differentiation depends on c-Kit [9–10], there is no direct evidence that bone marrow cells from c-Kit mutant mice show defective T cell differentiation in vitro. To determine the effect of SCF on T cell differentiation in vitro, we utilized the feeder-free T-cell culture system with DLL1-Fc [23]. When purified Prl2+/+ LMPPs were cultured on DLL1-Fc (R&D Systems)-coated plate in the presence of graded dose of SCF, total cells generated were increased in a SCF dosage-dependent manner (Fig. 3E). While SCF promoted differentiation of LMPPs toward the DN2/3 stage, even at low doses (1 ng/ml), the fold increase was small (Supplementing information Fig. S4A and S4B). T cell differentiation was also accompanied by increased myeloid differentiation (Supplementing information Fig. S4A). In contrast, Prl2−/− LMPPs, ETPs, and DN2 cells showed severely decreased expansion in DLL1-Fc cultures in the presence of increased levels of SCF (Fig. 3E), suggesting that PRL2 mediates SCF/c-Kit signaling in early T cell progenitors. These data demonstrated that PRL2 is more important for early stage (c-Kit positive) than later stage T cell development.
Notch signaling regulates PRL2 expression during T cell differentiation
When we assessed PRL2 expression in purified KSLs, multipotent progenitors (MPPs), common lymphoid progenitors (CLPs), ETPs, DN2 and DN3 thymocytes by quantitative real-time PCR, we found that PRL2 was highly expressed in DN2 thymocytes (Fig. 4A), suggesting that PRL2 may be a downstream target of active Notch signaling. We then examined PRL2 expression following Delta ligand engagement. We plated purified LMPPs or DN2 cells on DLL1-Fc cultures and measured the change in PRL2 mRNA levels over time. PRL2 mRNA was upregulated during DLL1 mediated T cell differentiation but not by control IgG (Fig. 4B). We also found that ectopic expression of constitutive active Notch1, intracellular domain of Notch1 (Notch-ICN1), increased PRL2 protein levels in Prl2+/+ Lin−Sca-1+ cells (Fig. 4C). RBPJ/CSL transcription factor is the key mediator of Notch signaling in the thymus [1–2], and we found that both mouse and human PRL2 promoter regions have RBPJ/CSL and HES binding motifs (Supplementing information Fig. S5A). To determine whether RBPJ/CSL associates with the promoter regions of PRL2, we performed chromatin immunoprecipitation (ChIP) assays in a human T cell progenitor cell line Tail-7 [24]. We found that RBPJ/CSL was associated with several regions of the PRL2 promoter (Fig. 4D). Genome-wide ChIP-seq analysis also showed Notch binding to the PRL2 (PTP4A2) locus in human T cell leukemia cell line CUTLL1 (Supplementing information Fig. S5B) [25]. In luciferase reporter assays, we found that Notch-ICN1 activated the PRL2 promoter but not the PRL2 promoter containing mutations of CSL binding sites (Fig. 4E and Supplementing information Fig. S5C). These findings demonstrate that PRL2 is a direct target of Notch/RBPJ in T cell progenitors.
Figure 4. Notch signaling regulates PRL2 expression during T cell differentiation.

(A) Quantitative real-time PCR analysis of PRL2 expression in sorted MPPs, CLPs, ETPs, DN2, DN3, DN4, DP, and CD4+ thymocytes, presented relative to expression in KSLs, set as 1. PRL2 is highly expressed in DN2 cells. n = three biological replicates. (B) Sorted Prl2+/+ LMPPs were cultured in the presence or the absence of DLL1-Fc. Cells were collected at each time point and PRL2 expression was determined by quantitative real-time PCR analysis. **P<0.01, n = three biological replicates. (C) Notch-ICN1 was introduced into Prl2+/+ Lin−Sca1+ cells and the levels of PRL2 protein were determined by western blot analysis. (D) Chromatin-bound DNAs from Tail-7 cells were immunoprecipitated with a RBPJ-antibody or normal mouse IgG. Quantitative real-time PCR amplification was performed on corresponding templates using primers for PRL2 gene. (E) Notch-ICN1 transactivates PRL2 promoter. 293 cells were transfected with PRL2 promoter driven luciferase reporter plasmids containing either RBPJ binding sites or mutant RBPJ binding sites. Luciferase activity was assayed 24 hours after transfection. *P<0.05, n=3.
Notch/RBPJ pathway upregulates c-Kit expression in ETPs
Similar to PRL2 promoter, c-Kit promoter has multiple putative RBPJ/CSL and HES binding sites (Supplementing information Fig. S6A), suggesting that Notch/RBPJ pathway may regulate c-Kit expression in T cell progenitors. We observed increased fluorescent intensity of c-Kit in LMPPs treated with DLL1-Fc compared with cells treated with IgG (Fig. 5A). We then examined the kinetics of c-Kit transcription following Delta/Notch engagement and found that DLL1-Fc increased the level of c-Kit mRNAs, which was maintained for several days (Fig. 5B). Consistent with this observation, we found that Lin−Sca-1+ cells expressing Notch-ICN1 showed increased level of c-Kit proteins compared to control cells (Fig. 5C). To determine whether upregulation of c-Kit was mediated by Notch/RBPJ pathway, we examined c-Kit expression in RBPJ-deficient T cell progenitors [26]. When we evaluated RBPJfl/flCre+ mice 18 days after pI:pC injection, we found that the fluorescent intensity of c-Kit was markedly reduced in ETPs of the RBPJfl/flCre+(KO) thymus compared to that of the RBPJfl/flCre− (WT) thymus (Fig. 5D). Interestingly, we detected very few c-Kit+ DN2 cells in the RBPJ KO thymus (Supplementing information Fig. S6B). In addition, KSLs and HSC-enriched populations (CD48−KSLs) isolated from the RBPJ KO BM showed decreased fluorescent intensity of c-Kit compared to that of the RBPJ WT BM cells (Fig. 5E). To determine whether RBPJ/CSL associates with the promoter regions of KIT, we performed chromatin immunoprecipitation (ChIP) assays in Tail-7 cells and found no association of RBPJ with KIT promoter (data not shown). Genome-wide ChIP-seq analysis in CUTLL1 cells did not show Notch binding to KIT promoter (Supplementing information Fig. S6C) [25], suggesting that Notch/RBPJ may indirectly regulate KIT expression in T cell progenitors. To determine whether increased c-Kit expression was specific to the Delta/Notch engagement in the thymic environment, we compared the abilities of DLL1-Fc and Jagged1-Fc (Jag1) to drive c-Kit expression in LMPP cells [26]. Sorted LMPPs were cultured with control IgG, DLL1-Fc, or Jag1-Fc. While DN2/3 cells were efficiently produced from DLL1-treated WT LMPPs, only low numbers of DN2/3 cells were generated from Jag1-treated WT LMPPs (Fig. 5F). Importantly, we found that RBPJ KO LMPPs failed to maintain high c-Kit expression by any stimulation (Fig. 5F) and that level of c-Kit mRNAs was decreased in RBPJ KO LMPPs regardless of any ligand (Fig. 5G). These findings indicate both Delta (Thymic environmental factor) and Jagged (BM environmental factor) ligands utilize the canonical Notch/RBPJ pathway to upregulate and/or maintain c-Kit expression for the appropriate cell differentiation program.
Figure 5. PRL2 sustains Notch mediated c-Kit expression in T cell progenitors.

(A) Expression of c-Kit was induced by DLL1-Fc during T cell differentiation, assessed by flow cytometry. (B) Sorted Prl2+/+ LMPPs and DN2 cells were cultured in the presence or the absence of DLL1-Fc. Cells were collected at each time point and c-Kit expression was determined by quantitative real-time PCR analysis, presented relative to expression in sorted LMPPs. **P<0.01, n = three biological replicates. (C) Notch-ICN1 was introduced into wild type Lin−Sca1+ cells and the levels of c-Kit protein were determined by western blot analysis. (D, E) Flow cytometry analysis of c-Kit expression in primary RBPJ-KO thymus (D) or BM (E) at 3 weeks post pI:pC injection. The mean fluorescent intensity (MFI) of c-Kit was greatly reduced in RBPJ-deficient cells compared with that of the WT cells. (F) T cell induction from RBPJ-KO or WT LMPPs. Sorted LMPPs were cultured with control IgG, DLL1, or Jag1. The MFI of c-Kit was measured at day 7. (G) Kinetic study of c-Kit expression in T cell cultures of LMPPs isolated from WT and RBPJ-KO mice. Relative c-Kit mRNA levels were measured by quantitative real-time PCR analysis. **P<0.01, n = three biological replicates.
PRL2 sustains Notch-mediated c-Kit expression in T cell progenitors
Given that dysfunctional c-Kit signaling impaired T cell differentiation, we measured c-Kit intensity in DN1 cells using OP9-DL1 cultures. While most cells within the Prl2+/+ DN1 population were c-Kitbright ETPs, more than half of the Prl2−/− DN1 cells failed to maintain high c-Kit expression (Fig. 6A). A similar phenotype was observed in DLL1-Fc cultures. Notably, c-Kit mRNA was upregulated in freshly isolated Prl2−/− LMPPs and in subsequent cultures (Supplementing information Fig. S7A and S7B), suggesting that decreased c-Kit protein seen in Prl2−/− T cell progenitors may be due to accelerated turnover. We also found that the primary Prl2−/− thymus had lower level of c-Kit protein compared to the Prl2+/+ thymus (Figure 6B), ruling out potential artifacts caused by cell cultures. Notably, Prl2−/− LMPPs treated with DLL1 differentiated into myeloid cells (Fig. 6C), demonstrating that PRL2 deficiency and low level of SCF have similar impact on T cell progenitor differentiation (Supplementing information Fig. S3A). Consistent with these findings, sorted Prl2−/− ETPs retained myeloid colony forming capabilities (Fig. 6D). To determine the impact of PRL2 on KIT activation, we stimulated Prl2+/+ and Prl2−/− hematopoietic progenitor cells with SCF and monitored KIT phosphorylation over time. We found that Prl2−/− hematopoietic progenitor cells showed decreased KIT phosphorylation at tyrosine 703 compared to Prl2+/+ cells following SCF stimulation (Fig. 6E). In addition, we found that PRL2 was associated with KIT in 293 cells (Fig. 6F). These data demonstrate that PRL2 is a key modulator of KIT signaling.
Figure 6. PRL2 sustains Notch-mediated c-Kit expression in T cell progenitors.

(A) Expression of c-Kit in DN1-gated Prl2+/+ and Prl2−/− cells in OP9-DL1 cultures, assessed by flow cytometry. Right panels show the mean fluorescent intensity (MFI) of c-Kit in DN1 cells. **P<0.01, n=3. (B) Expression of c-Kit in DN1 thymocytes isolated from Prl2+/+ and Prl2−/− mice, assessed by flow cytometry. Right panels show the mean fluorescent intensity (MFI) of c-Kit in ETP gated thymocytes. **P<0.01, n=10. (C) Myeloid differentiation was evaluated in DLL1 cultures of LMPPs. Prl2−/− LMPPs differentiated into CD11b+/Gr1+ myeloid cells at day 16 (0.5±0.3 vs 11.5±4.1 %, **P<0.01, n=3). (D) Myeloid progenitors were quantified by methylcellulose culture using ETPs from Prl2+/+ and Prl2−/− mice. (E) PRL2 is important for KIT activation following SCF stimulation. Immunoblot analysis of KIT phosphorylation in WT and Prl2 null hematopoietic progenitor cells following SCF stimulation. (F) PRL2 interacts with KIT in 293 cells.
To understand the molecular basis of PRL2 deficiency on T cell differentiation, we performed gene expression profiling assays using sorted ETPs. Interestingly, we found that Tcf-1 and Bcl-2 mRNAs were decreased in Prl2−/− ETPs compared to Prl2+/+ ETPs. Several Notch downstream targets, including Hes1 and Fbxw7, were modestly increased in Prl2−/− ETPs (Supplementing information Fig. S7C). We also observed increased levels of Cebpα in Prl2−/− ETPs and LMPPs compared to Prl2+/+ cells (Supplementing information Fig. S7D). Although Prl2−/− DN1 cells did not exhibit enhanced apoptosis in steady state, Prl2−/− ETPs harvested from OP9-DL1 cultures showed increased apoptosis (Supplementing information Fig. S8A and S8B), indicating that PRL2 affects ETP expansion and survival after in vitro cultures. Thus, sustaining high level of c-Kit expression by PRL2 is crucial for ETP expansion, survival, and commitment during early stage T cell development (Fig. 7).
Figure 7. Protein tyrosine phosphatase PRL2 mediates Notch and Kit signals in early T cell progenitors.

(A) Working model. Cooperation of PRL2 phosphatase with Notch and c-Kit signals is critical for early T cell differentiation. Delta/Notch engagement induces the expression of both c-Kit and PRL2 in ETPs. PRL2 then sustains and/or potentiates Kit signaling to facilitate DN1-DN2 transition. In the absence of PRL2, c-Kit expression is decreased and decreased c-Kit expression promotes ETP differentiation toward myeloid lineages. Thus, sustaining high level of c-Kit expression by PRL2 is crucial for ETP expansion, survival, and commitment during early stage T cell development.
DISCUSSION
Notch signals provided by the thymic environment are critical for ETP maintenance and T cell differentiation [1–2]. One of the unique features of ETPs is the expression of high levels of c-Kit [1]. However, whether Notch signals link to c-Kit expression remains elusive. While one previous study showed a possible connection between Notch and c-Kit expression in B cell biased progenitors [14], recent studies indicate that BM derived LMPPs are the major source of ETPs [27–28]. Thus, it is important to investigate c-Kit regulation at LMPP and ETP stages in order to clarify this outstanding question. We found that canonical Notch signaling-mediated c-Kit expression is critical for early T cell differentiation, underscoring the importance of the c-Kit “bright” phenotype of ETPs. Further, we demonstrate that PRL2 participates in this process by sustaining Notch-driven c-Kit signaling activity. Thus, the Notch/RBPJ pathway regulates early T cell development, at least in part, through upregulating PRL2 and c-Kit expression as well as sustaining higher c-Kit activity (Fig. 7).
SCF/c-Kit signaling is important for T cell differentiation. In fact, c-Kit bright DN1 cells can produce more T cells than c-Kit low DN1 cells [24]. One of the important findings in the various c-Kit LOF mutant mice strains is that T cell defects are more dramatic than the defects of the other lineages [11–12]. Given that hematopoietic defects seen in the c-Kit mutant mice may be due to a cumulative effect from HSCs to progenitors in all lineages [5–8], it has been difficult to specify which stage of T cell development requires c-Kit expression. Our work indicates that there is a “functional window” for c-Kit dependency at the ETP stage and that Delta/Notch engagement is essential for c-Kit upregulation. Our data provide new evidence that Delta/Notch engagement in the thymus drives higher c-Kit expression in early T cell progenitors and promotes T cell differentiation.
Recent studies have shown that HSCs that express an intermediate level of c-Kit and HSCs that express a high level of c-Kit (c-Kithigh) had distinct self-renewal capability, indicating that c-Kit surface expression and activity are tightly regulated in HSCs to ensure the proper balance between self-renewal and terminal differentiation [29–30]. While the heterozygous c-KitW/+ mutant mice had no or modest hematopoietic defects in steady state, HSC self-renewal was dramatically impaired following transplantation of c-Kit mutant cells into lethally-irradiated recipient mice [13], suggesting that the effect of reduced c-Kit expression and activity on HSCs is more profound under stress conditions. Therefore, our observations that show differences between homeostatic and stress T cell development in PRL2 null mice are consistent with these reports. These findings also suggest that appropriate levels of c-Kit can be maintained in different tissues. It is important to maintain low levels of c-Kit in the BM to enhance HSC self-renewal, whereas sustaining high levels of c-Kit in the thymus facilitates the effective DN1-to-DN2 transition.
While Jagged-1/Notch2 association plays certain roles in postnatal HSC maintenance [31–32], the canonical Notch1/RBPJ pathway is dispensable for adult HSC function [26, 33]. In fact, either Jagged-1 or Notch2 mutation results in multiple musculoskeletal and liver abnormalities, but none of these mutations affects T cells [34]. However, we found that both DLL1/Notch1 and Jag1/Notch2 utilized the canonical Notch/RBPJ pathway to regulate c-Kit expression in T cell progenitors. While both Jagged-1 and DLL1 can induce c-Kit expression [31], it is not clear why Jagged-1/Notch2 showed decreased T cell potential compared with DLL1/Notch1. It could be due to multiple factors, including differences in ligand density between thymic Delta-like 4 and Jagged in the BM niche [23, 35], different signaling [36–37], or differential regulators, such as lunatic fringe, which modulates Delta/Notch specificity [38].
We found that c-Kit mRNA was upregulated in freshly isolated Prl2−/− LMPPs whereas the fluorescence intensity of c-Kit in Prl2−/− LMPPs was decreased, suggesting that PRL2 may regulate c-Kit turnover in T cell progenitors. PRL2 deficient cells and c-KitY823F mutant cells appear to share similar features, including increased c-Kit turnover and inability to sustain c-Kit activity [39]. Given that the c-KitY823F mutant affects cell survival [39], the enhanced apoptosis of PRL2 null ETPs in OP9-DL1 culture may, at least in part, be due to inappropriate function of the second TK under higher demand of c-Kit activity.
PRLs are highly expressed in human cancer cells, including ovarian, lung, breast and liver cancers [15–17]. Recent findings suggest that PRLs may play important roles in the pathogenesis of hematological malignancies [19]. PRL3 is highly expressed in BCR-ABL positive acute lymphoblastic leukemia (ALL), which may be responsible for the aggressive phenotype of the disease [40]. PRL2 is located on human chromosome 1p35, a region often rearranged or amplified in malignant lymphoma and B-cell chronic lymphocytic leukemia (B-CLL) [41]. The leukemia initiating cells (LICs) in the Notch-ICN1 induced leukemia were recently characterized as c-Kit+ cells [42]. We found that expression of Notch-ICN1 in Lin−Sca1+ cells upregulated c-Kit and sustained DN2 cells without stromal support. However, PRL2 null cells expressing Notch-ICN1 failed to maintain DN2 cells. Recently, we reported that PRL2 plays a critical role in the pathogenesis of T-ALL [43]. We discovered that genetic and pharmacological inhibition of PRL2 impaired the proliferation and survival of human T-ALL cells in vitro and deletion of PRL2 inhibited the initiation and maintenance of oncogenic NOTCH1-induced T-ALL development in vivo. Mechanistically, NOTCH1 signaling upregulated PRL2 to maintain the leukemogenic potential of c-Kit+ leukemia initiating cells [43]. Our data suggest that PRL2 is important for DN1-DN2 transition during normal T cell development but maintains c-Kit+ LICs to promote the pathogenesis of T-ALL.
CONCLUSIONS
Delta/Notch engagement induces the expression of both PRL2 and c-Kit in ETPs. PRL2 sustains and/or potentiates Kit signaling to facilitate DN1-DN2 transition. In the absence of PRL2, c-Kit expression is decreased. Decreased c-Kit expression promotes ETP differentiation toward myeloid lineages. Thus, sustaining high level of c-Kit expression by PRL2 is crucial for ETP expansion, survival and commitment during early stage T cell development (Fig. 7).
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grant CA69202 (Z.-Y. Z.), Department of Defense Grant W81XWH-13-1-0187 (Y. L.), a St. Baldrick’s Foundation Scholar Award (Y. L.), an Elsa Pardee Foundation New Investigator Award (Y. L.), Leukemia Research Foundation New Investigator Award (Y. L.), an Alex’s Lemonade Stand Grant (Y. L.), a Children’s Leukemia Research Association Grant (Y. L.), and American Cancer Society Institutional Research Grants (Y. L. and M. K.).
This work was supported by a Project Development Team within the ICTSI NIH/NCRR Grant Number UL1TR001108. We would like to thank Mrs. Marilyn Wales for helping in the preparation of the manuscript.
Footnotes
AUTHOR CONTRIBUTIONS
M. K., Y. B., Z.-Y. Z., and Y. L. designed the research; M. K., S.N., Y. B., M. Y., R. G., S. C., C. Y., Y. D., L. Z., S. R., and Y.Y-O. performed the research; M. K. and Y. L. analyzed the data and performed the statistical analysis; W.S.P., N. C., M.C.Y., R. K., M.H. K., and H.D. L. provided reagents to the study; M. K., Z.-Y. Z. and Y. L. wrote the manuscript.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors declared that no conflict interest exists.
References
- 1.Rothenberg EV. Transcriptional control of early T and B cell developmental choices. Annu Rev Immunol. 2014;32:283–321. doi: 10.1146/annurev-immunol-032712-100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nat Rev Immunol. 2008;8:9–21. doi: 10.1038/nri2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuzaki Y, Gyotoku J, Ogawa M, et al. Characterization of c-kit positive intrathymic stem cells that are restricted to lymphoid differentiation. J Exp Med. 1993;178:1283–1292. doi: 10.1084/jem.178.4.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godfrey DI, Kennedy J, Suda T, Zlotnik A. A developmental pathway involving four phenotypically and functionally distinct subsets of CD3-CD4-CD8- triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J Immunol. 1993;150:4244–4252. [PubMed] [Google Scholar]
- 5.Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c-Kit(W/W) mutants reveal pivotal role for c-kit in the maintenance of lymphopoiesis. Immunity. 2002;17:277–288. doi: 10.1016/s1074-7613(02)00386-2. [DOI] [PubMed] [Google Scholar]
- 6.Lennartsson J, Ronnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev. 2012;92:1619–1649. doi: 10.1152/physrev.00046.2011. [DOI] [PubMed] [Google Scholar]
- 7.Agosti V, Corbacioglu S, Ehlers I, et al. Critical role for Kit-mediated Src kinase but not PI 3-kinase signaling in pro T and pro B cell development. J Exp Med. 2004;199:867–878. doi: 10.1084/jem.20031983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kissel H, Timokhina I, Hardy MP, et al. Point mutation in kit receptor tyrosine kinase reveals essential roles for kit signaling in spermatogenesis and oogenesis without affecting other kit responses. EMBO J. 2000;19:1312–1326. doi: 10.1093/emboj/19.6.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massa S, Balciunaite G, Ceredig R, Rolink AG. Critical role for c-kit (CD117) in T cell lineage commitment and early thymocyte development in vitro. Eur J Immunol. 2006;36:526–532. doi: 10.1002/eji.200535760. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Pierce LJ, Spangrude GJ. Distinct roles of IL-7 and stem cell factor in the OP9-DL1 T-cell differentiation culture system. Exp Hematol. 2006;34:1730–1740. doi: 10.1016/j.exphem.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeda S, Shimizu T, Rodewald HR. Interactions between c-kit and stem cell factor are not required for B-cell development in vivo. Blood. 1997;89:518–525. [PubMed] [Google Scholar]
- 12.Thoren LA, Liuba K, Bryder D, et al. Kit regulates maintenance of quiescent hematopoietic stem cells. J Immunol. 2008;180:2045–2053. doi: 10.4049/jimmunol.180.4.2045. [DOI] [PubMed] [Google Scholar]
- 13.Sharma Y, Astle CM, Harrison DE. Heterozygous kit mutants with little or no apparent anemia exhibit large defects in overall hematopoietic stem cell function. Exp Hematol. 2007;35:214–220. doi: 10.1016/j.exphem.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoflinger S, Kesavan K, Fuxa M, et al. Analysis of Notch1 function by in vitro T cell differentiation of Pax5 mutant lymphoid progenitors. J Immunol. 2004;173:3935–3944. doi: 10.4049/jimmunol.173.6.3935. [DOI] [PubMed] [Google Scholar]
- 15.Stephens BJ, Han H, Gokhale V, Von Hoff DD. PRL phosphatases as potential molecular targets in cancer. Mol Cancer Ther. 2005;4:1653–1661. doi: 10.1158/1535-7163.MCT-05-0248. [DOI] [PubMed] [Google Scholar]
- 16.Rios P, Li X, Kohn M. Molecular mechanisms of the PRL phosphatases. FEBS J. 2012;280:505–524. doi: 10.1111/j.1742-4658.2012.08565.x. [DOI] [PubMed] [Google Scholar]
- 17.Campbell AM, Zhang ZY. Phosphatase of regenerating liver: a novel target for cancer therapy. Expert Opin Ther Targets. 2014;18:555–569. doi: 10.1517/14728222.2014.892926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong Y, Zhang L, Zhang S, et al. Phosphatase of regenerating liver 2 (PRL2) is essential for placental development by down-regulating PTEN (Phosphatase and Tensin Homologue Deleted on Chromosome 10) and activating Akt protein. J Biol Chem. 2012;287:32172–32179. doi: 10.1074/jbc.M112.393462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi M, Bai Y, Dong Y, et al. PRL2/PTP4A2 phosphatase is important for hematopoietic stem cell self-renewal. Stem Cells. 2014;32:1956–1967. doi: 10.1002/stem.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi M, Chen S, Gao R, et al. Phosphatase of regenerating liver in hematopoietic stem cells and hematological malignancies. Cell Cycle. 2014;13:2827–2835. doi: 10.4161/15384101.2014.954448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong Y, Zhang L, Bai Y, et al. Phosphatase of regenerating liver 2 (PRL2) deficiency impairs Kit signaling and spermatogenesis. J Biol Chem. 2013;289:3799–3810. doi: 10.1074/jbc.M113.512079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshimoto M, Porayette P, Glosson NL, et al. Autonomous murine T-cell progenitor production in the extra-embryonic yolk sac before HSC emergence. Blood. 2012;119:5706–5714. doi: 10.1182/blood-2011-12-397489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dallas MH, Varnum-Finney B, Delaney C, Kato K, Bernstein ID. Density of the Notch ligand Delta1 determines generation of B and T cell precursors from hematopoietic stem cells. J Exp Med. 2005;201:1361–1366. doi: 10.1084/jem.20042450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barata JT, Boussiotis VA, Yunes JA, et al. IL-7-dependent human leukemia T-cell line as a valuable tool for drug discovery in T-ALL. Blood. 2004;103:1891–1900. doi: 10.1182/blood-2002-12-3861. [DOI] [PubMed] [Google Scholar]
- 25.Yashiro-Ohtani Y, Wang H, Zang C, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci U S A. 2014;111:E4946–53. doi: 10.1073/pnas.1407079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Zhang H, Rodriguez S, Cao L, et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-kappaB-dependent manner. Cell Stem Cell. 2014;15:51–65. doi: 10.1016/j.stem.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allman D, Sambandam A, Kim S, et al. Thymopoiesis independent of common lymphoid progenitors. Nat Immunol. 2003;4:168–174. doi: 10.1038/ni878. [DOI] [PubMed] [Google Scholar]
- 28.Zlotoff DA, Sambandam A, Logan TD, et al. CCR7 and CCR9 together recruit hematopoietic progenitors to the adult thymus. Blood. 2010;115:1897–1905. doi: 10.1182/blood-2009-08-237784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grinenko T, Arndt K, Portz M, et al. Clonal expansion capacity defines two consecutive developmental stages of long-term hematopoietic stem cells. J Exp Med. 2014;211:209–215. doi: 10.1084/jem.20131115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin JY, Hu W, Naramura M, Park CY. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J Exp Med. 2014;211:217–231. doi: 10.1084/jem.20131128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varnum-Finney B, Halasz LM, Sun M, et al. Notch2 governs the rate of generation of mouse long- and short-term repopulating stem cells. J Clin Invest. 2011;121:1207–1216. doi: 10.1172/JCI43868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poulos MG, Guo P, Kofler NM, et al. Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep. 2013;4:1022–1034. doi: 10.1016/j.celrep.2013.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maillard I, Koch U, Dumortier A, et al. Canonical notch signaling is dispensable for the maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2008;2:356–366. doi: 10.1016/j.stem.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zanotti S, Canalis E. Notch signaling in skeletal health and disease. Eur J Endocrinol. 2013;168:R95–103. doi: 10.1530/EJE-13-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hozumi K, Mailhos C, Negishi N, et al. Delta-like 4 is indispensable in thymic environment specific for T cell development. J Exp Med. 2008;205:2507–2513. doi: 10.1084/jem.20080134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong CT, Cheng HT, Chang LW, et al. Target selectivity of vertebrate notch proteins. Collaboration between discrete domains and CSL-binding site architecture determines activation probability. J Biol Chem. 2006;281:5106–5119. doi: 10.1074/jbc.M506108200. [DOI] [PubMed] [Google Scholar]
- 37.Lehar SM, Dooley J, Farr AG, Bevan MJ. Notch ligands Delta 1 and Jagged1 transmit distinct signals to T-cell precursors. Blood. 2005;105:1440–1447. doi: 10.1182/blood-2004-08-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Visan I, Yuan JS, Tan JB, Cretegny K, Guidos CJ. Regulation of intrathymic T-cell development by Lunatic Fringe- Notch1 interactions. Immunol Rev. 2006;209:76–94. doi: 10.1111/j.0105-2896.2006.00360.x. [DOI] [PubMed] [Google Scholar]
- 39.Agosti V, Corbacioglu S, Ehlers I, et al. Critical role for Kit-mediated Src kinase but not PI 3-kinase signaling in pro T and pro B cell development. J Exp Med. 2004;199:867–878. doi: 10.1084/jem.20031983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Climent JA, Alizadeh AA, Segraves R, et al. Transformation of follicular lymphoma to diffuse large cell lymphoma is associated with a heterogeneous set of DNA copy number and gene expression alterations. Blood. 2003;101:3109–17. doi: 10.1182/blood-2002-07-2119. [DOI] [PubMed] [Google Scholar]
- 41.Mohamed AN, Palutke M, Eisenberg L, Al-Katib A. Chromosomal analyses of 52 cases of follicular lymphoma with t(14;18), including blastic/blastoid variant. Cancer Genet Cytogenet. 2001;126:45–51. doi: 10.1016/s0165-4608(00)00383-6. [DOI] [PubMed] [Google Scholar]
- 42.Schubbert S, Cardenas A, Chen H, et al. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014;74:7048–7059. doi: 10.1158/0008-5472.CAN-14-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi M, Bai Y, Chen S, et al. Phosphatase PRL2 promotes oncogenic Notch-induced T cell leukemia. Leukemia. 2016 Dec 9; doi: 10.1038/leu.2016.340. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.