

Abstract
Background
Delayed gastric emptying in diabetic mice and humans is associated with changes in macrophage phenotype and loss of interstitial cells of Cajal (ICC) in the gastric muscle layers. In diabetic mice, classically activated M1 macrophages are associated with delayed gastric emptying whereas alternatively activated M2 macrophages are associated with normal gastric emptying. This study aimed to determine if secreted factors from M1 macrophages could injure mouse ICC in primary culture.
Methods
Cultures of gastric ICC were treated with conditioned medium (CM) from activated bone marrow-derived macrophages (BMDMs) and the effect of CM was quantified by counting ICC per high-powered field.
Key Results
BMDMs were activated to a M1 or M2 phenotype confirmed by qRT-PCR. CM from M1 macrophages reduced ICC numbers by 41.1%, while M2-CM had no effect as compared to unconditioned, control media. Immunoblot analysis of 40 chemokines/cytokines found 12 were significantly increased in M1-CM, including tumor necrosis factor alpha (TNFα). ELISA detected 0.697 ± 0.03 ng mL−1 TNFα in M1-CM. Recombinant mouse TNFα reduced Kit expression and ICC numbers in a concentration-dependent manner (EC50 = 0.817 ng mL−1). Blocking M1-CM TNFα with a neutralizing antibody preserved ICC numbers. The caspase inhibitor Z-VAD.fmk partly preserved ICC numbers (cells/field; 6.63±1.04, 9.82±1.80 w/Z-VAD.fmk, n=6, P < 0.05).
Conclusions & Inferences
This work demonstrates that TNFα secreted from M1 macrophages can result in Kit loss and directly injure ICC in vitro partly through caspase-dependent apoptosis and may play an important role in ICC depletion in diabetic gastroparesis.
Keywords: diabetic gastroparesis, caspase-mediated apoptosis, cell death, cytokines, gastrointestinal motility
Abbreviated abstract
In diabetic mice, M1 macrophages are associated with delayed gastric emptying whereas M2 macrophages are associated with normal gastric emptying. TNFα, a factor present in M1 conditioned medium, reduced Kit-positive ICC numbers and TNFα neutralizing antibodies blocked the effect of M1 medium. TNFα derived from M1 macrophages injures ICC in vitro and TNFα may be important in diseases like diabetic gastroparesis.
Introduction
Interstitial cells of Cajal (ICC) are multi-functional cells that contribute to normal gastrointestinal function including generation of the electrical slow wave, mediation of neuromuscular signaling, setting membrane voltage gradients across muscle wall and mechanosensation.1–3 Damage to ICC or a reduction in ICC has been described in many gastrointestinal motility disorders.2, 4–8 Gastroparesis, defined as delayed emptying of stomach contents in the absence of a mechanical obstruction,9–12 is one well-studied motility disorder where changes to ICC networks have been reported.13, 14 It is a significant cause of morbidity in a subset of people with diabetes and animal models of diabetes develop delayed gastric emptying, a main feature of gastroparesis. Loss of Kit-positive ICC is associated with the development of delayed gastric emptying in both humans and diabetic mice.13, 15–17
The mechanism for damage to ICC networks in diabetic gastroparesis has not been established. However there is a close association between changes to ICC and alteration of the number and type of macrophages in the gastric muscularis propria in mice and humans with delayed gastric emptying.18, 19 Recent data suggest that macrophages can directly affect the integrity of ICC networks in diseases like diabetes.20 There is significant diversity in the patterns of macrophage activation21, 22 but they can be broadly classified into two main groups: the pro-inflammatory, classically activated macrophages (M1) and the anti-inflammatory, alternatively activated macrophages (M2). Activation of macrophages results in a phenotypic change affecting their physiology, expression of surface proteins, and secretome profile.23 Macrophages in the gastrointestinal muscularis propria of mice have been characterized extensively.24 In response to disease, injury or infection, resident macrophages in the muscularis propria are replaced by activated macrophages.
The phenotypic changes that occur to macrophages in the gastric muscularis propria when delayed gastric emptying develops in diabetic humans and mice are consistent with a role for those macrophages in directly affecting ICC networks.18, 19 In diabetic mice with normal ICC networks and normal gastric emptying, mannose receptor (CD206, MRC1) positive M2 macrophages express cytoprotective factors including heme oxygenase-1 and interleukin-10 (IL-10).18 Whereas, in mice with damaged ICC networks and delayed gastric emptying, increased levels of inducible nitric oxide synthase (iNOS, NOS2), a marker of M1 macrophages, are found.18 The factors generated by M1 macrophages that contribute to ICC network damage and the mechanism by which ICC networks are damaged in diabetic mice with delayed gastric emptying have not been determined. It is known that CD206-negative macrophages are required for development of delayed gastric emptying in diabetic mice, as diabetic osteopetrotric (CSF1op/op) mice, which lack gastric macrophages due to a mutation in the macrophage colony stimulating factor (CSF1), never develop delayed gastric emptying and have normal ICC networks.17 These data suggest that macrophages are needed to induce damage to ICC networks, resulting in delayed gastric emptying.
This study aimed to determine if secreted molecules from bone-marrow derived macrophages (BMDMs) activated to an M1 phenotype could injure dissociated mouse primary ICC in vitro, to identify factors responsible and determine the mechanisms for their effects.
Materials and Methods
Animals
Mice were maintained and the experiments were done with approval from the Institutional Animal Care and Use Committee of the Mayo Clinic. BALB/c mice (Harlan Sprague-Dawley; Indianapolis, IN, USA) were used for all experiments except cell lineage tracing. For cell lineage tracing of ICC, R26mT-mG/+ (Jackson Lab, Bar Harbor, ME, stock#007576) were interbred with KitCreERT2/+ (Gift from Dr. Dieter Saur) knockin line as previously described.25 All mice were killed by CO2 inhalation followed by cervical dislocation.
Bone marrow-derived macrophages
Bone marrow-derived macrophages (BMDM) were generated as previously described.26 This protocol yields macrophages that are >90% pure and have the ability to respond to exogenous stimuli. Briefly, BMDM were obtained from femurs and tibias using aseptic techniques. Marrow cores were flushed into sterile tubes using syringes fitted with 25 gauge needles and filled with Dulbecco’s phosphate-buffered saline without Ca2+/Mg. Cells were washed once in media then plated and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with F12 (Invitrogen; Carlsbad, CA, USA) supplemented with 1% penicillin/streptomycin (Life Technologies; Carlsbad, CA, USA), 10 mM L-glutamine (Invitrogen), 10% characterized fetal bovine serum (Hyclone; Logan, UT, USA), and 40 ng mL−1 recombinant mouse M-CSF (ProSpec; East Brunswick, NJ, USA). Isolated BMDM were cultured for 7 days then split and plated at 0.5×106 cells per well (9.5 cm2) and exposed to either 40 ng mL−1 recombinant mouse interferon-γ (IFN-γ) (R&D Systems; Minneapolis, MN, USA), or to 20 ng mL−1 recombinant human IL-10 (Gibco; Carlsbad, CA, USA) and 10 ng mL−1 recombinant human transforming growth factor β (TGFβ) (Gibco). Control cells were not treated with IFN-γ, IL-10 or TGFβ. Macrophages were treated with exogenous stimuli for 24 hours. Macrophages were then harvested for quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis to determine macrophage phenotype or used to generate macrophage conditioned medium (CM).
Quantitative RT-PCR
Reverse transcription reactions were conducted using the SuperScript VILO cDNA Synthesis Kit (Invitrogen). 500 ng of RNA for each sample was used. cDNA was then used for real-time PCR using the LightCycler 480 system with SYBR Green I Master Mix (Roche, Indianapolis, IN, USA). The program used for all reactions was 95°C for 10 min, followed by 45 cycles at 95°C for 10 s, 60°C for 10 s, and 72°C for 20 s. Gene expression was normalized to β-actin expression and assessed using the Δ(ΔCt) calculation method. Primers were designed with the Primer3 software and obtained from Integrated DNA Technologies (Coralville, IA, USA) or commercially predesigned primers were obtained. In house designed primer sequences are listed in Supplemental Table 1. IL-4 (cat# PPM03013F), IL-5 (cat# PPM03014F) and IL-10 (cat# PPM03017C) primers were obtained from Qiagen (Valencia, CA).
Interstitial cells of Cajal Primary Culture
Primary ICC cultures enriched for gastric ICC were obtained by enzymatic dissociation of the mouse gastric body, fundus, and pylorus based on a previously described protocol for small bowel ICC dissociations.27 Utilizing a collagenase-based dissociation cocktail, tissues were dissociated and then plated on a feeder layer of fibroblasts expressing the membrane bound, m248 form of Kit ligand (steel factor), which is important to maintain ICC phenotype. M199 media (Invitrogen) with 1% antibiotic/antimycotic (Gibco) medium was used for optimal ICC growth. Primary ICC were allowed to attach and recover for 24 hours before treatment. For conditioned macrophage media experiments, 50% of the culture medium was replaced with macrophage-derived conditioned medium from activated BMDM. Each experimental condition was run in duplicate. Cultures were immunolabeled for Kit, a marker of ICC, using the rat monoclonal anti-Kit antibody, ACK2 (eBioscience; San Diego, CA, USA), or a goat anti-Kit polycloncal antibody (R&D). Nuclei were counterstained with 4, 6-diamidino-2-phenylindole, dihydrochloride, DAPI (Molecular Probes; Eugene, OR, USA) as previously described.28 An Olympus BX51WI fluorescence microscope with a 20X objective (NA 0.5) was used to count the number of Kit–positive cells per high-power field (0.94 mm2). At least 35 high-power fields were counted per culture as this is the optimal number to minimize field-to-field variation while not over-counting fields. TNFR1 and TNFR2 localization was analyzed by co-labeling untreated primary ICC cultures with primary antibody for Kit and primary antibodies for either TNFR1 and TNFR2 (Supplemental Table 2).
Conditioned Media
Macrophages activated with either IFNγ or IL-10/TGF-β were used to generate macrophage CM. After activation of BMDM with the exogenous stimuli for 24 hours, the stimulating factors were removed and macrophages were washed three times with 1X PBS. The medium used for primary ICC cultures (M199 with 1% antibiotic/antimycotic) was then added. Activated macrophages were allowed to secrete soluble factors into the media for 20 hours. Media were then removed, filtered with a 0.2 μm filter, flash frozen in liquid N2 and stored at −80°C until use.
Fractionation of the macrophage conditioned media
Conditioned media were fractioned by molecular weight using size separating centrifugal filters. Aliquots of macrophage-conditioned media were filtered using 3 kDa Amicon Ultra centrifugal filter units as described by the manufacturer (Millipore; Billerica, MA, USA). Following fractionation, each fraction was reconstituted to the original volume to return the factors back to the original concentration.
Cytokine Array
For the cytokine array, the Proteome Profiler Mouse Cytokine Array Kit, Panel A from R&D Systems was used and 100 μg of total protein was incubated with the blots according to the manufacturer’s specifications. Densitometric analysis of immunoblot intensity was performed using Adobe Photoshop 6.0 software (Adobe Systems; San Jose, CA, USA).
ELISA
Macrophage-conditioned media were tested for presence of TNFα. Sandwich ELISA for mouse TNFα was done according to the manufacturer’s instructions (eBioscience). This ELISA has a sensitivity of 8 pg mL−1 and a standard curve range of 8 – 1,000 pg mL−1. Each biological replicate was run in duplicate.
Detection of Senescence by Immunohistochemistry
A rabbit polyclonal antibody targeting γ-H2AX was used to detect double-strand breaks in DNA to serve as a marker for cellular senescence. Cells were exposed to TNFα for 24-hours before being processed for immunohistochemistry and analysis of γ-H2AX immunoreactivity. Cells that had γ-H2AX-positive nuclei marking persistent DNA damage foci were only counted if the cell was also positive for Kit immunoreactivity. As noted above, 35 high-power fields were counted for 2 coverslips for each condition. A list of antibodies and optimized concentrations used are listed in Supplemental Table 2.
Cell Lineage Tracing of ICC in Primary Culture
R26mT-mG/+;KitCreERT2/+ mice, in which CreERT2 is expressed in ICC and can be activated via tamoxifen administration, were used for cell lineage tracing experiments. To activate CreERT2, primary ICC cultures were incubated with a low dose of 4-hydroxytamoxifen (100 nM) for 24 h.29 Following 4-hydroxytamoxifen treatment, cells were incubated with 2.25 ng/ml TNFα for 24 h followed by immunohistochemistry for Kit. Cells of ICC lineage which underwent recombination were identified by GFP positivity. Total GFP-positive cells were assessed as for each condition as previously described above.
Confocal Microscopy
To show localization of TNFR1 and TNFR2, images of immunolabeling were collected using an Olympus FV1000 laser scanning confocal microscope. Confocal images were collected with ×60 1.2-NA water objective. All images were prepared for individual figures using Adobe Photoshop CS. No 3D reconstructions, deconvolution, surface or volume rendering, or gamma adjustments were performed for the final images.
Reagents
Recombinant TNFα (R&D Systems) was used at various concentrations as indicated in the results section and figure legends. For TNFα neutralizing experiments, a goat IgG anti-TNFα neutralizing antibody (10.0 μg/ml, R&D Systems, AF-410-NA) 30 was incubated with conditioned medium for 1 hour prior to treatment of cells along with control conditions using a goat IgG isotype control (10.0 μg/ml, R&D Systems, AB-108-C). Z-VAD.fmk (Promega; Madison, WI, USA) was used at a concentration of 40 μM.31 Z-VAD.fmk was added to primary culture at the same time as TNFα and incubated with the ICC for a total of 24 hours. Necrostatin-1 (nec-1) in both active and inactive (Millipore) forms was used at a concentration of 30 μM.32, 33 Nec-1 was added to ICC primary cultures 1 hour before the addition of TNFα and incubated with ICC for a total of 24 hours.
Statistical analysis
Data are expressed as mean with the standard error of the mean. The significance of differences between groups was examined using either Student t test or one-way ANOVA, as appropriate. P < 0.05 (*) was considered statistically significant in all analyses.
Results
Macrophage Phenotype
The phenotype of activated BMDMs was determined following treatment with IFNγ or IL-10/TGFβ. Expression of iNOS and IL-6 RNAs were used as indicators of a classically-activated M1 macrophage phenotype (Fig. 1). Expression of Arg1, FIZZ, IL-4, IL-5, and IL-10 were used as indicators of an alternatively-activated M2 macrophage phenotype (Fig. 1). Data were normalized to expression of β-actin. Macrophages activated with IFNγ showed significantly higher expression of iNOS and IL-6 mRNA, significantly lower expression of IL-4 and IL-5, and there was no significant difference in expression of Arg1, FIZZ and IL-10 mRNA compared to cells that were not treated (Fig. 1). In contrast, macrophages activated with IL-10/TGFβ expressed significantly less iNOS, IL-6, and IL-10 mRNA than unstimulated cells, but significantly higher expression of Arg1, FIZZ, and IL-4 mRNA (Fig. 1). There was no significant change in the expression of IL-5. We concluded that treatment of BMDMs with IFNγ yields an activated macrophage with a phenotype similar to that of classically-activated M1 macrophages, while IL-10/TGFβ treatment activates macrophages into a phenotype similar to that of alternatively-activated M2 macrophages. For this study, BMDM activated by IFNγ will be referred to as M1 macrophages, BMDM activated by IL-10/TGFβ will be referred to as M2 macrophages, and BMDM that were not treated will be referred to as M(Unst.).
Figure 1. Bone marrow-derived macrophages can be activated to a classically-activated M1 or alternatively-activated M2 macrophage phenotype by external stimuli.

Expression of iNOS and IL-6 RNA, markers of classically-activated M1 macrophages and Arg1, FIZZ, IL-4, IL-5, and IL-10 RNA, markers of alternatively-activated M2 macrophages in bone marrow-derived macrophages following treatment with IFNγ or IL-10/TGFβ. Data were normalized to expression in unstimulated control macrophages (Unst). Beta-actin was used as a housekeeping control. Data are the means ± SEM of n = 5 independent experiments, * indicates P < 0.05 vs unstimulated cells, one-sample t-test.
M1-conditioned media reduced ICC numbers in primary culture
Macrophages secrete a variety of soluble chemokines and cytokines that can affect the survival and fate of adjacent cells in tissues. We tested whether soluble factors secreted by activated macrophages would affect ICC in primary culture. Gastric ICC were dissociated and enriched in our primary culture system and then incubated for 24 hours in the presence of media conditioned by incubation with activated M1 or M2 macrophages. Incubation of ICC with M1-CM caused a decrease in the number of Kit-positive ICC whereas there was no effect of M2-CM as shown in the representative images in Fig. 2A. This effect was reproducible with M1-CM causing a 41.1% decrease (P < 0.05) in ICC numbers (Fig. 2B). In order to identify the active molecule in M1-CM that decreased Kit-positive ICC numbers, we used size-separation spin columns to fractionate the components in the media by molecular weight. M1-CM media separated into greater-than 3 kDa and less-than 3 kDa fractions had the same effects as the complete M1-CM medium on ICC numbers (Fig 2C). This indicated that more than one active molecule was present in M1-CM. To test whether there was a non-specific effect of fractionating the medium, due to an effect such as dilution of important growth factors, we tested M2-CM fractionated by the same method. The fractions of M2-CM had no effect on ICC numbers (Fig 2C). Mixing M1-CM and M2-CM and applying it to ICC cultures resulted in loss of the negative effect of M1-CM on ICC numbers (2D).
Figure 2. M1-conditioned medium (M1-CM) has large and small molecular weight bioactive components that reduce ICC numbers in primary culture.

Effect of conditioned medium (CM) on ICC in primary culture. M1-CM but not M2-CM caused a decrease in the number of Kit-positive ICC after 24 hours of exposure to the medium. (A) Representative images showing Kit-positive ICC treated with unconditioned medium as a control (left panel), M1-CM (middle panel) and M2-CM (right panel). Note that fewer ICC are seen in the middle panel. (DAPI nuclear counterstain is shown in blue, scale bar = 50 μm). (B) Quantification of the effects of the media on ICC numbers. Data are shown as ICC per high-powered field. M1-CM caused a significant reduction in ICC numbers (n = 7 independent experiments, * indicates P <0.05, 1way ANOVA with Dunnett’s Post Test). (C) The activity of the M1-CM was retained after fractionating the medium into >3 kDa and <3 kDa fractions. Size separation did not alter the effect of M2-CM on ICC numbers (n = 4 independent experiments, * indicates P < 0.05, 1way ANOVA with Dunnett’s Post Test vs unconditioned medium). (D) Mixing of M1-CM and M2-CM prevented the loss of ICC numbers (n = 4 independent experiments, * indicates P < 0.05, 1way ANOVA with Dunnett’s Post Test).
Identification of pro-inflammatory cytokines and chemokines in M1-conditioned media
To identify possible bioactive components present in M1-CM, an immunoblot was done that probed for 40 different pro-inflammatory cytokines and chemokines. Representative images of the immunoblot show the presence of numerous factors present in M1-CM while there were few pro-inflammatory factors present in the M2-CM (Fig. 3A). Of the factors that were present in M1-CM, 12 were significantly increased as compared to M2-CM (Fig. 3B, n = 5 independent experiments, adjusted P < 0.05). The factors that were significantly enriched in M1-CM were known markers of a pro-inflammatory phenotype, including the interleukin IL-6, the chemokines; chemokine (C-C motif) ligand 2 (CCL2), the chemokine (C-C motif) ligand 3 (CCL3), the chemokine (C-C motif) ligand 4 (CCL4), the chemokine (C-C motif) ligand 5 (CCL5), the chemokine (C-X-C motif) ligand 1 (CXCL1), the chemokine (C-X-C motif) ligand 2 (CXCL2) and the chemokine (C-X-C motif) ligand 10 (CXCL10), as well as TNF-α. The anti-inflammatory interleukin-1 receptor antagonist (IL-1Ra) was also detected as well as IFNγ and M-CSF, which were used to activate the BMDM cells into M1 macrophages. These data demonstrate that the BMDM treated with IFNγ were macrophages activated towards an M1 phenotype and expressed well known pro-inflammatory markers.
Figure 3. M1-CM contains numerous pro-inflammatory cytokines and chemokines.

Detection of cytokine and chemokine expression in M1-CM and M2-CM by immunoblotting. (A) Representative blots showing expression of cytokines and chemokines in M1-CM (upper panel) and M2-CM (lower panel). (B) Relative expression of the identified proteins as determined by densitometry. Data are the means ± SEM of n = 5 independent experiments, * indicates adj P < 0.05, 2way ANOVA with Bonferroni’s correction for multiple comparisons.
Recombinant TNFα decreased Kit-positive ICC numbers and neutralizing TNFα in M1-conditioned media preserved Kit-positive ICC numbers
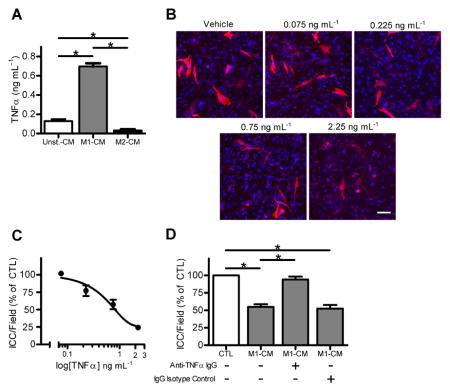
The presence of TNFα in M1-CM was of interest as this cytokine is a known mediator of cell death and injury in diseases associated with inflammation and TNFα had one of the highest pixel densities of the immunoblot. Therefore we determined the concentration of TNFα in Unst-CM, M1-CM and M2-CM by ELISA. M1-CM had contained 0.697 ± 0.03 ng mL−1 of TNFα, which was significantly higher than the levels of TNFα in the other two media (Fig 4A, n = 7, P < 0.05). We then tested the effect of recombinant mouse TNFα on primary ICC cultures. TNFα caused a concentration dependent decrease in Kit-positive ICC numbers at concentrations of TNFα from 0.075 ng mL−1 to 2.25 ng mL−1 (Fig. 4B). The EC50 value for the effect of TNFα was 0.817 ng mL−1 (Fig. 4C), which was similar to the concentration in the M1-CM. Lastly, we tested whether blocking TNFα signaling by incubating the M1-CM with a neutralizing antibody against TNFα would inhibit the effect of the medium on Kit-positive ICC numbers. The larger fraction of M1-CM (> 3 kDa) was used as TNFα (17 kDa TNF monomer, 51 kDa trimer) would be retained within this fraction. The TNFα neutralizing antibody significantly inhibited the effect of M1-CM (> 3 kDa) on Kit-positive ICC numbers (Fig 4D). The effect of M1-CM (> 3 kDa) was not altered by incubation with IgG isotype control (Fig 4D).
Figure 4. rmTNFα reduced Kit-positive ICC numbers in a concentration-dependent manner and neutralizing TNFα blocks the effect of M1-CM on Kit-positive ICC numbers.

(A) M1-CM contains significantly more TNFα than Unst-CM and M2-CM by ELISA. Data are means ± SEM, n = 7 independent experiments, * indicates P < 0.05, 1way ANOVA with Dunnett’s Post Test. (B) Images of Kit+ ICC after being treated for 24 hours with the indicated concentrations of rmTNFα (DAPI nuclear counterstain shown in blue, scale bar = 50 μm). (C) Concentration response curve for the effect of rmTNFα on ICC in primary culture. Fitted line reports an EC50 value of 0.817 ng mL−1 (n = 4). (D) A TNFα-neutralizing antibody inhibits the effect of M1-CM (> 3 KDa fraction) on ICC in primary culture. IgG isotype control had no effect. Data are the means ± SEM of n = 4 independent experiments, * indicates P < 0.05, ANOVA with Dunnett’s Post Test.
Reduced Kit-positive ICC numbers in primary culture by TNFα is partially mediated by caspase-dependent apoptosis
TNFα exerts a broad range of biological effects via two primary transmembrane receptors; TNFR1 and TNFR2. Immunohistochemistry on primary ICC cultures showed that TNFR1 colocalizes with Kit-positive ICC (Fig. 5A) while TNFR2 was not found to be present on ICC (Fig. 5A). These findings are in agreement with previous reports showing that TNFR1 is expressed on almost all cell types, while TNFR2 is primarily expressed in cells of the immune system.34–36 TNFα signaling through TNFR1 leads to initiation of cell death pathways including apoptosis37, 38 and necroptosis38, 39 and TNFα has also been linked to the promotion of cellular senescence.40–42 To test whether the effect of TNFα on Kit-positive ICC numbers was due to caspase-dependent apoptosis, we used the pan-caspase inhibitor Z-VAD.fmk. Incubation of ICC cultures with medium containing Z-VAD.fmk and a maximally effective concentration of TNFα that was 3-fold higher than the EC50 value (2.25 ng mL−1), significantly but not completely reduced the effect of TNFα on Kit-positive ICC numbers (Fig. 5B, cells/field; 6.63±1.04, 9.82±1.80 w/Z-VAD.fmk, n=6, P < 0.05) consistent with a role for caspase-dependent apoptosis in the reduction in ICC numbers by TNFα. In order to test whether caspase-independent cell death contributed to the effects of TNFα on Kit-positive ICC numbers, we used necrostatin 1, a small molecule that allosterically blocks the kinase activity of RIP1, which is essential for caspase-independent necroptosis.32, 43, 44 We found that both active necrostatin 1 and the inactive form of the compound did not alter the effect of TNFα on Kit-positive ICC numbers and had no effect when added alone (Fig. 5C). To examine whether TNFα increased cellular senescence in ICC, we used the senescence marker γH2AX, a phosphorylated histone variant which is associated with double-strand breaks,45 and found no difference in expression of γH2AX-positive foci in Kit-positive ICC in control and TNFα-treated conditions (Fig. 5D, 5E). This indicates that there is no apparent role for cellular senescence in the reduction of Kit-positive ICC numbers with TNFα treatment. We next quantified the total number of DAPI-positive nuclei per field. We found that there was no significant difference in total nuclei in control conditions as compared to TNFα-treated conditions (Fig. 5F). This indicated that the stem cell factor secreting fibroblast feeder layer and other cells surviving from the dissociated gastric muscle were not depleted by TNFα treatment. Lastly, we used an ICC lineage tracing model to track the fate of ICC in primary culture after treatment with TNFα. Immunohistochemistry for Kit showed that in the TNFα-treated condition there are many GFP+ cells remaining, however, they are largely Kit-negative (5G). Quantification of GFP+ cells showed no significant difference in GFP+ cells in the TNFα-treated condition as compared to control (5H), suggesting that the Kit-negative cells remain in the culture.
Figure 5. TNFα-mediated loss of Kit-positive ICC numbers is partially due to a caspase-dependent apoptotic mechanism.

(A) Single confocal images showing the ICC marker Kit (green, top left panel) and TNFR1 (red, top middle panel). The merged image (top right panel) indicates the presence of TNFR1 on ICC. Images showing the ICC marker Kit (green, bottom left panel) and TNFR2 (red, bottom middle panel). The merged image (bottom right panel) indicated that TNFR2 is not localized to ICC (scale bar = 30 μm). Cells were stained with nuclear counterstain DAPI (blue). Dissociated cells from three separate primary cultures were examined for the presence of TNFR1 and TNFR2. (B) Reduced effect of TNFα on Kit-positive ICC numbers following incubation with the pan-caspase inhibitor Z-VAD.fmk (40 μM) (Means ± SEM of n = 6 independent experiments, * indicates P <0.05, ANOVA with Dunnett’s Post Test). (C) Necrostatin-1 (30 μM), an inhibitor of caspase-independent necroptosis, did not significantly affect TNFα-treated Kit-positive ICC numbers (Means ± SEM of n = 4 independent experiments, ANOVA with Dunnett’s Post Test). (D) Immunohistochemistry for γH2AX, a marker of cellular senescence. DAPI nuclear counterstain is shown in blue (top left panel), ICC labeled for Kit are shown in red (top right panel), and γH2AX is shown in green (bottom left panel). The merged image (bottom right panel) shows γH2AX-positive foci in the nucleus of a Kit-positive ICC (arrow), while there are also ICC without γH2AX-positive foci in the nucleus (arrowheads) (scale bar = 30 μm). (E) γH2AX-positive ICC were unaltered in TNFα-treated Kit-positive ICC as compared to untreated control (Means ± SEM of n = 4 independent experiments, one-sample t-test). (F) Total nuclei per high-powered field were quantified to determine if there was a loss of the feeder cell fibroblasts. Total nuclei/field were unaltered in TNFα-treated condition as compared to control (Means ± SEM of n = 5 independent experiments, one-sample t-test). (G) ICC lineage tracing. GFP+ cells having ICC origin (green, left panel), and immunohistochemistry for Kit (pseudo-colored red, middle panel). The merged image (right panel) shows cells that are Kit-positive and GFP-negative (arrow heads) and cells that are GFP-positive and Kit-negative (arrows) (scale bar 50 μm). (H) Total GFP-positive cells were quantified and there was no significant difference between the TNFα-treated condition compared to vehicle control condition (Means ± SEM of n = 3 independent experiments, one-sample t-test).
Discussion
Our primary finding was that TNFα derived from classically-activated M1 macrophages reduced the number of Kit-positive ICC in vitro via caspase mediated apoptosis and loss of Kit expression, a marker of functional ICC. This suggests that in diabetic gastrointestinal tissues where activated macrophages lie in close proximity to ICC, there is a potential role for M1 macrophages to alter ICC networks and potentially other cell types and thus alter gastrointestinal contractility. Previous studies have shown that in diabetic mouse models of gastroparesis, animals that retain normal gastric emptying have normal ICC networks and CD206-positive, HO1-positive M2 macrophages in the muscularis propria, while in animals that developed delayed gastric emptying, ICC networks were damaged and the macrophages were classically activated M1 macrophages, based on being CD206-negative and HO1-negative with increased levels of iNOS mRNA in the tissue.16, 18 Furthermore, we recently reported that diabetic CSF1op/op mice, which lack macrophages, never develop delayed gastric emptying and have normal ICC networks.17 Based on these previous observations, we hypothesized that activated M1 macrophages secrete injurious pro-inflammatory mediators that can damage ICC. In this study, we provided evidence supporting the hypothesis that activated M1 macrophages can directly alter ICC survival and contribute to the pathophysiology of motility disorders such as diabetic gastroparesis where damage to ICC networks is observed.13, 14, 19
For this study we used M-CSF-cultured macrophages from mouse bone marrow, which remains the most commonly used in vitro system for generating macrophages. We ensured that the macrophages were activated towards a M1 and a M2 phenotype. IFNγ treated macrophages showed high levels of iNOS and IL-6 while IL-10/TGFβ treated macrophages had high levels of Arg1, FIZZ and IL-4, confirming activation towards a M1 and M2 phenotype, respectively.46–48 Macrophage polarization status is diverse and alternative methods, such as treating with IL-4 to polarize to M2 macrophages, have been previously used to obtain M2 macrophage phenotypes. We chose a method that resulted in a macrophage phenotype more closely resembling the M2C phenotype as that phenotype was associated with preservation of ICC numbers and gastric emptying in NOD diabetic mice.18 This study focused on injurious factors from M1 macrophages, and did not determine which factors from M2 macrophages are protective or how M2 macrophages might protect ICC from depletion by M1 macrophages. The primary culture system used here was chosen because of the capacity to do comparatively high throughput experiments on regulators of ICC proliferation and survival, which were validated as representative of the changes found in whole animals and in organotypic cultures.27, 49–51 Therefore, we predict that these results are representative of what M1 macrophages might do in more intact systems including organotypic cultures or, more importantly, in our animal models of delayed gastric emptying in diabetic mice and that it will be important to test for these effects in the in vivo preclinical models.
As hypothesized, conditioned media from M1 macrophages decreased Kit-positive ICC numbers in vitro suggesting that a soluble bioactive factor is released into the medium by the activated macrophages. Importantly, conditioned medium from M2 macrophages had no effect on Kit-positive ICC numbers in vitro although mixing M2 media with M1 media eliminated the effect of M1 media, further supporting the hypothesis that M1 macrophages are injurious to ICC while M2 macrophages are not, but can protect against injury when present. The premise that soluble macrophage factors can alter cell survival has been investigated previously. Soluble factors in conditioned media from activated microglia, resident macrophages found in the brain, can trigger apoptosis in hippocampal neurons 52 and conditioned medium from M1 macrophages derived from human blood monocytes reduced cell numbers in the colon cancer cell lines HT-29 and CACO-2.53
Identification of bioactive factors present in complex media can be challenging as the secretome of macrophages is large and diverse. Meissner et al. found that activated bone marrow-derived macrophages had the capacity to release at least 775 proteins, of which 364 were proteins with annotated extracellular functions and 52 were known cytokines.54 Our initial approach to identifying the active factor that reduced Kit-positive ICC numbers in our culture system was to determine the approximate molecular weight of the bioactive factor. Size separation of the M1-conditioned media showed that bioactive factors were present in both a small molecular fraction (< 3kDa) and a large molecular fraction (> 3kDa). For this study, we focused on identifying the larger molecule, which we hypothesized was a cytokine or chemokine. We have not yet identified the smaller molecule or molecules.
M1 and M2-conditioned media were studied using a protein array for expression of 40 of the most commonly investigated pro-inflammatory cytokines and chemokines. We found that 12 were increased in M1 conditioned media as compared to M2-conditioned media. The M1 conditioned media had significant amounts of the pro-inflammatory cytokines IFNγ, IL-6, and TNFα as well as chemokines involved in immune cell recruitment including CCL2, CCL3, CCL4, CCl5, CXCL1, CXCL2, and CXCL10. The presence of these mediators further strengthens the conclusions that the macrophages used in this study were of a M1 phenotype. Of the 12 factors identified in M1 conditioned media, TNFα was of immediate interest, as it is a secreted molecule associated with a number of diseases including cancer, rheumatoid arthritis, type II diabetes, Crohn’s disease, and multiple sclerosis.55
We tested TNFα as a potential bioactive factor by adding it directly to primary ICC cultures. We found that rmTNFα reduced Kit-positive ICC numbers in a dose dependent manner. The EC50 value for the effect of TNFα (0.817 ng mL−1) on Kit-positive ICC numbers in vitro is relatively low in comparison to concentrations of TNFα used in other studies on other cell types. Studies on human and rat vascular and intestinal smooth muscle found that 100 ng mL−1 TNFα did not reduce cell numbers but rather induced proliferation of these cell types.56–58 In addition, a concentration of 40 ng mL−1 of TNFα was needed to reduce tyrosine-hydoxylase-positive neurons by 50% in rat primary culture59 and in rat enteric neuron primary cultures the number of neurons was unchanged after treatment with 50 ng mL−1 TNFα.60 The apparently higher sensitivity of ICC to TNFα exposure as compared to other cell types is of interest because in diabetic gastroparesis the predominant cellular defect noted among humans and animal models is damaged ICC networks more than other cell types. The relatively low concentrations of TNFα used in our study had profound effects on Kit-positive ICC numbers in primary culture and suggests that ICC may be especially susceptible to loss of Kit and damage by TNFα secreted from M1 macrophages, especially because macrophages lie in close proximity to ICC.61
Further supporting a role for TNFα in reducing Kit-positive ICC numbers in culture is that depleting TNFα in the large molecular weight fraction of conditioned media using a neutralizing antibody prevented the effect of M1-CM on Kit-positive ICC numbers. This indicates that TNFα was the large molecular weight factor that was necessary and sufficient for the effect of M1-CM on ICC. Given the presence of many other pro-inflammatory mediators in M1 conditioned media, we were surprised that targeted depletion of TNFα fully preserved Kit-positive ICC numbers. Data from neutralizing TNFα in M1 conditioned media, along with the effect of rmTNFα on ICC numbers are both strong evidence that TNFα can alter the survival of Kit-positive ICC and understanding the mechanism of action is important.
We found that ICC in vitro express TNFR1, but not TNFR2. Therefore we explored the possibility that the effects of TNFα on ICC were due to signaling through TNFR1. TNFα signaling via TNFR1 commonly results in cell death through caspase-mediated apoptosis or necroptosis,62, 63 but other studies have suggested that TNFα may induce cellular senescence via the NF-κB pathway.40–42 We looked at these different pathways by utilizing the pan-caspase inhibitor Z-VAD.fmk, the necroptosis inhibitor necrostatin-1, and a commonly used marker of cellular senescence, γH2AX. We found that the reduction in Kit-positive ICC in our primary system is partly due to caspase-mediated apoptosis, as the pan-caspase inhibitor partly blocked the effect of TNFα on Kit-positive ICC numbers in vitro. We assume that the incomplete inhibition of the effects of the caspase inhibitor are due to our inability to either completely block caspase activation with the inhibitor, the saturating concentration of TNFα that was used in these experiments or the transformation of ICC into Kit-negative cells that are resistant to apoptosis. Furthermore, we found no change in Kit-positive ICC numbers after blocking necroptosis and found no change in γH2AX-positive ICC after TNFα exposure, suggesting that necroptosis and cellular senescence play no role in the reduction of ICC in this model.
Gibbons et al. found that 1–2% of ICC are dead or dying by apoptosis in the normal human colon,64 suggesting that ICC have the ability to undergo caspase-mediated apoptotic cell death and that regeneration is required for maintenance of normal ICC networks even in healthy tissue. Small perturbations to the microenvironments surrounding ICC, such as increased levels of TNFα, could lead to increased rates of apoptotic ICC and contribute to the development of motility disorders.
ICC lineage tracing showed that a majority of ICC exposed to TNFα lost Kit immunoreactivity. Kit is essential for the ICC phenotype.28, 65 It’s been previously shown that hypomorphic mutations in Kit protein (W/Wv mice)66 or Kit ligand (Sl/Sld mice),67 or block of Kit signaling with neutralizing antibodies,68 result in disrupted ICC networks and defects in gastrointestinal motility. Damage to Kit-positive ICC or a reduction in Kit-positive ICC population via the chronic effects of low concentrations of macrophage-derived TNFα could alter motility. Others have shown effects of acute administration of TNFα at high concentrations on gastrointestinal smooth muscle contractility.69 Our data indicate that long-term administration of low concentrations of TNFα would also change contractility given the levels of Kit-positive ICC loss seen in our culture system, but it will require further experimentation to confirm this.
In summary, our data demonstrate that TNFα derived from activated M1 macrophages reduces Kit-positive ICC in vitro and its effects are largely a result of the loss of Kit and partly mediated through caspase-dependent apoptosis. These observations indicate that activated M1 macrophages associated with diseases such as diabetic gastroparesis can secrete factors that have the ability to alter ICC network integrity and function. While we have identified TNFα as a bioactive factor that can alter Kit-positive ICC numbers in vitro, there remains an unidentified smaller component or components that will need to be identified. In addition, the injurious effect of the M1 conditioned media after transfer from the macrophages to the ICC cultures may be underestimated because some toxic mediators released by macrophages are labile with short half-lives (e.g. nitrogen oxide free radicals). Regardless, we show that TNFα injures Kit-positive ICC in vitro and warrants further investigation into its role in the pathophysiology of diseases such as diabetic gastroparesis. If in vivo experiments confirm a role for TNFα in the development of human diabetic gastroparesis, available therapeutics targeting this molecule may be of use.
Supplementary Material
Key Points.
In diabetic mice, M1 macrophages are associated with delayed gastric emptying whereas M2 macrophages are associated with normal gastric emptying. We determined whether secreted factors from M1 macrophages injured interstitial cells of Cajal (ICC) in vitro.
Conditioned medium from M1 macrophages reduced ICC numbers. TNFα, a factor present in M1 medium, reduced Kit expression and ICC numbers and TNFα neutralizing antibodies blocked the effect of M1 medium.
TNFα derived from M1 macrophages injures ICC in vitro and TNFα may be important in diseases like diabetic gastroparesis.
Acknowledgments
We thank Mr. Gary Stoltz and Mrs. Kristy Zodrow for their technical assistance. We thank Dr. Dieter Saur and Dr. Sabine Klein for the gift of the KitCreERT2/+ mice.
Grant Support: This study was funded by National Institutes of Health grant NIH P01 DK 68055 and Grant NIH RO1 DK 057061.
Abbreviations
- M2
Alternatively-activated macrophage
- Arg1
arginase-1
- BMDM
bone marrow-derived macrophages
- M1
classically-activated macrophage
- CM
conditioned medium
- FIZZ
found in inflammatory zone
- iNOS
inducible nitric oxide synthase
- IFN-γ
interferon-γ
- IL-4
interleukin 4
- IL-5
interleukin 5
- IL-6
interleukin 6
- IL-10
interleukin 10
- ICC
interstitial cells of Cajal
- CD206
mannose receptor
- TGFβ
transforming growth factor β
- TNFα
tumor necrosis factor alpha
Footnotes
Author contributions: S.T.E, S.J.G., P.J.V., G.C., D.S., and G.F. conception and design of research; S.T.E. and P.J.V. performed experiments; S.T.E., S.J.G., P.J.V. and G.F. analyzed data; S.T.E., S.J.G., P.J.V., G.C. and G.F. interpreted results of experiments; S.T.E. and S.J.G. prepared figures; S.T.E., S.J.G., and G.F. drafted manuscript; S.T.E., S.J.G., P.J.V., G.C., and G.F. edited and revised manuscript; S.T.E., S.J.G., P.J.V., G.C., D.S., and G.F. approved final version of manuscript.
Disclosures: STE, SJG, PJV, GC, DS, and GF declare that they have no conflict of interest to disclose.
References
- 1.Ward SM, Burns AJ, Torihashi S, Sanders KM. Mutation of the proto-oncogene c-kit blocks development of interstitial cells and electrical rhythmicity in murine intestine. J Physiol. 1994;480(Pt 1):91–7. doi: 10.1113/jphysiol.1994.sp020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373:347–9. doi: 10.1038/373347a0. [DOI] [PubMed] [Google Scholar]
- 3.Farrugia G. Interstitial cells of Cajal in health and disease. Neurogastroenterol Motil. 2008;20(Suppl 1):54–63. doi: 10.1111/j.1365-2982.2008.01109.x. [DOI] [PubMed] [Google Scholar]
- 4.Faussone-Pellegrini MS, Cortesini C. The muscle coat of the lower esophageal sphincter in patients with achalasia and hypertensive sphincter. An electron microscopic study. J Submicrosc Cytol. 1985;17:673–85. [PubMed] [Google Scholar]
- 5.He CL, Burgart L, Wang L, Pemberton J, Young-Fadok T, Szurszewski J, Farrugia G. Decreased interstitial cell of cajal volume in patients with slow-transit constipation. Gastroenterology. 2000;118:14–21. doi: 10.1016/s0016-5085(00)70409-4. [DOI] [PubMed] [Google Scholar]
- 6.He CL, Soffer EE, Ferris CD, Walsh RM, Szurszewski JH, Farrugia G. Loss of interstitial cells of cajal and inhibitory innervation in insulin-dependent diabetes. Gastroenterology. 2001;121:427–34. doi: 10.1053/gast.2001.26264. [DOI] [PubMed] [Google Scholar]
- 7.Feldstein AE, Miller SM, El-Youssef M, Rodeberg D, Lindor NM, Burgart LJ, Szurszewski JH, Farrugia G. Chronic intestinal pseudoobstruction associated with altered interstitial cells of cajal networks. J Pediatr Gastroenterol Nutr. 2003;36:492–7. doi: 10.1097/00005176-200304000-00016. [DOI] [PubMed] [Google Scholar]
- 8.Zarate N, Mearin F, Wang XY, Hewlett B, Huizinga JD, Malagelada JR. Severe idiopathic gastroparesis due to neuronal and interstitial cells of Cajal degeneration: pathological findings and management. Gut. 2003;52:966–70. doi: 10.1136/gut.52.7.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farrell FJ, Keeffe EB. Diabetic gastroparesis. Dig Dis. 1995;13:291–300. doi: 10.1159/000171509. [DOI] [PubMed] [Google Scholar]
- 10.Talley NJ, Verlinden M, Jones M. Can symptoms discriminate among those with delayed or normal gastric emptying in dysmotility-like dyspepsia? Am J Gastroenterol. 2001;96:1422–8. doi: 10.1111/j.1572-0241.2001.03683.x. [DOI] [PubMed] [Google Scholar]
- 11.Vittal H, Farrugia G, Gomez G, Pasricha PJ. Mechanisms of disease: the pathological basis of gastroparesis--a review of experimental and clinical studies. Nat Clin Pract Gastroenterol Hepatol. 2007;4:336–46. doi: 10.1038/ncpgasthep0838. [DOI] [PubMed] [Google Scholar]
- 12.Camilleri M, Grover M, Farrugia G. What are the important subsets of gastroparesis? Neurogastroenterol Motil. 2012;24:597–603. doi: 10.1111/j.1365-2982.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grover M, Farrugia G, Lurken MS, Bernard CE, Faussone-Pellegrini MS, Smyrk TC, Parkman HP, Abell TL, et al. Cellular changes in diabetic and idiopathic gastroparesis. Gastroenterology. 2011;140:1575–85. e8. doi: 10.1053/j.gastro.2011.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grover M, Bernard CE, Pasricha PJ, Lurken MS, Faussone-Pellegrini MS, Smyrk TC, Parkman HP, Abell TL, et al. Clinical-histological associations in gastroparesis: results from the Gastroparesis Clinical Research Consortium. Neurogastroenterol Motil. 2012;24:531–9. e249. doi: 10.1111/j.1365-2982.2012.01894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ordog T, Takayama I, Cheung WK, Ward SM, Sanders KM. Remodeling of networks of interstitial cells of Cajal in a murine model of diabetic gastroparesis. Diabetes. 2000;49:1731–9. doi: 10.2337/diabetes.49.10.1731. [DOI] [PubMed] [Google Scholar]
- 16.Choi KM, Gibbons SJ, Nguyen TV, Stoltz GJ, Lurken MS, Ordog T, Szurszewski JH, Farrugia G. Heme oxygenase-1 protects interstitial cells of Cajal from oxidative stress and reverses diabetic gastroparesis. Gastroenterology. 2008;135:2055–64. 64 e1–2. doi: 10.1053/j.gastro.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cipriani G, Gibbons SJ, Verhulst PJ, Choi KM, Eisenman ST, Hein SS, Ordog T, Linden DR, et al. Diabetic mice lacking macrophages are protected against the development of delayed gastric emptying. Cell Mol Gastroenterol Hepatol. 2016;2:40–7. doi: 10.1016/j.jcmgh.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi KM, Kashyap PC, Dutta N, Stoltz GJ, Ordog T, Shea Donohue T, Bauer AJ, Linden DR, et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology. 2010;138:2399–409. 409 e1. doi: 10.1053/j.gastro.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernard CE, Gibbons SJ, Mann IS, Froschauer L, Parkman HP, Harbison S, Abell TL, Snape WJ, et al. Association of low numbers of CD206-positive cells with loss of ICC in the gastric body of patients with diabetic gastroparesis. Neurogastroenterol Motil. 2014;26:1275–84. doi: 10.1111/nmo.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neshatian L, Gibbons SJ, Farrugia G. Macrophages in diabetic gastroparesis--the missing link? Neurogastroenterol Motil. 2015;27:7–18. doi: 10.1111/nmo.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cipriani G, Gibbons SJ, Kashyap PC, Farrugia G. Intrinsic Gastrointestinal Macrophages: Their Phenotype and Role in Gastrointestinal Motility. Cell Mol Gastroenterol Hepatol. 2016;2:120–30. e1. doi: 10.1016/j.jcmgh.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein S, Seidler B, Kettenberger A, Sibaev A, Rohn M, Feil R, Allescher HD, Vanderwinden JM, et al. Interstitial cells of Cajal integrate excitatory and inhibitory neurotransmission with intestinal slow-wave activity. Nat Commun. 2013;4:1630. doi: 10.1038/ncomms2626. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14):1. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wouters MM, Gibbons SJ, Roeder JL, Distad M, Ou Y, Strege PR, Szurszewski JH, Farrugia G. Exogenous serotonin regulates proliferation of interstitial cells of Cajal in mouse jejunum through 5-HT2B receptors. Gastroenterology. 2007;133:897–906. doi: 10.1053/j.gastro.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Rich A, Miller SM, Gibbons SJ, Malysz J, Szurszewski JH, Farrugia G. Local presentation of Steel factor increases expression of c-kit immunoreactive interstitial cells of Cajal in culture. Am J Physiol Gastrointest Liver Physiol. 2003;284:G313–20. doi: 10.1152/ajpgi.00093.2002. [DOI] [PubMed] [Google Scholar]
- 29.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 30.Saika S, Ikeda K, Yamanaka O, Flanders KC, Okada Y, Miyamoto T, Kitano A, Ooshima A, et al. Loss of tumor necrosis factor alpha potentiates transforming growth factor beta-mediated pathogenic tissue response during wound healing. Am J Pathol. 2006;168:1848–60. doi: 10.2353/ajpath.2006.050980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cowburn AS, White JF, Deighton J, Walmsley SR, Chilvers ER. z-VAD-fmk augmentation of TNF alpha-stimulated neutrophil apoptosis is compound specific and does not involve the generation of reactive oxygen species. Blood. 2005;105:2970–2. doi: 10.1182/blood-2004-07-2870. [DOI] [PubMed] [Google Scholar]
- 32.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol. 2015;16:618–27. doi: 10.1038/ni.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brockhaus M, Schoenfeld HJ, Schlaeger EJ, Hunziker W, Lesslauer W, Loetscher H. Identification of two types of tumor necrosis factor receptors on human cell lines by monoclonal antibodies. Proc Natl Acad Sci U S A. 1990;87:3127–31. doi: 10.1073/pnas.87.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ksontini R, MacKay SL, Moldawer LL. Revisiting the role of tumor necrosis factor alpha and the response to surgical injury and inflammation. Arch Surg. 1998;133:558–67. doi: 10.1001/archsurg.133.5.558. [DOI] [PubMed] [Google Scholar]
- 36.Higuchi Y, McTiernan CF, Frye CB, McGowan BS, Chan TO, Feldman AM. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation. 2004;109:1892–7. doi: 10.1161/01.CIR.0000124227.00670.AB. [DOI] [PubMed] [Google Scholar]
- 37.Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15:362–74. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 39.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Herbert BS, Rajashekhar G, Ingram DA, Yoder MC, Clauss M, Rehman J. Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. Faseb J. 2009;23:1358–65. doi: 10.1096/fj.08-110296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 42.Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012;122:2601–12. doi: 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 44.Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727–36. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- 45.Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppe JP, Campeau E, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124:68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nair MG, Cochrane DW, Allen JE. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunol Lett. 2003;85:173–80. doi: 10.1016/s0165-2478(02)00225-0. [DOI] [PubMed] [Google Scholar]
- 47.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–9. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- 48.Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, Olszewski MA. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio. 2013;4:e00264–13. doi: 10.1128/mBio.00264-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi KM, Gibbons SJ, Roeder JL, Lurken MS, Zhu J, Wouters MM, Miller SM, Szurszewski JH, et al. Regulation of interstitial cells of Cajal in the mouse gastric body by neuronal nitric oxide. Neurogastroenterol Motil. 2007;19:585–95. doi: 10.1111/j.1365-2982.2007.00936.x. [DOI] [PubMed] [Google Scholar]
- 50.Stanich JE, Gibbons SJ, Eisenman ST, Bardsley MR, Rock JR, Harfe BD, Ordog T, Farrugia G. Ano1 as a regulator of proliferation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G1044–51. doi: 10.1152/ajpgi.00196.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wouters MM, Roeder JL, Tharayil VS, Stanich JE, Strege PR, Lei S, Bardsley MR, Ordog T, et al. Protein kinase C{gamma} mediates regulation of proliferation by the serotonin 5-hydroxytryptamine receptor 2B. J Biol Chem. 2009;284:21177–84. doi: 10.1074/jbc.M109.015859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flavin MP, Coughlin K, Ho LT. Soluble macrophage factors trigger apoptosis in cultured hippocampal neurons. Neuroscience. 1997;80:437–48. doi: 10.1016/s0306-4522(97)00078-x. [DOI] [PubMed] [Google Scholar]
- 53.Engstrom A, Erlandsson A, Delbro D, Wijkander J. Conditioned media from macrophages of M1, but not M2 phenotype, inhibit the proliferation of the colon cancer cell lines HT-29 and CACO-2. Int J Oncol. 2014;44:385–92. doi: 10.3892/ijo.2013.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meissner F, Scheltema RA, Mollenkopf HJ, Mann M. Direct proteomic quantification of the secretome of activated immune cells. Science. 2013;340:475–8. doi: 10.1126/science.1232578. [DOI] [PubMed] [Google Scholar]
- 55.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 56.Davis R, Pillai S, Lawrence N, Sebti S, Chellappan SP. TNF-alpha-mediated proliferation of vascular smooth muscle cells involves Raf-1-mediated inactivation of Rb and transcription of E2F1-regulated genes. Cell Cycle. 2012;11:109–18. doi: 10.4161/cc.11.1.18473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rastogi S, Rizwani W, Joshi B, Kunigal S, Chellappan SP. TNF-alpha response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012;19:274–83. doi: 10.1038/cdd.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nair DG, Miller KG, Lourenssen SR, Blennerhassett MG. Inflammatory cytokines promote growth of intestinal smooth muscle cells by induced expression of PDGF-Rbeta. J Cell Mol Med. 2014;18:444–54. doi: 10.1111/jcmm.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGuire SO, Ling ZD, Lipton JW, Sortwell CE, Collier TJ, Carvey PM. Tumor necrosis factor alpha is toxic to embryonic mesencephalic dopamine neurons. Exp Neurol. 2001;169:219–30. doi: 10.1006/exnr.2001.7688. [DOI] [PubMed] [Google Scholar]
- 60.Gougeon PY, Lourenssen S, Han TY, Nair DG, Ropeleski MJ, Blennerhassett MG. The pro-inflammatory cytokines IL-1beta and TNFalpha are neurotrophic for enteric neurons. J Neurosci. 2013;33:3339–51. doi: 10.1523/JNEUROSCI.3564-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mikkelsen HB. Interstitial cells of Cajal, macrophages and mast cells in the gut musculature: morphology, distribution, spatial and possible functional interactions. J Cell Mol Med. 2010;14:818–32. doi: 10.1111/j.1582-4934.2010.01025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 63.Naude PJ, den Boer JA, Luiten PG, Eisel UL. Tumor necrosis factor receptor cross-talk. Febs J. 2011;278:888–98. doi: 10.1111/j.1742-4658.2011.08017.x. [DOI] [PubMed] [Google Scholar]
- 64.Gibbons SJ, De Giorgio R, Faussone Pellegrini MS, Garrity-Park MM, Miller SM, Schmalz PF, Young-Fadok TM, Larson DW, et al. Apoptotic cell death of human interstitial cells of Cajal. Neurogastroenterol Motil. 2009;21:85–93. doi: 10.1111/j.1365-2982.2008.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanders KM, Ordog T, Koh SD, Torihashi S, Ward SM. Development and plasticity of interstitial cells of Cajal. Neurogastroenterol Motil. 1999;11:311–38. doi: 10.1046/j.1365-2982.1999.00164.x. [DOI] [PubMed] [Google Scholar]
- 66.Ward SM, Beckett EA, Wang X, Baker F, Khoyi M, Sanders KM. Interstitial cells of Cajal mediate cholinergic neurotransmission from enteric motor neurons. J Neurosci. 2000;20:1393–403. doi: 10.1523/JNEUROSCI.20-04-01393.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ward SM, Burns AJ, Torihashi S, Harney SC, Sanders KM. Impaired development of interstitial cells and intestinal electrical rhythmicity in steel mutants. Am J Physiol. 1995;269:C1577–85. doi: 10.1152/ajpcell.1995.269.6.C1577. [DOI] [PubMed] [Google Scholar]
- 68.Maeda H, Yamagata A, Nishikawa S, Yoshinaga K, Kobayashi S, Nishi K. Requirement of c-kit for development of intestinal pacemaker system. Development. 1992;116:369–75. doi: 10.1242/dev.116.2.369. [DOI] [PubMed] [Google Scholar]
- 69.Shi XZ, Sarna SK. Homeostatic and therapeutic roles of VIP in smooth muscle function: myo-neuroimmune interactions. Am J Physiol Gastrointest Liver Physiol. 2009;297:G716–25. doi: 10.1152/ajpgi.00194.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.