

Abstract
Background and Purpose
Amyotrophic lateral sclerosis (ALS) is a fatal and rapidly progressing motor neuron disease without effective treatment. The complement system is up‐regulated in ALS, with recent studies indicating that the activation product C5a accelerates disease progression via the C5a1 receptor (C5aR1). We therefore examined the therapeutic effect of C5a1 receptor antagonism in hSOD1G93A mice, the most widely used preclinical model of ALS.
Experimental Approach
The selective and orally active C5a1 receptor antagonist, PMX205, was administered to hSOD1G93A mice in drinking water, both pre‐ and post‐disease onset. Blood, brain and spinal cord pharmacokinetics were performed using LC–MS/MS methods. Effects of PMX205 on hSOD1G93A disease progression was determined using body weight, hindlimb grip strength, survival time and blood analysis.
Key Results
PMX205 entered the intact CNS at pharmacologically active concentrations, with increased entry observed in hSOD1G93A mice as the disease progressed, in line with augmented blood–brain barrier breakdown. hSOD1G93A mice treated with PMX205 before disease onset had significantly improved hindlimb grip strength, slower disease progression and extended survival, compared with vehicle treatment. These improvements were associated with reductions in pro‐inflammatory monocytes and granulocytes and increases in T‐helper lymphocytes in peripheral blood. PMX205 treatment beginning 3 weeks following disease onset also attenuated disease progression, significantly extending survival.
Conclusion and Implications
These results confirm that C5a1 receptors play a pathogenic role in hSOD1G93A mice, further validating the C5a‐C5a1 receptor signalling axis as a potential therapeutic target to slow disease progression in ALS.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- BBB
blood brain barrier
- BSB
blood spinal cord barrier
- WT
wild‐type
Tables of Links
| TARGETS |
|---|
| GPCRs |
| C5a1 receptor |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to Pharmacology (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/2016 (Alexander et al., 2015).
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal and rapidly progressing motor neuron disease that is characterised by selective loss of corticospinal neurons within the motor cortex and α‐motor neurons of the spinal cord and brainstem (Bruijn et al., 2004). The progressive loss of motor neurons results in atrophy of skeletal muscles and symptoms of muscle weakness, ultimately leading to paralysis and death due to the failure of respiratory muscles (Cozzolino et al., 2008). The mechanisms leading to ALS are still unclear, but there is compelling evidence to suggest that neuroinflammation and innate immunity may contribute to the progression of disease (Lee et al., 2013). The complement system has long been implicated in the pathogenesis of ALS, with numerous clinical and animal studies demonstrating strong up‐regulation of complement activation fragments in the blood, skeletal muscle, cerebrospinal fluid and neurological tissue of ALS patients and similarly in animal models of ALS (Lee et al., 2012).
One key factor produced following complement activation is the potent pro‐inflammatory complement activation peptide C5a. C5a exerts its inflammatory effects through the GPCR C5a1 receptor, also known as C5aR1, which is widely expressed on inflammatory cells such as granulocytes and monocytes/macrophages, as well as within the CNS, predominantly on glia (Woodruff et al., 2010). Similar to other complement fragments, C5a1 receptor is also dramatically up‐regulated in ALS (Lee et al., 2012; Lee et al., 2013). Given the role of C5a as a potent inflammatory mediator in other models of neurodegenerative diseases (Woodruff et al., 2006; Fonseca et al., 2009), it has also become a potential therapeutic target for ALS. In support of this, we have previously demonstrated that therapeutic inhibition of C5a1 receptor in hSOD1G93A transgenic rats delays the onset of motor symptoms and increases survival compared with untreated animals (Woodruff et al., 2008). Furthermore, we have shown that deletion of the C5ar1 gene increases survival in hSOD1G93A mice (Woodruff et al., 2014).
In the present study, we aimed to extend our earlier findings by examining the effect of C5a1 receptor antagonism on disease progression in hSOD1G93A mice, the most widely used preclinical model of ALS. We used the potent and selective, cyclic peptide C5a1 receptor antagonist, PMX205, to therapeutically block C5a1 receptors in hSOD1G93A mice. We found that orally administered PMX205 could enter the CNS at pharmacologically active concentrations and significantly improve hindlimb grip strength, slow disease progression and extend survival in hSOD1G93A mice. These improvements were associated with reductions in pro‐inflammatory (M1) monocytes and granulocytes and increased T‐helper lymphocyte numbers in circulating blood. In summary, our results further validate the C5a‐ C5a1 receptor signalling axis as a potential therapeutic target to slow disease progression in ALS.
Methods
Ethical statement
All experimental procedures were approved by the University of Queensland Animal Ethics Committee and complied with the policies and regulations regarding animal experimentation. They were conducted in accordance with the Queensland Government Animal Research Act 2001, associated Animal Care and Protection Regulations (2002 and 2008), and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 8th Edition. Animal studies are reported in compliance with the ARRIVE guideline (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Animals
Transgenic hSOD1G93A mice (B6‐Cg‐Tg (SOD1‐G93A) 1Gur/J) expressing the high copy number (~25 copies) of mutant human SOD1 on a C57BL/6J background were initially obtained from The Jackson Laboratory (Bar Harbor, ME, USA). A breeding colony was maintained at the University of Queensland Biological Resources Animal Facilities under specific pathogen‐free conditions. For all therapeutic efficacy studies, female hSOD1G93A littermates were used and separated using a simple randomization procedure (coin toss) to receive either vehicle or drug treatment (i.e. litter‐matched) in a double‐blinded manner. Female hSOD1G93A mice were used because of the higher availability of litter‐matched controls of this gender; our prior studies in hSOD1G93A x C5aR−/− mice indicate no gender difference in C5a1 receptor‐mediated disease pathology (Woodruff et al., 2014). All animals were singly housed under identical conditions in a 12 h light/dark cycle (lights on at 0630 h) with free access to food and water.
Drug treatment
PMX205 (hydrocinnamate‐[OP(D‐Cha)WR]) is an orally active cyclic hexapeptide that has been extensively described pharmacologically (March et al., 2004; Woodruff et al., 2005). It was synthesized as previously described (Woodruff et al., 2005) and purified by reverse‐phase HPLC. For early treatment, PMX205 was administered to mice via their drinking water (20 or 60 μg·mL−1). Based on drinking water measurements taken in hSOD1G93A mice, these dosages equated to 3 and 9 mg·kg−1·day−1 respectively. Litter‐matched female hSOD1G93A transgenic mice were treated with PMX205 or distilled water alone (vehicle) from 35 days postnatal (termed ‘pre‐onset’). For late treatment, PMX205 or vehicle was administered from 91 days postnatal, which is approximately 21 days after initial motor deficit symptoms (termed ‘post‐onset’; Lee et al., 2013). All drug treatments were continued daily throughout until the end‐stage of disease (i.e. point of euthanasia). Prior to study commencement, a power analysis was performed on data obtained from previous hSOD1G93A x C5aR1−/− mice survival studies (Woodruff et al., 2014), resulting in a sample size of n = 13 (power 80%, α 0.05, effect size 5%), which was used in the efficacy trials. In the post‐onset group, two mice from each group died during the trial due to non‐ALS pathologies. According to the ALS preclinical guidelines (Ludolph et al., 2007; Scott et al., 2008), the matched littermate was also removed from the trial, thus resulting in a final n = 9 for these groups. To avoid any potential bias, all drug and vehicle treatments were coded and then administered and subsequently analysed by a researcher (J.D.L.) blinded to the treatment groups. Decoding only occurred after each animal experiment was completed.
Disease progression and survival analysis
The rate of disease progression in hSOD1G93A mice was determined by age at which maximal grip strength declined by 25, 50, 75 and 100%. Survival was determined by the inability of the animal to right itself within 15–30 s if laid on either side. This is a widely accepted end point for lifespan studies in ALS mice (Ludolph et al., 2007; Scott et al., 2008) and guarantees that the animal is killed before it is unable to reach food or water. Mice were killed by cervical dislocation following anaesthesia (i.p. injection of tiletamine 50 mg·kg−1, zolazepam 50 mg·kg−1 and xylazine 12 mg·kg−1).
Weight measurements and hindlimb grip strength test
Vehicle‐ and PMX205‐treated hSOD1G93A mice were weighed weekly at the same time of day (1600 h), from 42 days of age until the defined end‐stage (loss of righting reflex). A digital force gauge (Ugo Basile) was used to measure maximal hindlimb muscle grip strength as described previously (Lee et al., 2013). Mice were held by their tail and lowered until their hindlimbs grasped the T‐bar connected to the digital force gauge. The tail was then lowered until the body was horizontal with the apparatus, and the mice were pulled away from the T‐bar with a smooth steady motion until both of their hindlimbs released the bar. The strength of the grip was measured in gram force. Each mouse was given 10 attempts and the maximum grip strength from these attempts recorded.
Blood, brain and spinal cord pharmacokinetics of PMX205 following i.v. and drinking water administration
For the determination of the bioavailability of PMX205 in the brain and spinal cord, PMX205 was administered via tail vein injection or in the drinking water. hSOD1G93A and wild‐type (WT) mice at three different stages of disease progression (onset =70 days, mid‐symptomatic =130 days and end‐stage =155 days; Lee et al., 2013) were anaesthetized (i.p. injection of tiletamine 50 mg·kg−1, zolazepam 50 mg·kg−1 and xylazine 12 mg·kg−1) and injected i.v. with PMX205 at a dose of 1 mg·kg−1 via the tail vein. Blood samples were then taken precisely 2.5 min following compound injection via cardiac puncture. In a separate study, hSOD1G93A and WT mice, aged 91 days, were also orally dosed in the drinking water with PMX205 at a dose of 60 μg·mL−1 for 5 days, followed by anaesthesia (i.p. injection of tiletamine 50 mg·kg−1, zolazepam 50 mg·kg−1 and xylazine 12 mg·kg−1) and immediate blood collection via cardiac puncture. Blood samples from all these animals were then centrifuged to obtain plasma. Immediately following blood collection from each cohort, mice were transcardially perfused with 50 mL of sterile saline to remove circulating blood from brain and spinal cord vasculature. Whole brain and spinal cord samples were then obtained and homogenized in an equal volume of MilliQ water. Next, 100 μL of tissue homogenate or 50 μL of plasma sample was mixed with 10 μL internal standard (PMX53; 200 ng·mL−1 in 50% methanol/water) and deproteinized with 1:3 ice‐cold LC–MS grade acetonitrile. The supernatant was removed after centrifugation and dried by vacuum concentration at room temperature. Samples were resuspended in 50 μL of 75% methanol/water, and 10 μL was analysed with an API 3200 triple quadrupole Q TRAP LC–MS/MS system with Turbo V ion source coupled with an Agilent 1200 series HPLC system. Chromatographic analysis was carried out with a Agilent Poroshell 120 SB‐C18 analytical column (150 × 2.1 mm; 2.7 μm) under binary gradient conditions. For quantitative purposes, one m/z transition per analyte, that is, 420.32‐70.0 for PMX205 and 448.86‐120 for the internal standard PMX53, was monitored. PMX205 levels in the brain and spinal cord were expressed as ng·g−1 of tissue, with results corrected for process efficiency, extraction efficiency and recovery relative to the internal standard. The limit of detection for PMX205 using this method was 55 pg with an S/N ratio of 3.3.
BBB and BSB permeability assessments
For assessment of the blood–brain barrier (BBB) and blood–spinal cord barrier (BSB) permeability changes throughout disease progression, hSOD1G93A and WT mice were injected with 100 μL of 10% sodium fluorescein via tail vein injection. After 15 min of injection, blood samples were collected via cardiac puncture for serum extraction. Mice were perfused with PBS followed by collection of the motor cortex and spinal cord. Samples were then homogenized in an equal volume of milliQ water followed by protein precipitation using acetonitrile. The supernatant of these samples was dried and reconstituted in equal weight volume of Tris buffer (pH 7.4). Fluorescence was determined in 10 μL of reconstituted samples analysed with 200 μL of Tris buffer (pH 7.4) using a plate reader with λex 460 nm and λem 515 nm. Fluorescent uptake is the ratio of fluorescence in motor cortex and spinal cord relative to serum. Final results are expressed as relative fluorescence uptake in motor cortex and spinal cord samples of hSOD1G93A mice compared with fluorescence uptake in WT controls.
Flow cytometry
Flow cytometry was performed to examine and quantify select immune cell populations in blood samples of vehicle‐ and PMX205‐treated hSOD1G93A mice at three stages of disease progression (onset, mid‐symptomatic and end‐stage) (Brennan et al., 2015). In brief, mice were anaesthetized using methoxyflurane, and blood samples (100 μL) were collected through retro‐orbital eye bleeds. The samples were diluted 1:4 with anticoagulant buffer (4 mM EDTA in 1 × PBS, pH 7.4) and subsequently lysed with red blood cell lysis buffer (0.85% NH4Cl in double distilled H2O, pH 7.4) for 5 min at room temperature. Following centrifugation at 500× g for 5 min at 4°C, the cells were resuspended in flow cytometry blocking buffer (0.5% BSA, 2 mM EDTA in 1 × PBS, pH 7.4) followed by incubation with rat anti‐CD16/32 (1:200; BD Biosciences, North Ryde, NSW, Australia) for 10 min at 4°C to block FC receptors. Cells were immunolabelled with rat anti‐Ly6G‐BUV395 (1:100; BioLegend, San Diego, CA, USA), rat anti‐Ly6C‐V450 (1:200; BioLegend, San Diego, CA, USA), rat anti‐CD3‐Alexa488 (1:200; BD Biosciences, North Ryde, NSW, Australia), rat anti‐CD4‐BV786 (1:200; BioLegend, San Diego, CA, USA) and rat anti‐CD11b‐PE (1:300; BioLegend, San Diego, CA, USA) for 1 h at room temperature. All antibodies were diluted in the blocking buffer (0.5% BSA, 2 mM EDTA in 1 × PBS, pH 7.4). For the exclusion of dead cells, samples were also incubated with near‐infrared conjugated viability dye Zombie NIR (1:100; BioLegend). Stained samples were then analysed using an LSR II flow cytometer (BD Biosciences) and FlowJo analysis software. After acquisition, compensation was applied to remove PE/Alexa 488 spectral overlap. Cell doublets and triplets were excluded based on FSC‐A/FSC‐H linearity. Gating strategies were applied as per Table 1.
Table 1.
Flow cytometry gating strategy
| Indicative Cell Type | Gating Utilised |
|---|---|
| Granulocytes | Neutrophils: CD11b+Ly6G+Ly6C+SSCmed |
| Eosinophils: CD11b+Ly6G+Ly6C+SSChi | |
| Monocytes | M1: CD11b+Ly6G‐Ly6Chi/medSSClo |
| M2: CD11b+Ly6G‐Ly6CloSSClo | |
| T Lymphocytes | CD4 + T‐cells: CD3+CD4+ |
| CD4 ‐ T‐cells (eg. CD8 cells): CD3+CD4‐ |
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as mean ± SEM and all analyses were performed using GraphPad Prism 6.0 (San Diego, CA, USA). The statistical difference for survival analyses between vehicle‐ and PMX205‐treated hSOD1G93A mice was analysed using a log‐rank (Mantel–Cox) test. The statistical difference between vehicle‐ and PMX205‐treated hSOD1G93A mice for body weight, hindlimb grip strength and disease progression were analysed using a two‐way ANOVA and a post hoc Fisher's least significant difference (LSD) test for each time point when F value for drug treatments was significant (P ≤ 0.05). For the results from flow cytometry, statistical difference between vehicle‐ and PMX205‐treated hSOD1G93A mice were also analysed using a two‐way ANOVA and a post hoc Fisher's LSD test for each stages of disease progression. The statistical difference for levels of PMX205 and fluorescence uptake between WT and hSOD1G93A mice was analysed using a two‐tailed student t‐test for each stage of disease progression. Differences between group means were considered significant when P ≤ 0.05.
Materials
PMX205 was synthesized and purified as described previously (Woodruff et al., 2005). Tiletamine, zolazepam, xylazine were obtained from Lyppard Australia Pty Ltd, Melbourne, VIC, Australia.
Results
PMX205 concentrations in the brain and spinal cord of hSOD1G93A mice correlate with BBB and BSB breakdown
We have previously shown the C5a1 receptor to be progressively up‐regulated on reactive microglia in hSOD1G93A mice (Lee et al., 2013). Here, we used a validated quantitative LC–MS/MS method to assess entry of the selective C5a1 receptor antagonist PMX205 into the brain and spinal cord at various stages of the disease progression. Plasma PMX205 levels were similar in WT and hSOD1G93A mice and did not alter throughout disease progression (Figure 1A). By contrast, the levels of PMX205 progressively increased in the brain and spinal cord of hSOD1G93A mice, reaching threefold to fourfold (brain) and threefold to fivefold (spinal cord) that of age‐matched WT mice (Figure 1B, C). Interestingly, PMX205 levels in the brain were increased very early in the disease (Figure 1B), suggesting an early disruption of the BBB in these mice.
Figure 1.
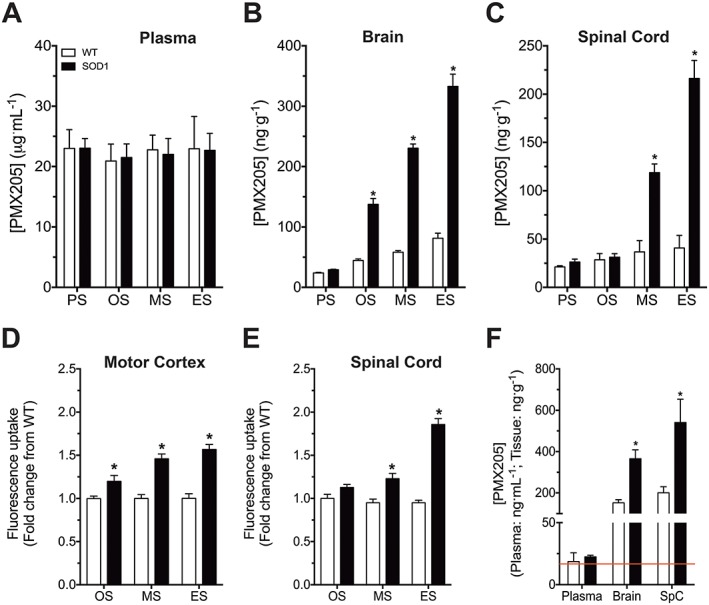
PMX205 concentrations in the brain and spinal cord of hSOD1G93A and WT mice alongside a BBB and BSB permeability analysis. hSOD1G93A and WT mice were injected i.v. with the selective C5a1 receptor antagonist PMX205 at four different stages of disease progression (1 mg·kg−1). (A–C) Plasma, brain and spinal cord PMX205 levels at pre‐symptomatic (PS; P30), onset (OS; P70), mid‐symptomatic (MS; P130) and end‐stage (ES; P150) in WT and hSOD1G93A mice (n = 5). *P < 0.05, significantly different from WT; Student's t‐test at each stage. (D, E) BBB and BSB permeability in WT and hSOD1G93A mice at OS, MS and ES (n = 4). *P < 0.05, significantly different from WT; Student's t‐test at each stage. hSOD1G93A and WT mice were also orally dosed with PMX205 at 91 days of age over a period of 5 days (9 mg·kg−1·day−1). (F) PMX205 levels in plasma, brain and spinal cord after this oral dosing in WT and hSOD1G93A mice (n = 5). *P < 0.05, significantly different from WT; Student's t‐test for each tissue. The red line indicates the IC50, 16.7 ng·mL−1. Data are expressed as mean ± SEM.
To investigate if the increased penetration of PMX205 in hSOD1G93A brains and spinal cords during disease correlated with progressive breakdown of the BBB or BSB, we used fluorescein uptake as a measure of barrier permeability. We observed a progressively increasing uptake of fluorescein in the motor cortex and lumbar spinal cord with disease in hSOD1G93A mice, compared with WT mice (Figure 1D, E). The temporal pattern of fluorescein uptake mirrored that of PMX205 brain and spinal cord levels (Figures 1B, C), collectively suggesting that increased entry of PMX205 in the brain and spinal cord of hSOD1G93A mice indeed correlated with increased BBB and BSB permeability.
Finally, we aimed to identify whether our proposed therapeutic dosing regimen (i.e. oral drinking water PMX205 administration) could result in therapeutically active levels of PMX205 in the CNS tissues of hSOD1G93A mice. We thus allowed hSOD1G93A and WT mice to consume PMX205 via the drinking water (60 μg·mL−1) ad libitum over a period of 5 days. Using this dosage regime, we found that PMX205 could effectively enter the brain and spinal cord at levels substantially above the expected IC50 for C5a1 receptor inhibition (March et al., 2004; Woodruff et al., 2005). Furthermore, similar to our i.v. dosing studies, we identified increased levels of PMX205 in the CNS of hSOD1G93A mice compared with WT mice (Figure 1F). Together, this demonstrates that oral dosing of PMX205 in the drinking water at 60 μg·mL−1 is able to reach the CNS at therapeutically relevant concentrations, and this route and dose of administration were thus used in our follow‐up studies on efficacy.
C5a1 receptor antagonist PMX205 treatment extends survival, improves hindlimb grip strength and slows disease progression in hSOD1G93A mice
In this first efficacy study, a cohort of litter‐matched hSOD1G93A mice were treated with PMX205 or vehicle from 35 days of age onwards. PMX205 treatment at 60 μg·mL−1 in the drinking water resulted in an average dose of 9 mg·kg−1·day−1 based on drinking volume measurements throughout the study (3–4 mL·day−1). hSOD1G93A mice treated with PMX205 from this ‘pre‐onset’ age had a significant extension in survival time when compared with litter‐matched untreated hSOD1G93A mice (Figure 2A). The weight of vehicle‐ and PMX205‐treated hSOD1G93A mice both reached the maximum at 126 days of age; however, there was no difference in body weight loss between vehicle‐ and PMX205‐treated hSOD1G93A mice (Figure 2B). Motor deficits were also assessed in these animals using hindlimb grip strength, a sensitive marker of neuromotor performance (Lee et al., 2013). PMX205 treatment significantly counteracted the loss of hindlimb grip strength from 105 days of age (Figure 2C), with treated mice reaching approximately double the strength of diseased vehicle hSOD1G93A controls from 126 days onwards. Looking at overall disease progression, that is, the age at which the maximal grip strength declined by 25, 50, 75 and 100%, we found that PMX205 treatment at this dose also significantly slowed disease course, delaying the progressive loss of motor function (Figure 2D).
Figure 2.
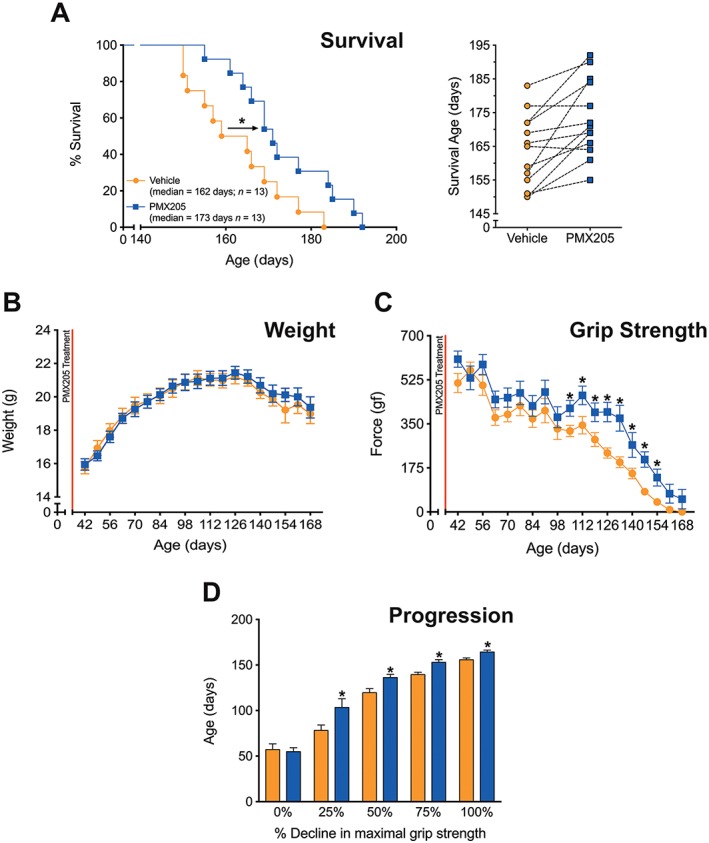
‘Pre‐onset’ PMX205 treatment at 9 mg·kg−1·day−1 in hSOD1G93A transgenic mice. hSOD1G93A mice were orally dosed with the selective C5a1 receptor antagonist PMX205 at 35 days of age (9 mg·kg−1·day−1 in drinking water through to end‐stage). (A, left panel) A Kaplan–Meier plot of ages (in days) in which hSOD1G93A mice treated with dH2O (vehicle) or PMX205 reached end‐stage of disease (complete hindlimb paralysis and an inability to right itself once placed on its back (n = 13). * P < 0.05, significantly different from vehicle; log‐rank test. (A, right panel) The end‐stage survival age for each litter‐matched pair of vehicle‐ and PMX205‐treated hSOD1G93A mice. (B) Body weight in vehicle‐ and PMX205‐treated hSOD1G93A mice (n = 13). P > 0.05, significantly different from vehicle; two‐way ANOVA. (C) Hindlimb grip strength in hSOD1G93A mice treated with either vehicle or PMX205 (n = 13) *P < 0.05, significantly different from vehicle; two‐way ANOVA with Fisher's LSD test at each age. (D) Disease progression (determined by age at which maximal grip strength decline at 25, 50, 75 and 100%) in hSOD1G93A mice treated with PMX205 when compared with vehicle‐treated hSOD1G93A mice at 25, 50, 75 and 100% decline in maximal grip strength (n = 13). * P < 0.05, significantly different from vehicle;two‐way ANOVA with Fisher's LSD test at each quartile. Data are expressed as mean ± SEM.
Post‐onset PMX205 treatment extends survival and slows disease progression but does not significantly impact motor performance in hSOD1G93A mice
We next determined if inhibition of C5a1 receptors at a later stage of disease could still reduce ALS pathology in mice. This is particularly important as ALS patients generally present in the clinic with manifest disease, where motor neuron loss has already progressed. Thus, this experiment provides a model that would more closely follow a therapeutic intervention for human ALS patients. hSOD1G93A mice were therefore orally dosed with PMX205 (9 mg·kg−1·day−1) at 91 days of age, when there is already considerable decline in motor performance and motor neuron loss in the hSOD1G93A mouse model (Lee et al., 2013). Importantly, PMX205 treatment in hSOD1G93A mice from this post‐onset disease age also resulted in an increase in survival time compared with litter‐matched vehicle‐treated hSOD1G93A mice (Figure 3A). Similar to the pre‐onset treatment group, post‐onset PMX205 treatment did not affect body weight loss in hSOD1G93A mice (Figure 3B). Post‐onset PMX205 treatment also did not significantly improve grip strength loss in hSOD1G93A mice (Figure 3C). Importantly, however, post‐onset PMX205 treatment significantly slowed disease progression at the later stages of disease, with delays to the loss of maximal grip strength (Figure 3D). This demonstrates that treatment of ALS mice with PMX205, well into their disease progression, can still have therapeutic benefits, albeit to a lesser degree than the earlier treatment regimen.
Figure 3.
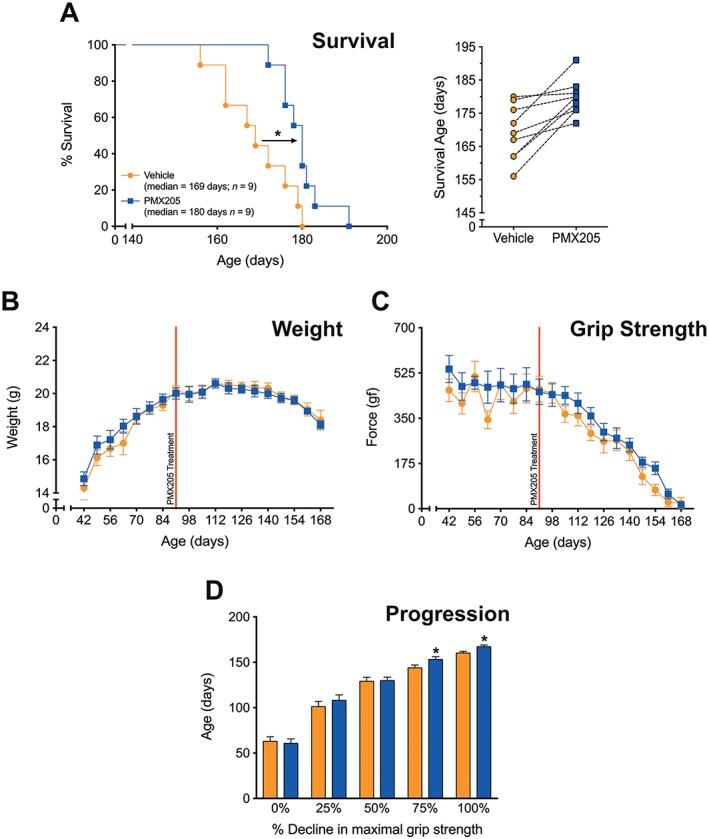
‘Post‐onset’ PMX205 treatment at 9 mg·kg−1·day−1 in hSOD1G93A transgenic mice. hSOD1G93A mice were orally dosed with the selective C5a1 receptor antagonist PMX205 at 91 days of age (9 mg·kg−1·day−1 in drinking water through to end‐stage). (A, left panel) A Kaplan–Meier plot of ages (in days) in which hSOD1G93A mice treated with dH2O (vehicle) or PMX205 reached end‐stage of disease (complete hindlimb paralysis and an inability to right itself once placed on its back (n = 9). *P < 0.05, significantly different from vehicle; log‐rank test). (A, right panel) The end‐stage survival age for each litter‐matched pair of vehicle‐ and PMX205‐treated hSOD1G93A mice. (B, C) Body weight and hindlimb grip strength in vehicle‐ and PMX205‐treated hSOD1G93A mice (n = 9). *P > 0.05, significantly different from vehicle; two‐way ANOVA. (D) Disease progression (determined by age at which maximal grip strength decline at 25, 50, 75 and 100%) in hSOD1G93A mice treated with vehicle or PMX205 (n = 9). *P < 0.05, significantly different from vehicle, two‐way ANOVA with Fisher's LSD test at each quartile. Data are expressed as mean ± SEM.
PMX205 treatment reduces circulating blood M1 monocytes and granulocytes, while increasing CD4+ T‐cells in hSOD1G93A mice
Circulating immune cells have previously been shown to contribute to ALS pathology (Beers et al., 2008; Butovsky et al., 2012) and may provide important peripheral biomarkers of efficacy for future clinical studies. We therefore investigated the major immune cell populations (monocytes, granulocytes and T lymphocytes) in hSOD1G93A mice following vehicle or PMX205 (9 mg·kg−1·day−1; pre‐onset) treatment at three different stages of disease progression. PMX205 treatment significantly reduced total monocytes at the mid‐symptomatic stage, when compared with vehicle‐treated hSOD1G93A mice (Figure 4A). This reduction was attributed predominantly to monocytes classically defined as M1 (CD11b+Ly6G−Ly6Chi/medSSClo), rather than M2 (Figure 4B, C). PMX205 treatment similarly reduced neutrophil granulocyte (CD11b+Ly6G+Ly6C+SSCmed) numbers at mid‐symptomatic stage of disease progression, when compared with vehicle‐treated hSOD1G93A mice (Figure 4D). Interestingly, eosinophil granulocyte (CD11b+Ly6G+Ly6C+SSChi) numbers were initially increased in PMX205‐treated hSOD1G93A mice at onset stage but then significantly decreased at the mid‐symptomatic stage compared with vehicle‐treated hSOD1G93A mice (Figure 4E). By contrast to monocytes and granulocytes, helper T‐cell (CD3+CD4+) numbers were increased at end‐stage disease (Figure 4F). Non‐helper/cytotoxic T‐cell (CD3+CD4−) numbers were significantly decreased at the mid‐symptomatic stage compared with vehicle‐treated hSOD1G93A mice (Figure 4G). Overall, these flow cytometry studies indicate a reduced pro‐inflammatory leukocyte phenotype in the blood of hSOD1G93A mice treated with PMX205 at mid‐symptomatic stage of disease.
Figure 4.
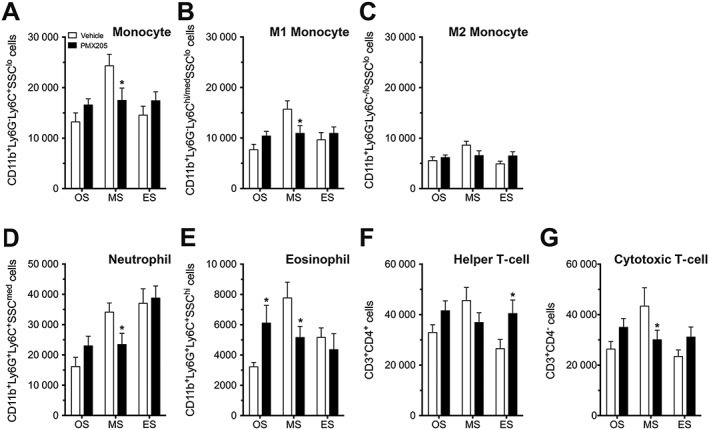
Circulating blood monocytes, granulocytes and T lymphocytes in vehicle‐ and PMX205‐treated hSOD1G93A mice. hSOD1G93A mice were orally dosed with the selective C5a1 receptor antagonist PMX205 at 35 days of age (9 mg·kg−1·day−1 in drinking water through to end‐stage). Major immune cell populations (monocytes, granulocytes and T lymphocytes) in vehicle‐ and PMX205‐treated hSOD1G93A mice were investigated at three different stages of disease progression using flow cytometry. (A–C) Total (CD11b+Ly6G−Ly6C+SSClo), M1 (CD11b+Ly6G−Ly6Chi/medSSClo) and M2 (CD11b+Ly6G−Ly6C−/loSSClo) monocyte numbers in vehicle‐ and PMX205‐treated hSOD1G93A mice at onset (OS; P70), mid‐symptomatic (MS; P130) and end‐stage of disease (ES; P150) (n = 6). *P < 0.05, significantly different from vehicle; two‐way ANOVA with Fisher's LSD test at each stage. (D, E) Neutrophil (CD11b+Ly6G+Ly6C+SSCmed) and eosinophil (CD11b+Ly6G+Ly6C+SSChi) granulocyte numbers at OS, MS and ES in vehicle‐ and PMX205‐treated hSOD1G93A mice (n = 6). * P < 0.05, significantly different from vehicle; two‐way ANOVA with Fisher's LSD test at each stage. (F, G) Helper T‐cell (CD3+CD4+) and non‐helper/cytotoxic T‐cell (CD3+CD4−) numbers in vehicle‐ and PMX205‐treated hSOD1G93A mice (n = 6) *P < 0.05, significantly different from vehicle; two‐way ANOVA with Fisher's LSD test at each stage. Data are expressed as mean ± SEM.
Discussion
Currently, there is no effective treatment for ALS. Although the pathogenesis of ALS is still unclear, there is convincing evidence that inflammation, both peripheral and central, may contribute substantially to disease. The complement system is emerging as a potential key innate immune system that is activated in multiple neurological diseases, promoting neuroinflammation and disease progression (Brennan et al., 2016). Previous studies have demonstrated that several complement factors, including C1q, factor B, C3, C4 and C5a1 receptors, are significantly increased during ALS progression in hSOD1G93A mice (Lee et al., 2013). However, genetic deletion of C1q, C3 and C4 in hSOD1 transgenic mouse models has not shown any significant beneficial effects on disease progression (Chiu et al., 2009; Lobsiger et al., 2013), although it should be noted that this strategy does not eliminate direct activation of C5 via the extrinsic pathway (Huber‐Lang et al., 2006). Consistent with this, our prior studies showed that genetic deletion of C5a1 receptors in hSOD1G93A mice extends survival (Woodruff et al., 2014). In addition, we also previously showed that chronic administration of C5a1 receptor antagonists to hSOD1G93A transgenic rats delayed the onset of motor symptoms and increased survival compared with untreated diseased animals (Woodruff et al., 2008). Collectively, these findings show that complement effector mechanisms downstream of C5 do contribute to disease progression and need to be directly targeted. In the present study, we have extended our previous work by conducting a detailed assessment of PMX205 pharmacokinetics with disease progression and by testing the benefits of pharmacological C5a1 receptor inhibition before and after disease onset in hSOD1G93A mice, the most commonly used preclinical model of ALS.
The present study demonstrates that blockade of C5a1 receptors via oral administration of the selective, non‐competitive, cyclic peptide C5a1 receptor antagonist, PMX205, significantly reduced pathology in hSOD1G93A mice at a dose of 9 mg·kg−1·day−1. Our results therefore demonstrate that increased C5a1 receptor activation in transgenic hSOD1G93A mouse models actively contributes to pathogenesis. Importantly, treatment of hSOD1G93A mice with a lower dose of PMX205 (3 mg·kg−1·day−1) did not extend survival time, improve motor performance or slow disease progression (Figure S1), indicating a dose‐dependency of the beneficial effects of PMX205. A similar dose‐dependency has been observed in a transgenic mouse model of Alzheimer's disease where a dose of 3 mg·kg−1·day−1 was found ineffective but an oral drinking water dose equivalent to 6 mg·kg−1·day−1 did achieve efficacy (Jain et al., 2013). Furthermore, a recent study demonstrated that an 8 mg·kg−1·day−1 oral dose of PMX205 was required to achieve efficacy in a mouse model of inflammatory bowel disease, while a 4 mg·kg−1·day−1 dose showed no effect (Jain et al., 2013). These findings underline the importance of optimal dose selection for PMX205, which may explain the lack of efficacy at lower doses of PMX205, as reported by others (Klos et al., 2013).
Drug treatment experiments included hSOD1G93A mice dosed from a very early age (day 35) to determine the maximum effect of C5a1 receptor blockade, as well as at a later time point, clearly after the onset of motor deficits (day 91). Our results showed that there was no significant difference in survival time extension between the two dosing groups, suggesting that the pathogenic involvement of C5a was more likely to occur subsequent to day 91 in this model. This is in agreement with our previous finding that up‐regulation of C5a1 receptors occurs late in the disease course, that is, at 130 and 175 days in hSOD1G93A mice (Lee et al., 2013). These results also indicate that C5a‐ C5a1 receptor signalling exacerbates disease pathology but does not trigger the onset of disease. It should be noted, however, that CNS uptake of PMX205 also increases with disease progression (Figure 1) and that this in turn may account for the similar survival efficacy between pre‐ and post‐onset groups. Although there was no difference in the increased survival time between pre‐ and post‐onset treatments, initiation of PMX205 treatment after disease onset did not significantly improve hindlimb grip strength. These findings are consistent with the fact that the onset of motor deficits, as measured by hindlimb grip strength, is closely correlated with the decline in lumbar spinal cord motor neuron numbers, which normally occurs prior to 70 days in hSOD1G93A mice (Lee et al., 2013). Hence, treating these animals at day 91 when there is already significant decline in motor neuron numbers would only be expected to show minimal improvement in neuromotor function. Post‐onset PMX205 treatment did, however, still have a benefit by delaying the age at which hSOD1G93A mice reached 75% and 100% decline in their maximal grip strength (i.e. mice did have a slower disease progression).
Our studies also demonstrated that there is an early disruption of BBB and BSB permeability in hSOD1G93A mice based on fluorescein extravasation. These findings support previous studies demonstrating damage to BBB and BSB integrity in hSOD1G93A mice via ultrastructural capillary alterations and leakage in the lumber spinal cord at both early and late stages of disease (Garbuzova‐Davis et al., 2007a,b; Zhong et al., 2008). Collectively, these findings suggest that there are functional impairments in barrier integrity very early in disease, potentially preceding significant motor neuron death (Zhong et al., 2008). Interestingly, the progressive increase in barrier permeability in hSOD1G93A mice during the disease course mirrored the increase in PMX205 levels in the brain and spinal cord. This has clear implications for any potential future clinical trials using PMX205, as there may be enhanced uptake of this drug by target tissues (i.e. motor cortex and spinal cord) that are undergoing degeneration in ALS with disease progression.
Another interesting finding from this study was the demonstration of higher CNS tissue distribution of PMX205 following oral drinking water dosing compared with i.v. dosing where much higher blood concentrations were observed. Prior studies have demonstrated that this class of molecules are relatively lipophilic and display rapid tissue distribution following dosing (Woodruff et al., 2005). PMX205 also has a relatively low oral bioavailability (Woodruff et al., 2005), which is in line with the low blood levels observed following oral dosing in this study. It is possible that there was slower elimination of PMX205 from the CNS compared with the blood circulation, resulting in increased levels of drug observed within the CNS over the 5‐day dosing period. Importantly, as this class of C5a1 receptor antagonists are insurmountable antagonists with an extremely low off‐rate, they effectively act as pseudo‐irreversible inhibitors of C5a‐ C5a1 receptor signalling (Paczkowski et al., 1999). Therefore, despite low circulating blood levels following drinking water dosing, widespread and prolonged blockade of C5a1 receptors would still be expected (Seow et al., 2016). Additional studies are warranted to further explore the nature of PMX205 pharmacokinetics and pharmacodynamics in CNS disease and how this can potentially be modified to improve therapeutic outcomes.
In addition to microglia activation, it is now also well accepted that peripheral immune cells, including monocytes and T lymphocytes, can modulate motor neuron death in ALS animal models (Beers et al., 2008; Butovsky et al., 2012). In our study, we found that there was a significant reduction in pro‐inflammatory M1 monocyte numbers in the blood of PMX205‐treated hSOD1G93A mice at mid‐symptomatic stage of disease compared with untreated mice. This supports studies demonstrating that disease progression and neuronal damage can be attenuated in hSOD1G93A mice by depleting Ly6C‐positive monocytes (Butovsky et al., 2012). We also found that blockade of C5a1 receptors in hSOD1G93A mice decreased circulating CD4− T lymphocytes, while the number of CD4+ T lymphocytes increased. These findings suggest an overall dampening of the inflammatory response with disease progression, which are also consistent with previous reports showing that helper and regulatory T lymphocytes are neuroprotective, whilst cytotoxic T lymphocytes are pathogenic in hSOD1G93A mice (Beers et al., 2008). Finally, we observed that there was a significant decrease in neutrophil and eosinophil granulocyte numbers in PMX205‐treated hSOD1G93A mice at mid‐stage of disease. The role of granulocytes in the disease progression of ALS is not well studied, although recent data suggest these cell types may play active roles in neurodegenerative disease (Zenaro et al., 2015). Additional research, including examining the effects of PMX205 on neuropathology and immune cell infiltration into the CNS, is thus warranted to further explore the mechanisms of PMX205 neuroprotection in SOD1G93A mice.
In summary, the present study demonstrates for the first time that oral administration of the selective and CNS‐permeable C5a1 receptor antagonist, PMX205, both pre‐ and post‐onset, can significantly attenuate disease in hSOD1G93A mice. In the blood, pro‐inflammatory M1 monocytes, granulocytes and CD4− T lymphocyte numbers were all decreased at mid‐stage of disease, while the number of CD4+ helper T lymphocytes was increased following PMX205 treatment. Together, these findings indicate that blockade of C5a1 receptors significantly attenuates hSOD1G93A disease progression and associated inflammation, which in turn transiently affects the number of circulating pro‐inflammatory leukocytes. Whether this effect on immune cells in the blood is the cause or consequence of PMX205 efficacy is currently under investigation. Overall, our studies affirm the view that blocking the C5a1 receptor, the receptor for the C5 activation fragment, C5a, is a useful therapeutic strategy to modify disease course in ALS.
Author contributions
J.D.L., P.G.N. and T.M.W. conceived the study. J.D.L. performed all mouse efficacy studies and flow cytometry with assistance from J.N.F. V.K. performed the pharmacokinetic LC–MS/MS and permeability assays, and M.J.R. assisted in flow cytometry design and efficacy data interpretation. Experiments were performed in the laboratories of P.G.N., T.M.W. and UQ's core facilities. J.D.L. and T.M.W. wrote the paper, with all other authors contributing edits and ideas. All authors approved the final version.
Conflict of interest
T.M.W. consults for Alsonex Pty Ltd, which is commercially developing PMX205 for ALS treatment. He holds no stocks, shares or other commercial interest in this company.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 “Pre‐Onset” PMX205 treatment at 3 mg kg−1 day−1 in hSOD1G93A transgenic mice. hSOD1G93A mice were orally dosed with the selective C5a1 receptor antagonist PMX205 at 35 d of age (red line; 3 mg kg−1 day−1 in drinking water through to end‐stage). A shows a Kaplan–Meier plot of ages (in days) in which hSOD1G93A mice treated with distilled H2O (vehicle; orange line) or PMX205 (blue line) reached end‐stage of disease (complete hind‐limb paralysis and an inability to right itself once placed on its back; n = 6, P = 0.89, log‐rank test). B and C shows body weight and hind‐limb grip strength in vehicle (orange line) and PMX205 (blue line) treated hSOD1G93A mice (n = 6, P > 0.05, two‐way ANOVA). D shows disease progression (determined by age at which maximal grip strength decline at 25, 50, 75 and 100%) in hSOD1G93A mice treated with vehicle (orange bar) and PMX205 (blue bar) (n = 6, two‐way ANOVA). Data are expressed as mean ± SEM.
Acknowledgements
The authors would like to sincerely thank Professor Stephen M. Taylor for the initial discussions related to this study and Dr Faith Brennan, Mrs Virginia Nink and Mr Geoffrey Osborne for their technical support on flow cytometry. J.D.L. holds a Motor Neuron Disease Research Institute of Australia (MNDRIA) Postdoctoral Fellowship, and the research was funded by grants from the MNDRIA (to T.M.W. and P.G.N.) and the National Health and Medical Research Council (NHMRC) project grant (APP1082271 to TMW and MJR). TMW is supported by a NHMRC Career Development Fellowship (APP1105420).
Lee, J. D. , Kumar, V. , Fung, J. N. T. , Ruitenberg, M. J. , Noakes, P. G. , and Woodruff, T. M. (2017) Pharmacological inhibition of complement C5a‐C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. British Journal of Pharmacology, 174: 689–699. doi: 10.1111/bph.13730.
References
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Appel SH (2008). CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A 105: 15558–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FH, Gordon R, Lao HW, Biggins PJ, Taylor SM, Franklin RJ et al. (2015). The complement receptor C5aR controls acute inflammation and astrogliosis following spinal cord injury. J Neurosci 35: 6517–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FH, Lee JD, Ruitenberg MJ, Woodruff TM (2016). Therapeutic targeting of complement to modify disease course and improve outcomes in neurological conditions. Semin Immunol 28: 292–308. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW (2004). Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27: 723–749. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G et al. (2012). Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest 122: 3063–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Phatnani H, Kuligowski M, Tapia JC, Carrasco MA, Zhang M et al. (2009). Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci U S A 106: 20960–20965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino M, Ferri A, Carri MT (2008). Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxid Redox Signal 10: 405–443. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM et al. (2009). Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol 183: 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzova‐Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, Sanberg PR (2007a). Ultrastructure of blood–brain barrier and blood–spinal cord barrier in SOD1 mice modeling ALS. Brain Res 1157: 126–137. [DOI] [PubMed] [Google Scholar]
- Garbuzova‐Davis S, Saporta S, Haller E, Kolomey I, Bennett SP, Potter H et al. (2007b). Evidence of compromised blood–spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS One 2: e1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber‐Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR et al. (2006). Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 12: 682–687. [DOI] [PubMed] [Google Scholar]
- Jain U, Woodruff TM, Stadnyk AW (2013). The C5a receptor antagonist PMX205 ameliorates experimentally induced colitis associated with increased IL‐4 and IL‐10. Br J Pharmacol 168: 488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klos A, Wende E, Wareham KJ, Monk PN (2013). International Union of Basic and Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev 65: 500–543. [DOI] [PubMed] [Google Scholar]
- Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD et al. (2013). Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation 10: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JD, Lee JY, Taylor SM, Noakes PG, Woodruff TM (2012). Innate immunity in ALS In: Maurer MH. (ed). Amyotrophic Lateral Sclerosis. InTech: Croatia. [Google Scholar]
- Lobsiger CS, Boillee S, Pozniak C, Khan AM, McAlonis‐Downes M, Lewcock JW et al. (2013). C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc Natl Acad Sci U S A 110: E4385–E4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludolph AC, Bendotti C, Blaugrund E, Hengerer B, Loffler JP, Martin J et al. (2007). Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: report on the 142nd ENMC international workshop. Amyotroph Lateral Scler 8: 217–223. [DOI] [PubMed] [Google Scholar]
- March DR, Proctor LM, Stoermer MJ, Sbaglia R, Abbenante G, Reid RC et al. (2004). Potent cyclic antagonists of the complement C5a receptor on human polymorphonuclear leukocytes. Relationships between structures and activity. Mol Pharmacol 65: 868–879. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paczkowski NJ, Finch AM, Whitmore JB, Short AJ, Wong AK, Monk PN et al. (1999). Pharmacological characterization of antagonists of the C5a receptor. Br J Pharmacol 128: 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N et al. (2008). Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler 9: 4–15. [DOI] [PubMed] [Google Scholar]
- Seow V, Lim J, Cotterell AJ, Yau MK, Xu W, Lohman RJ et al. (2016). Receptor residence time trumps drug‐likeness and oral bioavailability in determining efficacy of complement C5a antagonists. Sci Rep 6: 24575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM (2010). The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Med 12: 179–192. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Costantini KJ, Crane JW, Atkin JD, Monk PN, Taylor SM et al. (2008). The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol 181: 8727–8734. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Crane JW, Proctor LM, Buller KM, Shek AB, de Vos K et al. (2006). Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J 20: 1407–1417. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Lee JD, Noakes PG (2014). Role for terminal complement activation in amyotrophic lateral sclerosis disease progression. Proc Natl Acad Sci U S A 111: E3–E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff TM, Pollitt S, Proctor LM, Stocks SZ, Manthey HD, Williams HM et al. (2005). Increased potency of a novel complement factor 5a receptor antagonist in a rat model of inflammatory bowel disease. J Pharmacol Exp Ther 314: 811–817. [DOI] [PubMed] [Google Scholar]
- Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S et al. (2015). Neutrophils promote Alzheimer's disease‐like pathology and cognitive decline via LFA‐1 integrin. Nat Med 21: 880–886. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O'Banion MK et al. (2008). ALS‐causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11: 420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 “Pre‐Onset” PMX205 treatment at 3 mg kg−1 day−1 in hSOD1G93A transgenic mice. hSOD1G93A mice were orally dosed with the selective C5a1 receptor antagonist PMX205 at 35 d of age (red line; 3 mg kg−1 day−1 in drinking water through to end‐stage). A shows a Kaplan–Meier plot of ages (in days) in which hSOD1G93A mice treated with distilled H2O (vehicle; orange line) or PMX205 (blue line) reached end‐stage of disease (complete hind‐limb paralysis and an inability to right itself once placed on its back; n = 6, P = 0.89, log‐rank test). B and C shows body weight and hind‐limb grip strength in vehicle (orange line) and PMX205 (blue line) treated hSOD1G93A mice (n = 6, P > 0.05, two‐way ANOVA). D shows disease progression (determined by age at which maximal grip strength decline at 25, 50, 75 and 100%) in hSOD1G93A mice treated with vehicle (orange bar) and PMX205 (blue bar) (n = 6, two‐way ANOVA). Data are expressed as mean ± SEM.